

Abstract
Background
Individuals with idiopathic pulmonary arterial hypertension (PAH) display reduced oral glucose tolerance. This may involve defects in pancreatic function or insulin sensitivity, but this hypothesis has not been tested; moreover, fasting nutrient metabolism remains poorly described in PAH. Thus, we aimed to characterize fasting nutrient metabolism and investigated the metabolic response to hyperglycemia in PAH.
Methods
Twelve participants (6-PAH, 6-Controls) were administered a hyperglycemic clamp, while 52 (21-PAH, 31-Controls) underwent plasma metabolomic analysis. Glucose, insulin, C-peptide, free fatty acids and acylcarnitines were assessed from the clamp. Plasma metabolomics was conducted on fasting plasma samples.
Results
The clamp verified a reduced insulin response to hyperglycemia in PAH (−53% vs Control), but with similar pancreatic insulin secretion. Skeletal muscle insulin sensitivity was unexpectedly greater in PAH. Hepatic insulin extraction was elevated in PAH (+11% vs Control). Plasma metabolomics identified 862 metabolites: 213 elevated, 145 reduced in PAH (p<0.05). In both clamp and metabolomic cohorts, lipid oxidation and ketones were elevated in PAH. Insulin sensitivity, fatty acids, acylcarnitines and ketones correlated with PAH severity, while hepatic extraction and fatty acid:ketone ratio correlated with longer 6-minute walk distance.
Conclusion
Poor glucose control in PAH could not be explained by pancreatic ß-cell function or skeletal muscle insulin sensitivity. Instead, elevated hepatic insulin extraction emerged as an underlying factor. In agreement, nutrient metabolism in PAH favors lipid and ketone metabolism at the expense of glucose control. Future research should investigate the therapeutic potential of reinforcing lipid and ketone metabolism on clinical outcomes in PAH.
INTRODUCTION
Pulmonary arterial hypertension (PAH) is a debilitating disease characterized by remodeling of the pulmonary vascular bed leading to elevated pulmonary arterial pressure requiring progressive compensation by the right ventricle, and culminates in right heart failure. Despite recent clinical advances, prognosis remains poor with estimated 3-year survival rates of 55–69% in newly diagnosed patients(1, 2). The pathobiology of PAH is incompletely understood and none of the currently available treatment modalities directly target the underlying pulmonary vascular remodeling(3). As such, there remains a dire need to identify the underlying pathophysiology of PAH to improve disease management.
Recent epidemiologic observations have identified a disproportionate prevalence of obesity and diabetes comorbidities in individuals with PAH; diseases hallmarked by insulin resistance and poor glycemic control(4, 5). Corroborating data from in vitro, pre-clinical and biological experiments with humans have implicated metabolic disease in PAH pathogenesis(6). We previously reported metabolic abnormalities in lipid(7) and carbohydrate metabolism(8, 9) which were associated with poorer clinical outcomes. Importantly, patients with PAH exhibited overt glucose intolerance concomitant with reduced circulating insulin levels in response to an oral glucose tolerance test, suggesting impaired pancreatic ß-cell function (i.e. reduced insulin secretion) in response to hyperglycemia(9).
Additionally, ketone metabolism has been associated with the metabolic abnormalities in preclinical models of PAH(10) and humans(11). Ketogenesis is primarily controlled by the liver in response to hormonal, sympathetic and nutritional input, which regulates circulating ketone concentrations, predominantly ß-hydroxybutyrate (ßOHB). Another upstream regulator of ßOHB is the delivery of lipids to the liver to be oxidized and subsequently converted into ßOHB. This regulation of ßOHB is particularly relevant in PAH pathophysiology due to the characteristic progression of right heart failure, as the failing heart increasingly relies on ßOHB for fuel(12). Yet, plasma ßOHB during fasting and in response to hyperglycemia has not been characterized in PAH.
Further, methodologically rigorous approaches investigating metabolism are lacking in the PAH field. Poor oral glucose control has been reported in PAH(9, 13); the underlying mechanisms remain to be defined, but may involve pancreatic ß-cell function, skeletal muscle insulin sensitivity, ketogenesis or lipid metabolism. Thus, we utilized a combination of the state-of-the-art hyperglycemic clamp procedure together with unbiased plasma metabolomics to assess ß-cell function, insulin sensitivity and nutrient metabolism in two independent cohorts of PAH and Controls. We hypothesized that individuals with PAH would present with impaired pancreatic function and elevated ketone and lipid metabolism.
METHODS AND MATERIALS
Subjects
Sixty-four subjects were recruited as patients of the Cleveland Clinic’s Respiratory Institute or from the greater Cleveland area; twelve received a hyperglycemic clamp to assess pancreatic ß-cell function, while 52 underwent untargeted metabolomics. PAH designation was based on pre-existing diagnosis(9). This research was reviewed and approved by the Cleveland Clinic’s Institutional Review Board and informed consent was obtained prior to initiating study procedures.
Pancreatic ß-cell Insulin Secretion
Participants reported to the Cleveland Clinic’s Clinical Research Unit following stringent control procedures to minimize the influence of diet and physical activity on metabolic testing(14). A hyperglycemic clamp was performed in 6 PAH and 6 Controls(15) to assess the pancreatic response to hyperglycemia. Briefly, hyperglycemia (180mg/dl) was rapidly achieved with a primed infusion of intravenous glucose (Dextrose, 20%) and maintained by variable rate infusion for 3 hours. Blood glucose was measured every 5 minutes (YSI 2300; STAT Plus, Yellow Springs, OH) and the glucose infusion rate was adjusted according to the algorithm by DeFronzo(16). With hyperglycemia, the plasma insulin response is well-documented to be biphasic with an initial spike and nadir during the first 10 minutes (0–10min, 1st phase) followed by a linear increase over time (10–180min, 2nd phase). During the 1st phase, blood was collected every 2min; during the 2nd phase, blood was collected every 15min. Samples were processed and stored at −80°C until analysis.
Plasma Analytes
Insulin and C-peptide were assessed via radio-immunoassay (#HI-14K and #HCP-20K; Millipore Corporation, Billerica, MA). The insulin:C-peptide molar ratio was calculated to assess hepatic insulin extraction. ßOHB was quantified via colorimetric assay (#700190, Cayman Chemical, Ann Arbor, MI). Free fatty acids (FFA) and acylcarnitines were quantified by LC/MS/MS. Untargeted metabolomic analysis (Metabolon Inc., Durham, NC) was performed on fasting plasma samples. Additional details on the LC/MS/MS and metabolic approaches are available in the Online Methods Supplement.
Glucose Disposal and Insulin Sensitivity
Glucose infusion rates (GIR) were calculated from the glucose required to maintain hyperglycemia during steady-state (150–180min) of the clamp. Insulin sensitivity was estimated as glucose metabolism (M; mg/kg/min) normalized to the prevailing insulin concentration (M/I; mg/kg/min/μU.ml) during steady-state(15). Indirect calorimetry was performed with an automated system (Vmax Encore, Viasys HealthCare, Yorba Linda, CA) in a semi-darkened, thermoneutral (22±1°C) environment.
Statistical Analysis
Statistical analyses were performed using PRISM7 (GraphPad, La Jolla, CA). Group differences were assessed by two-way repeated measures ANOVA (Group × Time). Post hoc multiple comparison tests were optimized for each analysis (detailed in figure legends). Area under the curve (AUC) was calculated using the trapezoidal method. AUC and baseline comparisons were evaluated by unpaired Student’s t-test. Pearson’s correlation assessed relationships among outcome measures of interest. Normality was determined by the Shapiro-Wilk test (alpha=0.05). Non-normal data was log-transformed. Data expressed as mean±SD. Significance accepted at p<0.05.
Metabolomic data are presented as fold-change and box-and-whiskers plots of scaled intensity values. Missing values were imputed with the minimum observed value for each compound. Following log-transformation, one-way ANOVA identified metabolites that differed between groups. To account for multiple comparisons, an estimate of the false discovery rate (q-value) was calculated. Significance accepted at q<0.1.
RESULTS
Hyperglycemic Clamp
Glucose, Insulin and C-peptide
Participants (matched for age, BMI and sex) displayed similar fasting glucose, insulin and C-peptide concentrations (Table 1). Basal metabolic rate was elevated in PAH, when normalized for height, weight, age and sex, consistent with previous findings(9). The insulin response to hyperglycemia was reduced by 53% in PAH (Figure 1b). C-peptide, a marker of pancreatic ß-cell function and insulin secretion, was similar between groups (Figure 1c). The glucose infusion rate required to maintain hyperglycemia was similar for both groups (Figure 1d). Insulin sensitivity (M/I) was elevated by 92% in PAH compared to Controls (Figure 1e).
TABLE 1.
Subject Characteristics
| Control | PAH | p-value | |
|---|---|---|---|
| N (sex) | 6 (5 F) | 6 (5 F) | - |
| Age (years) | 40±15 | 49±12 | 0.32 |
| Height (cm) | 171±7 | 167±8 | 0.36 |
| Weight (kg) | 90±18 | 87±12 | 0.75 |
| BMI (kg/m2) | 31±4 | 31±5 | 0.73 |
| Baseline Glucose (mg/dl) | 90±11.6 | 86±3.4 | 0.40 |
| Baseline Insulin (μU/ml) | 16±7.1 | 11±2.9 | 0.18 |
| Baseline C-peptide (ng/ml) | 2.2±0.5 | 2.0±0.6 | 0.44 |
| Baseline ßOHB (mM) | 0.16±0.03 | 0.22±0.03 | 0.02 |
| Baseline FFA (μM) | 857±245 | 961±408 | 0.61 |
| HOMA-IR | 3.7±2.0 | 2.4±0.5 | 0.17 |
| BMR (kcals/day) | 1484±317 | 1519±88 | 0.82 |
| Baseline RER | 0.80±0.03 | 0.79±0.03 | 0.81 |
| SBP (mmHg) | 130±4 | 120±8 | 0.28 |
| DBP (mmHg) | 76±8 | 69±10 | 0.25 |
| Heart Rate (bpm) | 69±10 | 81±13 | 0.11 |
| O2 Saturation (%) | 99.2±1.3 | 94.5±2.1 | <0.01 |
| Liver Panel | |||
| Total Protein (g/dl) | 6.8±0.5 | 6.6±0.5 | 0.65 |
| Albumin (g/dl) | 3.9±0.3 | 4.0±0.3 | 0.84 |
| Total Bilirubin (mg/dl) | 0.45±0.14 | 0.52±21 | 0.54 |
| ALP (U/L) | 55±14 | 72±20 | 0.12 |
| AST (U/L) | 19±8 | 14±4 | 0.20 |
| ALT (U/L) | 24±13 | 14±7 | 0.15 |
Data represent mean±SD.
PAH, subjects with pulmonary arterial hypertension; BMI, body mass index; ßOHB, ß-hydroxybutyrate; FFA, free fatty acids; HOMA-IR, homeostatic model assessment of insulin resistance; BMR, basal metabolic rate; RER, respiratory exchange ratio, VO2/VCO2; SBP, systolic blood pressure; DBP, diastolic blood pressure; O2 Saturation; oxygen saturated hemoglobin relative to total hemoglobin; ALP, alkaline phosphatase; AST, aspartate transaminase; ALT, alanine transaminase.
Figure 1. Hyperglycemic Clamp.
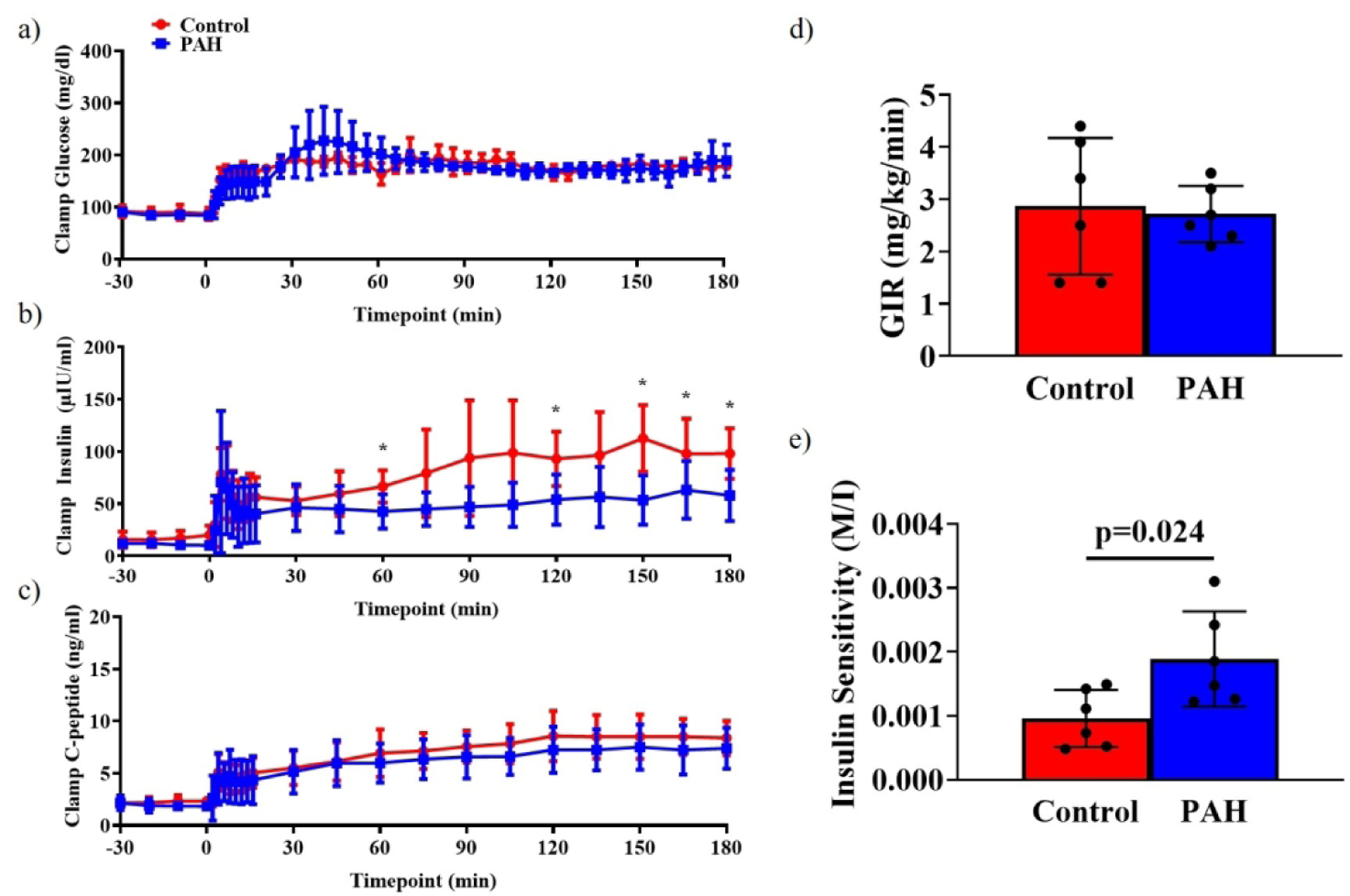
Hyperglycemic clamp results for glucose (a), insulin (b) and C-peptide (c). Research participants were effectively clamped at 180 mg/dl glucose. a) 150–180 min, PAH: 177±4 mg/dl, Control: 179±1 mg/dl; ns. Insulin concentrations were reduced in PAH compared to Control. b) 150–180 min, PAH: 55.2±10.3 μIU/ml, Control: 102.8±12.2 μIU/ml; 2-way repeated measures ANOVA (Group × Time), significant effect of Time and Interaction. C-peptide concentrations were similar between groups. c) 150–180 minutes, PAH: 7.4±0.9 ng/ml, Control: 8.5±0.8 ng/ml; 2-way repeated measures ANOVA (Group × Time), significant effect of Time. *, Tukey’s post hoc test, p<0.05. d) Glucose infusion rate was similar between groups (PAH: 2.7±0.2 mg/kg/min, Control: 2.9±0.5 mg/kg/min). e) Insulin sensitivity (M/I) was elevated in PAH compared to Controls (M/I, 150–180 minute; PAH: 0.025±0.007, Control: 0.013±0.008, p=0.024. PAH, pulmonary arterial hypertension; ßOHB, ß-hydroxybutyrate; M/I, mg glucose/kg body weight/min/μU insulin.ml.
Ketogenesis (ßOHB) and Hepatic Insulin Extraction
Baseline ßOHB was elevated in PAH (+40%, p=0.018) while both groups reached similar levels during hyperglycemia (Figure 2a). Reductions in ßOHB only reached significance in PAH (60min, 120min, 180min, all p<0.02), and ßOHB remained greater in PAH throughout the clamp (ßOHB AUC; PAH: 13.0±0.7AU; Control: 10.4±0.5AU, p=0.016). Hepatic insulin extraction was greater in PAH during the 1st and 2nd phase insulin response (Figure 2bc).
Figure 2. ß-Hydroxybutyrate and Hepatic Insulin Extraction.
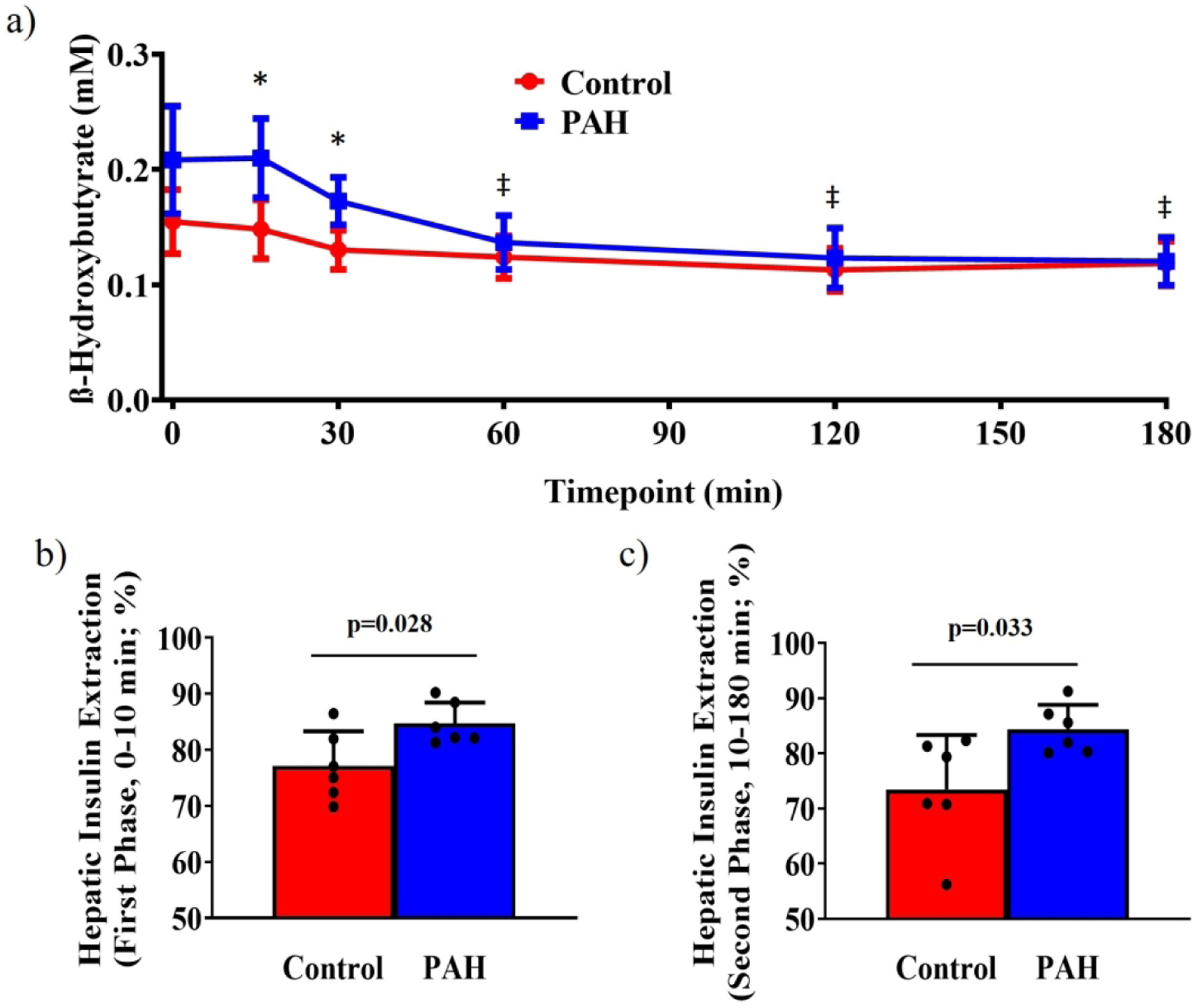
a) The ketone, ß-hydroxybutyrate was elevated in PAH through 30 minutes of hyperglycemia, reaching similar levels as Controls by 60 minutes. (PAH: Baseline 0.22±0.02 mM, 180 minute 0.12±0.01 mM; Control: Baseline 0.16±0.01 mM, 180 minute 0.12±0.01 mM; 2-way repeated measures ANOVA, Group p=0.018, Time p<0.001, Interaction p<0.001). ßOHB, ß-hydroxybutyrate; *, different between groups, Tukey’s post hoc test, p<0.05; ‡, different from baseline within PAH group, Dunnett’s multiple comparisons test, p<0.02. Hepatic insulin extraction was significantly greater in PAH compared to Controls during the 1st phase insulin response b) 0–10 minutes; PAH: 84.7±1.5%, Control: 77.1±2.5%, p=0.028 and through prolonged hyperglycemia c) 10–180 minutes; PAH: 84.4±1.8%, Control: 73.5±2.5%, p=0.033; significance determined by unpaired Student’s t-test. PAH, pulmonary arterial hypertension; hepatic insulin extraction, molar ratio of plasma insulin/plasma C-peptide expressed as a percentage.
Free Fatty Acids (FFA) and Acylcarnitines
Total FFAs were similar between groups at baseline and decreased equivalently throughout the clamp (Figure 3a). Palmitate (C16:0) was the most prevalent lipid species, while palmitoleate (C16:1) was elevated in PAH at baseline (Figure 3b). Acetylcarnitine was greater at baseline in PAH while both groups decreased with hyperglycemia. The reduction was greater in PAH (delta; PAH: −6.3±0.4 μM, Control: −4.7±0.5 μM, p=0.039; Figure 3c).
Figure 3. Free Fatty Acids and Lipid Oxidation.
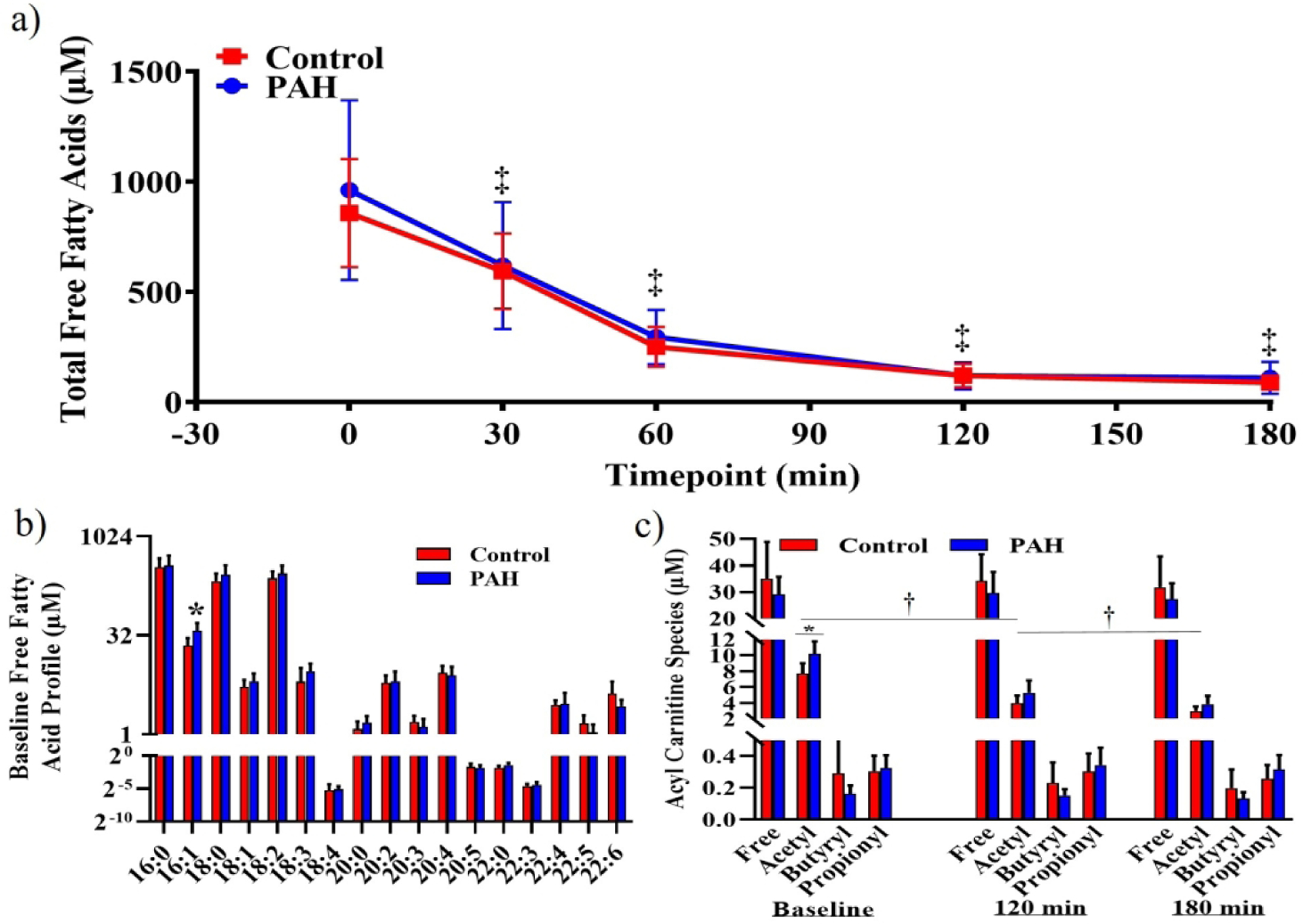
a) Total plasma free fatty acids were similar between groups at baseline (PAH: 961±167 μM; Control: 857±100 μM) and decreased equally with hyperglycemia (180 minute; PAH: 111±30 μM, Control: 89±13 μM; 2-way repeated measures ANOVA, Time p<0.001). b) Lipidomic profiling of free fatty acids revealed elevated palmitoleate (16:1) in PAH (PAH: 37.3±4.9 μM; Control: 22.1±2.9 μM; p=0.023). c) Free-, butyryl- and propionyl-carntines were similar between groups and remained unchanged at 120 and 180 minutes of hyperglycemia. Acetylcarnitine was elevated in PAH. Both groups decreased acetylcarnitine similarly in response to hyperglycemia. (PAH: Baseline 10.2±0.7 μM, 120 minute 5.3±0.6 μM, 180 minute 3.9±0.4 μM; Control Baseline 7.7±0.5 μM, 120 minute 4.0±0.4 μM, 180 minute 3.0±0.2 μM, 2-way repeated measures ANOVA, Group p=0.033, Time p<0.001, Interaction p<0.001, Tukey’s multiple comparisons tests, within group (Baseline vs 120 minute vs 180 minute), all p<0.05).*, different between groups, p<0.05; †, different from baseline, p<0.05; ‡, Sidak’s multiple comparison test, different from Baseline, p<0.05.
Correlational Analysis with Metabolic Measures
Given these novel and unintuitive nutrient metabolism findings, we investigated relationships between the major metabolic pathways: insulin sensitivity, ketogenesis and hepatic extraction. Insulin sensitivity (M/I) correlated with hepatic extraction (Figure 4a) and ßOHB throughout the clamp (Figure 4b). These relationships remained significant when using 1st phase hepatic extraction or fasting ßOHB as predictive factors for clamp-derived M/I (Figure S1ab). Further, the ratio of fasting free fatty acids to ketones (FFA:ßOHB) predicted clamp-derived M/I when controlling for BMI and PAH status (r2=0.614, p=0.045; multiple linear regression); the presence of PAH contributed significantly to the model (p=0.044) while BMI did not. ßOHB correlated with 2nd phase hepatic extraction at baseline (Figure 4c) and throughout the clamp (Figure 4d). In agreement with known physiology of lipid oxidation and ketogenesis, acetylcarnitine correlated with ßOHB (Figure 4e).
Figure 4. Correlational Analysis with Metabolic Measures.

To gain insight into nutrient metabolism in PAH, we interrogated relationships between the primary metabolic findings. Insulin sensitivity (M/I) correlated strongly with the a) hepatic insulin extraction (2nd phase, r2=0.498, p=0.010) and b) ßOHB (r2=0.504, p=0.010) during the clamp. ßOHB concentrations correlated with 2nd phase hepatic extraction at c) baseline (r2=0.402, p=0.027) and d) throughout the clamp (r2=0.467, p=0.014). ßOHB expectedly correlated with e) acetylcarnitine (r2=0.452, p=0.017) and additionally with f) palmitoleate (r2=0.527, p=0.008). PAH, pulmonary arterial hypertension; ßOHB, ß-hydroxybutyrate; M/I, mg glucose/kg body weight/min/μU insulin.ml.
Palmitate (C16:0) correlated strongly with total FFAs (r2=0.943, p<0.001, Figure S2a), suggesting palmitate increases in proportion with total FFA concentrations. In contrast, palmitoleate (C16:1) did not correlate with total FFAs (r2=0.051, p=0.481, Figure S2b), suggesting alternate regulation of palmitoleate outside of global FFA concentrations. Further, palmitoleate was strikingly well-correlated with ßOHB (Figure 4f) and M/I (Figure S1cd), implicating a potential relationship with nutrient metabolism in agreement with emerging literature on palmitoleate as a bio-active lipid(17).
Correlational Analysis with Clinical Measures
Table 2 details clinical characteristics of PAH participants. Given the unexpected metabolic findings and the prevailing hypothesis that aberrant metabolism is inherent to PAH pathophysiology, we assessed whether the metabolic abnormalities were associated with functional capacity (6-minute walk distance) or measures of right heart dysfunction. 6-minute walk distance correlated with both the 1st (Figure S3a) and 2nd phase (Figure S3b) hepatic extraction, and with FFA:ßOHB (Figure S3c). Presence of pericardial effusion correlated with greater ßOHB during the clamp (Figure S3d). Presence of right atrial dilation correlated with lower hepatic extraction (Figure S3e). FFAs during the clamp were associated with higher resting heart rate (Figure S3f), while baseline FFAs were related to lower O2 saturation (Figure S3g). M/I correlated with both right ventricular systolic pressure (Figure S3h) and NT-proBNP (Figure S3i).
TABLE 2.
Clinical Characteristics of PAH Patients (clamp study, n=6)
| Measured Value | |
|---|---|
| NYHA Class | |
| II | 5 |
| III | 1 |
| Echocardiography | |
| RV dilation present | 5 |
| RV dysfunction present | 2 |
| RA dilation present | 3 |
| RVSP (mmHg) | 71.3±12.8 |
| Pericardial effusion present | 3 |
| NT-proBNP (pg/ml) | 80.8±24.4 |
| Medications | |
| sildenafil | 1 |
| taldalafil | 3 |
| bosentan | 1 |
| macitentan | 2 |
| treprostinil | 5 |
| 6MWT (m) | 411.7±25.6 |
| Right heart catheter data | |
| RAP (mmHg) | 8.2±1.3 |
| mPAP (mmHg) | 48.2±4.8 |
| PAWP (mmHg) | 11.5±2.4 |
| Cardiac Index (L/min/m2) | 3.08±0.18 |
| PVR (Wood units) | 6.2±0.7 |
Data represent count or mean±SEM.
PAH, pulmonary arterial hypertension; NYHA, New York Heart Association functional class; RV, right ventricle; RA, right atrium; RVSP, right ventricular systolic pressure; NT-proBNP, N-terminal pro b-type natriuretic peptide; 6MWT, 6-minute walk test; RAP, right atrial pressure; mPAP, mean pulmonary arterial pressure; PAWP, pulmonary arterial wedge pressure; PVR, pulmonary vascular resistance.
Plasma Metabolomics
To validate the clamp metabolic findings and clinical associations in a larger cohort, untargeted metabolomic analysis was conducted on 21 idiopathic PAH patients and 31 age-, BMI- and sex-matched Controls. Subject characteristics of the metabolomics cohort and scaled intensity values of all 862 targets are available elsewhere(11). We identified 862 metabolites, of which 358 were different in PAH (213 elevated; 145 reduced). A volcano plot illustrating the metabolite responses are displayed in Figure 5A. A heat map displaying the top 30 differentially expressed metabolites is illustrated in Figure 5B. Notably, ßOHB was 5-fold higher in PAH (q=0.001, Figure 5C). Thirty metabolites represent fatty acid oxidation (acylcarnitines); 21 were elevated in PAH, including acetylcarnitine (1.7-fold, q<0.001, Figure 5D). Mirroring clamp data, ßOHB and acetylcarnitine strongly correlated (Figure S4a). Of 38 fatty acid metabolites, 19 were elevated in PAH, including palmitate (Figure S4b), while palmitoleate showed the greatest increase (Figure S4c). Seven amino acids have ketogenic capacity of which six were reduced in PAH (trp, tyr, ile, thr, lys, leu, all q<0.10, Figure S4d). Correlation analysis was conducted between metabolites of interest identified from the clamp study and clinical measures. Right ventricular systolic pressure was associated with ßOHB (Figure S5a), palmitate (Figure S5b) and acetylcarnitine (Figure S5c). Grade of tricuspid regurgitation correlated with ßOHB (Figure S5d) and palmitate (Figure S5e). Palmitate was negatively associated with MVO2 (Figure S5f). Total free fatty acids were not available for this metabolomics dataset, thus palmitate was used as a surrogate comparison due to its known strong correlation to total FFAs(18) and independent validation in the clamp cohort in this report (Figure S2a).
Figure 5. Untargeted Plasma Metabolomics.

a) Volcano plot (excluding metabolites from drug or environmental exposure) representing fold-change of log scaled data, q<0.10. b) Heat map of top 30 differentially expressed metabolites. Both c) ßOHB and d) acetylcarnitine were elevated in PAH compared to Control. Box-and-whiskers plots presented using the Tukey method. PAH, pulmonary arterial hypertension; ßOHB, ß-hydroxybutyrate.
DISCUSSION
This is the first work to use the hyperglycemic clamp to investigate ß-cell function in individuals with idiopathic pulmonary arterial hypertension. We confirmed a decreased insulin response to hyperglycemia in PAH, corroborating our findings from an oral glucose tolerance test(9). The novel contribution of this work is that the reduced insulin response to hyperglycemia was not attributable to reduced pancreatic ß-cell insulin secretion or skeletal muscle insulin sensitivity compared to age-, BMI- and sex-matched Controls. Rather, hepatic insulin extraction was significantly elevated in PAH, suggesting hepatic insulin extraction may contribute to the poor oral glucose tolerance in observed PAH pathophysiology. Another primary contribution of this work was defining preferential lipid and ketone metabolism associated with PAH clinical measures, along with replicating these findings in a larger cohort using a plasma metabolomics approach. Fatty acid oxidation (acetylcarnitine) and ketogenesis (ßOHB) were elevated in PAH, while maintaining a greater propensity to convert available free fatty acids to ketones (defined by the FFA:ßOHB ratio) was associated with improved exercise endurance, an important clinical marker in PAH. These data suggest an adaptive shift in nutrient metabolism towards fatty acid and ketone utilization at the expense of glucose control in PAH.
Our interpretations are based upon the framework that glucose control is primarily regulated by the hormone insulin – governed by 3 critical control points: pancreatic insulin secretion, hepatic insulin extraction, and skeletal muscle insulin sensitivity. Upon induction of hyperglycemia, insulin is secreted by the pancreatic ß-cells. Prior to reaching arterial circulation, insulin is partially sequestered by hepatic insulin extraction, modifying the circulating insulin concentration. Finally, circulating insulin acts on peripheral tissues to facilitate tissue-specific glucose disposal, skeletal muscle being the largest consumer, whereby the insulin sensitivity of skeletal muscle partially dictates the magnitude of this response. By this manner, these 3 primary tissues are factors underlying glucose control in PAH: pancreatic insulin secretion, hepatic insulin extraction and skeletal muscle insulin sensitivity. An integrated working model of our findings at these critical control points are illustrated in Figure 6, including equivalent pancreatic insulin secretion, elevated hepatic insulin extraction and increased skeletal muscle insulin sensitivity in PAH compared to age-, BMI- and sex-matched Controls.
Figure 6. Summary of Metabolic Findings.

The underlying physiology of PAH is characterized by elevated blood pressure in the arteries of the lungs, which puts stress on the right ventricle, culminating in progressive heart failure. Poor oral glucose control in PAH has been observed in the literature, but the underlying factors remain to be fully elucidated. Importantly, glucose control is primarily regulated by the hormone insulin, which dictates the disposal of glucose from the blood into peripheral tissues. The impact of insulin-mediated glucose control in response to hyperglycemia is primarily regulated by 3 critical control points: pancreatic ß-cell insulin secretion, hepatic insulin extraction and skeletal muscle insulin sensitivity. Here, we assessed fasting metabolism in PAH along with the insulin response to hyperglycemia induced by the gold-standard hyperglycemic clamp technique. In PAH, fasting metabolism was shifted towards utilization of ketones and lipids, despite similar insulin levels compared to Controls. During experimentally induced hyperglycemia, we observed reduced circulating insulin in PAH (Figure 1b). We further observed that (1) pancreatic ß-cell insulin secretion was equivalent between PAH and Controls, (2) hepatic insulin extraction was elevated in PAH and (3) skeletal muscle insulin sensitivity was increased in PAH. PAH, pulmonary arterial hypertension.
Liver – Insulin Extraction and Ketogenesis
Important to the interpretation of these results, the Control group was overweight/obese and sedentary and presented with classic signs of early stages of insulin resistance: elevations in FFAs and insulin secretion with reductions in fasting ßOHB(19, 20) and hepatic insulin extraction(21, 22). Pancreatic ß-cell function (i.e. insulin secretion) was similar between groups, suggesting PAH patients are progressing along the natural history of diabetes. Typically, hepatic extraction is progressively reduced along this continuum, thus, the finding of greater hepatic insulin extraction in PAH was unexpected. To our knowledge, this is the first report suggesting PAH presents with elevated hepatic insulin extraction in response to hyperglycemia. Given neither pancreatic ß-cell insulin secretion nor skeletal muscle insulin sensitivity were indicative of poor glucose control, it remains plausible that the elevation in hepatic insulin extraction plays a role in the poor oral glucose control observed in PAH(23). Future research should utilize more direct approaches to assess hepatic insulin extraction, such as stable isotopic tracers, invasive portal vein sampling or rigorous modeling calculations in combination with an oral glucose tolerance test(24), to further pursue this line of inquiry in PAH metabolism.
Another important role of the liver in metabolic health is hepatic ketogenesis. Fasted circulating ßOHB reflects hepatic ketogenesis(25), thus the elevated ßOHB observed in the PAH group is indicative of increased ketogenesis. This agrees with the literature on the metabolism of heart failure(12, 26) where ketones are preferentially utilized by cardiac tissue as an adaptive response to declining heart function(27), as ketones are a more energetically favorable fuel source(28). Fasting hepatic ketogenesis also positively correlated with hepatic insulin extraction in this population. This relationship between these two hepatic processes is both novel and intriguing because ketogenesis is partly regulated by insulin. Yet, it remains unknown whether the insulin extracted by hepatocytes remains inert prior to degradation, or instead, first induces local insulin signaling within hepatocytes (e.g. inhibiting ketogenesis).
An additional regulator of ketogenesis is delivery of FFAs to the liver via adipose tissue lipolysis(29), whereby increased FFAs are associated with increased ketones in healthy physiology. To address whether the observed elevation in fasting hepatic ketogenesis was merely a reflection of elevated FFAs, we measured fasting free fatty acids, showing no differences between groups. To address whether the ability to convert prevailing free fatty acids to ketones was related to clinical outcomes in PAH, we calculated a ratio of free fatty acids to ketones (FFA:ßOHB) and assessed correlations with primary clinical measures. Independently, FFA or ßOHB are associated with poorer clinical measures in this study. In contrast, the FFA:ßOHB ratio was negatively associated with clinical severity, suggesting that a greater ability to convert available FFAs to ketones is indicative of better clinical health in PAH. Although ketones have previously been viewed as a mere byproduct of metabolism, recent research has implicated ketones as a regulating factor in glucose and lipid metabolism(30), along with possessing independent signaling properties(31). Taken together, the FFA:ßOHB ratio may provide a novel characterization of metabolism in both PAH and isolated metabolic disease. Further, the physiologic relationship between ßOHB concentrations and hepatic insulin extraction have, to our knowledge, not been previously reported. This provides incentive to investigate the relationship between hepatic ketogenesis and hepatic insulin extraction in other clinical settings.
Skeletal Muscle – Insulin Sensitivity
Glucose infused during the clamp is actively transported into skeletal muscle, and in the case of the hyperglycemic clamp, provides a proxy estimate of skeletal muscle insulin sensitivity (M/I)(15). This measure of insulin sensitivity is classically reduced in concert with oral glucose tolerance, where individuals with poor oral glucose tolerance are expected to also have poor insulin sensitivity. Ourselves and others have previously identified poor oral glucose tolerance in PAH(9, 13), thus it was an unexpected to observe increased skeletal muscle insulin sensitivity in PAH compared to Controls. Several explanations for elevated insulin sensitivity in PAH may exist. First, pulmonary arterial hypertension increases basal metabolic rate(9), which was observed in this study when controlling for height, weight, age and sex. We therefore performed an additional analysis to control insulin sensitivity for basal metabolic rate, with unremarkable effects (Figure S6a). Another explanation may be an effect of hypoxia in PAH or the overweight/obese Control group, which may enhance(32) or impair(33) insulin sensitivity. It is also possible that medications specific to PAH increase insulin sensitivity or blood flow, however, we did not find any relationships with medications and insulin sensitivity within this small cohort.
The FFA:ßOHB ratio was associated with improved clinical measures and predicted M/I after controlling for BMI and PAH status. This suggests the hepatic capacity to convert FFAs to ketones may be a novel factor in skeletal muscle insulin sensitivity. Insulin sensitivity was also predicted by hepatic extraction, which is in agreement with recent human literature(34) and instills additional confidence in the validity of these unexpected results. Reductions in ketogenic amino acids in concert with elevated ketones is particularly noteworthy, as fasted-state amino acids primarily originate from muscle tissue and loss of muscle mass is common in advanced PAH and heart failure. This observation begs the question as to whether exogenous provision of ketogenic precursors could protect PAH-induced muscle catabolism and should be pursued further.
Ultimately, larger studies utilizing clamp-derived insulin sensitivity are warranted to verify the observation of elevated skeletal muscle insulin sensitivity in PAH compared to age, BMI and sex-matched Controls. The addition of a lean, healthy control group, representative of normal skeletal muscle insulin sensitivity, would substantially strengthen the study design. Importantly, identifying the metabolism underlying poor oral glucose tolerance in the context of increased skeletal muscle insulin sensitivity would be an impactful contribution to our understanding of PAH pathophysiology.
Free Fatty Acids and Fat Oxidation
Fasting FFAs were similar between groups, but notably, higher than levels typically reported for healthy, non-obese individuals(35, 36). PAH and Controls reduced FFAs similarly during the clamp and, given FFA concentrations are primarily driven by rates of adipose tissue lipolysis, this suggests the adipose tissue of PAH and Controls respond similarly to insulin. To gain deeper insight into fatty acid metabolism, we performed short-chain acylcarnitine analysis. Acetylcarnitine, an indicator of complete fatty acid oxidation, was elevated in PAH and was associated with ßOHB in both clamp and metabolomics cohorts, supporting the interpretation of preferential lipid and ketone metabolism in PAH. An interesting finding was elevated palmitoleate in PAH, which has been reported to impart beneficial effects on insulin sensitivity and cardiovascular health(37), providing a potential mechanism for the elevated insulin sensitivity in PAH and adding to the growing interest of palmitoleate in metabolic research.
Clinical Correlates and Implications
Distance walked during a 6-minute walk test is an important predictor of mortality and clinical outcomes in PAH, and in the current study was correlated with hepatic insulin extraction, implicating a role for nutrient metabolism in PAH clinical outcomes, not just metabolic health. In both clamp and metabolomic cohorts, FFAs and ßOHB were independently associated with poorer clinical measures, while the FFA:ßOHB ratio correlated with improved clinical measures. In the context of the literature, these results suggest increased FFAs and ßOHB are an early metabolic adaptation in response to the heart failure associated with PAH.
A prioritization of FFA and ßOHB fuel sources requires circulating insulin to be maintained at lower concentrations. In physiologic agreement, we observed markedly increased skeletal muscle insulin sensitivity in PAH, permitting equivalent glucose disposal with less than half the insulin concentrations of Controls. The factors responsible for maintaining reduced insulin in PAH physiology are unclear, though analysis of c-peptide and insulin relationships implicates elevated hepatic insulin extraction. The regulation of hepatic extraction is not well understood(38, 39), but several possibilities exist, including liver congestion(40) caused by elevated right atrial pressure, autonomic control of the liver(41), changes in liver blood flow(29) or side effects of PAH medications(42).
It also remains to be resolved if these metabolic observations are regulated in coordinated fashion or merely coincidental findings related to the complex physiology and pharmacotherapy in PAH. However, we speculate that elevated hepatic insulin extraction may be a physiologic means to maintain low insulin levels in the context of elevated peripheral insulin sensitivity and thus support ketone metabolism to supply fuel-efficient ketones to cardiac tissue(27, 28) – an adaptation that may be necessary to compensate for declining heart function, but which inadvertently contributes to poor glucose control in individuals with PAH and overweight/obesity.
These data agree with the left heart failure literature, evidencing sustained ß-cell function with elevated hepatic insulin extraction(43). However, the left heart failure literature primarily uses oral glucose testing and typically shows lower fasting insulin levels in populations with more severe heart failure compared to our study population. The novelty in this study is the use of the hyperglycemic clamp, which by design, creates hyperglycemia while avoiding the contribution of gut factors (i.e. gut peptides) and gastric emptying present with oral glucose procedures. The observed reductions in circulating insulin and elevated hepatic insulin extraction in response to hyperglycemia support the repurposing of some anti-diabetic agents for consideration in future PAH research. Special interest should be noted for the sodium glucose co-transporter-2 (SGLT2) inhibitors or glucagon-like peptide 1 (GLP1) receptor agonists, as they improve glucose control while having independent cardioprotective effects(44). Similarly, research is needed to determine the impact of medical nutrition therapy to reduce glucose excursions and support lipid and ketone metabolism on PAH-related outcomes.
Limitations
Due to complexity and patient burden, the hyperglycemic clamp was limited to a sample size of 12 participants, yet we show strong reproducibility of results with prior work. Further, we validated the primary findings in a separate cohort of 52 participants using unbiased plasma metabolomics. Our interpretations on nutrient metabolism were based on plasma concentrations and thus, we cannot address nutrient flux (production vs disposal) or other potentially confounding factors like endogenous glucose production, glucose effectiveness or lipid turnover, which require stable isotope tracer approaches. Hepatic/extrahepatic extraction is difficult to directly quantify without portal vein sampling, thus we utilized a commonly reported surrogate measure (molar ratio of insulin:C-peptide)(45, 46); additional considerations and limitations on this calculation of hepatic insulin extraction are available elsewhere(47, 48). M/I assumes a linear response of glucose disposal to increasing insulin concentrations, which has been evidenced within the insulin concentrations observed in this study and remains commonly reported from hyperglycemic clamps(49, 50). Hyperinsulinemic-euglycemic clamps in combination with oral glucose tolerance testing should be used to verify these results. The clamp cohort was primarily female, and although this is consistent with the predominantly female population of individuals with PAH, we conducted a separate analysis removing the male participants. This analysis revealed unremarkable effects (Table S1). Finally, elevated basal metabolic rate is common to PAH pathology and may impact nutrient metabolism, including insulin sensitivity and ketogenesis. We therefore performed an additional analysis to adjust M/I and ßOHB for basal metabolic rate, again showing unremarkable effects (Figure S6ab). Despite these limitations, this research provides novel insight into the underlying metabolism present in PAH pathophysiology.
Conclusion
This research provides finer detail of the metabolic underpinnings of PAH, and suggests that the poor oral glucose control is impacted by elevated hepatic insulin extraction rather than defects in pancreatic ß-cell function or skeletal muscle insulin sensitivity. This is the first work to utilize the highly technical hyperglycemic clamp to assess ß-cell function in PAH, isolating the insulin response to hyperglycemia per se. Finally, these data support the narrative that the shift in nutrient metabolism (preferential lipid and ketone utilization at the expense of glucose control) is a beneficial adaptation to compensate for the failing right heart. The metabolic implications of this work may help inform future trials investigating therapeutic approaches to improve PAH outcomes, including supporting lipid and ketone metabolism and minimizing glucose excursions through dietary, lifestyle or pharmacologic strategies.
Supplementary Material
ACKNOWLEDGEMENTS
We thank all our research participants, our funding sources, the Clinical Research Unit staff for assistance with subject screening, clinical visits and hyperglycemic clamp support and Drs. Charles Hoppel and Henri Brunengraber for expert technical advice and analysis.
Support: This research was supported in part by the following grants: K23HL125697 (GAH), R01HL130209 (RAD), UL1RR024989, U54GM104940 (JPK) and T32AT004094 (JTM – trainee).
Footnotes
Publisher's Disclaimer: This manuscript has recently been accepted for publication in the European Respiratory Journal. It is published here in its accepted form prior to copyediting and typesetting by our production team. After these production processes are complete and the authors have approved the resulting proofs, the article will move to the latest issue of the ERJ online.
Conflict of Interest:
The authors declare that they have no conflicts of interest with this work.
REFERENCES
- 1.Humbert M, Sitbon O, Yaici A, Montani D, O’Callaghan DS, Jais X, Parent F, Savale L, Natali D, Gunther S, Chaouat A, Chabot F, Cordier JF, Habib G, Gressin V, Jing ZC, Souza R, Simonneau G, French Pulmonary Arterial Hypertension N. Survival in incident and prevalent cohorts of patients with pulmonary arterial hypertension. Eur Respir J. 2010;36(3):549–55. Epub 2010/06/22. doi: 10.1183/09031936.00057010. [DOI] [PubMed] [Google Scholar]
- 2.Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Muros-Le Rouzic E, Romero AJ, Benton WW, Elliott CG, McGoon MD, Benza RL. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest. 2015;148(4):1043–54. Epub 2015/06/13. doi: 10.1378/chest.15-0300. [DOI] [PubMed] [Google Scholar]
- 3.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR, Rabinovitch M. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J. 2019;53(1). Epub 2018/12/14. doi: 10.1183/13993003.01887-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Badesch DB, Raskob GE, Elliott CG, Krichman AM, Farber HW, Frost AE, Barst RJ, Benza RL, Liou TG, Turner M, Giles S, Feldkircher K, Miller DP, McGoon MD. Pulmonary arterial hypertension: baseline characteristics from the REVEAL Registry. Chest. 2010;137(2):376–87. Epub 2009/10/20. doi: 10.1378/chest.09-1140. [DOI] [PubMed] [Google Scholar]
- 5.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, Howard LS, Pepke-Zaba J, Sheares KK, Corris PA, Fisher AJ, Lordan JL, Gaine S, Coghlan JG, Wort SJ, Gatzoulis MA, Peacock AJ. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med. 2012;186(8):790–6. Epub 2012/07/17. doi: 10.1164/rccm.201203-0383OC. [DOI] [PubMed] [Google Scholar]
- 6.Harvey LD, Chan SY. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J Clin Med. 2017;6(4). Epub 2017/04/05. doi: 10.3390/jcm6040043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heresi GA, Aytekin M, Newman J, DiDonato J, Dweik RA. Plasma levels of high-density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182(5):661–8. Epub 2010/05/08. doi: 10.1164/rccm.201001-0007OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barnes JW, Tian L, Heresi GA, Farver CF, Asosingh K, Comhair SA, Aulak KS, Dweik RA. O-linked beta-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation. 2015;131(14):1260–8. Epub 2015/02/11. doi: 10.1161/CIRCULATIONAHA.114.013878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heresi GA, Malin SK, Barnes JW, Tian L, Kirwan JP, Dweik RA. Abnormal Glucose Metabolism and High-Energy Expenditure in Idiopathic Pulmonary Arterial Hypertension. Ann Am Thorac Soc. 2017;14(2):190–9. Epub 2016/12/07. doi: 10.1513/AnnalsATS.201608-605OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin T, Gu J, Huang C, Zheng S, Lin X, Xie L, Lin D. (1)H NMR-Based Analysis of Serum Metabolites in Monocrotaline-Induced Pulmonary Arterial Hypertensive Rats. Dis Markers. 2016;2016:5803031 Epub 2016/04/09. doi: 10.1155/2016/5803031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heresi GA, Mey JT, Bartholomew JR, Haddadin IS, Tonelli, Adriano, Dweik RA, Kirwan JP, Kalhan S. EXPRESS: Plasma Metabolomic Profile in Chronic Thromboembolic Pulmonary Hypertension. Pulmonary Circulation.0(ja):2045894019890553. doi: 10.1177/2045894019890553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation. 2016;133(8):698–705. Epub 2016/01/29. doi: 10.1161/CIRCULATIONAHA.115.017355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hemnes AR, Luther JM, Rhodes CJ, Burgess JP, Carlson J, Fan R, Fessel JP, Fortune N, Gerszten RE, Halliday SJ, Hekmat R, Howard L, Newman JH, Niswender KD, Pugh ME, Robbins IM, Sheng Q, Shibao CA, Shyr Y, Sumner S, Talati M, Wharton J, Wilkins MR, Ye F, Yu C, West J, Brittain EL. Human PAH is characterized by a pattern of lipid-related insulin resistance. JCI Insight. 2019;4(1). Epub 2019/01/11. doi: 10.1172/jci.insight.123611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haus JM, Solomon TP, Marchetti CM, Edmison JM, Gonzalez F, Kirwan JP. Free fatty acid-induced hepatic insulin resistance is attenuated following lifestyle intervention in obese individuals with impaired glucose tolerance. J Clin Endocrinol Metab. 2010;95(1):323–7. Epub 2009/11/13. doi: 10.1210/jc.2009-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirwan JP, Kohrt WM, Wojta DM, Bourey RE, Holloszy JO. Endurance exercise training reduces glucose-stimulated insulin levels in 60- to 70-year-old men and women. J Gerontol. 1993;48(3):M84–90. [DOI] [PubMed] [Google Scholar]
- 16.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol. 1979;237(3):E214–23. Epub 1979/09/01. doi: 10.1152/ajpendo.1979.237.3.E214. [DOI] [PubMed] [Google Scholar]
- 17.Frigolet ME, Gutierrez-Aguilar R. The Role of the Novel Lipokine Palmitoleic Acid in Health and Disease. Adv Nutr. 2017;8(1):173S–81S. Epub 2017/01/18. doi: 10.3945/an.115.011130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mittendorfer B, Liem O, Patterson BW, Miles JM, Klein S. What does the measurement of whole-body fatty acid rate of appearance in plasma by using a fatty acid tracer really mean? Diabetes. 2003;52(7):1641–8. Epub 2003/06/28. doi: 10.2337/diabetes.52.7.1641. [DOI] [PubMed] [Google Scholar]
- 19.Vice E, Privette JD, Hickner RC, Barakat HA. Ketone body metabolism in lean and obese women. Metabolism. 2005;54(11):1542–5. Epub 2005/10/29. doi: 10.1016/j.metabol.2005.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Elia M, Stubbs RJ, Henry CJ. Differences in fat, carbohydrate, and protein metabolism between lean and obese subjects undergoing total starvation. Obes Res. 1999;7(6):597–604. [DOI] [PubMed] [Google Scholar]
- 21.Polonsky KS, Given BD, Hirsch L, Shapiro ET, Tillil H, Beebe C, Galloway JA, Frank BH, Karrison T, Van Cauter E. Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest. 1988;81(2):435–41. Epub 1988/02/01. doi: 10.1172/JCI113338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cobelli C, Toffolo GM, Dalla Man C, Campioni M, Denti P, Caumo A, Butler P, Rizza R. Assessment of beta-cell function in humans, simultaneously with insulin sensitivity and hepatic extraction, from intravenous and oral glucose tests. Am J Physiol Endocrinol Metab. 2007;293(1):E1–E15. Epub 2007/03/08. doi: 10.1152/ajpendo.00421.2006. [DOI] [PubMed] [Google Scholar]
- 23.Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, Paulus WJ, Rossignol P, Taegtmeyer H, Bauersachs J, Bayes-Genis A, Brutsaert D, Bugger H, Clarke K, Cosentino F, De Keulenaer G, Dei Cas A, Gonzalez A, Huelsmann M, Iaccarino G, Lunde IG, Lyon AR, Pollesello P, Rena G, Riksen NP, Rosano G, Staels B, van Laake LW, Wanner C, Farmakis D, Filippatos G, Ruschitzka F, Seferovic P, de Boer RA, Heymans S. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Heart J. 2018;39(48):4243–54. Epub 2018/10/09. doi: 10.1093/eurheartj/ehy596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moller JB, Dalla Man C, Overgaard RV, Ingwersen SH, Tornoe CW, Pedersen M, Tanaka H, Ohsugi M, Ueki K, Lynge J, Vasconcelos NM, Pedersen BK, Kadowaki T, Cobelli C. Ethnic differences in insulin sensitivity, beta-cell function, and hepatic extraction between Japanese and Caucasians: a minimal model analysis. J Clin Endocrinol Metab. 2014;99(11):4273–80. Epub 2014/08/15. doi: 10.1210/jc.2014-1724. [DOI] [PubMed] [Google Scholar]
- 25.Nosadini R, Avogaro A, Trevisan R, Duner E, Marescotti C, Iori E, Cobelli C, Toffolo G. Acetoacetate and 3-hydroxybutyrate kinetics in obese and insulin-dependent diabetic humans. Am J Physiol. 1985;248(5 Pt 2):R611–20. Epub 1985/05/01. doi: 10.1152/ajpregu.1985.248.5.R611. [DOI] [PubMed] [Google Scholar]
- 26.Puchalska P, Crawford PA. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017;25(2):262–84. Epub 2017/02/09. doi: 10.1016/j.cmet.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, Recchia FA, Kelly DP. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4(4). Epub 2019/01/23. doi: 10.1172/jci.insight.124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lestan B, Walden K, Schmaltz S, Spychala J, Fox IH. beta-Hydroxybutyrate decreases adenosine triphosphate degradation products in human subjects. J Lab Clin Med. 1994;124(2):199–209. [PubMed] [Google Scholar]
- 29.Hevener AL, Bergman RN, Donovan CM. Novel glucosensor for hypoglycemic detection localized to the portal vein. Diabetes. 1997;46(9):1521–5. Epub 1997/09/01. doi: 10.2337/diab.46.9.1521. [DOI] [PubMed] [Google Scholar]
- 30.d’Avignon DA, Puchalska P, Ercal B, Chang Y, Martin SE, Graham MJ, Patti GJ, Han X, Crawford PA. Hepatic ketogenic insufficiency reprograms hepatic glycogen metabolism and the lipidome. JCI Insight. 2018;3(12). Epub 2018/06/22. doi: 10.1172/jci.insight.99762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Veech RL, Bradshaw PC, Clarke K, Curtis W, Pawlosky R, King MT. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life. 2017;69(5):305–14. Epub 2017/04/04. doi: 10.1002/iub.1627. [DOI] [PubMed] [Google Scholar]
- 32.Azevedo JL Jr., Carey JO, Pories WJ, Morris PG, Dohm GL. Hypoxia stimulates glucose transport in insulin-resistant human skeletal muscle. Diabetes. 1995;44(6):695–8. Epub 1995/06/01. doi: 10.2337/diab.44.6.695. [DOI] [PubMed] [Google Scholar]
- 33.Thomas A, Belaidi E, Moulin S, Horman S, van der Zon GC, Viollet B, Levy P, Bertrand L, Pepin JL, Godin-Ribuot D, Guigas B. Chronic Intermittent Hypoxia Impairs Insulin Sensitivity but Improves Whole-Body Glucose Tolerance by Activating Skeletal Muscle AMPK. Diabetes. 2017;66(12):2942–51. Epub 2017/09/09. doi: 10.2337/db17-0186. [DOI] [PubMed] [Google Scholar]
- 34.Utzschneider KM, Kahn SE, Polidori DC. Hepatic insulin extraction in NAFLD is related to insulin resistance rather than liver fat content. J Clin Endocrinol Metab. 2018. Epub 2018/12/20. doi: 10.1210/jc.2018-01808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karpe F, Dickmann JR, Frayn KN. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes. 2011;60(10):2441–9. Epub 2011/09/29. doi: 10.2337/db11-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergman BC, Cornier MA, Horton TJ, Bessesen DH. Effects of fasting on insulin action and glucose kinetics in lean and obese men and women. Am J Physiol Endocrinol Metab. 2007;293(4):E1103–11. Epub 2007/08/09. doi: 10.1152/ajpendo.00613.2006. [DOI] [PubMed] [Google Scholar]
- 37.Functional Dietary Lipids: Food Formulation, Consumer Issues and Innovation for Health. Sanders TAB, editor: Woodhead Publishing; 2016. 332 p. [Google Scholar]
- 38.Tamaki M, Fujitani Y, Hara A, Uchida T, Tamura Y, Takeno K, Kawaguchi M, Watanabe T, Ogihara T, Fukunaka A, Shimizu T, Mita T, Kanazawa A, Imaizumi MO, Abe T, Kiyonari H, Hojyo S, Fukada T, Kawauchi T, Nagamatsu S, Hirano T, Kawamori R, Watada H. The diabetes-susceptible gene SLC30A8/ZnT8 regulates hepatic insulin clearance. J Clin Invest. 2013;123(10):4513–24. Epub 2013/09/21. doi: 10.1172/JCI68807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piccinini F, Polidori DC, Gower BA, Bergman RN. Hepatic but Not Extrahepatic Insulin Clearance Is Lower in African American Than in European American Women. Diabetes. 2017;66(10):2564–70. Epub 2017/07/16. doi: 10.2337/db17-0413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hadengue A, Benhayoun MK, Lebrec D, Benhamou JP. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology. 1991;100(2):520–8. [DOI] [PubMed] [Google Scholar]
- 41.Jensen KJ, Alpini G, Glaser S. Hepatic nervous system and neurobiology of the liver. Compr Physiol. 2013;3(2):655–65. Epub 2013/05/31. doi: 10.1002/cphy.c120018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osei K, Gaillard T, Schuster D. Thiazolidinediones increase hepatic insulin extraction in African Americans with impaired glucose tolerance and type 2 diabetes mellitus. A pilot study of rosiglitazone. Metabolism. 2007;56(1):24–9. Epub 2006/12/13. doi: 10.1016/j.metabol.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 43.Melenovsky V, Benes J, Franekova J, Kovar J, Borlaug BA, Segetova M, Tura A, Pelikanova T. Glucose Homeostasis, Pancreatic Endocrine Function, and Outcomes in Advanced Heart Failure. J Am Heart Assoc. 2017;6(8). Epub 2017/08/09. doi: 10.1161/JAHA.116.005290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Home P Cardiovascular outcome trials of glucose-lowering medications: an update. Diabetologia. 2019;62(3):357–69. Epub 2019/01/05. doi: 10.1007/s00125-018-4801-1. [DOI] [PubMed] [Google Scholar]
- 45.Bril F, Barb D, Portillo-Sanchez P, Biernacki D, Lomonaco R, Suman A, Weber MH, Budd JT, Lupi ME, Cusi K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology. 2017;65(4):1132–44. Epub 2016/12/17. doi: 10.1002/hep.28985. [DOI] [PubMed] [Google Scholar]
- 46.Bril F, Lomonaco R, Orsak B, Ortiz-Lopez C, Webb A, Tio F, Hecht J, Cusi K. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology. 2014;59(6):2178–87. Epub 2014/04/30. doi: 10.1002/hep.26988. [DOI] [PubMed] [Google Scholar]
- 47.Polonsky KS, Rubenstein AH. C-peptide as a measure of the secretion and hepatic extraction of insulin. Pitfalls and limitations. Diabetes. 1984;33(5):486–94. Epub 1984/05/01. doi: 10.2337/diab.33.5.486. [DOI] [PubMed] [Google Scholar]
- 48.Polonsky K, Frank B, Pugh W, Addis A, Karrison T, Meier P, Tager H, Rubenstein A. The limitations to and valid use of C-peptide as a marker of the secretion of insulin. Diabetes. 1986;35(4):379–86. Epub 1986/04/01. doi: 10.2337/diab.35.4.379. [DOI] [PubMed] [Google Scholar]
- 49.De Bernardi Rodrigues AM, da Silva Cde C, Vasques AC, Camilo DF, Barreiro F, Cassani RS, Zambon MP, Antonio MA, Geloneze B, Brazilian Metabolic Syndrome Study I. Association of Sleep Deprivation With Reduction in Insulin Sensitivity as Assessed by the Hyperglycemic Clamp Technique in Adolescents. JAMA Pediatr. 2016;170(5):487–94. Epub 2016/03/22. doi: 10.1001/jamapediatrics.2015.4365. [DOI] [PubMed] [Google Scholar]
- 50.Shah SS, Ramirez CE, Powers AC, Yu C, Shibao CA, Luther JM. Hyperglycemic clamp-derived disposition index is negatively associated with metabolic syndrome severity in obese subjects. Metabolism. 2016;65(6):835–42. Epub 2016/05/14. doi: 10.1016/j.metabol.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.