

INTRODUCTION:
Eosinophilic esophagitis (EoE) is a T-helper 2 (Th2), eosinophilic disease associated with pathologic tissue remodeling that leads to end-organ dysfunction. During early-stage disease, inflammation and subepithelial fibrosis are coupled and reversible, but in late-stage or therapy-resistant disease, there can be uncoupling of these features with progressive esophageal rigidity and strictures contributing to clinical dysphagia and food impactions. No current pharmacotherapeutic interventions directly target esophageal fibrosis. Based on the ability of the thiazolidinediones (TZD) to regulate intestinal and hepatic fibrosis, we tested the antifibrotic effects of the TZDs, rosiglitazone and pioglitazone, in preclinical studies using primary human esophageal fibroblasts.
METHODS:
Primary fibroblasts isolated from normal or EoE esophagi were treated with transforming growth factor (TGF)-β1 in the absence or presence of TZDs and, in some experiments, without or with budesonide and analyzed by quantitative real-time PCR and immunoblotting. Immunohistochemical analysis of human esophageal biopsies was performed.
RESULTS:
EoE esophageal biopsies and esophageal fibroblasts expressed higher levels of the TZD receptor, peroxisome proliferator-activated receptor-γ (PPAR-γ), than normal controls. PPAR-γ was inducible by the Th2 cytokine, interleukin 4 (IL-4). TZD significantly reduced TGF-β1-induced myofibroblast and fibrotic gene and protein expression preferentially in EoE, but not normal esophageal fibroblasts. In esophageal fibroblasts, TGF-β1 increased phosphorylated Smad2/3 and p38, but TZDs preferentially inhibited p38 phosphorylation, suggesting signaling pathway-specific effects. The TZDs were more potent than budesonide at decreasing collagen-1α1 expression.
DISCUSSION:
The TZDs preferentially exert antifibrotic effects in TGF-β1-activated EoE fibroblasts and provide a preclinical foundation for further investigation of the potential of the TZDs in EoE pathologic remodeling.
INTRODUCTION
Eosinophilic esophagitis (EoE) is a chronic, T-helper 2 (Th2), antigen-driven eosinophilic disease of the esophagus in children and adults (1–5). Pathologic tissue remodeling results in esophageal rigidity and fibrostenosis that manifest clinically as dysphagia and food impactions (6–9). Unbridled or suboptimally controlled EoE-associated inflammation can lead to persistent or recurrent esophageal strictures (10–13). In its earlier stages, esophageal inflammation and remodeling are coupled in EoE. However, over time, uncontrolled fibrosis and rigidity can dissociate from inflammation and demonstrate pharmacotherapeutic resistance (14,15). The ensuing diminished esophageal distensibility, rather than the degree of esophageal eosinophilia or mucosal inflammation, is a predictor for EoE-related food impaction in adult EoE (16).
Submucosal fibrosis is a prominent histologic feature of EoE and believed to represent a histologic marker of esophageal dysfunction (17,18). Medical and elimination diet therapies can reverse esophageal subepithelial fibrosis in pediatric EoE and improve esophageal distensibility in adults (19–21). Although most children and many adults respond to standard EoE therapy, disease response is not universal and is often neither absolute nor sustained (22–24). Subgroups of patients, such as those with a narrowed esophagus at diagnosis, are often resistant to standard EoE anti-inflammatory therapies, but these patients are the most clinically in need of therapeutic interventions (19,20,25,26). Inflammation-independent esophageal remodeling is believed to occur, especially in long-standing disease, given the observations that a physically rigid extracellular environment induces mechanotransduction and alters esophageal smooth muscle and fibroblast hypertrophy and gene expression (27,28). Furthermore, persistent activation of fibroblasts can be observed even after resolution of inflammation (27). These issues underscore the significant unmet need for novel antifibrotic therapies in EoE.
The thiazolidinediones (TZD), such as the FDA-approved antidiabetic drugs rosiglitazone and pioglitazone, function as agonists for the ligand-activated nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ). PPAR-γ is a key regulator of lipid and glucose metabolism expressed in multiple cell types that also regulates antifibrotic, proadipogenic, and anti-inflammatory responses (29–32). For example, rosiglitazone monotherapy is effective in the treatment of mild-to-moderately active ulcerative colitis (33,34). Pioglitazone, a more clinically favorable TZD, improves liver fibrosis in adults with nonalcoholic steatohepatitis (NASH), achieving a resolution of NASH in up to 51% of patients (35–37). Importantly, pioglitazone is reportedly well tolerated and without major drug-related adverse events (35,38).
Given the natural trajectory of EoE toward pathologic remodeling with fibrosis, we hypothesized that the TZDs might have therapeutic activity in the esophagus to decrease fibrotic gene and protein expression in esophageal fibroblasts. Herein, we document the differential expression of PPAR-γ in EoE vs normal esophagi and the ability of the TZDs to reduce transforming growth factor (TGF)-β1-induced fibrotic and myofibroblast gene and protein expression preferentially in the EoE-derived esophageal fibroblasts, setting a foundation for further evaluation of their potential utility as new therapeutic agents to treat the fibrotic esophagus in EoE.
METHODS
Reagents, human esophageal tissues, and primary human esophageal fibroblast isolation
TGF-β1 (5 ng/mL; R&D Systems, Minneapolis, MN), rosiglitazone (20 μM; Abcam, Boston, MA), pioglitazone (20 μM; Sigma-Aldrich, St. Louis, MO), budesonide (0.1 μM; St. Louis, MO), and IL-4 (10 ng/mL; R&D Systems, Minneapolis, MN) were used in cell culture experiments. Inactive EoE, defined as less than 15 eosinophils per high-power field (hpf), was achieved with EoE-directed therapy of topical fluticasone or budesonide, proton pump inhibitor, and/or elimination diet. Active EoE was defined as greater than or equal to 15 eosinophils per hpf. Human organ transplant donor esophagi served as normal controls and were provided by the National Disease Research Interchange and the Arkansas Regional Organ Recovery Agency Control. All histopathology slides, prepared from esophageal tissues from patients with EoE and normal non-EoE controls (Table 1), were reviewed separately by an internal pathologist who was blinded to therapy. All experiments were conducted with the approval from the UCSD institutional review board (IRB). Patients with EoE and their parents were assented/consented for data and biopsy collection per IRB protocol. Primary human esophageal fibroblasts (FBL) were isolated from patients with EoE and non-EoE controls (Table 2), as previously described (39).
Table 1.
Characteristics of archived slides of patients with EoE and normal non-EoE controls used for immunohistochemical staining

Table 2.
Clinical characteristics of normal non-EoE controls and patients with EoE used for experiments

Cell culture and stimulation
Human esophageal fibroblast cells were matched as closely as possible for sex, race, and passage. At subconfluence, the cells were serum starved overnight and treated with a TZD, budesonide, their combination, or vehicle for 2–3 hours, followed by incubation with TGF-β1 or vehicle.
Preparation of total RNA and cDNA and quantitative PCR
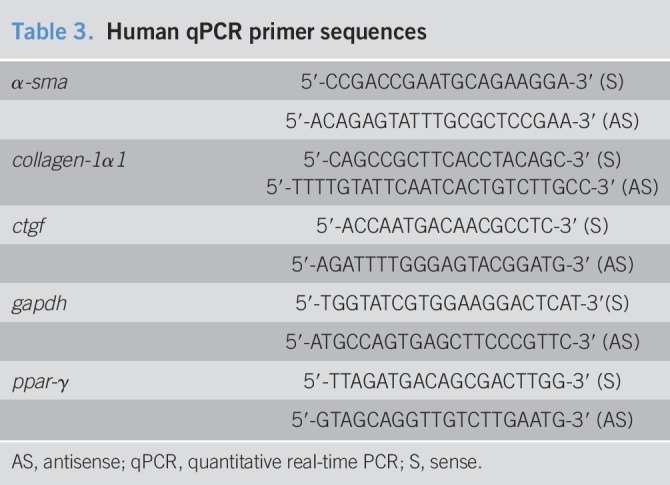
Total RNA from fibroblast cultures was extracted using RNA STAT-60 (Tel-Test, Friendswood, TX) protocol, and oligo(dT)-primed cDNA was synthesized, as previously described using the Qiagen real time-PCR kit and manufacturer's instructions (40). Quantitative RT PCR primers are listed in Table 3; quantitative real-time PCR was carried out as previously described using the appropriate gene-specific primers, and relative gene expression was calculated using the 2−ΔΔCt method, with glyceraldehyde 3-phosphate dehydrogenase (gapdh) as the housekeeping gene (40).
Table 3.
Human qPCR primer sequences
Western immunoblot analysis
Adherent cells were washed with ice-cold phosphate-buffered saline containing 1 mM sodium orthovanadate (Na3VO4) and lysed in ice-cold RIPA lysis buffer freshly prepared and supplemented with 1 mM Na3VO4, complete protease inhibitor cocktail, and 2 mM phenylmethylsulfonyl fluoride. Whole cell lysates were centrifuged at 14,000g for 15 minutes at 4 °C. Equivalent amounts of total proteins were loaded into each well and electrophoresed on NuPAGE 4–12% Bis-Tris gels (Life Technologies, Grand Island, NY), transferred to polyvinylidene difluoride membranes, blocked with 5% bovine serum albumin, incubated with primary antibodies overnight (1:1,000) at 4 °C, detected using species-appropriate horseradish peroxidase-conjugated secondary antibodies, and quantified as previously described (40).
Immunostaining and histologic assessment
Tissue sections (5 μm) were deparaffinized and hydrated before immunostaining, as previously described (41). After antigen retrieval, the slides were incubated with anti-PPAR-γ (1:800; AbCam, Cambridge, MA) or isotype control. The samples were processed for immunohistochemistry using the appropriate species-specific secondary antibodies, as previously described (40). All images were analyzed under identical light setting including magnification, gain, camera position, and background illumination.
Statistical analysis
Using GraphPad PRISM v8.2.0 (GraphPad Software, San Diego, CA), the analysis of variance and t-tests were performed to assess statistical significance, defined as P-values < 0.05.
Ethics approval
Approved by the Institutional Review Board (IRB).
RESULTS
Rosiglitazone inhibits profibrotic gene expression dose-dependently in EoE-derived esophageal fibroblasts
To test the effects of TZDs on profibrotic gene expression in human esophageal fibroblasts, we treated cells with TGF-β1 in the absence or presence of rosiglitazone. TGF-β1 induced mRNA expression of α-smooth muscle actin (α-sma) in primary esophageal fibroblasts derived from healthy donors and patients with EoE (Figure 1a). At the concentrations tested at up to 20 μM, as guided by our dose-response studies in EoE cells, rosiglitazone exerted insignificant activities on basal and TGF-β1-induced α-sma gene expression in esophageal fibroblasts derived from healthy donors (Figure 1a). By contrast, rosiglitazone significantly inhibited TGF-β1-induced mRNA expression of α-sma, collagen-1α1, and connective tissue growth factor (ctgf) in EoE esophageal fibroblasts (Figure 1a–d). A dose-dependent effect of rosiglitazone was observed (Figure 1e,f). A lack of reduction in the housekeeper gene expression and on responses in normal fibroblasts demonstrated that rosiglitazone effects were not due to toxicity at the concentrations used herein.
Figure 1.

EoE-derived primary esophageal fibroblasts respond selectively to rosiglitazone: mRNA analysis. Human primary esophageal fibroblasts (FBL) derived from normal donors or patients with EoE were stimulated with rosiglitazone (20 μM for a–d; 10 μM or 20 μM for e and f) or vehicle (dimethylsulfoxide) for 3 hours and then treated with TGF-β1 (TGF, 5 ng/mL) for 24 hours in the presence of vehicle or rosiglitazone. mRNA expression was determined by quantitative real-time PCR and expressed as mean ± SD. (a) Representative of 3 normal controls and 4 separate patients with EoE. (b–d) Representative of 4 separate patients with EoE. (e and f) Representative of 2 separate experiments in fibroblasts from 1 patient with EoE. EoE, eosinophilic esophagitis; Med, medium; ns, not significant; rosi, rosiglitazone. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
TZDs inhibit profibrotic protein expression in EoE-derived esophageal fibroblasts
TGF-β1 induced protein expression of collagen-1α1 and α-sma in human primary esophageal fibroblasts (Figures 2 and 3). In esophageal fibroblasts isolated from patients with EoE, rosiglitazone reduced basal collagen-1 (Figure 2a,c; n = 8; P = 0.0496) and inhibited TGF-β1-induced collagen-1 protein level (Figure 2c–e; n = 8; P = 0.0192). By contrast, rosiglitazone did not inhibit basal protein expression of α-sma but did exhibit a trend toward reducing TGF-β1-driven α-sma protein in EoE-derived esophageal fibroblasts (Figure 2a,g,h; N = 7). This observation was validated by a significant reduction in biological replicates performed in a representative patient with EoE (Figure 2i; P = 0.0021). Consistent with its effects on mRNA expression, rosiglitazone did not inhibit TGF-β1-induced protein expression of α-sma and collagen-1α1 in normal esophageal fibroblasts (Figure 2a,b,f; N = 3).
Figure 2.

EoE-derived primary esophageal fibroblasts selectively respond to rosiglitazone: protein analysis. Human primary esophageal fibroblasts from normal donors or patients with EoE were stimulated with vehicle (dimethylsulfoxide) or rosiglitazone (20 μM) for 3 hours and then treated with TGF-β1 (5 ng/mL) for 24 hours in the presence or absence of rosiglitazone. A representative blot is shown (a). Protein expression of α-sma and collagen-1 was analyzed and quantified in 3 normal controls and in 7–8 patients with EoE (b–i). (e and i) Panels represent quantification of biological replicates in a patient with EoE. EoE, eosinophilic esophagitis; Med, medium; ns, not significant; rosi, rosiglitazone. *P < 0.05; **P < 0.01; ****P < 0.0001.
Figure 3.

EoE-derived primary esophageal fibroblasts respond to the TZDs. Human primary esophageal fibroblasts from a representative patient with EoE were stimulated with vehicle (dimethylsulfoxide), rosiglitazone (20 μM), or pioglitazone (20 μM) for 3 hours and then treated with TGF-β1 (5 ng/mL) for 24 hours in the presence or absence of vehicle, rosiglitazone, or pioglitazone (repeated in 3 separate experiments [a]). (b–d) Panels represent quantification of 3 separate experiments in cells derived from a representative patient with EoE. EoE, eosinophilic esophagitis; Med, medium; ns, not significant; pio, pioglitazone; rosi, rosiglitazone; TZD, thiazolidinediones. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we evaluated the effects of pioglitazone, a TZD with a more favorable clinical profile than rosiglitazone (30), in esophageal fibroblasts isolated from a representative patient with EoE who responded to rosiglitazone. Pioglitazone also significantly decreased TGF-β1-mediated collagen-1α1 and α-sma induction in EoE-derived esophageal fibroblasts (Figure 3a–c). However, the effects of rosiglitazone and pioglitazone on basal and TGF-β1-treated protein levels of fibronectin at the concentrations used herein were negligible (Figure 3d). Taken together, these data suggested that EoE might induce a reprogramming of esophageal fibroblasts, with one of the downstream effects resulting in preferential increased responsiveness of EoE-derived cells to treatment with the TZDs, rosiglitazone and pioglitazone. Furthermore, the protein-specific effects of the TZDs suggested pathway-specific modulation of TGF-β1 signaling.
Rosiglitazone inhibits TGF-β1-induced phosphorylation of p38, but not Smad2/3, in EoE-derived esophageal fibroblasts
To better understand the distinct pathways affected by TZDs in esophageal fibroblasts, we evaluated the effects on p38 and pSmad2/3. As expected, TGF-β1 induced phosphorylation of both p38 and Smad2/3 in EoE-derived fibroblasts (Figure 4a). Although rosiglitazone significantly inhibited p38 phosphorylation induced by TGF-β1, Smad2/3 phosphorylation was not affected (Figure 4a–c). Rosiglitazone inhibited TGF-β1-induced p38 phosphorylation dose-dependently (Figure 4d,e). Total p38 levels were unchanged basally, on stimulation with TGF-β1, or in the presence of rosiglitazone (data not shown). These results indicate that rosiglitazone may function, in part, via modulation of the noncanonical TGF-β1 signaling pathway leading to decreased p38 phosphorylation.
Figure 4.

Rosiglitazone inhibits p38 phosphorylation but has minimal effect on Smad2/3 phosphorylation in EoE-derived primary esophageal fibroblasts. Human primary esophageal fibroblasts from patients with EoE were treated with vehicle (dimethylsulfoxide) and rosiglitazone (20 μM) for 3 hours and then stimulated with TGF-β1 (5 ng/mL) for 30 minutes in the presence of vehicle or rosiglitazone. A representative blot is shown (a). (b and c) Panels represent quantification of blots of phospho-p38 and phospho-Smad2/3 in 4–5 different patients with EoE. A representative blot of a dose-response study using 2 or 20 μM rosiglitazone (d). (e) Panels represent quantification of blots of the dose-response study on phospho-p38 from 2 patients with EoE. EoE, eosinophilic esophagitis; Med, medium; R2, rosiglitazone (2 μM); R20, rosiglitazone (20 μM); TGF, TGF-β1. ns, not significant; *P < 0.05; **P < 0.01.
Comparison of rosiglitazone and budesonide in EoE fibroblasts
We compared the effects of rosiglitazone with budesonide, a commonly used topical EoE therapy. In esophageal fibroblasts from 2 separate patients with EoE, budesonide had no effects on TGFβ1-induced protein expression of collagen-1α1 (Figure 5). By contrast, rosiglitazone significantly reduced the expression of TGF-β1-induced collagen-1α1 protein level in EoE-derived fibroblasts. No synergistic effects were observed at the concentrations of budesonide and rosiglitazone used herein.
Figure 5.

Comparison of budesonide and rosiglitazone in EoE primary esophageal fibroblasts. EoE-derived esophageal fibroblasts from 2 patients were treated with vehicle, budesonide (0.1 μM), or rosiglitazone (20 μM) for 3 hours and then stimulated with TGF-β1 in the absence or presence of vehicle, budesonide, or rosiglitazone. Representative blot (a). (b) Quantification of blots from experiments from 2 separate patients with EoE. EoE, eosinophilic esophagitis; Med, medium; B, budesonide; R, rosiglitazone. ns, not significant; *P < 0.05.
PPAR-γ is enriched in EoE-derived esophageal biopsies and fibroblasts
Given the observation that EoE esophageal fibroblasts had a preferential response to TZD-mediated downregulation of TGF-β1 signaling as compared to normal fibroblasts, we evaluated the expression of PPAR-γ in normal and EoE esophageal biopsies and fibroblasts. PPAR-γ mRNA expression was higher in the EoE-derived fibroblasts as compared to normal (Figure 6a). We hypothesized that Th2 cytokines such as interleukin (IL)-4 might modulate the expression of PPAR-γ expression. Esophageal fibroblasts upregulated PPAR-γ mRNA expression on stimulation by IL-4 (Figure 6a).
Figure 6.

PPAR-γ expression in human esophageal fibroblasts (FBL) and human esophageal biopsies. PPAR-γ mRNA expression was quantified in normal (N = 2) and EoE-derived esophageal fibroblasts (N = 2) basally (left) and after IL-4 (10 ng/mL) stimulation for 6 hours (right) (a). Representative images of EoE (fibrosis score 1, left panel; fibrosis score 3, middle panel) and normal control (right panel) biopsies stained for PPAR-γ protein expression (b). Insets show detail (400×, original magnification) and isotype control (200×). Quantification of PPAR-γ-positive staining in active EoE (N = 11), inactive EoE (N = 5), and normal controls (N = 5) (c). EoE, eosinophilic esophagitis; IL-4, interleukin-4; ns, not significant; PPAR-γ, peroxisome proliferator-activated receptor-γ. *P < 0.05; **P < 0.01; ****P < 0.0001.
When assessing esophageal biopsies, we found that active EoE esophageal biopsies, but not normal esophageal biopsies, had PPAR-γ expression in the epithelium and subepithelial lamina propria (Figure 6b). Indeed, active EoE esophageal biopsies had higher expression of PPAR-γ than inactive EoE esophagi and normal healthy esophagi (N = 11 active EoE, 5 inactive EoE, and 5 normal biopsies, respectively) (Figure 6c). Taken together, these results suggest that the Th2 inflammatory milieu in EoE upregulates PPAR-γ expression, thereby rendering EoE cells more responsive to PPAR-γ agonists.
DISCUSSION
EoE is a chronic disease with progressive esophageal fibrostenosis when left untreated or when the disease is unresponsive to currently available therapies. Although inflammation is considered as the trigger for fibrosis, the inflammation-fibrosis connection may become uncoupled (42). Over time, florid inflammation can diminish while fibrosis continues to progress because of pathologic and uninhibited tissue remodeling, resulting in further tissue dysfunction. Standard treatments such as topical corticosteroids and antigen elimination diets resolve inflammation but have variable effects on remodeling, likely depending on the patient phenotype, genotype, and disease stage (23,43). Commonly used medications such as topical corticosteroids can reduce the onset of strictures and food impactions with chronic use in adults and children, especially when the disease duration is shorter. However, many patients, especially adults, are diagnosed after long-standing disease and thus can have uncoupling of inflammation and rigidity (14,15), thereby rendering their EoE suboptimally or poorly responsive to standard EoE therapy. Although topical corticosteroids or food elimination can reverse histologic fibrosis and epithelial remodeling, as well as esophageal rigidity in subsets of children and adults, the resolution of eosinophilia can be incomplete even in clinical responders, leaving nonresponders and even some corticosteroid-responsive patients vulnerable to progression toward esophageal narrowing (9,26,44). In addition, the fibrostenotic esophagus is often resistant to medical treatments (26). This situation leaves a pressing need for novel isolated or adjuvant antifibrotic therapies.
The TZDs activate PPAR-γ to promote anti-inflammatory and antifibrotic effects. We observed upregulation of PPAR-γ expression at the transcript and protein levels in EoE esophageal fibroblasts and biopsies, respectively, potentially suggesting a protective compensatory mechanism to protect the esophagus from further pathologic remodeling. This likely reflects the wound healing and protective nature of fibroblasts as they attempt to downregulate pathologic remodeling and potentially promote tissue softening. Importantly, this may present an opportunity to use an endogenously occurring wound healing mechanism via PPAR-γ agonism. The EoE epithelium and subepithelium had increased expression of PPAR-γ in the severe active state, with little to no detection of PPAR-γ expression in the normal state. Similarly, consistent with the concept of EoE being akin to an asthmatic type pathogenesis in the esophagus (9,45), PPAR-γ expression is elevated in the airway epithelium of asthma and allergic airway disease (46,47). PPAR-γ deficiency in airway epithelial cells exacerbated murine allergic airway disease (47), and PPAR-γ activation via agonist nebulization was protective, resulting in reduced airway hyperreactivity, allergic inflammation, and eosinophil activation (48). Furthermore, PPAR-γ agonism decreased airway collagen deposition and TGF-β expression in a murine asthma model (49). Our data suggest that EoE patients with active disease may have higher PPAR-γ expression and thus would be potential clinical candidates for additional therapies such as the TZDs. As such, TZD compounds may be useful as therapeutic options to preferentially target diseased tissues in EoE while potentially sparing healthy tissues.
PPAR-γ-positive CD4+ T cells were recently identified in EoE tissues (50). PPAR-γ has been described to promote Th2 immune responses (51,52). PPAR-γ was reported to promote Th2 cells that express IL-9 (53), a cytokine thought to be pathogenic in EoE (54). However, PPAR-γ inhibited T cell activation and CD4+ T cell effector function, including inhibition of IL-4 production (55–58). Furthermore, PPAR-γ positively regulated the tissue accumulation, phenotype and function of regulatory T cells to suppress tissue inflammation (59,60). Pioglitazone administration resulted in an enrichment of tissue regulatory T cells (59). The expression of PPAR-γ in T cells was protective against experimental colitis (61,62). PPAR-γ suppressed eosinophilic activation (48) and mast cell maturation and activation (63). Further investigation of the effects of the TZDs on EoE-derived immune cells will be required in future studies, but it is likely that activation of the PPAR-γ pathway could provide anti-inflammatory and antifibrotic benefits in patients with EoE.
The TZDs have been demonstrated to improve ulcerative colitis and NASH (33–37,64,65). Early progress in the development of TZDs for ulcerative colitis, however, was hampered by the potential cardiotoxicity profile of rosiglitazone, although the myocardial infarction risk was subsequently disproven (66). In contrast to rosiglitazone, pioglitazone has been shown to have a cardioprotective effect (66–69). In addition, given the inherent antifibrotic and proadipogenic effects of the TZDs, there is a concern for an effect on bone mineral density and the potential risk for fractures (70–75). However, recent placebo-controlled long-term studies evaluating pioglitazone for NASH reported that the drug was well tolerated, without major drug-related adverse events (35,38). It is of note that most of these studies used a higher dose of pioglitazone, ranging from 30 mg to 45 mg daily. A lower dose of pioglitazone of 7.5 mg orally daily has been proposed (76,77). Previous studies in ulcerative colitis used up to 8 mg per day with favorable clinical activities (33,34). In addition to dose reduction, it is possible that topical delivery of the TZDs might achieve directed beneficial therapeutic effects while sparing some of the potential adverse drug effects when given systemically. For example, topical delivery of rosiglitazone as an enema improved ulcerative colitis (34). In addition, a locally active, novel topical PPAR-γ agonist AS002 was recently developed and was demonstrated to induce PPAR-γ in human colonic biopsies stimulated ex vivo. In mice, AS002 prevented and reversed colitis in vivo in a murine model of colitis (78). The combination of reduced dosing and topical esophageal delivery of the TZDs might allow for directed therapy to the esophagus while minimizing potential systemic adverse effects.
The strengths of our study center on the elucidation of the effect of 2 different TZDs on TGF-β1 signaling and TGF-β1-induced mRNA and protein expression of markers of fibrosis and myofibroblast differentiation in primary human esophageal fibroblasts from patients with EoE and from normal controls. We sought to evaluate and use the lowest effective concentrations of the TZDs that would consistently inhibit diseased cells while potentially sparing normal cells. In other studies, much higher concentrations of rosiglitazone, up to 100 μM, have been described to exert antifibrotic effects in cultured normal cells of the gastrointestinal tract (79). At the concentrations used herein, up to 20 μM, as determined by dose-response studies in EoE cells, although normal esophageal fibroblasts were relatively resistant to the effects of the TZDs, fibroblasts derived from EoE biopsies were consistently sensitive to the TZDs at the mRNA and protein expression levels. The effect on p38 phosphorylation could be observed potently even at lower concentrations, e.g., 2 μM of rosiglitazone. The TZDs antagonized the noncanonical TGF-β1 pathway leading to diminished phosphorylated p38 levels while sparing the canonical TGF-β1-mediated Smad2/3 phosphorylation pathway. This observation for p38 phosphorylation is in congruence with previous reports that described the effects of the PPAR-γ ligands in fibroblasts from other human tissues (80,81). In addition, rosiglitazone had distinct effects on EoE esophageal fibroblasts as compared to budesonide. As such, it is possible that the TZD class of drugs could potentially function in vivo as adjuvant antifibrotic therapies in patients who have advanced, strictured, or steroid-resistant EoE.
Limitations of the current study include limited availability of normal controls and a finite number of patients with EoE from whom esophageal biopsies could be obtained for immunohistochemical analysis and for isolation and culturing of primary esophageal fibroblasts. Although EoE is rising both in incidence and prevalence, it remains a relatively rare disease. In addition, we directed our analysis of the effects of the TZDs on known mRNA, protein, and intracellular signaling targets. Future experiments expanding to unexplored transcriptomes and previously unidentified protein and signaling targets would provide a more comprehensive understanding of the effects of the TZDs, as would experiments evaluating drug effects on other cell types isolated and cultured from esophageal biopsies. Future studies systematically evaluating the effects of the TZDs on cells derived from different EoE phenotypes, e.g., inflammatory, fibrostenotic, or mixed disease, would provide a better understanding of the therapeutic utility of the TZDs.
In conclusion, our experiments suggest the potential application of TZDs in patients with EoE with concurrent fibrosis and/or those who are resistant to topical corticosteroid therapy. Targeting other profibrotic pathways such as lysyl oxidase and TNF-α in fibrostenotic EoE is also under development (82). Because the TZDs are already approved medications in other diseases, it may be of utility to consider the repurposing of these drugs to treat complicated EoE. Our data are compelling as a preclinical foundation for further translational and clinical investigation of the potential utility of TZDs in EoE.
CONFLICTS OF INTEREST
Guarantor of the article: Seema S. Aceves, MD, PhD.
Specific author contributions: Q.M.N. proposed the concept, designed the study and experiments, performed the experiments, analyzed the data, and wrote the manuscript. L.H. and L.D. assisted with the experiments. R.N. reviewed the histopathology. R.D. and R.K. procured the specimens and reviewed the manuscript. F.J.M. contributed to the overall study design and reviewed the manuscript. S.S.A. designed the study and experiments and supervised the project, tissue/cell procurement, and data analysis. S.S.A. oversaw the consenting process for all subjects and wrote the manuscript.
Financial support: NIH/NIAID AI 092135 (SSA), AI 135034 (SSA), NIH/NCATS CTSA 5 UL1 TR001114 (QMN), 5KL2 TR001112 (QMN), L30 TR002507 (QMN), a Scripps Clinic Medical Group Research & Education Award (QMN), and a CEGIR award (QMN). CEGIR (Consortium of Eosinophilic Gastrointestinal Disease Researchers; U54 AI117804) is part of the Rare Disease Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR), NCATS, and is funded through collaboration between NIAID, NIDDK, and NCATS. CEGIR is also supported by patient advocacy groups including APFED, CURED, and EFC.
Potential competing interests: S.S.A. and R.D. are coinventors of oral viscous budesonide which is patented by UCSD and licensed by Shire/Takeda pharmaceuticals. All remaining authors have no conflicts of interest to disclose.
Patient consent: Obtained.
Study Highlights.
WHAT IS KNOWN
✓ Pathologic tissue remodeling in EoE results in esophageal fibrostenotic disease.
✓ The fibrostenotic EoE esophagus is often resistant to medical and elimination diet therapies.
✓ There is an unmet need for novel antifibrotic therapies for EoE pathologic remodeling.
WHAT IS NEW HERE
✓ EoE esophageal biopsies and esophageal fibroblasts express higher levels of the TZD receptor PPAR-γ than normal controls.
✓ The TZDs preferentially exert antifibrotic and signaling effects in TGF-β1-activated EoE fibroblasts.
TRANSLATIONAL IMPACT
✓ Data suggest the potential application of TZDs in patients with EoE with concurrent fibrosis and/or those who are resistant to topical corticosteroid therapy.
ACKNOWLEDGMENTS
We thank Loan Duong and Renee Rawson for excellent technical assistance with cell culture.
References
- 1.Furuta GT, Katzka DA. Eosinophilic esophagitis. N Engl J Med 2015;373:1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Davis BP, Rothenberg ME. Mechanisms of disease of eosinophilic esophagitis. Annu Rev Pathol 2016;11:365–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothenberg ME. Molecular, genetic, and cellular bases for treating eosinophilic esophagitis. Gastroenterology 2015;148:1143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill DA, Spergel JM. The immunologic mechanisms of eosinophilic esophagitis. Curr Allergy Asthma Rep 2016;16:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O'Shea KM, Aceves SS, Dellon ES, et al. Pathophysiology of eosinophilic esophagitis. Gastroenterology 2018;154:333–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng E, Souza RF, Spechler SJ. Tissue remodeling in eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol 2012;303:G1175–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aceves SS. Remodeling and fibrosis in chronic eosinophil inflammation. Dig Dis 2014;32:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirano I, Aceves SS. Clinical implications and pathogenesis of esophageal remodeling in eosinophilic esophagitis. Gastroenterol Clin North Am 2014;43:297–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nhu QM, Aceves SS. Tissue remodeling in chronic eosinophilic esophageal inflammation: Parallels in asthma and therapeutic perspectives. Front Med (Lausanne) 2017;4:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mavi P, Rajavelu P, Rayapudi M, et al. Esophageal functional impairments in experimental eosinophilic esophagitis. Am J Physiol Gastrointest Liver Physiol 2012;302:G1347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dellon ES, Kim HP, Sperry SL, et al. A phenotypic analysis shows that eosinophilic esophagitis is a progressive fibrostenotic disease. Gastrointest Endosc 2014;79:577–85.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singla MB, Chehade M, Brizuela D, et al. Early comparison of inflammatory vs. Fibrostenotic phenotype in eosinophilic esophagitis in a multicenter longitudinal study. Clin Transl Gastroenterol 2015;6:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warners MJ, Oude Nijhuis RAB, de Wijkerslooth LRH, et al. The natural course of eosinophilic esophagitis and long-term consequences of undiagnosed disease in a large cohort. Am J Gastroenterol 2018;113:836–44. [DOI] [PubMed] [Google Scholar]
- 14.Pentiuk S, Putnam PE, Collins MH, et al. Dissociation between symptoms and histological severity in pediatric eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2009;48:152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Safroneeva E, Straumann A, Coslovsky M, et al. Symptoms have modest accuracy in detecting endoscopic and histologic remission in adults with eosinophilic esophagitis. Gastroenterology 2016;150:581–90.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nicodeme F, Hirano I, Chen J, et al. Esophageal distensibility as a measure of disease severity in patients with eosinophilic esophagitis. Clin Gastroenterol Hepatol 2013;11:1101–7.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chehade M, Sampson HA, Morotti RA, et al. Esophageal subepithelial fibrosis in children with eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2007;45:319–28. [DOI] [PubMed] [Google Scholar]
- 18.Li-Kim-Moy JP, Tobias V, Day AS, et al. Esophageal subepithelial fibrosis and hyalinization are features of eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2011;52:147–53. [DOI] [PubMed] [Google Scholar]
- 19.Lieberman JA, Morotti RA, Konstantinou GN, et al. Dietary therapy can reverse esophageal subepithelial fibrosis in patients with eosinophilic esophagitis: A historical cohort. Allergy 2012;67:1299–307. [DOI] [PubMed] [Google Scholar]
- 20.Carlson DA, Hirano I, Zalewski A, et al. Improvement in esophageal distensibility in response to medical and diet therapy in eosinophilic esophagitis. Clin Transl Gastroenterol 2017;8:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Runge TM, Eluri S, Woosley JT, et al. Control of inflammation decreases the need for subsequent esophageal dilation in patients with eosinophilic esophagitis. Dis Esophagus 2017;30:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straumann A, Schoepfer AM. Therapeutic concepts in adult and paediatric eosinophilic oesophagitis. Nat Rev Gastroenterol Hepatol 2012;9:697–704. [DOI] [PubMed] [Google Scholar]
- 23.Nhu QM, Aceves SS. Medical and dietary management of eosinophilic esophagitis. Ann Allergy Asthma Immunol 2018;121:156–61. [DOI] [PubMed] [Google Scholar]
- 24.Straumann A, Katzka DA. Diagnosis and treatment of eosinophilic esophagitis. Gastroenterology 2018;154:346–59. [DOI] [PubMed] [Google Scholar]
- 25.Eluri S, Runge TM, Cotton CC, et al. The extremely narrow-caliber esophagus is a treatment-resistant subphenotype of eosinophilic esophagitis. Gastrointest Endosc 2016;83:1142–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dellon ES. Management of refractory eosinophilic oesophagitis. Nat Rev Gastroenterol Hepatol 2017;14:479–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muir AB, Dods K, Henry SJ, et al. Eosinophilic esophagitis-associated chemical and mechanical microenvironment shapes esophageal fibroblast behavior. J Pediatr Gastroenterol Nutr 2016;63:200–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tkachenko E, Rawson R, La E, et al. Rigid substrate induces esophageal smooth muscle hypertrophy and eosinophilic esophagitis fibrotic gene expression. J Allergy Clin Immunol 2016;137:1270–2.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glass CK, Saijo K. Nuclear receptor transrepression pathways that regulate inflammation in macrophages and T cells. Nat Rev Immunol 2010;10:365–76. [DOI] [PubMed] [Google Scholar]
- 30.Ahmadian M, Suh JM, Hah N, et al. PPARgamma signaling and metabolism: The good, the bad and the future. Nat Med 2013;19:557–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Speca S, Dubuquoy L, Desreumaux P. Peroxisome proliferator-activated receptor gamma in the colon: Inflammation and innate antimicrobial immunity. J Clin Gastroenterol 2014;48(Suppl 1):S23–7. [DOI] [PubMed] [Google Scholar]
- 32.Derosa G, Sahebkar A, Maffioli P. The role of various peroxisome proliferator-activated receptors and their ligands in clinical practice. J Cell Physiol 2018;233:153–61. [DOI] [PubMed] [Google Scholar]
- 33.Lewis JD, Lichtenstein GR, Deren JJ, et al. Rosiglitazone for active ulcerative colitis: A randomized placebo-controlled trial. Gastroenterology 2008;134:688–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pedersen G, Brynskov J. Topical rosiglitazone treatment improves ulcerative colitis by restoring peroxisome proliferator-activated receptor-gamma activity. Am J Gastroenterol 2010;105:1595–603. [DOI] [PubMed] [Google Scholar]
- 35.Cusi K, Orsak B, Bril F, et al. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: A randomized trial. Ann Intern Med 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- 36.Musso G, Cassader M, Paschetta E, et al. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: A meta-analysis. JAMA Intern Med 2017;177:633–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bril F, Kalavalapalli S, Clark VC, et al. Response to pioglitazone in patients with nonalcoholic steatohepatitis with vs without type 2 diabetes. Clin Gastroenterol Hepatol 2018;16:558–66.e2. [DOI] [PubMed] [Google Scholar]
- 38.Sanyal AJ, Chalasani N, Kowdley KV, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beppu LY, Anilkumar AA, Newbury RO, et al. TGF-beta1-induced phospholamban expression alters esophageal smooth muscle cell contraction in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2014;134:1100–7.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rawson R, Yang T, Newbury RO, et al. TGF-beta1-induced PAI-1 contributes to a profibrotic network in patients with eosinophilic esophagitis. J Allergy Clin Immunol 2016;138:791–800.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aceves SS, Newbury RO, Dohil R, et al. Esophageal remodeling in pediatric eosinophilic esophagitis. J Allergy Clin Immunol 2007;119:206–12. [DOI] [PubMed] [Google Scholar]
- 42.Stramer BM, Mori R, Martin P. The inflammation-fibrosis link? A Jekyll and Hyde role for blood cells during wound repair. J Invest Dermatol 2007;127:1009–17. [DOI] [PubMed] [Google Scholar]
- 43.Shoda T, Wen T, Aceves SS, et al. Eosinophilic oesophagitis endotype classification by molecular, clinical, and histopathological analyses: A cross-sectional study. Lancet Gastroenterol Hepatol 2018;3:477–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rajan J, Newbury RO, Anilkumar A, et al. Long-term assessment of esophageal remodeling in patients with pediatric eosinophilic esophagitis treated with topical corticosteroids. J Allergy Clin Immunol 2016;137:147–56.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Virchow JC. Eosinophilic esophagitis: Asthma of the esophagus? Dig Dis 2014;32:54–60. [DOI] [PubMed] [Google Scholar]
- 46.Benayoun L, Letuve S, Druilhe A, et al. Regulation of peroxisome proliferator-activated receptor gamma expression in human asthmatic airways: Relationship with proliferation, apoptosis, and airway remodeling. Am J Respir Crit Care Med 2001;164:1487–94. [DOI] [PubMed] [Google Scholar]
- 47.Lakshmi SP, Reddy AT, Banno A, et al. Airway epithelial cell peroxisome proliferator-activated receptor gamma regulates inflammation and mucin expression in allergic airway disease. J Immunol 2018;201:1775–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woerly G, Honda K, Loyens M, et al. Peroxisome proliferator-activated receptors alpha and gamma down-regulate allergic inflammation and eosinophil activation. J Exp Med 2003;198:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Honda K, Marquillies P, Capron M, et al. Peroxisome proliferator-activated receptor gamma is expressed in airways and inhibits features of airway remodeling in a mouse asthma model. J Allergy Clin Immunol 2004;113:882–8. [DOI] [PubMed] [Google Scholar]
- 50.Wen T, Aronow BJ, Rochman Y, et al. Single-cell RNA sequencing identifies inflammatory tissue T cells in eosinophilic esophagitis. J Clin Invest 2019;129:2014–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen T, Tibbitt CA, Feng X, et al. PPAR-gamma promotes type 2 immune responses in allergy and nematode infection. Sci Immunol 2017;2:eaal5196. [DOI] [PubMed] [Google Scholar]
- 52.Nobs SP, Natali S, Pohlmeier L, et al. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J Exp Med 2017;214:3015–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Micosse C, von Meyenn L, Steck O, et al. Human "TH9" cells are a subpopulation of PPAR-gamma(+) TH2 cells. Sci Immunol 2019;4:eaat5943. [DOI] [PubMed] [Google Scholar]
- 54.Doshi A, Khamishon R, Rawson R, et al. Interleukin 9 alters epithelial barrier and E-cadherin in eosinophilic esophagitis. J Pediatr Gastroenterol Nutr 2019;68:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang XY, Wang LH, Chen T, et al. Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPARgamma) agonists. PPARgamma co-association with transcription factor NFAT. J Biol Chem 2000;275:4541–4. [DOI] [PubMed] [Google Scholar]
- 56.Clark RB, Bishop-Bailey D, Estrada-Hernandez T, et al. The nuclear receptor PPAR gamma and immunoregulation: PPAR gamma mediates inhibition of helper T cell responses. J Immunol 2000;164:1364–71. [DOI] [PubMed] [Google Scholar]
- 57.Chung SW, Kang BY, Kim TS. Inhibition of interleukin-4 production in CD4+ T cells by peroxisome proliferator-activated receptor-gamma (PPAR-gamma) ligands: Involvement of physical association between PPAR-gamma and the nuclear factor of activated T cells transcription factor. Mol Pharmacol 2003;64:1169–79. [DOI] [PubMed] [Google Scholar]
- 58.Park HJ, Kim DH, Choi JY, et al. PPARgamma negatively regulates T cell activation to prevent follicular helper T cells and germinal center formation. PLoS One 2014;9:e99127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cipolletta D, Feuerer M, Li A, et al. PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012;486:549–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cipolletta D, Cohen P, Spiegelman BM, et al. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: Age, diet, and PPARgamma effects. Proc Natl Acad Sci USA 2015;112:482–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guri AJ, Mohapatra SK, Horne WT, II, et al. The role of T cell PPAR gamma in mice with experimental inflammatory bowel disease. BMC Gastroenterol 2010;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hontecillas R, Bassaganya-Riera J. Peroxisome proliferator-activated receptor gamma is required for regulatory CD4+ T cell-mediated protection against colitis. J Immunol 2007;178:2940–9. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Y, Li X, Fang S, et al. Peroxisome proliferator-activated receptor gamma agonist suppresses mast cell maturation and induces apoptosis. Mol Med Rep 2017;16:1793–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lewis JD, Lichtenstein GR, Stein RB, et al. An open-label trial of the PPAR-gamma ligand rosiglitazone for active ulcerative colitis. Am J Gastroenterol 2001;96:3323–8. [DOI] [PubMed] [Google Scholar]
- 65.Bertin B, Dubuquoy L, Colombel JF, et al. PPAR-gamma in ulcerative colitis: A novel target for intervention. Curr Drug Targets 2013;14:1501–7. [DOI] [PubMed] [Google Scholar]
- 66.Rizos CV, Kei A, Elisaf MS. The current role of thiazolidinediones in diabetes management. Arch Toxicol 2016;90:1861–81. [DOI] [PubMed] [Google Scholar]
- 67.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive study (PROspective pioglitAzone clinical trial in macroVascular events): A randomised controlled trial. Lancet 2005;366:1279–89. [DOI] [PubMed] [Google Scholar]
- 68.Erdmann E, Dormandy JA, Charbonnel B, et al. The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction: Results from the PROactive (PROactive 05) study. J Am Coll Cardiol 2007;49:1772–80. [DOI] [PubMed] [Google Scholar]
- 69.Wilcox R, Kupfer S, Erdmann E, et al. Effects of pioglitazone on major adverse cardiovascular events in high-risk patients with type 2 diabetes: Results from PROspective pioglitAzone clinical trial in macro vascular events (PROactive 10). Am Heart J 2008;155:712–7. [DOI] [PubMed] [Google Scholar]
- 70.Dormandy J, Bhattacharya M, van Troostenburg de Bruyn AR, et al. Safety and tolerability of pioglitazone in high-risk patients with type 2 diabetes: An overview of data from PROactive. Drug Saf 2009;32:187–202. [DOI] [PubMed] [Google Scholar]
- 71.Derosa G. Efficacy and tolerability of pioglitazone in patients with type 2 diabetes mellitus: Comparison with other oral antihyperglycaemic agents. Drugs 2010;70:1945–61. [DOI] [PubMed] [Google Scholar]
- 72.Bray GA, Smith SR, Banerji MA, et al. Effect of pioglitazone on body composition and bone density in subjects with prediabetes in the ACT NOW trial. Diabetes Obes Metab 2013;15:931–7. [DOI] [PubMed] [Google Scholar]
- 73.Grey A, Bolland M, Fenwick S, et al. The skeletal effects of pioglitazone in type 2 diabetes or impaired glucose tolerance: A randomized controlled trial. Eur J Endocrinol 2014;170:255–62. [DOI] [PubMed] [Google Scholar]
- 74.Billington EO, Grey A, Bolland MJ. The effect of thiazolidinediones on bone mineral density and bone turnover: Systematic review and meta-analysis. Diabetologia 2015;58:2238–46. [DOI] [PubMed] [Google Scholar]
- 75.Pop LM, Lingvay I, Yuan Q, et al. Impact of pioglitazone on bone mineral density and bone marrow fat content. Osteoporos Int 2017;28:3261–9. [DOI] [PubMed] [Google Scholar]
- 76.Adachi H, Katsuyama H, Yanai H. The low dose (7.5 mg/day) pioglitazone is beneficial to the improvement in metabolic parameters without weight gain and an increase of risk for heart failure. Int J Cardiol 2017;227:247–8. [DOI] [PubMed] [Google Scholar]
- 77.Yanai H, Adachi H. The low-dose (7.5 mg/day) pioglitazone therapy. J Clin Med Res 2017;9:821–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Da Silva S, Keita AV, Mohlin S, et al. A novel topical PPARgamma agonist induces PPARgamma activity in ulcerative colitis mucosa and prevents and reverses inflammation in induced colitis models. Inflamm Bowel Dis 2018;24:792–805. [DOI] [PubMed] [Google Scholar]
- 79.Koo JB, Nam MO, Jung Y, et al. Anti-fibrogenic effect of PPAR-gamma agonists in human intestinal myofibroblasts. BMC Gastroenterol 2017;17:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeon KI, Kulkarni A, Woeller CF, et al. Inhibitory effects of PPARgamma ligands on TGF-beta1-induced corneal myofibroblast transformation. Am J Pathol 2014;184:1429–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nuwormegbe SA, Sohn JH, Kim SW. A PPAR-gamma agonist rosiglitazone suppresses fibrotic response in human pterygium fibroblasts by modulating the p38 MAPK pathway. Invest Ophthalmol Vis Sci 2017;58:5217–26. [DOI] [PubMed] [Google Scholar]
- 82.Kasagi Y, Dods K, Wang JX, et al. Fibrostenotic eosinophilic esophagitis might reflect epithelial lysyl oxidase induction by fibroblast-derived TNF-alpha. J Allergy Clin Immunol 2019;144:171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]