

Abstract
The most rapid path to discovering treatment options for the novel coronavirus SARS-CoV-2 is to find existing medications that are active against the virus. We have focused on identifying repurposing candidates for the transmembrane serine protease family member II (TMPRSS2), which is critical for entry of coronaviruses into cells. Using known 3D structures of close homologs, we created seven homology models. We also identified a set of serine protease inhibitor drugs, generated several conformations of each, and docked them into our models. We used three known chemical (non-drug) inhibitors and one validated inhibitor of TMPRSS2 in MERS as benchmark compounds and found six compounds with predicted high binding affinity in the range of the known inhibitors. We also showed that a previously published weak inhibitor, Camostat, had a significantly lower binding score than our six compounds. All six compounds are anticoagulants with significant and potentially dangerous clinical effects and side effects. Nonetheless, if these compounds significantly inhibit SARS-CoV-2 infection, they could represent a potentially useful clinical tool.
Keywords: SARS-CoV-2, COVID-19, Homology Modeling, Ligand Docking, Drug Repurposing
INTRODUCTION
A novel coronavirus, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), emerged in December 2019 and has since spread to 166 countries and territories around the world [1]. With over 190,000 cases and 7,800 deaths as of March 18, 2020, the discovery of effective treatments for the virus is an urgent public health need [2]. One promising drug target for SARS-CoV-2 is the transmembrane serine protease family member II (TMPRSS2). TMPRSS2 is a host protein that sits on the cell membrane and mediates the entry of pathogenic human coronaviruses into cells by cleaving and activating the viral Spike (S) protein [3–6]. TMPRSS2 is co-expressed in lung tissue with angiotensin converting enzyme 2 (ACE2) which acts as the cell surface receptor for SARS and SARS-CoV-2 [7,8]. Knockout of TMPRSS2 reduces the spread of severe acute respiratory syndrome (SARS-CoV) and Middle East respiratory syndrome (MERS-CoV) in the airway of a mouse model [9]. Kawase et al. showed that inhibition of TMPRSS2 by serine protease inhibitor Camostat blocks the entry of SARS-CoV in human Calu-3 airway epithelial cells at 10 micromolar concentrations [10]. A 2016 screen of 1,017 drugs found that the serine protease inhibitor Nafamostat inhibited TMPRSS2 and prevented S-protein mediated membrane fusion of MERS-CoV [11]. Recently, Hoffman et al. showed that SARS-CoV-2 also binds the ACE2 receptor, and that Camostat blocks the entry of SARS-CoV-2 into human lung cells [12]. There are several other small molecule inhibitors that have been reported to inhibit TMPRSS2 with low nanomolar affinity[13], but these are chemicals that have not been tested for safety in humans. Medications are urgently needed to treat SARS-CoV-2, and one of the most rapid pathways would be to find existing medications that inhibit TMPRSS2.
Computationally modeling protein structures and protein-ligand interactions can be an effective method for predicting the binding of small molecule drugs to protein targets. Previously, we have used protein structure modeling approaches to assess the functional utility of CASP predictions [14], identify novel off-target activities of kinase inhibitors [15], estimate the maximum therapeutic recommended dose of small molecule drugs [16], and show that selective estrogen modulators (SERMS) can interact with taxane binding sites and influence microtubule dynamics [17]. In this work, we use protein structure modeling to create seven plausible models of TMPRSS2, and computationally screen several approved small molecule serine protease inhibitors for activity against TMPRSS2. The advantage of screening for approved drugs is that they have guaranteed safety profile and can be readily deployed for clinical use. We identify a number of promising anticoagulant drugs currently on the market that have predicted binding energies close to the known chemical inhibitors, and thus might be suitable for experimental follow up.
METHODS
Ligand Library Preparation
We searched Pharos [18] for active ligands of TMPRSS2. We used the sequence of the TMPRSS2 homologs (e-value < 10**10, described in next section) to identify a set of serine protease inhibitor drugs that might also bind TMPRSS2. We did this by assessing likely pocket similarity through sequence alignment, reasoning that drugs that bind the most similar pockets are most likely to bind TMPRSS2. We then searched DrugBank [19] for these related serine protease targets and selected inhibitors that are currently marketed or discontinued in human trials. The list of these possible inhibitors is shown in Table 1. We obtained isomeric and canonical Simplified Molecular-Input Line-entry System (SMILES) strings for these molecules from DrugBank and PubChem [20] and generated conformers, tautomers, and ionization states at pH 7.0 ± 2.0 for each compound using LigPrep [21]. We generated all enantiomers in cases where chiral centers were present, but stereochemistry was unspecified. The resulting set of ligand conformations is our “ligand library.” Input parameter files for ligand preparation are included in the Supplemental Material.
Table 1.
A list of repurposing candidates by searching Drugbank for targets that were similar in sequence (BLAST e-value < 10**−10) and structure (PocketFEATURE similarity < −4.0). We chose compounds that had advanced into clinical trials. Most of the compounds are anticoagulants that target human serine proteases which are active in the clotting cascade.
| Drugbank ID | Name | Target |
|---|---|---|
| DB00278 | Argatroban | Prothrombin |
| DB04898 | Ximelagatran | Prothrombin |
| DB06228 | Rivaroxaban | Coagulation factor X |
| DB06605 | Apixaban | Coagulation factor X |
| DB06635 | Otamixaban | Coagulation factor X |
| DB06695 | Dabigatran | Prothrombin |
| DB09075 | Edoxaban | Coagulation factor X |
| DB12598 | Nafamostat | Coagulation factor X |
| DB12831 | Gabexate | Plasma kallikrein |
| DB12364 | Betrixaban | Coagulation factor X |
| DB06920 | Eribaxaban | Coagulation factor X |
| DB11984 | Letaxaban | Coagulation factor X |
| DB12863 | Sivelestat | Neutrophil Elastase |
| DB13729 | Camostat | Prostasin |
| DB12289 | Darexaban | Coagulation factor X |
Homology Ensemble Preparation
We downloaded the amino acid sequence for the TMPRSS2 catalytic domain from UniprotKB [Uniprot Accession O15393]. We used the following workflow using the Schrodinger Advanced Homology Modeling Interface: (1) we searched the Protein Data Bank (PDB) for template structures based on sequence similarity (using BLASTp); (2) we identified globally conserved residues in the sequence (HMMER/Pfam [22]); (3) we selected high scoring (used BLASTp e-value) templates from three closely related proteins (Human Plasma Kallikrein, Factor XIa, Hepsin); (3) we predicted and aligned secondary structure motifs using the single template alignment (STA) setting; and (4) we refined the resulting homology structures by optimizing hydrogen bond assignments and minimizing side chain energies, using the protocol of Ramachandran et al. [23]. Input parameter files for each step in our homology modeling workflow are included in the Supplemental Material.
Docking
We used the SiteMap tool [24], which scans protein structures for putative binding sites, to identify the active binding site regions in our homology model structures. To check the accuracy of the specified regions, we manually inspected the proximity of the predicted binding site to the serine protease catalytic triad (Ser-Asp-His). If no predicted binding sites were located near the catalytic triad of a homology model structure, we did not use that structure for docking. We docked the conformers in our ligand library with the active site of each homology model using Glide [25]. To ensure that compounds were docked in the correct site, we compared homology structures docked with active ligands to their original template structures by visual inspection and using PocketFEATURE. PocketFEATURE is a program that compares two 3D pockets in proteins and assesses their overall similarity with a score [26]. We used a PocketFEATURE cutoff of −4.0 (Supplementary Figure 2) as an indication that the pocket had not been greatly disrupted during modeling and docking. Input parameter files for each step in our docking workflow are included in the Supplemental Material.
RESULTS
Our ligand library consisted of 19 small molecules, each adopting an average of 1.89 steric conformations for a total library size of 36. We created seven homology models using our protocol to ensure that results were robust to anomalies that might arise from sources of variability such as differences between serine protease family members with similarly high e-values, or bound ligands that influence protein conformation and binding site geometry. The resulting seven models represent an ensemble of structures with an average root mean squared deviation (RMSD) of 1.27 Angstroms and a maximum RMSD (between model 1O5E and model 4NA8) of 1.675 Angstroms. Figure 1 summarizes the way in which these homology models differ, the location of their active site, and their energy. Table 2 shows their pairwise RMS distances.
Figure 1.
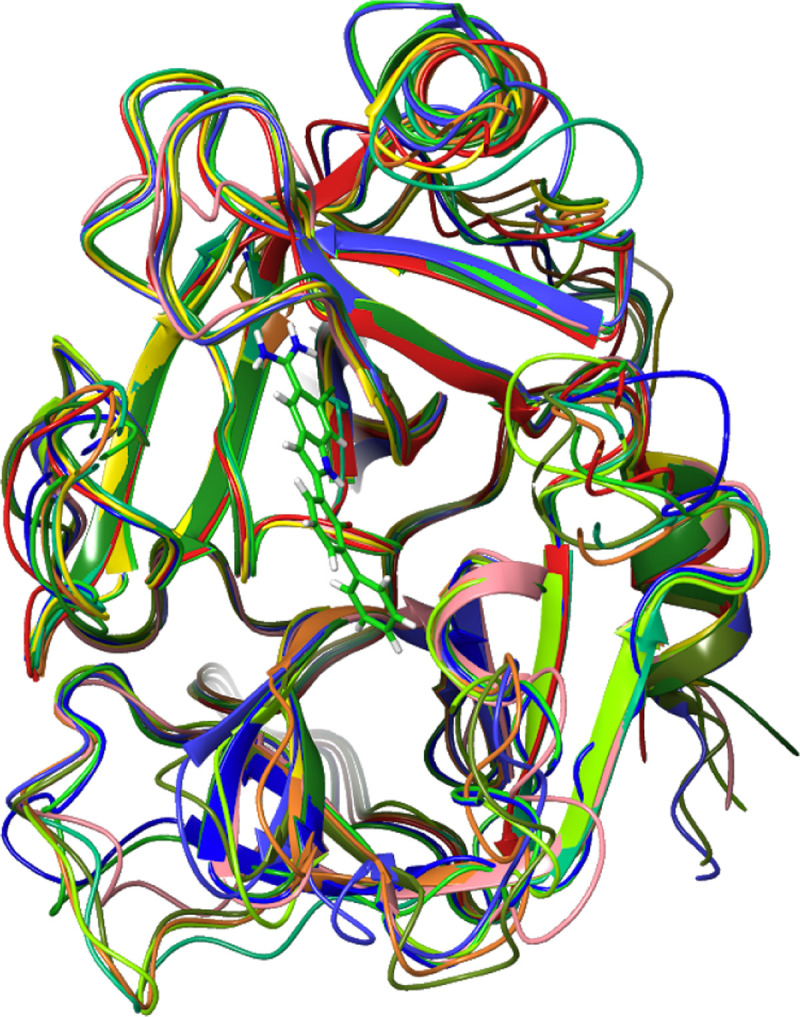
Aligned homology model structures with template structure 1O5E (Hepsin/TMPRSS1, not shown) to highlight the differences between the model structures and the active binding site. The model structures vary in loop regions outside of the active site. The large variation in loop at the three o’clock position (Res 83–88, NKTKSD) suggests that this region may be important for controlling the size of ligands that can enter the pocket. The average energy of the structures is −8573 ± 167 kcal/mol. The RMSD of the binding site prediction centroids is 2.4 Angstroms.
Table 2.
RMSD values were computed for each pair of homology model structures in our ensemble. The average RMSD was 1.27 Angstroms. The maximum RMSD was 1.675 Angstroms. This is close to the length of an alkane bond (1.54 Angstroms), and less than what is considered good resolution for a protein crystal structure (2.4 Angstroms).
| Template 1 | Template 2 | RMSD | Pocket Score1 |
|---|---|---|---|
| 105E | 2ANY | 1.350 | −11.63 |
| 1O5E | 4NA8 | 1.675 | −9.25 |
| 1O5E | 5CE1 | 0.648 | −7.94 |
| 1O5E | 5TJX | 1.527 | −8.90 |
| 1O5E | 6O1G | 1.565 | −7.47 |
| 1O5E | 3W94 | 1.144 | −6.26 |
| 2ANY | 4NA8 | 1.136 | −9.71 |
| 2ANY | 5CE1 | 1.337 | −10.13 |
| 2ANY | 5TJX | 0.923 | −10.84 |
| 2ANY | 6O1G | 1.086 | −9.19 |
| 2ANY | 3W94 | 1.252 | −6.96 |
| 4NA8 | 5CE1 | 1.582 | −9.51 |
| 4NA8 | 5TJX | 0.979 | −9.18 |
| 4NA8 | 6O1G | 1.055 | −7.59 |
| 4NA8 | 3W94 | 1.558 | −6.89 |
| 5CE1 | 5TJX | 1.322 | −8.92 |
| 5CE1 | 6O1G | 1.561 | −7.94 |
| 5CE1 | 3W94 | 1.127 | −7.42 |
| 5TJX | 6O1G | 0.955 | −9.35 |
| 5TJX | 3W94 | 1.454 | −8.32 |
| 6O1G | 3W94 | 1.515 | −6.84 |
PocketFeature computes the similarity of binding sites by comparing sets of protein microenvironments [15]. Scores below −4.0 indicate very high similarity (Supplementary Figure 2). Highly similar pockets are likely to bind the same ligands. We have previously used PocketFeature to identify novel off-target activity of kinase inhibitors and SERMs [15,17]. Here we use it to assess the similarity of homology model binding sites. The binding sites of our homology models exhibit extremely high similarity in a range which is typically observed for different crystal structures of the same protein.
Figure 2 shows a heatmap of the docking scores normalized by rank, with the most stable being dark (lowest energy) and the least stable light (high energy, unfavorable). The known non-drug inhibitors (45899577, 56677007, 56663319) all had at least one conformer that bound all the models with very low energy, serving as positive controls. Camostat, which was reported by Hoffman et al. to bind the protein in the 100 micromolar range [12], has a median binding energy. Nafamostat, which was reported by Yamamoto et al. to inhibit TMPRSS2 at 10-fold lower concentration than Camostat, ranked third. Several other human trial compounds and marketed drugs have a favorable low binding energy in many or all of the homology models. These include at least one conformer of Argatroban, Otamixaban, Letaxaban, Darexaban, and Edoxaban. Figure 3 shows a box-plot of the docking scores of these drug conformers to all seven homology models more quantitatively. Table 3 shows a summary of the predicted high-binding drugs, their lowest and average docking scores, their status as marketed or experimental, their marketed indication, and their most common side effects. Figure 4 shows key electrostatic interactions between the docked chemical structure of the best scoring ligand (Otamixaban) and residues in the binding pocket of a TMPRSS2 homology model.
Figure 2.

Docked conformers for known active ligands of TMPRSS2 and FDA-approved serine protease inhibitors to an ensemble of homology models generated from protein structure templates. The compounds are clustered by docking score rank. Darker cells indicate better docking scores. Columns correspond to homology models of TMPRSS2. Rows correspond to conformers. Molecules appended with numbers (i.e. dabigatran, dabigatran-2, etc) denote different conformers. Known active ligands (Pubmed CIDs 4689977, 56677007, 56663319) ranked highest across all homology models. Argatroban, Otamixaban, Letaxaban, Darexaban, Edoxaban, Betrixaban, and Nafamostat also ranked highly across a majority of model structures and clustered together with known active ligands. Nafamostat is reported to inhibit TMPRSS2 mediated cell fusion of MERS-CoV in vitro at high nanomolar concentrations.[11] Camostat, which was shown to inhibit SARS-CoV-2 entry in vitro at micromolar concentrations, was close to the median.[12]
Figure 3.

Docked conformers for known active ligands of TMPRSS2 and FDA approved serine protease inhibitors to an ensemble of homology models generated from protein structure templates. The box for each conformer shows the range of docking scores over all homology models. Different conformations of the same molecule are appended with numbers (i.e. dabigatran, dabigatran-2, etc). Lower scores indicate better docking and scores below −7.5 are considered promising.[27, 28] Known active ligands (Pubmed CIDs 4689977, 56677007, 56663319) had lowest docking scores. Agatroban, Nafamostat, Otamixaban, and Letaxaban scored below −7.5 for at least one model.
Table 3.
Six selected compounds with the highest minimum docking scores, annotated with their marketing approval status, indications, most frequently observed side effects, minimum docking score, and average docking score. Docking scores provide a rough estimation of binding energy (ΔG, kcal/mol) [27,28]. Lower scores indicate higher binding affinity. Docking scores may vary depending on the system, however generally scores less than −7.5 indicate high affinity binding.
| Name | Status | Indication1 | Side Effects2 | Min Score | Avg Score |
|---|---|---|---|---|---|
| Otamixaban | Experimental | Thrombosis | Catheter site hematoma | −10.81 | −7.29 |
| Argatroban | Marketed | Thrombosis | Increased INR | −8.45 | −6.29 |
| Nafamostat | Marketed | Disseminative blood vessel coagulation | Platelet count decreased | −8.14 | −5.08 |
| Letaxaban | Experimental | Thrombosis | Angina Pectoris | −7.60 | −6.26 |
| Darexaban | Experimental | Thrombosis | Bleeding events | −6.67 | −5.83 |
| Edoxaban | Marketed | Thromboembolism | Gastrointestinal hemorrhage | −6.62 | −5.43 |
Indications were compiled from FDA drug labels and clinical trial records.
Side effects were compiled from the FDA adverse event reporting system (FAERS) and clinical trial records.
Figure 4.

The interactions between our TMPRSS2 homology model and docked chemical structure for Otamixaban. Otamixaban blocks access to the catalytic triad (His 41, Asp 180, Ser 186) by forming a pi stacking interaction with His 41, salt bridges with Glu 44, Lys 45, and hydrogen bonds with Asp 180, Gln 183, and Gly 209. The hydrogen bonding interaction between the Asp 180 and the amidine group of Otamixiban is similar to the interaction between Benzamidine and Plasma Kallikrein in template structure 2ANY.
DISCUSSION
SARS-CoV-2 is a highly infective and often lethal virus. Medical treatments are urgently required while longer term approaches such as new targeted therapies and vaccines are being developed. Currently marketed drugs that may incidentally reduce the infectivity of the virus are the fastest path to potential treatments. In this report, we used computational methods to discover drug repurposing candidates for TMPRSS2, a target identified in previously published work for its role in infection by coronaviruses. We generated seven homology models and used a standard docking protocol to evaluate the binding of three known non-drug inhibitors of TMPRSS2 and other several serine protease inhibitors.
The known inhibitors provide a positive control in our work. These include the three chemicals reported as known inhibitors, but importantly also include Nafamostat, which has been shown to inhibit the MERS coronavirus through a TMPRSS2-mediated inhibition [11]. Nafamostat ranks third among our novel hits, and so makes the first two hits particularly interesting for followup. For these four positive controls, the uniformly low docking scores give confidence that our protocol is generating homology models and docking results that are consistent with previous work. In addition, Camostat’s relatively lower binding affinity is consistent with its placement in the middle of our list of candidate inhibitors. The lowest energy predictions may therefore be reasonable candidates for taking forward into in vitro validation experiments—and appropriate subsequent studies if these are positive.
All of the predicted low-energy binding molecules have anticoagulant activity. They also have a wide range of adverse events reported, including abnormal liver function tests, rash, anemia, and of course bleeding-related adverse effects. Therefore, even if they prove to be effective inhibitors of TMPRSS2 and useful for combating SARS-CoV-2 infections, they would need to be used carefully by physicians in order to trade off the risk of the viral infection with the risk of these drugs. Patients should clearly not use these prescription medications except under the supervision of a physician. Additionally, our observations are only a first small step in determining whether these drugs have activity against the virus and their potential clinical utility.
The search for a TMPRSS2 inhibitor need not find an extremely avid binder to be a useful medication in a clinical setting. Many diseases ultimately respond to several medications with different mechanisms of action, taken in combination. This strategy has two benefits. First, it makes it more difficult for the pathogen to evade treatment with a single mutation—multiple mutations would likely be required to develop resistance to multiple drugs. Second, by interfering with the pathogen at multiple points in its underlying pathways, the treatments can be more effective at eliminating the pathogen. The introduction of combined therapy in HIV was pivotal in changing the success rates of medication, and many other diseases have benefited from a polypharmacy approach. Thus, our results may provide a path for one part of a developing arsenal of medications that weaken or eliminate the virus.
We are seeking experimental confirmation of these findings through in vitro assays that can quantify the relative impact of both the known inhibitors and our predictions.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank current and former members of the Helix Lab for their helpful comments and suggestions. Some of the computing for this project was performed on the Sherlock cluster. We would like to thank Stanford University and the Stanford Research Computing Center for providing computational resources and support that contributed to these research results. This work was supported by NIH LM05652, GM102365, TR002515, T32_LM 012409, T15_LM 007033, and Chan-Zuckerberg Biohub.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- [1].Zhou Peng, et al. “A pneumonia outbreak associated with a new coronavirus of probable bat origin.” Nature 579 (2020): 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].World Health Organization. “Coronavirus disease 2019 (COVID-19) situation report 58”. https://www.who.int/docs/default-source/coronaviruse/situation-reports/20200318-sitrep-58-covid-19.pdf?sfvrsn=20876712_2 (2020)
- [3].Matsuyama Shutoku, et al. “Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2.” Journal of virology 84.24 (2010): 12658–12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Glowacka Ilona, et al. “Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response.” Journal of virology 85.9 (2011): 4122–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gierer Stefanie, et al. “The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies.” Journal of virology 87.10 (2013): 5502–5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bertram Stephanie, et al. “TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium.” Journal of virology 87.11 (2013): 6150–6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bertram Stephanie, et al. “Influenza and SARS-coronavirus activating proteases TMPRSS2 and HAT are expressed at multiple sites in human respiratory and gastrointestinal tracts.” PloS one 7.4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Heurich Adeline, et al. “TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein.” Journal of virology 88.2 (2014): 1293–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Iwata-Yoshikawa Naoko, et al. “TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection.” Journal of virology 93.6 (2019): e01815–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kawase Miyuki, et al. “Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry.” Journal of virology 86.12 (2012): 6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yamamoto Mizuki, et al. “Identification of nafamostat as a potent inhibitor of Middle East respiratory syndrome coronavirus S protein-mediated membrane fusion using the split-protein-based cell-cell fusion assay.” Antimicrobial agents and chemotherapy 60.11 (2016): 6532–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hoffmann Markus, et al. “SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor.” Cell (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sielaff Frank, et al. “Development of substrate analogue inhibitors for the human airway trypsin-like protease HAT.” Bioorganic & medicinal chemistry letters 21.16 (2011): 4860–4864. [DOI] [PubMed] [Google Scholar]
- [14].Liu Tianyun, et al. “Biological and functional relevance of CASP predictions.” Proteins: Structure, Function, and Bioinformatics 86 (2018): 374–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lo Yu-Chen, et al. “Computational analysis of kinase inhibitor selectivity using structural knowledge.” Bioinformatics 35.2 (2019): 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Liu T., et al. “Estimation of maximum recommended therapeutic dose using predicted promiscuity and potency.” Clinical and translational science 9.6 (2016): 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lo Yu-Chen, et al. “Pocket similarity identifies selective estrogen receptor modulators as microtubule modulators at the taxane site.” Nature communications 10.1 (2019): 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nguyen Dac-Trung, et al. “Pharos: collating protein information to shed light on the druggable genome.” Nucleic acids research 45.D1 (2017): D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wishart David S., et al. “DrugBank 5.0: a major update to the DrugBank database for 2018.” Nucleic acids research 46.D1 (2018): D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim Sunghwan, et al. “PubChem substance and compound databases.” Nucleic acids research 44.D1 (2016): D1202–D1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Release Schrödinger. “3: LigPrep.” Schrödinger, LLC, New York, NY: (2016). [Google Scholar]
- [22].Potter Simon C., et al. “HMMER web server: 2018 update.” Nucleic acids research 46.W1 (2018): W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ramachandran Balaji, Kesavan Sabitha, and Rajkumar Thangarajan. “Molecular modeling and docking of small molecule inhibitors against NEK2.” Bioinformation 12.2 (2016): 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Halgren Thomas. “Identifying and Characterizing Binding Sites and Assessing Druggability” Journal of Chemical Information and Modeling 49.2 (2009): 377–389. [DOI] [PubMed] [Google Scholar]
- [25].Release Schrödinger. “4: Glide.” Schrödinger, LLC, New York, NY: (2016). [Google Scholar]
- [26].Liu Tianyun and Altman Russ B.. “Using multiple microenvironments to find similar ligand-binding sites: application to kinase inhibitor binding.” PLoS computational biology 7.12 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Friesner Richard A., et al. “Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein– ligand complexes.” Journal of medicinal chemistry 49.21 (2006): 6177–6196. [DOI] [PubMed] [Google Scholar]
- [28].Balaji B. and Ramanathan M. “Prediction of estrogen receptor β ligands potency and selectivity by docking and MM-GBSA scoring methods using three different scaffolds.” Journal of enzyme inhibition and medicinal chemistry 27.6 (2012): 832–844. [DOI] [PubMed] [Google Scholar]
- [29].Desaphy Jérémy, et al. “sc-PDB: a 3D-database of ligandable binding sites—10 years on.” Nucleic acids research 43.D1 (2015): D399–D404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.