

Summary
Clonal evolution of osimertinib-resistance mechanisms in EGFR mutant lung adenocarcinoma is poorly understood. Using multi-region whole-exome and RNA sequencing of prospectively collected pre- and post-osimertinib-resistant tumors, including at rapid autopsies, we identify a likely mechanism driving osimertinib resistance in all patients analyzed. The majority of patients acquire two or more resistance mechanisms either concurrently or in temporal sequence. Focal copy-number amplifications occur subclonally and are spatially and temporally separated from common resistance mutations such as EGFR C797S. MET amplification occurs in 66% (n = 6/9) of first-line osimertinib-treated patients, albeit spatially heterogeneous, often co-occurs with additional acquired focal copy-number amplifications and is associated with early progression. Noteworthy osimertinib-resistance mechanisms discovered include neuroendocrine differentiation without histologic transformation, PD-L1, KRAS amplification, and ESR1-AKAP12, MKRN1-BRAF fusions. The subclonal co-occurrence of acquired genomic alterations upon osimertinib resistance will likely require targeting multiple resistance mechanisms by combination therapies.
Keywords: EGFR mutant lung cancer, non-small cell lung cancer, osimertinib, MET amplification, copy number ampplifications, neuroendocrine differentiation
Graphical Abstract
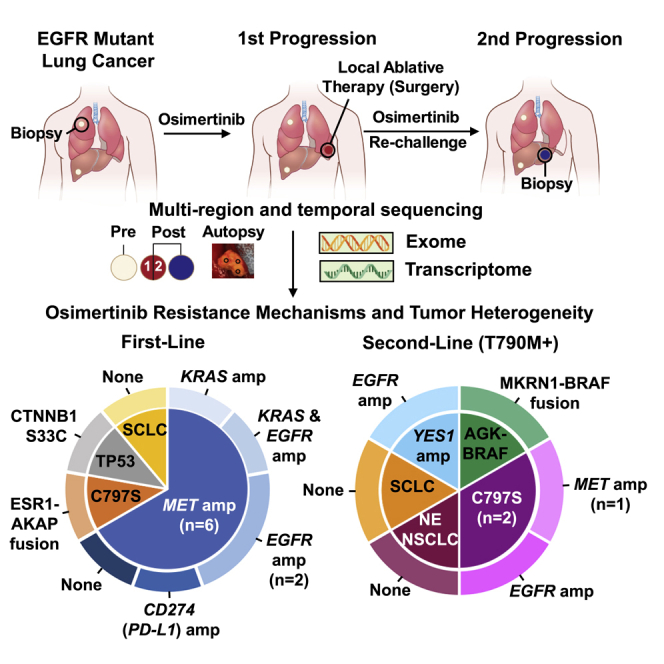
Highlights
Two or more subclonal genomic alterations are acquired upon osimertinib resistance
66% of first-line osimertinib-treated patients acquire MET amplification
Acquired focal copy-number alterations are associated with early progression
Neuroendocrine differentiation with NSCLC histology is revealed by RNA-seq analysis
Roper et al. perform multi-region whole-exome and RNA sequencing of pre- and post-resistant tumors from EGFR mutant lung cancer patients treated with osimertinib. They uncover the subclonal co-occurrence of acquired genomic alterations upon osimertinib resistance suggesting combination first-line therapies may prevent or delay key resistance mechanisms.
Introduction
Tyrosine kinase inhibitors (TKIs) have revolutionized the treatment of epidermal growth factor receptor (EGFR) mutant lung cancer as the vast majority of patients treated with EGFR TKIs derive clinical benefit. However, resistance to EGFR TKI therapy is inevitable. Acquired resistance mechanisms of first and –second generation EGFR TKIs have been extensively characterized.1 However, in a significant proportion of EGFR mutant lung cancer patients treated with osimertinib, a 3rd-generation EGFR TKI, a definite acquired resistance mechanisms has not been elucidated.2, 3, 4, 5, 6 Moreover, since osimertinib was recently approved by the FDA for the first-line treatment of patients with metastatic EGFR mutant lung cancer, there is a pressing need to understand acquired mechanisms of resistance to osimertinib in such patients. The studies to date assessing acquired resistance mechanisms among first-line osimertinib-treated patients have largely employed targeted sequencing platforms of circulating tumor DNA from plasma,7 which may not define the full genomic landscape of alterations that likely occur upon resistance to osimertinib.
Herein, we present results of an ongoing clinical trial assessing mechanisms of acquired osimertinib resistance in EGFR mutant lung cancer using multi-region whole-exome and RNA sequencing (RNA-seq) of biopsies and surgeries of pre- and post-osimertinib-resistant tumors as well as metastases at rapid autopsy. We dissected the clonal evolution of high-confidence somatic mutations and focal copy-number amplifications in cancer-related genes that likely mediate osimertinib resistance. We found the majority of patients treated with osimertinib exhibited two or more acquired resistance mechanisms. Among first-line osimertinib-treated patients, those with early progression developed MET amplification as a major resistance mechanism. In one affected individual, although exome sequencing did not reveal a resistance mechanism, RNA-seq uncovered neuroendocrine (NE) differentiation without histologic transformation as a possible mechanism of osimertinib resistance.
Results
We enrolled 34 patients with histologically confirmed EGFR mutant advanced lung adenocarcinoma in a prospective phase II clinical trial to evaluate osimertinib treatment and the use of local ablative therapy (LAT) for oligoprogressive disease (NCT02759835) in EGFR mutant non-small cell lung cancer (NSCLC) from April 2016 until data cutoff in September 2019. Patients were not selected based on oligometastatic status; rather, 32/34 patients had multiple organ involvement, including lung, liver, adrenal, brain, and bone. Biopsies or surgeries were performed pre-osimertinib treatment and at first progression on osimertinib. A subset of patients also underwent biopsies at second progression while being treated with osimertinib and following end-of-life in-patient hospice, and rapid autopsy upon death was performed8 (Figure 1A). 63% (n = 15/24) of first-line osimertinib and 50% (n = 5/10) of second-line osimertinib-treated patients had RECIST-defined partial response (Figure 1B). Overall, 21 patients had RECIST-defined first progression while receiving osimertinib (Figure 1B). Two patients on this trial discontinued treatment and were lost to follow-up. Of the 21 patients who had RECIST-defined progression, 14 patients underwent LAT (8 first-line and 6 s-line) (Figure 1B). Twelve patients had paired pre- and post-osimertinib-resistant tumors and three had post-osimertinib-resistant tumors available for analysis (Figure 1B; Table S1). Four patients (LAT002, LAT006, LAT014, and LAT021) consented for a rapid autopsy protocol,8 and rapid autopsy was performed after death (Table S1).
Figure 1.

Study Schema, Clinical Responses to Osimertinib, and Mechanisms of Resistance to Osimertinib
(A) Schematic diagram of the clinical protocol and sample analysis. Biopsies and/or surgeries were performed pre-treatment, at first progression on osimertinib treatment, and at the second progression on osimertinib. Tumor tissue DNA and corresponding germline DNA were analyzed using WES. RNA-seq was also performed for select samples with sufficient material. PDXs were generated from post-osimertinib-resistant tumor tissue, when available.
(B) Swimmer’s plot indicating line and length of osimertinib treatment, current treatment status, and whether pre- or post-osimertinib resistance sequencing was performed for individual patients.
(C and D) Heatmaps depict the distribution of non-silent somatic mutations among pre- and post-osimertinib-resistant tumors for (C) first-line and (D) previously treated patients. Select COSMIC cancer gene mutations are listed to the left of each heatmap (all mutations are listed in Table S3). The total number of non-silent mutations and the percentage of non-truncal mutations are shown below each heatmap. The bars to the right of each heatmap summarize intra- and inter-metastatic heterogeneity; mutations present in all regions (purple), in more than one but not all (green), or only in one region (brown). Biological replicates at post-osimertinib resistance for individual patients are shown.
(E) Table outlining osimertinib-resistance mechanisms among all patients, indicating whether paired pre- and post-osimertinib or post-osimertinib only tumor tissue was analyzed.
Two or More Osimertinib-Resistance Mechanisms in Individual Patients
To elucidate acquired osimertinib-resistance mechanisms, we performed whole-exome sequencing (WES), targeted sequencing, and RNA-seq on 66 tumors and matched germline DNA from a total of 15 patients. Median tumor purity across all samples analyzed was 48% (Table S2). A range of 111–435 non-silent somatic mutations were identified per patient by exome sequencing (Table S3). We found significant mutational heterogeneity within pre- and post-osimertinib-resistant tumors from each subject with non-truncal, non-silent variants ranging from 56% to 96% of all mutations (Figures 1C and 1D). We considered all high-confidence somatic mutations, focal copy-number amplifications (defined as greater than 10 or more copies, less than 5 megabases in length, in COSMIC cancer-related genes in the post- compared to pre-osimertinib tumors after adjusting for tumor ploidy and purity), and gene fusions for potential resistance mechanisms (Tables S3, S4 and S5). Focal copy-number deletions (defined as homozygous deletions, less than 5 megabases in length, in COSMIC cancer-related genes in post-compared to pre-osimertinib tumors after adjusting for tumor ploidy and purity) were also assessed but not included as resistance mechanisms because focal deletions in potential tumor suppressors were not detected in this cohort of patients (Table S5). Remarkably, we found an acquired osimertinib-resistance mechanism in 93% (n = 14/15) of patients (Figure 1E). Two or more resistance mechanisms co-existed in 73% (n = 11/15) of patients (Figure 1E). Importantly, a mechanism of acquired resistance was identified in all first-line osimertinib-treated patients (n = 9). Two patients, LAT016 and LAT022, had significant gene fusions; AGK-BRAF and MKRN1-BRAF in two separate progressive tumors in LAT016 and ESR1-AKAP12 in LAT022 that were discovered by Oncomine assays (Table S4). Interestingly, for LAT016, an AGK-BRAF fusion was acquired in a lung tumor at first progression but not in the adrenal tumor that progressed concurrently. Instead, a MKRN1-BRAF fusion was identified in the adrenal tumor at first progression and also identified from the liver biopsy of a tumor at second progression (Table S4). We were unable to identify an acquired osimertinib-resistance mechanism in one affected individual, LAT011, using exome and targeted sequencing (Figure 1E).
Clonal Evolution of EGFR Mutant NSCLC Treated with Osimertinib
While EGFR mutant NSCLCs is known to harbor genetically diverse cell populations,9,10 little is known about how such tumors evolve during treatment. Therefore, to investigate the spatial and temporal evolution of somatic mutations and focal copy-number amplifications in patients treated with osimertinib, we employed phylogenetic analysis of pre- and post-osimertinib-resistant as well as post-osimertinib-resistant only tumors from each subject. Overall, our phylogenetic analysis revealed between 6 and 23 subclones per subject (Figures 2 and 3; Figure S1). Oncogenic EGFR mutations (L858R, deletion 19 and exon 20 insertion), were generally clonal (defined as present in all subclones). One affected individual, LAT009, carried a germline EGFR T790M mutation and developed an EGFR L858R mutation later in tumor evolution consistent with previous reports inherited susceptibility of lung cancer and heterogeneous development of sensitizing EGFR mutations in these patients.11 All RB1 mutations (frameshift and nonsense) and nearly all PIK3CA mutations (N1044K and K111E) were clonal. Clonal RB1 mutations only occurred in two patients with small cell histologic transformation consistent with prior reports.12 TP53 mutations were not universally clonal and predicted loss-of-function TP53 mutations occurred subclonally in three first-line osimertinib-treated patients (LAT006, TP53 A24fs; LAT009, TP53 Y124H; LAT028, TP53 Y124C) suggesting these mutations may be an acquired osimertinib-resistance mechanism in select patients.
Figure 2.

Clonal Evolution of Pre- and Post-osimertinib-Resistant Tumors from First-Line Osimertinib-Treated Patients
(A–H) Phylogenetic trees representing the clonal architecture present in samples collected pre-osimertinib resistance and at first progression on osimertinib for patients (A) LAT001, (B) LAT006, (C) LAT009, (D) LAT010, (E) LAT014, (F) LAT021, (G) LAT0022, and (H) LAT028. Mutations and focal copy-number amplifications occurring in COSMIC cancer-related genes in each branch are indicated with arrows. Focal copy-number amplifications are highlighted in red. Clinical timeline from diagnosis of metastatic disease to progression on osimertinib for each subject is summarized below each phylogenetic tree. Anatomic locations of pre- and post-osimertinib-resistant tumors are shown for each subject. Color of circles in phylogenetic trees signal relative time point in tumor evolution: red (clonal), blue and yellow (early subclonal), and dark green, orange, and silver (late subclonal). Bar plots for each subject indicate the cancer cell fraction (CCF) of subclones for each tumor used to generate the phylogenetic trees. Biological replicates at post-osimertinib resistance for individual patients are shown.
(I) Dual EGFR/MET in first progression tumor from subject LAT006.
(J) Dual EGFR/KRAS FISH in progressive tumor from subject LAT014. Yellow arrows represent cells with amplification of EGFR/MET or KRAS/EGFR. White arrow represents cells with normal copies of EGFR/MET or KRAS/EGFR. WBRT, whole-brain radiation therapy; SRS, stereotactic radiosurgery; Osi, osimertinib; amp, amplification.
Figure 3.

Clonal Evolution of Pre- and Post-osimertinib-Resistant Tumors from Patients with Prior Treatment with an EGFR Tyrosine Kinase Inhibitor
Phylogenetic trees were constructed from WES of pre- and post-osimertinib-resistant tumors of patients (A) LAT003, (B) LAT011, (C) LAT016, and (D) LAT017. Mutations and CNAs in cancer-related genes in each branch are indicated with arrows. Clinical timeline from diagnosis of metastatic disease to progression on osimertinib for each subject is summarized below each phylogenetic tree. Anatomic locations of pre- and post-osimertinib-resistant tumors are shown for each subject. Color of circles in phylogenetic trees signal relative time point in tumor evolution: red (clonal), blue and yellow (early subclonal), and green, orange, silver (late subclonal). Biological replicates at post-osimertinib resistance for individual patients are shown. Osi, osimertinib; amp, amplification. Amplifications are shown in red. Bar plots for each subject indicate the cancer cell fraction (CCF) of subclones for each tumor used to generate the phylogenetic trees.
All focal copy-number amplifications occurred subclonally and most occurred earlier in tumor evolution (Figures 2 and 3; Figure S1). Among patients with two or more focal copy-number amplifications at the time of first progression while taking osimertinib (LAT006, LAT014, and LAT017), focal copy-number amplifications occurred in the same early subclone suggesting multiple, focal copy-number amplifications in genes such as EGFR, MET, KRAS, and YES1 can occur subclonally, and possibly simultaneously in the same subclone (Figures 2 and3; Figure S1). To validate our phylogenetic data, we performed dual EGFR/MET and KRAS/EGFR fluorescence in situ hybridization (FISH) on tumors obtained at first progression while receiving osimertinib from patients LAT006 and LAT014, respectively. We found that focal amplifications in EGFR/MET and KRAS/EGFR occurred concurrently and subclonally; they were present together in some cells of the progressive tumor (Figures 2I and 2J). Taken together, these data suggest multiple focal amplifications occur at specific spatial and temporal sequence during the development of acquired osimertinib resistance.
In addition to focal copy-number amplifications, all patients acquired high-confidence somatic mutations in cancer genes and a subset of patients had somatic mutations implicated in EGFR TKI resistance. In two patients, focal copy-number amplifications and somatic mutations occurred at different evolutionary time points and locations. For example, in subject LAT003, EGFR amplification was subclonal and was acquired in a clone that became highly prevalent in the post-osimertinib resistant compared to the pre-treatment tumor. In contrast, an acquired, subclonal EGFR C797S mutation occurred in a separate phylogenetic branch in a clone with similar prevalence in both pre- and post-osimertinib-resistance tumors (Figure 3A). Likewise, in subject LAT015, MET amplification occurred in a subclone that was present only at second progression while receiving osimertinib and was in a separate phylogenetic branch from both EGFR C797S and EGFR T790M mutations. These acquired EGFR mutations occurred in a common subclone in both sequenced tumors at first progression while receiving osimertinib, but both were absent in the tumor at second progression. (Figure S1C). Subject LAT022 developed an acquired EGFR C797S mutation and ESR1-AKAP12 fusion, but these occurred in only 1 of the 2 post-osimertinib-resistant samples that were sequenced (Figure 2G; Table S4). Thus, in the same affected individual, diverse osimertinib-resistance mechanisms comprising focal copy-number amplifications and acquired somatic mutations can be spatially and temporally separated.
Multiple Acquired Focal Copy-Number Amplifications Are Associated with Short-Term Response to First-Line Osimertinib-Treated Patients
While the median progression-free survival (PFS) of first-line treatment of EGFR mutant lung cancer with osimertinib is significantly longer than with earlier-generation EGFR TKIs (18.9, CI 15.2 to 21.4 versus 10.2, CI 9.6 to 11.1 months),13 there are nonetheless patients treated with first-line osimertinib who have early progression. We hypothesized that the unbiased genomic sequencing performed on pre-treatment and post-osimertinib-resistant tumors would provide insight into the variation in osimertinib treatment response. Using WES data, we analyzed the differences in focal copy-number amplifications between pre- and post-osimertinib tumor tissues. Greater number and magnitude of focal copy number amplifications was associated with a short-term osimertinib response (defined as <12 months’ PFS) among first-line osimertinib-treated patients (Figures 4A and 4B). The most common focal copy-number amplification emerged in the EGFR gene in post-osimertinib-resistance patients (n = 5 patients). Two patients, in addition to EGFR amplification, had focal copy-number amplifications in oncogenes KRAS/UBR5 (LAT014) and MET (LAT006) (Table S5). Subject LAT001 had MET amplification together with focal copy-number amplifications in KRAS, CBL, DDB2, and ARHGEF12 genes. Subject LAT021 had focal copy-number amplifications in PSIP1, MLLT3 genes, and cytoband 9p24.1, which contains the genes CD274 (PD-L1), PD-L2, JAK2, and NFIB (Table S5). Thus, our results demonstrate that multiple focal copy-number amplifications are acquired with osimertinib resistance, and the development of such amplifications, presumably due to increased genomic instability, is associated with a shorter time to progression while receiving osimertinib in patients without prior treatment.
Figure 4.

Multiple Acquired Focal Copy-Number Amplifications Are Associated with Short-Term Response to First-Line Osimertinib-Treated Patients
(A and B) Number (A) and magnitude (B) of acquired focal copy-number amplifications in patients with short versus long PFS. Short PFS: <12 months; long PFS: ≥12 months’ progression-free survival. Focal copy-number amplifications are defined as >10 copies between post- and pre-osimertinib treatment tumors adjusted for tumor purity and within a region spanning ≤10 megabases. Statistical significance was calculated using the Mann-Whitney test. Calculated p values are shown. Biological replicates for individual patients are shown.
Multi-region and Longitudinal Sampling Reveal Clonal Heterogeneity in MET Amplification upon Acquired Resistance to Osimertinib in Patients without Prior Therapy
Given previous evidence of intra-tumor heterogeneity in MET copy-number gain prior to TKI therapy,14 we hypothesized that there may be spatial and temporal heterogeneity in the development of MET copy-number gain and/or amplification upon resistance to osimertinib. Using multi-region and/or longitudinal sampling of osimertinib-resistant tumor tissue at first, second, or subsequent progressions, including at rapid autopsy, we focused on six first-line osimertinib-treated patients, LAT001, LAT002, LAT006, LAT014, LAT021, and LAT028, who had evidence of MET amplification after osimertinib as a first-line therapy (Figure 5; Figure S2). All six patients had a partial response to osimertinib (Figure 5; Figure S2; Table S1). Four of these patients (LAT001, LAT006, LAT021, and LAT028) were eligible for LAT. LAT001, LAT006, and LAT028 had surgery to remove progressive tumors, whereas LAT021 had radiation therapy.
Figure 5.

Heterogeneity of MET Amplification in the Development of Acquired Resistance to Osimertinib in Three Patients without Prior Therapy
(A) Treatment timeline from diagnosis to death for subject LAT001. The subject began osimertinib treatment under this study upon the diagnosis of EGFR mutant metastatic lung adenocarcinoma. Imaging at first restaging showed a treatment response in the posterior liver (green circle). Upon first progression at 7 months, the subject underwent LAT (posterior liver excision, green arrows). After a second progression on osimertinib (red arrows), the subject began a clinical trial of osimertinib plus savolitinib. The subject responded to osimertinib plus savolitinib at day 43 (red arrows). The subject subsequently progressed on day 77 (yellow circle) and was found to harbor MET D1246N mutation. Block arrows represent focal amplifications in areas without cancer-related genes. Yellow: 5q11.2; purple: 6p21.1; blue: 8q21.3; magenta: 6q24.1; green: 11p11.2, 11p24.2, 11q23.3, 12p12.1. Red text signifies anatomic sites of biopsies.
(B) Treatment timeline from diagnosis to death for subject LAT006. The subject started osimertinib therapy for metastatic EGFR mutant NSCLC. Three months after starting osimertinib, the tumor in the top-right lobe continued to respond (green circles), but the tumor in the bottom-left lobe progressed (red circles). The subject underwent LAT (bottom-left lung lobectomy, red arrow) and then osimertinib was reinitiated. At a second progression receiving osimertinib (yellow circle), the subject underwent stereotactic radiosurgery to the brain then started erlotinib followed by chemotherapy (carboplatin plus pemetrexed) followed by whole-brain radiation therapy and subsequently chemotherapy along with pembrolizumab. Upon progression (yellow circle), the subject had stereotactic radiosurgery to another brain lesion. The subject was then started on combination osimertinib and crizotinib to which the subject responded (yellow circle). Upon diagnosis of leptomeningeal disease, osimertinib was increased to 160 mg. Rapid autopsy was performed upon expiration.
(C) Treatment timeline from diagnosis to death for subject LAT021. After the diagnosis of metastatic EGFR mutant NSCLC, the subject was initiated on osimertinib. First on-trial imaging demonstrated a partial response (green circles). At first progression on osimertinib, the subject underwent LAT (proton therapy) and then reinitiated osimertinib. At a second progression while receiving osimertinib, WES of the first progressive tumor showed PDL1 amplification therefore pembrolizumab was started. There was mixed response on pembrolizumab with green arrows showing a site of response and yellow arrows showing sites of progression. The second progressive tumor had MET in addition to PDL1 amplification; therefore, the subject was treated with osimertinib and crizotinib, and the subject had resolution of PET-avid disease in multiple metastatic sites (yellow arrows). Red arrow signifies a new PET-avid site. FACETS copy-number plots from tumor exome sequencing and FISH for MET for each subject are shown. Only cancer-related genes within focal copy-number amplifications are displayed. Biological replicates at post-osimertinib resistance for individual subjects are shown. RT, radiation therapy; IO, immunotherapy; Osi, osimertinib; Criz, crizotinib; Savo, savolinitib
In subjects LAT001 and LAT006, multi-region WES of excised tumors upon first progression while receiving osimertinib showed MET amplification in 2 of 2 regions and in 1 of 2 regions, respectively (Figures 5A and 5B). By MET FISH, five of six regions from LAT001 were MET amplified by increased MET/CEP7 ratio (>2) and one region displayed MET polysomy (copy number [CN] >4) with a normal MET/CEP7 ratio (Figure 5A). In contrast, in LAT006, one of four regions was MET amplified by MET/CEP7 ratio, one region displayed MET polysomy (CN >5), and two regions were negative for both (Figure 5B). At second progression while receiving an osimertinib rechallenge, subject LAT001 enrolled in a phase I clinical trial of osimertinib and savolitinib combination15 and achieved a partial response with normalization of liver enzymes. Unfortunately, LAT001 developed resistance to the combination therapy within 2.5 months, and a repeat biopsy from a progressive tumor in the liver demonstrated a kinase domain mutation in MET, D1246N, that has been reported as an acquired resistance mutation to MET inhibitors16 (Figure 5A). A repeat biopsy at second progression while receiving osimertinib for subject LAT006 did not show MET amplification, but only MET polysomy (CN >4) was observed (Figure 5B). The affected individual received combination chemotherapy and immunotherapy with carboplatin, pemetrexed, and pembrolizumab. Interestingly, after progression on this regimen, a biopsy of the progressive tumor showed MET amplification by ratio (Figure 5B). Subject LAT006 then enrolled in the phase I clinical trial osimertinib and savolitinib combination; however, he could not continue after the first cycle due to grade 3 elevation of liver enzymes. Subsequently, he started on combination of osimertinib and crizotinib and responded until he developed leptomeningeal disease. WES of spinal metastases obtained by rapid autopsy did not show evidence of MET or other focal amplifications (Figure 5B). In summary, extensive clonal heterogeneity of MET amplification was revealed as a result of the multi-region and longitudinal WES that was further validated in additional samples using MET FISH testing, which may impact clinical outcomes of MET-directed therapy with osimertinib.
In contrast to subjects LAT001 and LAT006, subject LAT028 did not show evidence of heterogeneity in MET amplification upon first progression while receiving osimertinib as all five regions of tumor at first progression while receiving osimertinib were positive for MET amplification by copy number (CN >8) (Figure 6A). We therefore suspected that MET amplification may be an early or clonal event in this affected individual. Indeed, MET FISH testing on the pre-osimertinib-resistant tumor from LAT028 showed MET amplification by copy number (CN > 7) (Figure 6A). We subsequently performed MET FISH testing on all available pre-osimertinib-resistant tumors and uncovered pre-existing MET amplification by increased MET/CEP7 ratio in subject LAT014 (Figure 6B). WES and MET FISH of tumors (n = 5) of subject LAT014 acquired by rapid autopsy showed high MET amplification (ratio >5) in all tumors further supporting the early clonal origin of MET amplification in this affected individual (Figure 6B). In contrast, subject LAT002, who denied further treatment after first progression while receiving osimertinib had tumors obtained at rapid autopsy with significant MET heterogeneity; three sites were negative for MET amplification or polysomy (left frontal and left temporal lobes, right adrenal), three were positive for MET amplification by copy number (CN >7) (Li2, L2c, and L4a), one site was positive for MET amplification by MET/CEP7 ratio (Li1), and three sites were positive for MET polysomy with a normal MET/CEP7 ratio (CN >4) (L2b, L2e, B2a, L10a) (Figure S2B). Thus, our results suggest that MET amplification can either be a clonal or subclonal driver of osimertinib resistance in first-line osimertinib-treated EGFR mutant patients.
Figure 6.

Heterogeneity of MET Amplification in the Development of Acquired Resistance to Osimertinib in Subjects LAT014 and LAT028 without Prior Therapy
(A) Treatment timeline from diagnosis to death for subject LAT028. The subject began treatment with osimertinib under this study upon the diagnosis of EGFR mutant metastatic lung adenocarcinoma. Imaging at first restaging showed a response to treatment in the posterior liver (green and red circle). Upon first progression (red circle), the subject underwent LAT (top-left lobectomy).
(B) Treatment timeline from diagnosis to death for subject LAT014. The subject initiated treatment with osimertinib under this study upon the diagnosis of EGFR mutant metastatic lung adenocarcinoma. Imaging at first restaging showed a response to treatment in the right lung (green circle). At first progression, the subject had a new right pleural effusion and was not eligible for LAT. The subject underwent chemotherapy until progression in the liver (green arrow). At death, a rapid autopsy was performed. Red text signifies anatomic sites of biopsies. FACETS copy-number plots from tumor exome sequencing and FISH for MET for each subject are shown. Biological replicates at post-osimertinib resistance for individual subjects are shown. Only cancer-related genes within focal CNAs are shown. RT, radiation therapy; Osi, osimertinib; Surg, surgery
Unlike subjects LAT001, LAT002, LAT006, LAT014, and LAT028, subject LAT021 developed MET polysomy (CN >4), not amplification, upon first progression while receiving osimertinib (Figure 5C). At first progression, WES showed a single focal copy-number amplification at cytoband 9p24.1 (Figure 5C), which contains the genes for CD274 (PD-L1), PD-L2, JAK2, and NFIB. Immunohistochemical analysis demonstrated high PD-L1 expression and the presence of tumor infiltrating CD3+ and CD8+ T cells in the post-osimertinib-resistant tumor and a patient-derived xenograft (PDX) generated at first progression while receiving osimertinib (Figure S3). After a second progression while receiving osimertinib, given previous evidence of response to immunotherapy for patients with focal amplification in PD-L1,17 the affected individual was treated with pembrolizumab. Objective tumor response was noted (Figure 5C); however, progression in multiple bone metastases was observed after three treatment cycles. At second progression while receiving osimertinib (prior to pembrolizumab treatment), WES and MET FISH showed MET amplification (MET/CEP7 ratio = 12.0) in addition to PD-L1 amplification (Figure 5C). The affected individual was subsequently started on a combination osimertinib and crizotinib. She was then dose reduced on crizotinib for grade 3 neutropenia, but PET imaging at first re-staging in 6 weeks showed significant reduction in metabolic activity in most metastatic lesions (Figure 5C). The combination treatment was continued as “palliative treatment” while she enrolled in in-patient hospice for worsening performance status and finally succumbed to a possible sepsis. Rapid autopsy upon death showed viable tumor in only one of the many bone metastatic sites. No tumor was found in the lungs. Similarly, in subject LAT015, multi-region WES at first progression while receiving osimertinib did not show MET amplification (Figure S4A). MET FISH testing showed polysomy (CN >5) in 3 sites and amplification by ratio in two sites (MET/CEP7 ratio = 2.7 and 3.1) (Figure S4B). At second progression on osimertinib, there was MET amplification both by WES and MET FISH (MET/CEP7 ratio = 20.3) (Figures S4A and S4C).
To further validate our WES and MET FISH results, we compared MET expression in all patients with available pre-post- or post-only osimertinib RNA-seq data (n = 13). Expression of MET prior to osimertinib treatment was similar in all patients, even among subjects LAT014 and LAT028 who had evidence of pre-existing MET amplification (MET RPKM range 13–64) (Figure S5). Upon osimertinib resistance, all five first-line osimertinib-treated patients with evidence of MET amplification displayed increased MET expression (Figure S5). A post-osimertinib-resistant biopsy from subject LAT002 also showed elevated MET expression (Figure S5). The other patients did not have high MET expression upon osimertinib resistance (Figure S5). Interestingly, MET expression was high in all LAT006 post-osimertinib samples (Figure S5), despite the existing heterogeneity in MET amplification by WES and MET FISH. Overall, our multi-region and longitudinal sampling of progressive tumors demonstrates significant spatial and temporal heterogeneity in MET amplification and polysomy upon acquired resistance to osimertinib among first-line osimertinib-treated patients.
Amplification of Mutant EGFR Is a Common Mechanism of Resistance to Osimertinib
EGFR amplification frequently occurs together with EGFR TKI sensitizing mutations.18 Thus, when analyzing only post-treatment liquid or tissue biopsies2,6,7 it is difficult to ascertain whether EGFR amplification is an intrinsic or an acquired osimertinib-resistance mechanism. In our cohort, 67% (n = 8/12) of patients with paired pre- and post-osimertinib treatment biopsies had EGFR amplification prior to osimertinib treatment (Figure S6A; Table S6). Upon developing osimertinib resistance, 42% (n = 5/12) of all patients and 63% (n = 5/8) of the patients with pre-existing EGFR amplification showed further EGFR amplification (CN range 19–86 copies) (Figure S6B). 3/5 patients with further EGFR amplification and available pre- and post-treatment RNA-seq had an increase in EGFR expression (LAT014, LAT017, and LAT028), and a fourth had stable EGFR expression (LAT006) (Figure S6C). The remaining patients without evidence of EGFR amplification upon osimertinib resistance had decreased EGFR expression (Figure S6C). Additionally, using both targeted and exome sequencing, we found the mutant EGFR allele, rather than the wild-type allele, was further amplified upon osimertinib resistance (Figures S6D–S6G). Thus, these analyses suggest that, despite frequent pre-existing EGFR amplification, further amplification of the mutant allele of EGFR is a common mechanism of acquired resistance to osimertinib.
NE Differentiation with and without Histologic Transformation upon Osimertinib Resistance
Exome, targeted, and fusion analysis of paired pre- and post-osimertinib-resistant tumors uncovered mechanisms of osimertinib resistance in all patients except subject LAT011. To identify potential spatially heterogeneous acquired resistance mechanisms, we performed additional exome sequencing of eight distinct samples from the progressive tumor acquired by surgery at the time of first progression. These analyses did not reveal a known acquired resistance mechanism. Furthermore, there was no histologic evidence of transformation to SCLC, squamous cell carcinoma, or epithelial mesenchymal transition (EMT). Upon developing osimertinib resistance, subject LAT011 had rapid clinical progression, with multiple-resistant tumors appearing in the liver, did not respond to chemotherapy, and died 2 months after osimertinib was discontinued (Figure 7A).
Figure 7.

Neuroendocrine Differentiation with and without Histologic Transformation upon Osimertinib Resistance
(A) Treatment timeline from diagnosis to death for subject LAT011. After receiving erlotinib, osimertinib treatment was initiated under this study. First, on-trial imaging demonstrated a reduction in tumor volume (green circles). First progression of disease occurred after 10 months in the bottom-right lobe. The subject then underwent LAT (bottom-right lobectomy) followed by re-initiation of osimertinib. A second progression occurred in the liver. Chemotherapy was initiated, but progression at multiple liver sites occurred, and the subject died soon after this rapid progression.
(B) Volcano plot of reactome pathways enriched between post- and pre-osimertinib-resistant tumors across all LAT subjects.
(C) Pre- and post-osimertinib-resistant RPKM values of ASCL1 and HES1 of all LAT subjects. Only post-osimertinib RNA-seq data were available for subject LAT005.
(D) ssGSEA neuroendocrine differentiation scores of pre- and post-osimertinib-resistant LAT tumors.
(E) Hematoxylin and eosin (H&E), synaptophysin, and chromogranin immunohistochemical staining of tumors from subjects LAT005, LAT010, and LAT011 before treatment and upon acquired osimertinib resistance. LAT011 PDXs derived from lung (first progression) and liver (second progression) are shown. Biological replicates at post-osimertinib resistance are shown. Scale bars, 100 μm. Osi, osimertinib; Chemo, chemotherapy.
Using available RNA-seq data from subjects LAT001, LAT005, LAT006, LAT009, LAT010, LAT011, LAT014, and LAT017, we performed differential expression analysis between all pre- and post-osimertinib-resistant tumors to decipher novel mechanisms of osimertinib resistance with a focus on LAT011. Pathway analysis of the post- versus pre-osimertinib transcript expression data revealed that NOTCH was one of the most significantly downregulated pathways (Figure 4B; Table S6). Expression of HES1, a downstream effector of NOTCH signaling, was decreased in all post-osimertinib compared to pre-osimertinib tumor tissue in LAT011 (Figure 7B). Subject LAT010, whose tumor displayed mixed NSCLC/SCLC histology, also had decreased expression of HES1 upon osimertinib resistance. The level of expression of HES1 was low in a post-osimertinib sample from subject LAT005, who had full SCLC transformation (Figure 7B). In contrast, subjects LAT001, LAT006, LAT009, LAT014, and LAT017 did not have a decrease in HES1 expression upon osimertinib resistance (Figure 4B). Given that inhibition of NOTCH signaling is known to lead to NE differentiation,19,20 we hypothesized that LAT011 developed NE differentiation upon osimertinib resistance despite the lack of histologic evidence of small cell transformation. To further substantiate this hypothesis, we examined the expression of ASCL1, a downstream effector of NE differentiation, in post-osimertinib compared to pre-osimertinib tumor samples. ASCL1 expression increased in post-osimertinib compared to pre-osimertinib tumor samples in subjects LAT011 (NE differentiation without SCLC transformation) and LAT010 (mixed NSCLC/SCLC transformation) and LAT005 (SCLC transformation) but not in other subjects (LAT001, LAT006, LAT009, LAT014, and LAT017 (Figure 7B).
We next compared overall NE differentiation in subject LAT011 to other patients using a 50-gene expression lung cancer-specific NE expression signature.21 Using a single-sample gene set enrichment analysis (ssGSEA) analysis of this NE signature, we found the post-osimertinib tumor in subject LAT005 displayed the highest NE differentiation consistent with SCLC transformation, while the pre- and post-osimertinib-resistant tumors of subjects LAT001, LAT006, LAT009, LAT014, and LAT017 displayed the least NE differentiation consistent with retained adenocarcinoma histology (Figure 7D). The post-osimertinib T4 tumor of subject LAT010 displayed intermediate levels of NE differentiation, as expected, for mixed NSCLC/SCLC histology (Figure 7D). For subject LAT011, the post-osimertinib-resistant liver tumor with a poorly differentiated NSCLC histology showed increased NE differentiation in comparison to the pre-osimertinib tumor and multiple post-osimertinib-resistant lung tumors that retained an adenocarcinoma histology (Figure 7D).
We further validated our RNA-seq findings using immunohistochemistry to detect common NE markers. As expected, the SCLC transformed post-osimertinib-resistant tumor from subject LAT005 and the regions of SCLC transformation in the post-osimertinib-resistant tumor from subject LAT010 showed high synaptophysin and chromogranin expression (Figure 7E). For subject LAT011, tumors at first and second progression on osimertinib and corresponding PDXs showed moderately positive staining for synaptophysin with variable expression of chromogranin (Figure 7E). Together, these results provide evidence for NE differentiation of osimertinib-resistant tumors without evidence of histological transformation in subject LAT011 likely mediated by decreased NOTCH signaling.
Discussion
Here, we describe the heterogeneity and evolution of multiple resistance mechanisms in individual osimertinib-treated EGFR mutant lung adenocarcinoma patients. Our ongoing prospective clinical study allows for the longitudinal sampling of progressive tumors, including at autopsy, to conduct multi-region/temporal exome and transcriptome sequencing of tumors from EGFR mutant NSCLC patients undergoing osimertinib treatment. The unbiased genomic analyses revealed a likely mechanism driving osimertinib resistance in all patients analyzed. Particularly, in this study we have reported the most comprehensive genomic analysis on prospectively collected multiple tumors obtained from patients undergoing first-line as well as second-line osimertinib treatment upon development of resistance. The majority of patients acquired two or more subclonal resistance mechanisms either concurrently or in temporal sequence.
Among first-line osimertinib-treated patients in our study, MET amplification was the most common mechanism of resistance, occurring in 66% of patients (n = 6/9). Acquired MET amplification, however, was subclonal and highly heterogeneous, which may explain why MET amplification was previously reported in a lower percentage of first-line osimertinib-treated patients.7 Acquisition of subclonal MET amplification may predict suboptimal response to osimertinib similar to observations for patients with pre-existing MET amplification with early-generation EGFR TKIs.14 Indeed, two patients in our study found to have pre-existing, clonal MET amplification had early progression on osimertinib similar to the patients in our study with acquired, subclonal MET amplification. It is plausible that first-line osimertinib treatment results in expansion of rare pre-existing MET amplified clones, as has been observed with first and second generation EGFR TKIs,22 resulting in quicker progression. Thus, while clinical trials are underway evaluating MET inhibitors upon osimertinib resistance,15 clinical testing of MET inhibitors in combination with osimertinib in treatment-naive patients will be critical as our study demonstrates the risk of acquiring MET amplified subclones in such patients is high.
The optimal treatment approach for patients upon osimertinib resistance is unclear, and treatment options are currently limited. Our clonal evolutionary analysis using multi-region/temporal sequencing revealed the subclonal co-occurrence of acquired alterations upon osimertinib resistance, suggesting treatment for patients after progression on osimertinib will likely require targeting dual resistance mechanisms. For example, in our study, subject LAT006 was found to have MET amplification upon first progression on osimertinib and was eventually treated with a MET inhibitor. However, our analyses revealed the amplification of both MET and EGFR, suggesting patients such as LAT006 may benefit from a combination of MET inhibitor together with drugs targeting the EGFR amplified clone apart from osimertinib. Similarly, subject LAT003 developed the EGFR C797S mutation concurrently but spatial and temporally separated from EGFR amplification suggesting the potential use of a C797S inhibitor23 would need to be combined with additional EGFR-pathway directed therapy. In contrast, although subject LAT016 developed two unique spatially and temporally acquired BRAF fusions, both are likely targetable by BRAF and MEK inhibitors. Thus, through clonal analysis of multi-region and temporal sequencing, our study suggests the number of osimertinib-resistant EGFR mutant NSCLC patients with concurrent multiple resistance mechanism is likely much higher than previously reported.24 These data also provide the rationale for LAT approaches and multiple longitudinal biopsies in the context of treatment of EGFR mutant NSCLC patients with osimertinib.
Our study demonstrates that many patients with acquired resistance to osimertinib remain dependent on EGFR signaling through additional EGFR amplification. Dual inhibition of EGFR signaling with a second-generation EGFR TKI together with monoclonal antibodies to the EGFR extracellular domain has shown clinical effectiveness in acquired resistance to first-generation EGFR TKIs.25 Whether such a strategy would be effective upon acquired resistance to osimertinib is unknown, although there is recent evidence that targeting EGFR amplification can be an effective clinical strategy in other cancer types.26 Examining the clinical activity of osimertinib with selumetinib (NCT03392246), an inhibitor of MEK among patients with EGFR amplification upon acquired resistance to osimertinib, would also be of interest.
EGFR mutant NSCLC tumors treated with TKIs can histologically transform to SCLC or develop tumors with mixed NSCLC/SCLC histology, both of which have a poor clinical outcome.12,27,28 We identified an EGFR mutant NSCLC subject with rapid clinical progression upon osimertinib resistance whose post-osimertinib-resistant tumors displayed evidence of NE transformation but lacked histologic evidence of SCLC transformation. Acquired osimertinib resistance with NE features but with retained NSCLC histology may represent a disease transition state, further substantiated by recent reports of a subset of metastatic prostate cancer patients treated with anti-androgen therapy who develop androgen resistance with NE differentiation and adenocarcinoma features.29 The use of immunohistochemistry for NE markers or RNA-seq upon acquired osimertinib resistance, in patients with rapid clinical progression, may uncover additional cases with evidence of NE differentiation but without SCLC transformation. Comparing treatment and clinical outcomes in such cases to patients with classic SCLC transformation will be important and will require larger cohorts.
In conclusion, our study, utilizing whole-exome and transcriptome sequencing, reveals the clonal heterogeneity and tumor evolution of metastatic EGFR mutant lung cancer treated with osimertinib. Since the majority of patients treated with osimertinib had two or more subclonal resistance mechanisms, our data suggest combination therapies will be required to overcome acquired resistance. A MET inhibitor will be an important part of such combination therapy, particularly among first-line osimertinib-treated patients with early progression.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Synaptophysin (human) | Roche | Cat# 790-4407; RRID: AB_2336016 |
| Anti-Chromogranin (human) | Roche | Cat# 760-2519; RRID: AB_2335955 |
| Anti-PD-L1 (human) | Spring Bioscience | Cat# MA4421 |
| Biological Samples | ||
| Pre- and post-osimertinib resistant tumors | National Cancer Institute, Bethesda, Maryland | https://clinicaltrials.gov/ct2/show/NCT02759835 |
| Metastases collected by rapid autopsy | National Cancer Institute, Bethesda, Maryland | https://clinicaltrials.gov/ct2/show/NCT04308226 |
| Critical Commercial Assays | ||
| NextSeq 500 system | Illumina | N/A |
| SureSelect Clinical Research Exome Kits | Agilent | N/A |
| TruSeq RNA Exome Library Prep | Illumina | N/A |
| Deposited Data | ||
| Processed data and code used | This paper | https://github.com/aleighbrown/pwgs_snakemake |
| Raw data (exome and transcriptome) | This paper | dbGaP: phs002001 |
| Software and Algorithms | ||
| FACETS | Shen et al., 2016; PMID: 27270079 | https://bioinformaticshome.com/tools/cnv/descriptions/FACETS.html |
| BWA | Li et al., 2009; PMID: 20080505 | http://bio-bwa.sourceforge.net/ |
| EdgeR | Robinson et al., 2010. PMID: 19910308 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| Limma voom | Law et al., 2014. PMID: 24485249 | https://bioconductor.org/packages/release/bioc/vignettes/limma/inst/doc/usersguide.pdf |
| GenePattern | Subramanian et al., 2005; PMID: 16199517 | https://software.broadinstitute.org/cancer/software/genepattern |
| PhyloWGS | Deshwar et al., 2015; PMID: 25786235 | https://github.com/morrislab/phylowgs |
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Udayan Guha (udayan.guha@nih.gov).
Materials Availability
This study did not generate new, unique reagents.
Data and Code Availability
The accession number for the sequencing data reported in this paper is dbGaP: phs002001 Data analysis pipeline is available at: https://github.com/aleighbrown/pwgs_snakemake.
Experimental Model and Subject Details
Biospecimen Acquisition and Study Design
Tissue samples were acquired from patients enrolled in a single-arm, single-institution, open-label phase II study of osimertinib treatment and local ablative therapy (LAT) upon progression on osimertinib in EGFR mutant metastatic lung adenocarcinoma. Eligible patients were treated with osimertinib 80 mg per day daily until disease progression. A higher dose of 160 mg per day was used in patients with leptomeningeal disease. At the time of progression, patients with oligoprogressive disease, defined as no more than 5 sites of progressive disease who are amenable to treatment with LAT (surgery, radiation therapy, radiofrequency ablation, or cryoablation) underwent LAT. Osimertinib was resumed after LAT and they are followed for second progression on osimertinib (PFS2). Tumor samples were obtained at baseline by a mandatory biopsy. At the time of first progression on osimertinib, if a patient was eligible for surgery as a form of LAT, then a tissue sample was obtained for genomic studies to identify mechanisms of acquired resistance. For patients who were not eligible for LAT or a form of LAT that was not surgery (radiation, radiofrequency ablation, cryoablation), then a mandatory biopsy was performed, if clinically safe, to obtain tissue for genomic studies. Patients were not selected based on oligometastatic status, rather patients had multiple organ involvement, including lung, liver, adrenal, brain, and bone. Patients undergoing LAT were subsequently re-challenged with osimertinib. Patients who progressed a second time on osimertinib following LAT or patients who were not LAT eligible were referred to other clinical studies or received standard of care second line treatments. Patients also underwent a repeat biopsy of the second progressive lesion when clinically feasible (Figure 1A). Eligible patients were at least 18 years old and had stage IV lung adenocarcinoma confirmed by pathology review by the Laboratory of Pathology at the National Cancer Institute (NCI). All patients provided written informed consent. Gender, and age is provided for all study participants with genomic data (Table S1). We estimated that a minimum of 10 patients would pre- and post-osimertinib resistant tumor samples would be needed to adequently assess mechanisms of resistance to osimertinib. The study was approved by the NCI Institutional Review Board with the local protocol number 16C0092. Following surgery or a biopsy, tissues were paraffin embedded by standard clinical protocols in the Laboratory of Pathology at the NCI. Paraffin-embedded tissue sections were then macrodissected to enrich for tumor cells with at least 20% tumor content. DNA and RNA were extracted using the QIAGEN AllPrep DNA/RNA FFPE Kit or separately with QIAamp DNA FFPE Tissue Kit and RNeasy FFPE Kit respectively. The extraction was performed on a semi-automated instrument, QIAcube (QIAGEN), according to the instructions of the manufacturer. The concentration of DNA and RNA were measured with Qubit. Blood sample from patient was used for normal (germline) sequencing.
Rapid autopsy samples were obtained from patients with EGFR mutant lung cancer who gave informed consent for IRB approved protocol 13-C-0131 (NCT01851395) entitled ‘‘A Pilot Study of Inpatient Hospice with Procurement of Tissue on Expiration in Thoracic Malignancies.’’ The study was approved by the NCI Institutional Review Board with the local protocol number 13C0131. All patients were previously treated at the NCI and with life expectancy less than 3 months were offered inpatient hospice treatment at the Clinical Center of the National Institutes of Health and upon death autopsies were initiated within 3 hours. Prioritization of lesions removed at autopsy was based on CT scan performed within one month before death. All tumors within each patient were removed by an experienced pathologist and macro dissected to remove surrounding non-neoplastic tissue. Punch biopsy needles were used to obtain spatially distinct cores from each tumor. One-third of each tissue core sample was fixed in 10% buffered formalin, one-third in optimal cutting temperature compound (OCT) and the remaining tissue was immediately flash frozen in liquid nitrogen and stored at 80°C. For each tissue sample, a 5-mm section was taken to create a hematoxylin and eosin slide to visualize neoplastic cellularity using a microscope. DNA and RNA was isolated from approximately 30 mg of snap-frozen tumor tissue using the All Prep DNA/RNA Mini Kit (QIAGEN).
Method Details
Exome and RNA Sequencing of Tumors
Formalin-fixed, paraffin-embedded (FFPE) tumor tissue samples were prepared for whole-exome sequencing (WES) and RNA sequencing (RNA-Seq). One hundred nanograms of DNA was sheared to approximately 200 base pairs (bp) by sonication (Covaris, Woburn, MA). Exome enrichment was performed using SureSelect Clinical Research Exome Kits according to the manufacturer’s instructions (Agilent, Santa Clara, CA) and RNA enrichment was performed using TruSeq RNA Exome Library Prep according to manufacturer’s instructions (Illumina, San Diego). Paired-end sequencing (2 × 75 bp) was performed on an Illumina NextSeq500 instrument. The sequences were compared to the human reference genome hg19 using internally developed ClinOmics somatic Bioinformatic Pipeline v3.1. Peripheral blood DNA extracted from individual patients was used for germline exome sequencing. In brief, raw sequencing data in FASTQ format were aligned against the reference human genome (hg19) with BWA30(31)(30)(Li and Durbin, 2009)(30)(30). The Genome Analysis Toolkit (GATK) and HaplotypeCaller (HAPLOC) were used for germline SNV and indel calling; whereas MuTect and Strelka were used for somatic single nucleotide variant (SNV) and small indel calling respectively. ANNOVAR was used to functionally annotate genetic variants. Tier 1 somatic variants were defined as protein coding on a hotspot codon or if on a non-hotspot codon then must consist of the following: reported as a somatic change in five or more individual tumors, loss of function in tumor suppressor gene in Cancer Gene Census or loss of function by a known mechanism in a non-tumor suppressor gene for Cancer Gene Census genes. Tier 1 somatic variants were considered “high-confidence” mutations. Other tiers were protein coding somatic variants not on a hotspot codon but a loss of function variant by an alternative mechanism (Tier 2), a rare/de-novo variant (Tier 3) or not on ClinOmics gene list (Tier 4). FACETS31 algorithm was used to determine total and allele-specific DNA copy number from WES. RNA was extracted from FFPE tumor cores using RNeasy FFPE kits according to the manufacturer’s protocol (QIAGEN, Germantown, MD). RNA-seq libraries were generated using TruSeq RNA Access Library Prep Kits (TruSeq RNA Exome kits; Illumina) and sequenced on NextSeq500 sequencers using 75bp paired-end sequencing method (Illumina, San Diego, CA). Each sample was processed through an RNA-seq data analysis pipeline where reads were mapped to the ENSEMBL human genome GRCh37 build 71 using TopHat2. Read counts for each gene between samples were normalized using TMM method implemented in edgeR and then transformed to RPKM.
Further analysis of RNA-seq data
Differential gene expression was conducted using limma voom32 between pre-treatment and post-treatment samples. Since we had multiple libraries/biopsies from the same patient within each group, we estimated consensus correlation among those libraries and incorporated it in the linear model to identify differentially expressed genes between each group. GSEA was subsequently performed on all genes and enrichment scores were generated for REACTOME pathways. Pathways with FDR less than 0.05 were considered significantly enriched. Single-sample GSEA (ssGSEA) from GenePattern was used to generated neuroendocrine scores for each tumor based on previously published neuroendocrine gene sets21.
Phylogenetic analysis
PhyloWGS33 was used to reconstruct tumor clonal and evolutionary history of both copy number, simple somatic mutations and indels. Allele-specific copy numbers from FACETS and simple somatic mutations, SNVs and indels, called by Strelka34 were used as input. For tumors which had more than one biopsy, PhyloWGS was run in both single sample mode and in multi-sample mode. The JSON results of PhyloWGS were parsed in order to find the SSMs and focal copy number amplifications associated with each subclone for the best predicted trees. Due to the constraints of the infinite sites assumption35, the PhyloWGS model does not account for copy number changing to multiple different aberrant states at the same genomic locus across samples. For these tumors, PhyloWGS was run multiple times with each run having the copy number artificially set to neutral in one sample. Trees were then manually curated from the parsed subclones from the single-sample, multi-sample, and artificially neutral sample runs. Focal copy number amplifications in cancer related genes were then assigned to phylogenetic nodes based on association with somatic mutations. Cancer Cell Fraction (CCF) proportion for every subclone is presented as a horizontal bar in each time point. Colors correspond to their subclone in the phylogeny tree. Cancer Cell Fraction (CCF) proportion for every subclone is presented as a horizontal bar in each time point. Colors correspond to their subclone in the phylogeny tree. The bar plot is depicted by Matplotlib library in Python. The idea is adopted from Figure 1 of the supplementary document in36.
Immunohistochemical Staining and Fluorescence in Situ Hybridization
Immunohistochemical stains for synaptophysin, chromogranin and PD-L1 were performed at NIH, Laboratory of Pathology, according to manufacturer’s instruction. IHC-stained slides were scanned using the 40x magnification on NanoZoomer S360 Hamamatsu slide scanner. Fluorescence in Situ Hybridization (FISH) for MET, EGFR and KRAS was performed by Chromosome Pathology Unit, Lab. of Pathology, NCI, NIH. MET was considered amplified when MET/CEP7 > = 2.0 or mean MET > = 6.0 copies/cell. MET polysomy was defiend by mean MET > = 3.0 and < 6.0 copies/cell. MET was considered negative when above criteria are not satisfied and indeterminate when technical issues prevented the test from being reported as positive, negative, or equivocal.
Quantification and Statistical Analysis
All figures and graphs were generated using the “ggplot2” package available through the R statistical program. Correlations and t tests were conducted though the R base packages. All tests were two-tailed and p values less than 0.05 were considered significant.
Additional Resources
The ClinicalTrials.gov identifier for this study is NCT02759835.
Acknowledgments
We thank the clinical and research nurses as well as palliative care and spiritual care teams for their clinical care of the patients in this study, particularly those who were enrolled in hospice prior to autopsy. We also thank Willie Young and the Pathology residents who assisted with the autopsies. A.-L.B. and A.R.P. thank Drs. Alexander Goncearenco and Alejandro Schaffer for helpful discussions. This study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH (https://hpc.nih.gov/docs/userguide.html). This research was supported by the NIH Intramural Research Program, Center of Cancer Research, and National Cancer Institute and Intramural Research Program of the U.S. National Library of Medicine at the National Institutes of Health. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author Contributions
N.R., C.K., and U.G. designed the study. N.R., A.-L.B., J.S.W., S.P., O.R., C.T., L.X., and J.K. assisted with sample preparation and data analysis. A.-L.B. and N.R. and performed the computational analysis. N.R. performed the biostatistical analyses. S.H. performed the autopsies, and N.R., A.V., and U.G. harvested tissues during autopsies. N.R., C.K., N.N., E.P., E.A., A.R., A.K., D.K.S., C.D.H., and U.G. were involved with clinical care of patients in the clinical trial. N.R. and U.G. wrote the manuscript. All authors reviewed, commented on, and approved the manuscript.
Declaration of Interests
C.K. receives research funding to his current institution (Georgetown University) from AstraZeneca, Novartis, and Tesaro and received one-time travel support from CARIS Life Science. C.T. owns stock in Gilead, Celgene, Exelixis, Clovis, and Trevena, and he is an employee of the Food and Drug Administration outside of the submitted work. Z.P. has served as a compensated consultant or received honoraria from AstraZeneca, Spectrum, Ariad/Takeda, Novartis, ImmunoGen, AbbVie, GuardantHealth, Genentech, Eli Lilly, InCyte, and Medtronic and receives institutional research funding from Novartis, Takeda, Spectrum, AstraZeneca, and Tesaro. U.G. has a clinical trial agreement (CTA) with AstraZeneca for the current study and receives research funding from AstraZeneca, Esanex, and Aurigene. U.G. is currently an employee of Bristol Myers Squibb. The other authors have no conflicts of interest to report.
Published: April 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.xcrm.2020.100007.
Contributor Information
Nitin Roper, Email: nitin.roper@nih.gov.
Javed Khan, Email: khanjav@mail.nih.gov.
Udayan Guha, Email: udayan.guha@nih.gov.
Supplemental Information
References
- 1.Chong C.R., Jänne P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oxnard G.R., Hu Y., Mileham K.F., Husain H., Costa D.B., Tracy P., Feeney N., Sholl L.M., Dahlberg S.E., Redig A.J. Assessment of Resistance Mechanisms and Clinical Implications in Patients With EGFR T790M-Positive Lung Cancer and Acquired Resistance to Osimertinib. JAMA Oncol. 2018;4:1527–1534. doi: 10.1001/jamaoncol.2018.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le X., Puri S., Negrao M.V., Nilsson M.B., Robichaux J., Boyle T., Hicks J.K., Lovinger K.L., Roarty E., Rinsurongkawong W. Landscape of EGFR -dependent and -independent resistance mechanisms to osimertinib and continuation therapy post-progression in EGFR-mutant NSCLC. Clin. Cancer Res. 2018;24:6195–6203. doi: 10.1158/1078-0432.CCR-18-1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z., Yang N., Ou Q., Xiang Y., Jiang T., Wu X., Bao H., Tong X., Wang X., Shao Y.W. Investigating Novel Resistance Mechanisms to Third-Generation EGFR Tyrosine Kinase Inhibitor Osimertinib in Non-Small Cell Lung Cancer Patients. Clin. Cancer Res. 2018;24:3097–3107. doi: 10.1158/1078-0432.CCR-17-2310. [DOI] [PubMed] [Google Scholar]
- 5.Shi P., Oh Y.T., Deng L., Zhang G., Qian G., Zhang S., Ren H., Wu G., Legendre B., Jr., Anderson E. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin. Cancer Res. 2017;23:6567–6579. doi: 10.1158/1078-0432.CCR-17-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Piotrowska Z., Isozaki H., Lennerz J.K., Gainor J.F., Lennes I.T., Zhu V.W., Marcoux N., Banwait M.K., Digumarthy S.R., Su W. Landscape of Acquired Resistance to Osimertinib in EGFR-Mutant NSCLC and Clinical Validation of Combined EGFR and RET Inhibition with Osimertinib and BLU-667 for Acquired RET Fusion. Cancer Discov. 2018;8:1529–1539. doi: 10.1158/2159-8290.CD-18-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramalingam S.S., Yang J.C., Lee C.K., Kurata T., Kim D.W., John T., Nogami N., Ohe Y., Mann H., Rukazenkov Y. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018;36:841–849. doi: 10.1200/JCO.2017.74.7576. [DOI] [PubMed] [Google Scholar]
- 8.Roper N., Gao S., Maity T.K., Banday A.R., Zhang X., Venugopalan A., Cultraro C.M., Patidar R., Sindiri S., Brown A.L. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019;26:2651–2666. doi: 10.1016/j.celrep.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blakely C.M., Watkins T.B.K., Wu W., Gini B., Chabon J.J., McCoach C.E., McGranahan N., Wilson G.A., Birkbak N.J., Olivas V.R. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017;49:1693–1704. doi: 10.1038/ng.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nahar R., Zhai W., Zhang T., Takano A., Khng A.J., Lee Y.Y., Liu X., Lim C.H., Koh T.P.T., Aung Z.W. Elucidating the genomic architecture of Asian EGFR-mutant lung adenocarcinoma through multi-region exome sequencing. Nat. Commun. 2018;9:216. doi: 10.1038/s41467-017-02584-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bell D.W., Gore I., Okimoto R.A., Godin-Heymann N., Sordella R., Mulloy R., Sharma S.V., Brannigan B.W., Mohapatra G., Settleman J., Haber D.A. Inherited susceptibility to lung cancer may be associated with the T790M drug resistance mutation in EGFR. Nat. Genet. 2005;37:1315–1316. doi: 10.1038/ng1671. [DOI] [PubMed] [Google Scholar]
- 12.Lee J.K., Lee J., Kim S., Kim S., Youk J., Park S., An Y., Keam B., Kim D.W., Heo D.S. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 2017;35:3065–3074. doi: 10.1200/JCO.2016.71.9096. [DOI] [PubMed] [Google Scholar]
- 13.Soria J.C., Ohe Y., Vansteenkiste J., Reungwetwattana T., Chewaskulyong B., Lee K.H., Dechaphunkul A., Imamura F., Nogami N., Kurata T., FLAURA Investigators Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 14.Lai G.G.Y., Lim T.H., Lim J., Liew P.J.R., Kwang X.L., Nahar R., Aung Z.W., Takano A., Lee Y.Y., Lau D.P.X. Clonal MET Amplification as a Determinant of Tyrosine Kinase Inhibitor Resistance in Epidermal Growth Factor Receptor-Mutant Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2019;37:876–884. doi: 10.1200/JCO.18.00177. [DOI] [PubMed] [Google Scholar]
- 15.Sequist L.V., Lee J.S., Han J.-Y. Abstract CT033: TATTON Phase Ib expansion cohort: Osimertinib plus savolitinib for patients (pts) with < em > EGFR < /em > -mutant, < em > MET < /em > -amplified NSCLC after progression on prior third-generation epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) Cancer Res. 2019;79(13, Supplement) CT033–CT033. [Google Scholar]
- 16.Li A., Yang J.J., Zhang X.C., Zhang Z., Su J., Gou L.Y., Bai Y., Zhou Q., Yang Z., Han-Zhang H. Acquired MET Y1248H and D1246N Mutations Mediate Resistance to MET Inhibitors in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2017;23:4929–4937. doi: 10.1158/1078-0432.CCR-16-3273. [DOI] [PubMed] [Google Scholar]
- 17.Goodman A.M., Piccioni D., Kato S., Boichard A., Wang H.Y., Frampton G., Lippman S.M., Connelly C., Fabrizio D., Miller V. Prevalence of PDL1 Amplification and Preliminary Response to Immune Checkpoint Blockade in Solid Tumors. JAMA Oncol. 2018;4:1237–1244. doi: 10.1001/jamaoncol.2018.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Network T., Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lim J.S., Ibaseta A., Fischer M.M., Cancilla B., O’Young G., Cristea S., Luca V.C., Yang D., Jahchan N.S., Hamard C. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature. 2017;545:360–364. doi: 10.1038/nature22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H.J., Poran A., Unni A.M., Huang S.X., Elemento O., Snoeck H.W., Varmus H. Generation of pulmonary neuroendocrine cells and SCLC-like tumors from human embryonic stem cells. J. Exp. Med. 2019;216:674–687. doi: 10.1084/jem.20181155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W., Girard L., Zhang Y.A., Haruki T., Papari-Zareei M., Stastny V., Ghayee H.K., Pacak K., Oliver T.G., Minna J.D., Gazdar A.F. Small cell lung cancer tumors and preclinical models display heterogeneity of neuroendocrine phenotypes. Transl. Lung Cancer Res. 2018;7:32–49. doi: 10.21037/tlcr.2018.02.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turke A.B., Zejnullahu K., Wu Y.L., Song Y., Dias-Santagata D., Lifshits E., Toschi L., Rogers A., Mok T., Sequist L. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell. 2010;17:77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jia Y., Yun C.H., Park E., Ercan D., Manuia M., Juarez J., Xu C., Rhee K., Chen T., Zhang H. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–132. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu H.A., Arcila M.E., Rekhtman N., Sima C.S., Zakowski M.F., Pao W., Kris M.G., Miller V.A., Ladanyi M., Riely G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013;19:2240–2247. doi: 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Janjigian Y.Y., Smit E.F., Groen H.J., Horn L., Gettinger S., Camidge D.R., Riely G.J., Wang B., Fu Y., Chand V.K. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov. 2014;4:1036–1045. doi: 10.1158/2159-8290.CD-14-0326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maron S.B., Alpert L., Kwak H.A., Lomnicki S., Chase L., Xu D., O’Day E., Nagy R.J., Lanman R.B., Cecchi F. Targeted Therapies for Targeted Populations: Anti-EGFR Treatment for EGFR-Amplified Gastroesophageal Adenocarcinoma. Cancer Discov. 2018;8:696–713. doi: 10.1158/2159-8290.CD-17-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sequist L.V., Waltman B.A., Dias-Santagata D., Digumarthy S., Turke A.B., Fidias P., Bergethon K., Shaw A.T., Gettinger S., Cosper A.K. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marcoux N., Gettinger S.N., O’Kane G., Arbour K.C., Neal J.W., Husain H., Evans T.L., Brahmer J.R., Muzikansky A., Bonomi P.D. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. 2019;37:278–285. doi: 10.1200/JCO.18.01585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aggarwal R., Huang J., Alumkal J.J., Zhang L., Feng F.Y., Thomas G.V., Weinstein A.S., Friedl V., Zhang C., Witte O.N. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018;36:2492–2503. doi: 10.1200/JCO.2017.77.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen R., Seshan V.E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshwar A.G., Vembu S., Yung C.K., Jang G.H., Stein L., Morris Q. PhyloWGS: reconstructing subclonal composition and evolution from whole-genome sequencing of tumors. Genome Biol. 2015;16:35. doi: 10.1186/s13059-015-0602-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saunders C.T., Wong W.S., Swamy S., Becq J., Murray L.J., Cheetham R.K. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics. 2012;28:1811–1817. doi: 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- 35.Kimura M. The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics. 1969;61:893–903. doi: 10.1093/genetics/61.4.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kockan C., Hach F., Sarrafi I., Bell R.H., McConeghy B., Beja K., Haegert A., Wyatt A.W., Volik S.V., Chi K.N. SiNVICT: ultra-sensitive detection of single nucleotide variants and indels in circulating tumour DNA. Bioinformatics. 2017;33:26–34. doi: 10.1093/bioinformatics/btw536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data reported in this paper is dbGaP: phs002001 Data analysis pipeline is available at: https://github.com/aleighbrown/pwgs_snakemake.