

Abstract
Context
In the last decade, genome-wide association studies (GWASs) have catalyzed our understanding of the genetics of height and have identified hundreds of regions of the genome associated with adult height and other height-related body measurements.
Evidence Acquisition
GWASs related to height were identified via PubMed search and a review of the GWAS catalog.
Evidence Synthesis
The GWAS results demonstrate that height is highly polygenic: that is, many thousands of genetic variants distributed across the genome each contribute to an individual’s height. These height-associated regions of the genome are enriched for genes in known biological pathways involved in growth, such as fibroblast growth factor signaling, as well as for genes expressed in relevant tissues, such as the growth plate. GWASs can also uncover previously unappreciated biological pathways, such as theSTC2/PAPPA/IGFBP4 pathway. The genes implicated by GWASs are often the same genes that are the genetic causes of Mendelian growth disorders or skeletal dysplasias, and GWAS results can provide complementary information about these disorders.
Conclusions
Here, we review the rationale behind GWASs and what we have learned from GWASs for height, including how it has enhanced our understanding of the underlying biology of human growth. We also highlight the implications of GWASs in terms of prediction of adult height and our understanding of Mendelian growth disorders.
This review discusses the findings of GWAS of height, including the identification of relevant biological pathways and implications for growth disorders and height prediction.
It has long been recognized that height is heritable, and height has been a subject of study for at least several centuries. In the 18th century, Francis Galton first formally established that a child’s height is on average strongly correlated with midparental height, with only mild regression to the mean, suggesting that genetics play a large role in height determination (i.e., high heritability) (1). The genetic basis of height continues to be the subject of intense interest owing to its role as a model genetic trait because it displays a nearly prototypical bell-shaped distribution, is highly heritable, and is easily studied in large numbers: It is simple to measure accurately, and adult height is quite stable over time. Understanding the genetic architecture of height—the number of genetic variants that contribute to variation in height, their frequencies in the population, and the magnitude of each genetic variant’s effect (i.e., effect sizes)—can teach us lessons about how to study the genetic underpinnings of other complex genetic traits and disease. Moreover, understanding the genetics of height can illuminate the underlying biology of human growth and provide insight into disorders of growth, such as short stature.
In this review, we summarize key findings of genome-wide association studies (GWASs) for height and growth-related traits, with particular focus on the biological insights that these studies have generated. We will discuss the implications of height GWAS results for clinicians in terms of adult height prediction and understanding Mendelian growth disorders.
GWAS Overview
The common-variant, common-disease hypothesis posited that the genetic basis for common diseases and traits (such as type 2 diabetes or height) is driven at least in part by common genetic variants—those shared by many individuals in a given population (2). In keeping with this hypothesis, one can partially elucidate the genetic basis of a given trait or common disease by “genotyping” each of these common variants in a study population and testing each variant for association with the trait or disease of interest (Fig. 1). In GWASs, because one is performing millions of these association tests (one for each variant), a strict statistical threshold must be set to establish significance at a genome-wide level. Traditionally,P < 5 × 10−8 has been used as this threshold because it roughly corresponds to aP value of 0.05 after correction for multiple testing (3). The associated variants then need to be connected to the underlying biology of the disease or trait of interest. Making this connection can be challenging because most variants highlighted by GWASs are noncoding and posited to affect levels or function of one or more nearby gene(s) that are relevant to the disease or trait.
Figure 1.

Design of a GWAS for height. Many individuals are collected and are measured for their height. Genotyping assays are then performed on the DNA for each individual to identify the genetic variants each individual carries. Each variant is then tested for correlation with height in the study population. Shown on the top right panel are sample box-and-whisker plots for a genetic variant that is not associated for height. As can be seen, the average height of individuals with the three different genotypes at this variant are approximately the same. On the bottom right are sample box-and-whisker plots for a genetic variant that is associated with height. Here, it can be seen that individuals who carry one or two copies of the A allele of this variant have decreased height. Note the small effect of this variant (0.5-cm reduction in height per A allele), which in reality would be a very strong effect for a GWAS-associated variant. Also, the associated variant is not deterministic, as the distributions of heights among the three genotypes are largely overlapping.
One of the implications of testing common genetic variants in a GWAS is that the effect sizes for the GWAS-associated variants are, in most cases, modest (4). Common variants in general have small effects on disease risk because of evolutionary selective pressures: If a variant had a large effect on an important biological pathway, as can be seen with the variants that cause most single-gene disorders, natural selection would strongly suppress its frequency in the population, and the variant would not become common. (There are a few notable exceptions—for example, variants that cause blood disorders are often common in malaria-endemic regions because the negative selection is balanced by the positive effects of resistance to malaria.)
The small effect sizes of GWAS-associated variants have also been misconstrued as indicating a lack of biological importance (5,6). However, this is a false equivalence of effect size with biological meaning. As an illustration of the fallacy of conflating small effect size with lack of biological importance, a common genetic variant with allele frequency of 39% identified by GWASs near the gene encoding hydroxymethyl glutaryl coenzyme A reductase (HMGCR) raises low-density lipoprotein (LDL) cholesterol by an average of 2.45 mg/dL (7). This is a fairly small effect of the variant on LDL cholesterol and would be deemed clinically insignificant. However,HMGCR is critically important for LDL biology because the protein encoded byHMGCR is the target for statin drugs, which have tremendous effects on LDL cholesterol and coronary artery disease risk (8).
GWAS-associated variants mostly reside in noncoding regions of the genome (9). Therefore, most of these variants do not directly affect the protein structure or function but instead likely modulate the expression of a nearby gene. Because GWAS-associated variants don’t lie directly in a gene and gene regulation can occur at distances of hundreds of thousands or even sometimes millions of bases, identifying the target gene for a GWAS-associated variant can be very challenging (10–12). Furthermore, most common variants are correlated (“in linkage disequilibrium”) with many other nearby common variants, which makes the identification of the relevant variant difficult and further complicates the identification of the relevant biological mechanisms that account for associations seen in GWASs.
The small effect sizes of the GWAS variants necessitate very large sample sizes to detect statistically significant signals. Thus, most successful GWASs have many thousands and often hundreds of thousands of study individuals. Typically, with increasing sample sizes, more and more association signals will be able to be detected because of greater statistical power (13). One advantage of having more signals is that computational methods can identify commonalities across associated loci to help illuminate the underlying biology. For example, if many associated variants (more than expected by chance) are located near genes in the same biological pathway or genes expressed in the same cell type, then the GWAS results collectively implicate that biological pathway or cell type. We will revisit this concept later in the Review.
GWASs of Human Height
GWASs for height have been some of the earliest and largest GWASs conducted to date. The large sample sizes reflect the ease with which data on height can be collected. Also, unlike studies of individual diseases, every individual in the population can contribute to a genetic study on height. Furthermore, height is often collected as part of data for other studies, and thus the genetic data from other studies can be repurposed for a GWAS of height.
The first GWAS of adult standing height was performed in 4921 individuals of European ancestry. The study identified a strongly associated genetic variant in the geneHMGA2, which had a previously recognized role in early developmental processes (14). Interestingly, mouse studies had demonstrated thatHmga2 is critical for growth, as thepygmy mouse (which is small) carries a loss-of-function mutation inHmga2 (15,16). Furthermore, humans with mutations of large effect inHMGA2 have been noted to have short stature (17) or tall stature (18) depending on whether the mutation abrogates or enhancesHMGA2 expression/function. This finding of an association atHMGA2 gave the first indication that GWASs for height could identify genes relevant for growth, including those in which mutations of larger effect could result in clinically apparent growth disorders.
The Genetic Investigation of ANthropometric Traits (GIANT) consortium was subsequently formed by research groups around the world to pool samples to conduct increasingly large GWASs for human anthropometric traits, including height. In 2010, the GIANT consortium published a GWAS of ∼180,000 individuals for height (19). The study identified 180 regions of the genome associated with height. As with other GWASs, the effect sizes of the associated variants were modest. The most strongly associated variant was near the geneZBTB38 with aP value of 3.2 × 10−158, but each height-increasing allele at this variant increases height by only ∼0.4 cm (that is, homozygotes for the height-increasing allele will be on average 0.8 cm taller than homozygotes for the height-decreasing allele). ZBTB38 is a regulator of gene expression, but its role in growth biology remains unknown (20).
The next round of GWASs for height by the GIANT consortium was published in 2014, with ∼250,000 samples of European ancestry (21). Remarkably, this round identified 697 independently associated variants that were distributed across 423 physical regions of the genome (loci). The number of independently associated variants is larger than the number of loci because many loci contained multiple different variants acting independently to influence height, with the presumption that many different variants can each independently affect the same gene at a locus. With the release of the UKBiobank and the continued expansion of GIANT, the total sample sizes available by 2019 will exceed 2 million, and initial analyses have already identified >3000 independently associated variants in >700 loci. With the discovery of a large number of associated regions, more detailed follow-up analyses could be performed connecting these genetic variants to specific tissues involved in growth, as well as biological pathways involved in growth biology (21). We will discuss the biological insights gained from these common GWAS variants in a later section of this review.
Traditional GWAS genotyping assays are very accurate for reasonably common variants (currently, GWASs can accurately assess variants with allele frequencies of ∼1% or greater, and new advances are pushing this limit closer to 0.1%). However, variants that are rarer in the population also clearly play a role (22). To better evaluate the role of rarer variants, at least those in protein-coding regions, the GIANT consortium leveraged a new genotyping platform called the exome chip to perform a genetic association study in a manner very similar to GWASs (23). The exome chip genotyping platform is specifically designed to study rare variants in the protein-coding region of genes (fairly comprehensively down to ∼0.1% allele frequency and including some variants below this limit). The study identified 83 protein-coding variants independently associated with height. These variants were all predicted to alter coding sequences of protein, so, unlike most GWAS variants, these associations are more likely to influence height by directly altering protein function. Given that the variants identified by the exome chip strongly implicate the actual genes (unlike GWAS variants, which may be quite far from the target gene), the study helped to refine the list of height-related genes and the biological processes in which these genes function. Note that because of correlation among nearby variants, it is possible that some of the coding variants are not themselves the causal variants but merely proxies for nearby noncoding causal variants. As such, even the coding variants do not always provide a “smoking gun” that can definitively implicate a specific gene, but the connections between variant and gene are certainly clearer than for GWASs. Reassuringly, there was a large overlap between the pathways implicated by coding variants and by noncoding variants. However, the coding variants discovered by the exome chip tended to more strongly implicate pathways more directly related to skeletal biology (such as extracellular matrix), whereas the noncoding, more common variants seen in GWASs more strongly highlighted earlier global developmental processes (such as transcription factor binding/chromatin regulation and embryonic growth). This suggests a hypothesis that variants that affect global developmental processes can influence height if they have tissue-specific effects on gene regulation, whereas coding variants may be found more often in genes whose function or expression is specific to growth plate chondrocytes or other cell types specific related to height. Furthermore, because the exome chip assays rarer variants, some of the associated variants have larger effects on height. For example, a variant inAR (encoding the androgen receptor) on the X chromosome is present at an allele frequency of 0.21%, and men who carry the rare, height-decreasing allele at this variant are on average 2 cm shorter than men who carry the more common allele.
Although most individuals studied by the GIANT consortium have been of European ancestry, other efforts have focused primarily on individuals of non-European ancestry, including East Asian– and African-derived populations (24,25). Some genetic variants are more frequent or even unique to these populations, which has led to the discovery of additional association signals not seen in studies of European-derived populations. For example, in a GWAS of ∼94,000 East Asian individuals, Heet al. (24) identified 98 regions of the genome associated with height. Eighty-one of these had also been seen in Europeans (19), and the remainder were uniquely seen in Asians because the associated variant was very rare in Europeans. Thus, most GWAS associations are shared across populations, and the population-specific associations tend to arise from differences in allele frequencies rather than differences in biology. These results suggest that the genetic and especially the biological regulation of height are similar across the world.
A key insight from GWASs of height is that height is highly polygenic, meaning that literally thousands of genetic variants operate in a single individual to collectively influence height. Recent studies have even suggested that perhaps every gene expressed in a height-related tissue can be influenced by genetic variation and (at least slightly) affect height (26), although it remains to be seen how many genes are actually implicated. Indeed, as sample sizes get larger, the number of variants is increasing much more quickly than the number of loci, suggesting that a limited (but still sizable) number of genes is affected by many variants associated with height. Although each of these variants makes a small contribution to height, in aggregate they can explain a substantial portion of an individual’s height, as will be discussed later. This polygenic architecture represents a substantial departure from the traditional Mendelian model of genetics, where, in any one person, mutations in a single gene are the main determinant of that person’s phenotype. However, the GWAS results are completely consistent with the polygenic model proposed for height by Fisher in 1918 (27).
Illumination of Height-Relevant Tissues and Biological Pathways
Genetic studies offer an unbiased window into the underlying biology of a disease or trait in humans. By uncovering the genes and variants associated with a trait such as height, we can then search for tissues, cell types, genes, and biological pathways that are influenced by these variants. There are many approaches for performing these analyses; here, we outline how three broad approaches of leveraging height GWAS data have helped uncover biological pathways and tissues involved in growth.
The first approach is to scrutinize, as a group, the genes that are near the GWAS-associated variants. This group of genes can be tested for enrichment in previously established pathways or examined for connections or commonalities between these genes (28–30). Using this general approach, studies have shown that genes at GWAS-associated regions show enrichment for signaling pathways with relevance to growth (19,21,23). For example, GWASs have shown strong enrichment for pathways such as the transforming growth factorβ or Hedgehog signaling pathways, which have been previously implicated in growth and growth plate development (21,31,32). As another example, there are GWAS associations at many major components of the growth hormone axis, including associations at genes encodingGH1 (growth hormone),GHR (growth hormone receptor),IGF1 (IGF-1), andIGF1R (IGF-1 receptor) (19,21,23,24,33).
The second approach leverages the observation that most GWAS-associated variants are noncoding and not within the genes themselves but reside in regulatory regions of the genome (9). Because of their noncoding regulatory nature, they are likely to be found in regulatory elements in the genome. Thus, we can examine GWAS-associated variants for their enrichment in regulatory elements specific to tissues of interest. These elements can be profiled by using technologies such as chromatin immunoprecipitation sequencing or Assay for Transposase-Accessible Chromatin using sequencing (34,35). For example, Guoet al. (36) performed the latter test on embryonic murine femoral growth plates to identify growth plate regulatory regions. They identified a strong enrichment of GWAS-associated variants that overlap these regulatory regions specifically in growth plates. Using CRISPR/Cas9 gene editing in human chondrocyte cell lines, they were able to show that one of these regulatory regions that overlaps a GWAS-associated variant regulates expression ofCHSY1, a gene previously implicated in skeletal growth and in growth disorders (36–39). Bernsteinet al. (34) and Trynkaet al. (40) overlapped height GWAS data with publicly available epigenetic data from the ENCODE project. The study identified a strong enrichment of GWAS-associated variants in DNaseI hypersensitivity sites (which reflect regulatory elements) in human embryonic stem cells, suggesting that many GWAS-associated genes act at early stages of tissue or fetal development.
The third approach leverages the hypothesis that GWAS-associated variants likely influence expression of genes that are expressed in relevant tissues (11,41). This is different from the epigenetics-oriented approach, which focuses on the annotations of elements containing the associated variants themselves, and instead focuses on the tissue expression patterns of the nearby genes. The expression patterns of genes differ across tissues but have been profiled extensively using technologies such as RNA-sequencing and microarrays. To demonstrate the specific enrichment of GWAS associations in tissues relevant for height, Luiet al. (42) performed gene expression profiling in dissected murine growth plates. They looked for genes that were spatially or temporally regulated within the growth plate, presuming that those genes are critical for chondrocyte development, a key regulatory process in growth (31). They found a strong enrichment of height GWAS-associated variants near genes that are differentially expressed at the growth plate. Woodet al. (21) and Perset al. (30) took a more comprehensive approach by examining enrichment of genes near height GWAS-associated variants in gene expression profiles of tissues generated across all publicly available gene expression data. Using results from the latest GWAS for height in ∼250,000 individuals, the study was able to prioritize height-relevant tissues, such as cartilage, joint capsule, and synovial membrane, and cell types, such as chondrocytes and mesenchymal stem cells.
GWASs can also help uncover unexpected pathways involved in growth. For example, the exome chip study identified two rare coding variants in the geneSTC2 that increase height, an effect validated by using biological assays of STC2 function (23). STC2 is known to bind and inhibit the proteinase pregnancy-associated plasma protein A (PAPP-A), which cleaves and reduces bioactive levels of IGF–binding protein 4 (IGFBP-4) (43). The rare variants inSTC2 decreased its function, causing a lack of inhibition of PAPP-A, which would presumably lead to increased cleavage of IGFBP-4. Lower levels of intact IGFBP-4 should lead to elevations in free IGF-1 and thus increased stature. Intriguingly,PAPPA andIGFBP4 have both been associated with height by GWASs (21). Moreover, rare mutations inPAPPA2 cause short stature (44). These results highlight how multiple lines of genetic evidence can converge and help implicate new biological pathways in growth. More detailed analyses in the future will likely uncover additional unappreciated pathways involved in human growth.
In addition, there are very likely tissues and pathways involved in growth that are currently poorly captured in terms of curated gene sets, gene expression signatures, or noncoding regulatory profiles. For example, some neuroendocrine tissues, such as the hypothalamus, are not captured as well in these datasets and have not been extensively tested for enrichment with GWAS association results. Undoubtedly, at least some genes expressed in these tissues play a role. For example,GH1 encodes growth hormone, is expressed in the pituitary, and, of course plays a prominent role in growth (33). There is a strong GWAS signal atGH1 (19), but current methods do not highlight the pituitary as a relevant tissue, perhaps because more of the relevant genetic variation affects genes acting in the growth plate. With ever-increasing numbers of loci, curated gene sets, gene expression profiles, epigenetic datasets, and improvement in current computational algorithms, additional tissues enriched for GWAS associations and greater refinement of these tissues will likely be uncovered.
Insights Into Mendelian Growth Disorders
In contrast to GWAS associations, Mendelian growth disorders are typically caused by rare genetic variants in a single gene, usually coding, that have very large effects on stature. The genes at GWAS-associated regions are enriched for genes known to cause human growth disorders and skeletal dysplasias (19,21), includingACAN (45,46),NPR2 (47), andIGF1R (33), among many others. Given that it is often unclear which is the target gene for a GWAS-associated variant, the presence of a Mendelian growth disorder gene at a GWAS-associated region can often help assign a target gene to a GWAS-associated variant. Conversely, the presence of a GWAS association for height can provide additional evidence to support a candidate Mendelian growth disorder gene. For example, using computational methods, Woodet al. (21) and Perset al. (30) were able to nominateCHSY1 as a gene that likely influences height because there were several GWAS-associated variants nearby. Mutations inCHSY1 have been found to cause Temtamy preaxial brachydactyly syndrome, which has growth retardation as a key clinical feature (39,48).
The overlap in regions highlighted by GWASs and Mendelian genetics illustrates an important aspect of the genetic architecture of height: The same genes can be subject to genetic variants of varying effect size and frequency in the population. A GWAS is designed to highlight common variants, which (because they are common) have only small effects on gene expression/function and ultimately height. On the opposite end of the spectrum, individuals with disorders of growth may harbor highly disruptive mutations in the same genes implicated by GWASs. As an example of this concept, at the chromosome 2q35 locus, a common variant (allele frequency ∼10%) ∼10,000 bases away from theIHH gene alters height by 0.041 SD (∼0.29 cm) per allele (19) (Fig. 2). From the exome chip rare variant association study (23), a separate rare missense variant (rs142036701) inIHH, present in 0.2% of people, decreases height by 0.294 SD (2.09 cm). At the extreme, individuals with the rare syndrome of acrocapitofemoral dysplasia carry private, highly deleterious mutations inIHH and have statures 2.3 to 8.6 SDs below the mean (49,50).IHH encodes Indian Hedgehog, a signaling protein with critical roles in chondrocyte development in the growth plate (51). This spectrum of variants and effects on height also reinforces the point that although GWAS variants have small effects on phenotype, the genes targeted by the GWAS variant can have very large biological effects.
Figure 2.

A spectrum of genetic variation atIHH influences height. On the top is theIHH gene, which produces abundant amounts of normally functioning protein (blue dots). The plot in the middle shows the relationship between frequency of variants and effects on height. The red dot in the plot shows an extremely rare variant that causes acrocapitofemoral dysplasia and leads to a 2.3- to 8.6-SD reduction in height. As shown on the bottom left, this mutation leads to a truncated protein with very low expression. The yellow dot in the plot shows a rare variant detected in the rare variant GWAS that has a frequency of approximately 0.2% and causes a 0.294-SD reduction in height. As shown in the bottom middle, this variant alters the protein sequence ofIHH and results in a protein with slightly different functional properties. The green dot in the plot shows a common GWAS variant with frequency of 10%, which leads to a 0.041-SD reduction in height. As shown on the bottom right, this variant is in a regulatory region (not within theIHH gene itself) and influences expression ofIHH.
Insights Into Shared Biology With Other Diseases
GWASs also allow us to probe what other traits and diseases might share a genetic basis with human growth. This could provide insight into how the mechanisms of human growth overlap with the underlying mechanisms of other diseases.
One interesting study evaluated the genetic overlap between height and coronary artery disease (CAD) (52). It had been observed that shorter individuals have higher CAD risk (53,54), but it was not understood whether there was a genetic basis for this observation. The authors used the 180 GWAS signals from Lango Allenet al. (19) and evaluated the effect of these height GWAS-associated variants on CAD risk. The study did indeed find that height GWAS-associated variants in aggregate could influence CAD risk (52). On the basis of statistical analyses, pathways such as transforming growth factorβ and IGF-1 signaling were proposed to underlie this influence of height GWAS-associated variants on CAD risk. The authors proposed that height GWAS variants might influence biological processes that also play a role in atherosclerosis development, resulting in this unexpected shared genetic basis between height and CAD.
Another method, called linkage disequilibrium score regression, allows geneticists to evaluate the proportion of the genetic basis shared by any two traits or diseases (i.e., cross trait heritability) (55). The authors applied this method across hundreds of traits and diseases, including height, and calculated measurements of genetic correlations between each pair of traits or diseases. The method detected expected genetic correlations of height with such traits as the timing of puberty and birthweight, but also with unexpected traits, such as educational attainment and cholesterol levels (Fig. 3). Because more GWASs are being performed with ever-increasing sample sizes and power, these methods should allow for detection of many more genetic overlaps between height and other traits and diseases.
Figure 3.

Cross-trait genetic correlations between adult height and selected (allP < 0.001) additional traits and diseases. GWAS data from Lango Allenet al. (19) used for height. They-axis shows the –log10(p-value) for each correlation. All results downloaded from LD Hub (http://ldsc.broadinstitute.org/).
These studies highlight how little we still know about the underlying basis of height and human growth and how the biological mechanisms of growth are shared by other traits and diseases. By better cataloging the genetic basis of height and performing biological and functional follow-up to better understand the mechanisms of action of the height-associated variants, we will be able to generate insights into how the mechanisms of human growth also influence a wide range of other traits and diseases of medical importance.
GWASs for Other Growth-Related Traits
Many large GWASs have been conducted for adult height; however, given the availability of large sample sizes, other efforts have studied additional aspects of human growth. Chanet al. (56) examined sitting height ratio (SHR; the ratio of upper body to lower body length). Although standing height reflects the cumulative effects of limb length and spine/head length, SHR distinguishes between these components of standing height, and studying the genetics of SHR can identify genes important for limb growth vs axial skeleton growth. This study uncovered six regions of the genome that are statistically significantly associated with SHR. Moreover, of the 180 height GWAS associations from Lango Allenet al. (19), 130 were at least nominally associated (P < 0.05) with SHR. Some of these standing height GWAS associations increase SHR, whereas others decrease SHR, suggesting that different standing height–associated variants preferentially affect limb length vs spine/head length. Interestingly, computational pathway analyses revealed that genes near height GWAS-associated variants that also influence SHR are enriched for expression in bone, cartilage, and growth plate. In contrast, genes near height GWAS associations that do not influence SHR are enriched for embryonic developmental pathways.
Another growth-related trait that has been widely studied is birth weight. The most recent GWAS for birthweight, conducted by the Early Growth Genetics consortium in ∼150,000 individuals, identified 60 associated regions of the genome (57). The study uncovered overlap with adult height, but intriguingly, the study also found a shared genetic basis for birth weight with type 2 diabetes risk, adult systolic blood pressure, and CAD risk. This may suggest that some of the biological mechanisms underlying prenatal growth might also influence risk for adult diseases.
Predicting Height
An important concern for many families is predicting the adult height of their children. Given the influence of genetics on height, it would appear that genetic profiling of patients would offer an ideal approach for predicting adult height. Here, we review what GWASs have informed us about how effective genetics may be for the prediction of height.
The heritability of height, or the proportion of variation in height in the population that can be attributed to genetic variation, is estimated to be 60% to 80%, making it one of the more heritable traits (58–60). However, using the results of the GWAS of 250,000 samples and ∼700 regions of the genome independently associated with height, we are able to explain only 17% of the total variation in height (21). Sophisticated genetic studies have demonstrated that common genetic variants explain much of the heritability of height, and variants that are too rare to be detected by GWAS are likely to contribute less to the proportion of variance in height explained by genetics, at least in the population of individuals with heights close to the normal range (60). GWASs with larger sample sizes and improved power will likely detect some of these additional height-associated variants and increase the proportion of variation in height that can be explained (23).
Chanet al. (61) sought to empirically evaluate how well GWAS variants could explain height. The study used the 180 height-associated GWAS variants from Lango Allenet al. (19) and examined the height predictions in very tall and very short individuals (the top and bottom 1.5th percentile of individuals) from an independent cohort. The study found that the height estimated by using genetic markers was directionally concordant with the actual height. That is, individuals who are tall had on average higher genetically predicted height. Interestingly, the genetic predictions did not perform as well for very short individuals. The authors suggested that on the short extremes of height, individuals may have short stature because of rare variants of larger effect (perhaps as Mendelian genetic causes of short stature) that are not captured well by the GWAS, or perhaps from other nongenetic causes.
To illustrate the limitations of current GWAS results for predicting height, we show the CIs of height predictions using genetics (modeled for a European-ancestry population, where to date the largest studies have been performed) (Fig. 4). Using only a child’s sex, we can predict height with a 95% CI of approximately ±13.9 cm. Leveraging all of the current GWAS results, which collectively explain 17% of variation in height (21), we can narrow the 95% CI of our prediction to ±12.7 cm. Thus, even with extensive information about a child’s genetics, current technologies do not provide much additional benefit in predictions of adult height on a population level, especially if the parental heights are known. In an ideal world where the genetic associations can explain fully the 80% of variation in height (58), we would be able to narrow this 95% CI to ±6.2 cm. However, future GWAS results may confer increasing predictive power, especially for individuals at the extremes of GWAS-predicted heights. Additionally, individuals whose GWAS-predicted height is discordant from their actual height might be more likely to have an undiagnosed underlying growth disorder or chronic disease.
Figure 4.
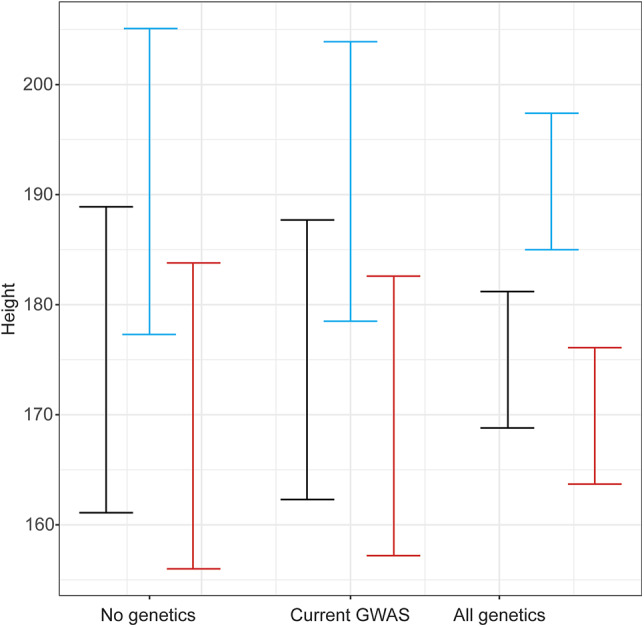
Efficacy of genetic data for the prediction of height. Displayed are the width of the 95% CI for height predictions if no genetics are taken into account, all current GWAS-associated variants (combining to explain 17% of variation) are used, or in an ideal setting where all of variants that influence height are known (combining to explain 80% of variation) and used to predict height. Calculations are based on an average height of 177 cm with an SD of 7.1 cm for men of European ancestry. The black bars are for an individual with an average predicted height (177 cm). The blue bars are for an individual whose predicted height is 2 SDs above the mean (191.2 cm). The red bars are for an individual whose predicted height is 1 SD below the mean (169.9 cm).
An important distinction must be made here. Although in the general population, common variants detected by GWAS are the primary contributors to variation in height, this does not preclude a role of rare variants in an individual. Individuals with extreme heights or those with additional features, such as skeletal anomalies of other congenital malformations, are more likely to have abnormal stature as a result of a severe mutation causing a growth disorder (62). In these individuals, technologies such as whole exome or genome sequencing may uncover mutations in genes that cause these growth disorders, with an appreciable diagnostic yield (63,64). However, the role of these technologies in individuals with extreme stature but without any syndromic features has not been rigorously and systematically explored.
Future Perspectives
Over the past decade, GWASs for height have been highly successful in identifying regions in the genome relevant for growth. They have generated remarkable biological insights, such as implicating the previously underappreciatedSTC2/PAPPA/IGFBP4 pathway. We have also learned about the genetic architecture of height and found that it is incredibly complex: Many thousands of genetic variants each influence height by just a small amount, but when combined, they generate the remarkable normal variation we observe in height, providing a clear model for how polygenic factors combine to regulate a human phenotype. Intriguing genetic overlaps between height and seemingly unrelated traits, such as CAD and educational attainment, have also been found.
There are many continued efforts to perform GWASs for height, in both European populations and other ethnic groups. The sample sizes in these efforts are ever-increasing in size and will likely identify many additional regions of the genome that influence height (26). Additional efforts will also focus on rarer variants, which as demonstrated by Marouliet al. (23) can help directly implicate causal genes because the associated variants are in the coding regions of the genes themselves. With these additional GWAS discoveries, we will likely be able to explain increasing proportions of the observed variation in height (60).
Improved predictive power of GWAS variants could in the near future make the application of GWAS data a useful clinical tool, as has been recently demonstrated with other diseases or traits, such as CAD (65). With improved predictive power, adding a child’s genetic information to parental heights and clinical information may improve the accuracy of predictions of adult height and could perhaps provide opportunities to identify those who may benefit from therapeutic intervention. Genomically predicted height could also assist in the evaluation of children presenting to the clinic with disorders of growth. For example, if a child with short stature has a genomically predicted height that is average or tall, this might suggest an underlying chronic disease that is impairing growth, or a rare genetic cause of short stature. In such a case, the patient would be a better candidate for more intensive clinical evaluation that could eventually include whole exome or genome sequencing. By contrast, if the genomically predicted height is short and thus consistent with the child’s short stature, additional evaluation might be of lower yield because the short stature can be explained by the child’s complement of naturally occurring common genetic variants. Testing whether incorporation of GWAS findings in the clinic is a useful tool represents an important area of future investigation.
An important but more distant clinical application of GWAS findings is the discovery of new therapeutics for growth disorders. GWASs for height have clearly identified genes, tissues, and pathways that influence growth. Many of these genes might serve as prime therapeutic targets (66). GWAS findings are already yielding dividends in the treatment of other Mendelian conditions, such as sickle cell anemia (67) and familial hypercholesterolemia (68). Whether similar success stories for growth disorders will emerge from GWASs for height remains to be seen.
Another major area of future investigation will be biological interrogation of height-associated genes. Functional follow-up of GWAS findings is challenging, especially for a phenotype such as height. Although overall tissue enrichments can be inferred as described above, the tissue implicated for each locus may still not be apparent. Because of the noncoding nature of GWAS variants, the causal gene for each locus may also be quite far from the GWAS-associated variants and thus difficult to discern (11,12). Growth is also a difficult phenotype to study functionally because many aspects of growth are not cell-autonomous, often necessitating expensive and time-consuming model organism studies, and robust and tractablein vitro models of the growth plate are not yet readily available. Despite these challenges, functional studies of height GWAS loci are beginning to yield dividends. For example, recent work using transgenic mouse lines and CRISPR/Cas9 genome editing showed how regulatory regions influence expression of the geneGDF5 and thereby affect growth plate development and ultimately height (69). Future work leveraging technologies such as CRISPR/Cas9, as well as improved gene expression and epigenetic profiles, will help scientists translate GWAS findings intonovel biological insights into human growth and development. These insights into the biology of growth could play an important role in the translation of genetic findings into therapies for disorders of growth.
Acknowledgments
Current Affiliation: A. Dauber’s current affiliation is the Division of Endocrinology, Children’s National Medical Center, Washington, DC 20010.
Disclosure Summary: The authors have nothing to disclose.
Glossary
Abbreviations:
- CAD
coronary artery disease
- GIANT
Genetic Investigation of ANthropometric Traits
- GWAS
genome-wide association study
- IGFBP-4
IGF–binding protein 4
- LDL
low-density lipoprotein
- PAPP-A
pregnancy-associated plasma protein A
- SHR
sitting height ratio
References
- 1. Galton F.Regression towards mediocrity in hereditary stature.J Anthropol Inst G B Irel.1886;15:246–263. [Google Scholar]
- 2. Altshuler D,Daly MJ,Lander E Genetic mapping in human disease.Science (80-.).2009;322(5903):881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pe’er I,Yelensky R,Altshuler D,Daly MJ. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants.Genet Epidemiol.2008;32(4):381–385. [DOI] [PubMed] [Google Scholar]
- 4. Visscher PM,Wray NR,Zhang Q,Sklar P,McCarthy MI,Brown MA,Yang J. 10 years of GWAS discovery: biology, function, and translation.Am J Hum Genet.2017;101(1):5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McClellan J,King M-C. Genetic heterogeneity in human disease.Cell.2010;141(2):210–217. [DOI] [PubMed] [Google Scholar]
- 6. Goldstein DB.Common genetic variation and human traits.N Engl J Med.2009;360(17):1696–1698. [DOI] [PubMed] [Google Scholar]
- 7. Teslovich TM,Musunuru K,Smith AV,Edmondson AC,Stylianou IM,Koseki M,Pirruccello JP,Ripatti S,Chasman DI,Willer CJ,Johansen CT,Fouchier SW,Isaacs A,Peloso GM,Barbalic M,Ricketts SL,Bis JC,Aulchenko YS,Thorleifsson G,Feitosa MF,Chambers J,Orho-Melander M,Melander O,Johnson T,Li X,Guo X,Li M,Shin Cho Y,Jin Go M,Jin Kim Y,Lee JY,Park T,Kim K,Sim X,Twee-Hee Ong R,Croteau-Chonka DC,Lange LA,Smith JD,Song K,Hua Zhao J,Yuan X,Luan J,Lamina C,Ziegler A,Zhang W,Zee RY,Wright AF,Witteman JC,Wilson JF,Willemsen G,Wichmann HE,Whitfield JB,Waterworth DM,Wareham NJ,Waeber G,Vollenweider P,Voight BF,Vitart V,Uitterlinden AG,Uda M,Tuomilehto J,Thompson JR,Tanaka T,Surakka I,Stringham HM,Spector TD,Soranzo N,Smit JH,Sinisalo J,Silander K,Sijbrands EJ,Scuteri A,Scott J,Schlessinger D,Sanna S,Salomaa V,Saharinen J,Sabatti C,Ruokonen A,Rudan I,Rose LM,Roberts R,Rieder M,Psaty BM,Pramstaller PP,Pichler I,Perola M,Penninx BW,Pedersen NL,Pattaro C,Parker AN,Pare G,Oostra BA,O’Donnell CJ,Nieminen MS,Nickerson DA,Montgomery GW,Meitinger T,McPherson R,McCarthy MI,McArdle W,Masson D,Martin NG,Marroni F,Mangino M,Magnusson PK,Lucas G,Luben R,Loos RJ,Lokki ML,Lettre G,Langenberg C,Launer LJ,Lakatta EG,Laaksonen R,Kyvik KO,Kronenberg F,König IR,Khaw KT,Kaprio J,Kaplan LM,Johansson A,Jarvelin MR,Janssens AC,Ingelsson E,Igl W,Kees Hovingh G,Hottenga JJ,Hofman A,Hicks AA,Hengstenberg C,Heid IM,Hayward C,Havulinna AS,Hastie ND,Harris TB,Haritunians T,Hall AS,Gyllensten U,Guiducci C,Groop LC,Gonzalez E,Gieger C,Freimer NB,Ferrucci L,Erdmann J,Elliott P,Ejebe KG,Döring A,Dominiczak AF,Demissie S,Deloukas P,de Geus EJ,de Faire U,Crawford G,Collins FS,Chen YD,Caulfield MJ,Campbell H,Burtt NP,Bonnycastle LL,Boomsma DI,Boekholdt SM,Bergman RN,Barroso I,Bandinelli S,Ballantyne CM,Assimes TL,Quertermous T,Altshuler D,Seielstad M,Wong TY,Tai ES,Feranil AB,Kuzawa CW,Adair LS,Taylor HA Jr,Borecki IB,Gabriel SB,Wilson JG,Holm H,Thorsteinsdottir U,Gudnason V,Krauss RM,Mohlke KL,Ordovas JM,Munroe PB,Kooner JS,Tall AR,Hegele RA,Kastelein JJ,Schadt EE,Rotter JI,Boerwinkle E,Strachan DP,Mooser V,Stefansson K,Reilly MP,Samani NJ,Schunkert H,Cupples LA,Sandhu MS,Ridker PM,Rader DJ,van Duijn CM,Peltonen L,Abecasis GR,Boehnke M,Kathiresan S. Biological, clinical and population relevance of 95 loci for blood lipids.Nature.2010;466(7307):707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tobert JA.Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors.Nat Rev Drug Discov.2003;2(7):517–526. [DOI] [PubMed] [Google Scholar]
- 9. Gusev A,Lee SH,Trynka G,Finucane H,Vilhjálmsson BJ,Xu H,Zang C,Ripke S,Bulik-Sullivan B,Stahl E,Kähler AK,Hultman CM,Purcell SM,McCarroll SA,Daly M,Pasaniuc B,Sullivan PF,Neale BM,Wray NR,Raychaudhuri S,Price AL;Schizophrenia Working Group of the Psychiatric Genomics Consortium . Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases.Am J Hum Genet.2014;95(5):535–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Raychaudhuri S.Mapping rare and common causal alleles for complex human diseases.Cell.2011;147(1):57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spain SL,Barrett JC. Strategies for fine-mapping complex traits.Hum Mol Genet.2015;24(R1):R111–R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gallagher MD,Chen-Plotkin AS. The post-GWAS era: from association to function.Am J Hum Genet.2018;102(5):717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Visscher PM,Brown MA,McCarthy MI,Yang J. Five years of GWAS discovery.Am J Hum Genet.2012;90(1):7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hammond SM,Sharpless NE. HMGA2, microRNAs, and stem cell aging.Cell.2008;135(6):1013–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhou X,Benson KF,Ashar HR,Chada K. Mutation responsible for the mouse pygmy phenotype in the developmentally regulated factor HMGI-C.Nature.1995;376(6543):771–774. [DOI] [PubMed] [Google Scholar]
- 16. Arlotta P,Tai AK,Manfioletti G,Clifford C,Jay G,Ono SJ. Transgenic mice expressing a truncated form of the high mobility group I-C protein develop adiposity and an abnormally high prevalence of lipomas.J Biol Chem.2000;275(19):14394–14400. [DOI] [PubMed] [Google Scholar]
- 17. Alyaqoub F,Pyatt RE,Bailes A,Brock A,Deeg C,McKinney A,Astbury C,Reshmi S,Shane KP,Thrush DL,Sommer A,Gastier-Foster JM. 12q14 microdeletion associated withHMGA2 gene disruption and growth restriction.Am J Med Genet A.2012;158A(11):2925–2930. [DOI] [PubMed] [Google Scholar]
- 18. Ligon AH,Moore SDP,Parisi MA,Mealiffe ME,Harris DJ,Ferguson HL,Quade BJ,Morton CC. Constitutional rearrangement of the architectural factor HMGA2: a novel human phenotype including overgrowth and lipomas.Am J Hum Genet.2005;76(2):340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lango Allen H,Estrada K,Lettre G,Berndt SI,Weedon MN,Rivadeneira F,Willer CJ,Jackson AU,Vedantam S,Raychaudhuri S,Ferreira T,Wood AR,Weyant RJ,Segrè AV,Speliotes EK,Wheeler E,Soranzo N,Park JH,Yang J,Gudbjartsson D,Heard-Costa NL,Randall JC,Qi L,Vernon Smith A,Mägi R,Pastinen T,Liang L,Heid IM,Luan J,Thorleifsson G,Winkler TW,Goddard ME,Sin Lo K,Palmer C,Workalemahu T,Aulchenko YS,Johansson A,Zillikens MC,Feitosa MF,Esko T,Johnson T,Ketkar S,Kraft P,Mangino M,Prokopenko I,Absher D,Albrecht E,Ernst F,Glazer NL,Hayward C,Hottenga JJ,Jacobs KB,Knowles JW,Kutalik Z,Monda KL,Polasek O,Preuss M,Rayner NW,Robertson NR,Steinthorsdottir V,Tyrer JP,Voight BF,Wiklund F,Xu J,Zhao JH,Nyholt DR,Pellikka N,Perola M,Perry JR,Surakka I,Tammesoo ML,Altmaier EL,Amin N,Aspelund T,Bhangale T,Boucher G,Chasman DI,Chen C,Coin L,Cooper MN,Dixon AL,Gibson Q,Grundberg E,Hao K,Juhani Junttila M,Kaplan LM,Kettunen J,König IR,Kwan T,Lawrence RW,Levinson DF,Lorentzon M,McKnight B,Morris AP,Müller M,Suh Ngwa J,Purcell S,Rafelt S,Salem RM,Salvi E,Sanna S,Shi J,Sovio U,Thompson JR,Turchin MC,Vandenput L,Verlaan DJ,Vitart V,White CC,Ziegler A,Almgren P,Balmforth AJ,Campbell H,Citterio L,De Grandi A,Dominiczak A,Duan J,Elliott P,Elosua R,Eriksson JG,Freimer NB,Geus EJ,Glorioso N,Haiqing S,Hartikainen AL,Havulinna AS,Hicks AA,Hui J,Igl W,Illig T,Jula A,Kajantie E,Kilpeläinen TO,Koiranen M,Kolcic I,Koskinen S,Kovacs P,Laitinen J,Liu J,Lokki ML,Marusic A,Maschio A,Meitinger T,Mulas A,Paré G,Parker AN,Peden JF,Petersmann A,Pichler I,Pietiläinen KH,Pouta A,Ridderstråle M,Rotter JI,Sambrook JG,Sanders AR,Schmidt CO,Sinisalo J,Smit JH,Stringham HM,Bragi Walters G,Widen E,Wild SH,Willemsen G,Zagato L,Zgaga L,Zitting P,Alavere H,Farrall M,McArdle WL,Nelis M,Peters MJ,Ripatti S,van Meurs JB,Aben KK,Ardlie KG,Beckmann JS,Beilby JP,Bergman RN,Bergmann S,Collins FS,Cusi D,den Heijer M,Eiriksdottir G,Gejman PV,Hall AS,Hamsten A,Huikuri HV,Iribarren C,Kähönen M,Kaprio J,Kathiresan S,Kiemeney L,Kocher T,Launer LJ,Lehtimäki T,Melander O,Mosley TH Jr,Musk AW,Nieminen MS,O’Donnell CJ,Ohlsson C,Oostra B,Palmer LJ,Raitakari O,Ridker PM,Rioux JD,Rissanen A,Rivolta C,Schunkert H,Shuldiner AR,Siscovick DS,Stumvoll M,Tönjes A,Tuomilehto J,van Ommen GJ,Viikari J,Heath AC,Martin NG,Montgomery GW,Province MA,Kayser M,Arnold AM,Atwood LD,Boerwinkle E,Chanock SJ,Deloukas P,Gieger C,Grönberg H,Hall P,Hattersley AT,Hengstenberg C,Hoffman W,Lathrop GM,Salomaa V,Schreiber S,Uda M,Waterworth D,Wright AF,Assimes TL,Barroso I,Hofman A,Mohlke KL,Boomsma DI,Caulfield MJ,Cupples LA,Erdmann J,Fox CS,Gudnason V,Gyllensten U,Harris TB,Hayes RB,Jarvelin MR,Mooser V,Munroe PB,Ouwehand WH,Penninx BW,Pramstaller PP,Quertermous T,Rudan I,Samani NJ,Spector TD,Völzke H,Watkins H,Wilson JF,Groop LC,Haritunians T,Hu FB,Kaplan RC,Metspalu A,North KE,Schlessinger D,Wareham NJ,Hunter DJ,O’Connell JR,Strachan DP,Wichmann HE,Borecki IB,van Duijn CM,Schadt EE,Thorsteinsdottir U,Peltonen L,Uitterlinden AG,Visscher PM,Chatterjee N,Loos RJ,Boehnke M,McCarthy MI,Ingelsson E,Lindgren CM,Abecasis GR,Stefansson K,Frayling TM,Hirschhorn JN. Hundreds of variants clustered in genomic loci and biological pathways affect human height.Nature.2010;467(7317):832–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Filion GJP,Zhenilo S,Salozhin S,Yamada D,Prokhortchouk E,Defossez P-A. A family of human zinc finger proteins that bind methylated DNA and repress transcription.Mol Cell Biol.2006;26(1):169–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wood AR,Esko T,Yang J,Vedantam S,Pers TH,Gustafsson S,Chu AY,Estrada K,Luan J,Kutalik Z,Amin N,Buchkovich ML,Croteau-Chonka DC,Day FR,Duan Y,Fall T,Fehrmann R,Ferreira T,Jackson AU,Karjalainen J,Lo KS,Locke AE,Mägi R,Mihailov E,Porcu E,Randall JC,Scherag A,Vinkhuyzen AA,Westra HJ,Winkler TW,Workalemahu T,Zhao JH,Absher D,Albrecht E,Anderson D,Baron J,Beekman M,Demirkan A,Ehret GB,Feenstra B,Feitosa MF,Fischer K,Fraser RM,Goel A,Gong J,Justice AE,Kanoni S,Kleber ME,Kristiansson K,Lim U,Lotay V,Lui JC,Mangino M,Mateo Leach I,Medina-Gomez C,Nalls MA,Nyholt DR,Palmer CD,Pasko D,Pechlivanis S,Prokopenko I,Ried JS,Ripke S,Shungin D,Stancáková A,Strawbridge RJ,Sung YJ,Tanaka T,Teumer A,Trompet S,van der Laan SW,van Setten J,Van Vliet-Ostaptchouk JV,Wang Z,Yengo L,Zhang W,Afzal U,Arnlöv J,Arscott GM,Bandinelli S,Barrett A,Bellis C,Bennett AJ,Berne C,Blüher M,Bolton JL,Böttcher Y,Boyd HA,Bruinenberg M,Buckley BM,Buyske S,Caspersen IH,Chines PS,Clarke R,Claudi-Boehm S,Cooper M,Daw EW,De Jong PA,Deelen J,Delgado G,Denny JC,Dhonukshe-Rutten R,Dimitriou M,Doney AS,Dörr M,Eklund N,Eury E,Folkersen L,Garcia ME,Geller F,Giedraitis V,Go AS,Grallert H,Grammer TB,Gräßler J,Grönberg H,de Groot LC,Groves CJ,Haessler J,Hall P,Haller T,Hallmans G,Hannemann A,Hartman CA,Hassinen M,Hayward C,Heard-Costa NL,Helmer Q,Hemani G,Henders AK,Hillege HL,Hlatky MA,Hoffmann W,Hoffmann P,Holmen O,Houwing-Duistermaat JJ,Illig T,Isaacs A,James AL,Jeff J,Johansen B,Johansson Å,Jolley J,Juliusdottir T,Junttila J,Kho AN,Kinnunen L,Klopp N,Kocher T,Kratzer W,Lichtner P,Lind L,Lindström J,Lobbens S,Lorentzon M,Lu Y,Lyssenko V,Magnusson PK,Mahajan A,Maillard M,McArdle WL,McKenzie CA,McLachlan S,McLaren PJ,Menni C,Merger S,Milani L,Moayyeri A,Monda KL,Morken MA,Müller G,Müller-Nurasyid M,Musk AW,Narisu N,Nauck M,Nolte IM,Nöthen MM,Oozageer L,Pilz S,Rayner NW,Renstrom F,Robertson NR,Rose LM,Roussel R,Sanna S,Scharnagl H,Scholtens S,Schumacher FR,Schunkert H,Scott RA,Sehmi J,Seufferlein T,Shi J,Silventoinen K,Smit JH,Smith AV,Smolonska J,Stanton AV,Stirrups K,Stott DJ,Stringham HM,Sundström J,Swertz MA,Syvänen AC,Tayo BO,Thorleifsson G,Tyrer JP,van Dijk S,van Schoor NM,van der Velde N,van Heemst D,van Oort FV,Vermeulen SH,Verweij N,Vonk JM,Waite LL,Waldenberger M,Wennauer R,Wilkens LR,Willenborg C,Wilsgaard T,Wojczynski MK,Wong A,Wright AF,Zhang Q,Arveiler D,Bakker SJ,Beilby J,Bergman RN,Bergmann S,Biffar R,Blangero J,Boomsma DI,Bornstein SR,Bovet P,Brambilla P,Brown MJ,Campbell H,Caulfield MJ,Chakravarti A,Collins R,Collins FS,Crawford DC,Cupples LA,Danesh J,de Faire U,den Ruijter HM,Erbel R,Erdmann J,Eriksson JG,Farrall M,Ferrannini E,Ferrières J,Ford I,Forouhi NG,Forrester T,Gansevoort RT,Gejman PV,Gieger C,Golay A,Gottesman O,Gudnason V,Gyllensten U,Haas DW,Hall AS,Harris TB,Hattersley AT,Heath AC,Hengstenberg C,Hicks AA,Hindorff LA,Hingorani AD,Hofman A,Hovingh GK,Humphries SE,Hunt SC,Hypponen E,Jacobs KB,Jarvelin MR,Jousilahti P,Jula AM,Kaprio J,Kastelein JJ,Kayser M,Kee F,Keinanen-Kiukaanniemi SM,Kiemeney LA,Kooner JS,Kooperberg C,Koskinen S,Kovacs P,Kraja AT,Kumari M,Kuusisto J,Lakka TA,Langenberg C,Le Marchand L,Lehtimäki T,Lupoli S,Madden PA,Männistö S,Manunta P,Marette A,Matise TC,McKnight B,Meitinger T,Moll FL,Montgomery GW,Morris AD,Morris AP,Murray JC,Nelis M,Ohlsson C,Oldehinkel AJ,Ong KK,Ouwehand WH,Pasterkamp G,Peters A,Pramstaller PP,Price JF,Qi L,Raitakari OT,Rankinen T,Rao DC,Rice TK,Ritchie M,Rudan I,Salomaa V,Samani NJ,Saramies J,Sarzynski MA,Schwarz PE,Sebert S,Sever P,Shuldiner AR,Sinisalo J,Steinthorsdottir V,Stolk RP,Tardif JC,Tönjes A,Tremblay A,Tremoli E,Virtamo J,Vohl MC,Amouyel P,Asselbergs FW,Assimes TL,Bochud M,Boehm BO,Boerwinkle E,Bottinger EP,Bouchard C,Cauchi S,Chambers JC,Chanock SJ,Cooper RS,de Bakker PI,Dedoussis G,Ferrucci L,Franks PW,Froguel P,Groop LC,Haiman CA,Hamsten A,Hayes MG,Hui J,Hunter DJ,Hveem K,Jukema JW,Kaplan RC,Kivimaki M,Kuh D,Laakso M,Liu Y,Martin NG,März W,Melbye M,Moebus S,Munroe PB,Njølstad I,Oostra BA,Palmer CN,Pedersen NL,Perola M,Pérusse L,Peters U,Powell JE,Power C,Quertermous T,Rauramaa R,Reinmaa E,Ridker PM,Rivadeneira F,Rotter JI,Saaristo TE,Saleheen D,Schlessinger D,Slagboom PE,Snieder H,Spector TD,Strauch K,Stumvoll M,Tuomilehto J,Uusitupa M,van der Harst P,Völzke H,Walker M,Wareham NJ,Watkins H,Wichmann HE,Wilson JF,Zanen P,Deloukas P,Heid IM,Lindgren CM,Mohlke KL,Speliotes EK,Thorsteinsdottir U,Barroso I,Fox CS,North KE,Strachan DP,Beckmann JS,Berndt SI,Boehnke M,Borecki IB,McCarthy MI,Metspalu A,Stefansson K,Uitterlinden AG,van Duijn CM,Franke L,Willer CJ,Price AL,Lettre G,Loos RJ,Weedon MN,Ingelsson E,O’Connell JR,Abecasis GR,Chasman DI,Goddard ME,Visscher PM,Hirschhorn JN,Frayling TM Electronic Medical Records and Genomics (eMEMERGEGE) ConsortiumMIGen ConsortiumPAGEGE ConsortiumLifeLines Cohort Study . Defining the role of common variation in the genomic and biological architecture of adult human height.Nat Genet.2014;46(11):1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Timpson NJ,Greenwood CMT,Soranzo N,Lawson DJ,Richards JB. Genetic architecture: the shape of the genetic contribution to human traits and disease.Nat Rev Genet.2018;19(2):110–124. [DOI] [PubMed] [Google Scholar]
- 23. Marouli E,Graff M,Medina-Gomez C,Lo KS,Wood AR,Kjaer TR,Fine RS,Lu Y,Schurmann C,Highland HM,Rüeger S,Thorleifsson G,Justice AE,Lamparter D,Stirrups KE,Turcot V,Young KL,Winkler TW,Esko T,Karaderi T,Locke AE,Masca NG,Ng MC,Mudgal P,Rivas MA,Vedantam S,Mahajan A,Guo X,Abecasis G,Aben KK,Adair LS,Alam DS,Albrecht E,Allin KH,Allison M,Amouyel P,Appel EV,Arveiler D,Asselbergs FW,Auer PL,Balkau B,Banas B,Bang LE,Benn M,Bergmann S,Bielak LF,Blüher M,Boeing H,Boerwinkle E,Böger CA,Bonnycastle LL,Bork-Jensen J,Bots ML,Bottinger EP,Bowden DW,Brandslund I,Breen G,Brilliant MH,Broer L,Burt AA,Butterworth AS,Carey DJ,Caulfield MJ,Chambers JC,Chasman DI,Chen YI,Chowdhury R,Christensen C,Chu AY,Cocca M,Collins FS,Cook JP,Corley J,Galbany JC,Cox AJ,Cuellar-Partida G,Danesh J,Davies G,de Bakker PI,de Borst GJ,de Denus S,de Groot MC,de Mutsert R,Deary IJ,Dedoussis G,Demerath EW,den Hollander AI,Dennis JG,Di Angelantonio E,Drenos F,Du M,Dunning AM,Easton DF,Ebeling T,Edwards TL,Ellinor PT,Elliott P,Evangelou E,Farmaki AE,Faul JD,Feitosa MF,Feng S,Ferrannini E,Ferrario MM,Ferrieres J,Florez JC,Ford I,Fornage M,Franks PW,Frikke-Schmidt R,Galesloot TE,Gan W,Gandin I,Gasparini P,Giedraitis V,Giri A,Girotto G,Gordon SD,Gordon-Larsen P,Gorski M,Grarup N,Grove ML,Gudnason V,Gustafsson S,Hansen T,Harris KM,Harris TB,Hattersley AT,Hayward C,He L,Heid IM,Heikkilä K,Helgeland Ø,Hernesniemi J,Hewitt AW,Hocking LJ,Hollensted M,Holmen OL,Hovingh GK,Howson JM,Hoyng CB,Huang PL,Hveem K,Ikram MA,Ingelsson E,Jackson AU,Jansson JH,Jarvik GP,Jensen GB,Jhun MA,Jia Y,Jiang X,Johansson S,Jørgensen ME,Jørgensen T,Jousilahti P,Jukema JW,Kahali B,Kahn RS,Kähönen M,Kamstrup PR,Kanoni S,Kaprio J,Karaleftheri M,Kardia SL,Karpe F,Kee F,Keeman R,Kiemeney LA,Kitajima H,Kluivers KB,Kocher T,Komulainen P,Kontto J,Kooner JS,Kooperberg C,Kovacs P,Kriebel J,Kuivaniemi H,Küry S,Kuusisto J,La Bianca M,Laakso M,Lakka TA,Lange EM,Lange LA,Langefeld CD,Langenberg C,Larson EB,Lee IT,Lehtimäki T,Lewis CE,Li H,Li J,Li-Gao R,Lin H,Lin LA,Lin X,Lind L,Lindström J,Linneberg A,Liu Y,Liu Y,Lophatananon A,Luan J,Lubitz SA,Lyytikäinen LP,Mackey DA,Madden PA,Manning AK,Männistö S,Marenne G,Marten J,Martin NG,Mazul AL,Meidtner K,Metspalu A,Mitchell P,Mohlke KL,Mook-Kanamori DO,Morgan A,Morris AD,Morris AP,Müller-Nurasyid M,Munroe PB,Nalls MA,Nauck M,Nelson CP,Neville M,Nielsen SF,Nikus K,Njølstad PR,Nordestgaard BG,Ntalla I,O’Connel JR,Oksa H,Loohuis LM,Ophoff RA,Owen KR,Packard CJ,Padmanabhan S,Palmer CN,Pasterkamp G,Patel AP,Pattie A,Pedersen O,Peissig PL,Peloso GM,Pennell CE,Perola M,Perry JA,Perry JR,Person TN,Pirie A,Polasek O,Posthuma D,Raitakari OT,Rasheed A,Rauramaa R,Reilly DF,Reiner AP,Renström F,Ridker PM,Rioux JD,Robertson N,Robino A,Rolandsson O,Rudan I,Ruth KS,Saleheen D,Salomaa V,Samani NJ,Sandow K,Sapkota Y,Sattar N,Schmidt MK,Schreiner PJ,Schulze MB,Scott RA,Segura-Lepe MP,Shah S,Sim X,Sivapalaratnam S,Small KS,Smith AV,Smith JA,Southam L,Spector TD,Speliotes EK,Starr JM,Steinthorsdottir V,Stringham HM,Stumvoll M,Surendran P,’t Hart LM,Tansey KE,Tardif JC,Taylor KD,Teumer A,Thompson DJ,Thorsteinsdottir U,Thuesen BH,Tönjes A,Tromp G,Trompet S,Tsafantakis E,Tuomilehto J,Tybjaerg-Hansen A,Tyrer JP,Uher R,Uitterlinden AG,Ulivi S,van der Laan SW,Van Der Leij AR,van Duijn CM,van Schoor NM,van Setten J,Varbo A,Varga TV,Varma R,Edwards DR,Vermeulen SH,Vestergaard H,Vitart V,Vogt TF,Vozzi D,Walker M,Wang F,Wang CA,Wang S,Wang Y,Wareham NJ,Warren HR,Wessel J,Willems SM,Wilson JG,Witte DR,Woods MO,Wu Y,Yaghootkar H,Yao J,Yao P,Yerges-Armstrong LM,Young R,Zeggini E,Zhan X,Zhang W,Zhao JH,Zhao W,Zhao W,Zheng H,Zhou W,Rotter JI,Boehnke M,Kathiresan S,McCarthy MI,Willer CJ,Stefansson K,Borecki IB,Liu DJ,North KE,Heard-Costa NL,Pers TH,Lindgren CM,Oxvig C,Kutalik Z,Rivadeneira F,Loos RJ,Frayling TM,Hirschhorn JN,Deloukas P,Lettre G EPIC-InterAct ConsortiumCHD Exome+ ConsortiumExomeBP ConsortiumT2D-Genes ConsortiumGoT2D Genes ConsortiumGlobal Lipids Genetics ConsortiumReproGen ConsortiumMAGIC Investigators . Rare and low-frequency coding variants alter human adult height.Nature.2017;542(7640):186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He M,Xu M,Zhang B,Liang J,Chen P,Lee JY,Johnson TA,Li H,Yang X,Dai J,Liang L,Gui L,Qi Q,Huang J,Li Y,Adair LS,Aung T,Cai Q,Cheng CY,Cho MC,Cho YS,Chu M,Cui B,Gao YT,Go MJ,Gu D,Gu W,Guo H,Hao Y,Hong J,Hu Z,Hu Y,Huang J,Hwang JY,Ikram MK,Jin G,Kang DH,Khor CC,Kim BJ,Kim HT,Kubo M,Lee J,Lee J,Lee NR,Li R,Li J,Liu J,Longe J,Lu W,Lu X,Miao X,Okada Y,Ong RT,Qiu G,Seielstad M,Sim X,Song H,Takeuchi F,Tanaka T,Taylor PR,Wang L,Wang W,Wang Y,Wu C,Wu Y,Xiang YB,Yamamoto K,Yang H,Liao M,Yokota M,Young T,Zhang X,Kato N,Wang QK,Zheng W,Hu FB,Lin D,Shen H,Teo YY,Mo Z,Wong TY,Lin X,Mohlke KL,Ning G,Tsunoda T,Han BG,Shu XO,Tai ES,Wu T,Qi L. Meta-analysis of genome-wide association studies of adult height in East Asians identifies 17 novel loci.Hum Mol Genet.2015;24(6):1791–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carty CL,Johnson NA,Hutter CM,Reiner AP,Peters U,Tang H,Kooperberg C. Genome-wide association study of body height in African Americans: the Women’s Health Initiative SNP Health Association Resource (SHARe).Hum Mol Genet.2012;21(3):711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boyle EA,Li YI,Pritchard JK. An expanded view of complex traits: from polygenic to omnigenic.Cell.2017;169(7):1177–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher RA.The correlation between relatives on the supposition of Mendelian inheritance.Trans R Soc Edinb.1918;52(2):399–433. [Google Scholar]
- 28. Segrè AV,Wei N,Altshuler D,Florez JC,Altshuler D,Florez JC;DIAGRAM Consortium; MAGIC Investigators . Pathways targeted by antidiabetes drugs are enriched for multiple genes associated with type 2 diabetes risk.Diabetes.2015;64(4):1470–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raychaudhuri S, Plenge RM, Rossin EJ, Ng ACY, Purcell SM, Sklar P, Scolnick EM, Xavier RJ, Altshuler D, Daly MJ, Daly MJ. Identifying relationships among genomic disease regions: predicting genes at pathogenic SNP associations and rare deletions. PLoS Genet. 2009;5(6):e1000534. [DOI] [PMC free article] [PubMed]
- 30. Pers TH,Karjalainen JM,Chan Y,Westra HJ,Wood AR,Yang J,Lui JC,Vedantam S,Gustafsson S,Esko T,Frayling T,Speliotes EK,Boehnke M,Raychaudhuri S,Fehrmann RSN,Hirschhorn JN,Franke L;Genetic Investigation of ANthropometric Traits (GIANT) Consortium . Biological interpretation of genome-wide association studies using predicted gene functions.Nat Commun.2015;6(1):5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mackie EJ,Tatarczuch L,Mirams M. The skeleton: a multi-functional complex organ: the growth plate chondrocyte and endochondral ossification.J Endocrinol.2011;211(2):109–121. [DOI] [PubMed] [Google Scholar]
- 32. Samsa WE,Zhou X,Zhou G. Signaling pathways regulating cartilage growth plate formation and activity.Semin Cell Dev Biol.2017;62:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baron J,Sävendahl L,De Luca F,Dauber A,Phillip M,Wit JM,Nilsson O. Short and tall stature: a new paradigm emerges.Nat Rev Endocrinol.2015;11(12):735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bernstein BE,Birney E,Dunham I,Green ED,Gunter C,Snyder M;ENCODE Project Consortium . An integrated encyclopedia of DNA elements in the human genome.Nature.2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Buenrostro JD,Giresi PG,Zaba LC,Chang HY,Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position.Nat Methods.2013;10(12):1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo M,Liu Z,Willen J,Shaw CP,Richard D,Jagoda E,Doxey AC,Hirschhorn J,Capellini TD. Epigenetic profiling of growth plate chondrocytes sheds insight into regulatory genetic variation influencing height.eLife.2017;6:e29329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wilson DG,Phamluong K,Lin WY,Barck K,Carano RAD,Diehl L,Peterson AS,Martin F,Solloway MJ. Chondroitin sulfate synthase 1 (Chsy1) is required for bone development and digit patterning.Dev Biol.2012;363(2):413–425. [DOI] [PubMed] [Google Scholar]
- 38. Tian J,Ling L,Shboul M,Lee H,O’Connor B,Merriman B,Nelson SF,Cool S,Ababneh OH,Al-Hadidy A,Masri A,Hamamy H,Reversade B. Loss of CHSY1, a secreted FRINGE enzyme, causes syndromic brachydactyly in humans via increased NOTCH signaling.Am J Hum Genet.2010;87(6):768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y,Laue K,Temtamy S,Aglan M,Kotan LD,Yigit G,Canan H,Pawlik B,Nürnberg G,Wakeling EL,Quarrell OW,Baessmann I,Lanktree MB,Yilmaz M,Hegele RA,Amr K,May KW,Nürnberg P,Topaloglu AK,Hammerschmidt M,Wollnik B. Temtamy preaxial brachydactyly syndrome is caused by loss-of-function mutations in chondroitin synthase 1, a potential target of BMP signaling.Am J Hum Genet.2010;87(6):757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Trynka G,Westra H-J,Slowikowski K,Hu X,Xu H,Stranger BE,Klein RJ,Han B,Raychaudhuri S. Disentangling the effects of colocalizing genomic annotations to functionally prioritize non-coding variants within complex-trait loci.Am J Hum Genet.2015;97(1):139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ongen H,Brown AA,Delaneau O,Panousis NI,Nica AC,Dermitzakis ET;GTEx Consortium . Estimating the causal tissues for complex traits and diseases.Nat Genet.2017;49(12):1676–1683. [DOI] [PubMed] [Google Scholar]
- 42. Lui JC,Nilsson O,Chan Y,Palmer CD,Andrade AC,Hirschhorn JN,Baron J. Synthesizing genome-wide association studies and expression microarray reveals novel genes that act in the human growth plate to modulate height.Hum Mol Genet.2012;21(23):5193–5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jepsen MR,Kløverpris S,Mikkelsen JH,Pedersen JH,Füchtbauer EM,Laursen LS,Oxvig C. Stanniocalcin-2 inhibits mammalian growth by proteolytic inhibition of the insulin-like growth factor axis.J Biol Chem.2015;290(6):3430–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dauber A,Muñoz-Calvo MT,Barrios V,Domené HM,Kloverpris S,Serra-Juhé C,Desikan V,Pozo J,Muzumdar R,Martos-Moreno GA,Hawkins F,Jasper HG,Conover CA,Frystyk J,Yakar S,Hwa V,Chowen JA,Oxvig C,Rosenfeld RG,Pérez-Jurado LA,Argente J. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability.EMBO Mol Med.2016;8(4):363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nilsson O,Guo MH,Dunbar N,Popovic J,Flynn D,Jacobsen C,Lui JC,Hirschhorn JN,Baron J,Dauber A. Short stature, accelerated bone maturation, and early growth cessation due to heterozygous aggrecan mutations.J Clin Endocrinol Metab.2014;99(8):E1510–E1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gleghorn L,Ramesar R,Beighton P,Wallis G. A mutation in the variable repeat region of the aggrecan gene (AGC1) causes a form of spondyloepiphyseal dysplasia associated with severe, premature osteoarthritis.Am J Hum Genet.2005;77(3):484–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bartels CF,Bükülmez H,Padayatti P,Rhee DK,van Ravenswaaij-Arts C,Pauli RM,Mundlos S,Chitayat D,Shih L-Y,Al-Gazali LI,Kant S,Cole T,Morton J,Cormier-Daire V,Faivre L,Lees M,Kirk J,Mortier GR,Leroy J,Zabel B,Kim CA,Crow Y,Braverman NE,van den Akker F,Warman ML. Mutations in the transmembrane natriuretic peptide receptor NPR-B impair skeletal growth and cause acromesomelic dysplasia, type Maroteaux.Am J Hum Genet.2004;75(1):27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Temtamy SA,Meguid NA,Ismail SI,Ramzy MI. A new multiple congenital anomaly, mental retardation syndrome with preaxial brachydactyly, hyperphalangism, deafness and orodental anomalies.Clin Dysmorphol.1998;7(4):249–255. [DOI] [PubMed] [Google Scholar]
- 49. Hellemans J,Coucke PJ,Giedion A,De Paepe A,Kramer P,Beemer F,Mortier GR. Homozygous mutations in IHH cause acrocapitofemoral dysplasia, an autosomal recessive disorder with cone-shaped epiphyses in hands and hips.Am J Hum Genet.2003;72(4):1040–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mortier GR,Kramer PPG,Giedion A,Beemer FA. Acrocapitofemoral dysplasia: an autosomal recessive skeletal dysplasia with cone shaped epiphyses in the hands and hips.J Med Genet.2003;40(3):201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kronenberg HM.Developmental regulation of the growth plate.Nature.2003;423(6937):332–336. [DOI] [PubMed] [Google Scholar]
- 52. Nelson CP,Hamby SE,Saleheen D,Hopewell JC,Zeng L,Assimes TL,Kanoni S,Willenborg C,Burgess S,Amouyel P,Anand S,Blankenberg S,Boehm BO,Clarke RJ,Collins R,Dedoussis G,Farrall M,Franks PW,Groop L,Hall AS,Hamsten A,Hengstenberg C,Hovingh GK,Ingelsson E,Kathiresan S,Kee F,König IR,Kooner J,Lehtimäki T,März W,McPherson R,Metspalu A,Nieminen MS,O’Donnell CJ,Palmer CNA,Peters A,Perola M,Reilly MP,Ripatti S,Roberts R,Salomaa V,Shah SH,Schreiber S,Siegbahn A,Thorsteinsdottir U,Veronesi G,Wareham N,Willer CJ,Zalloua PA,Erdmann J,Deloukas P,Watkins H,Schunkert H,Danesh J,Thompson JR,Samani NJ;CARDIoGRAM+C4D Consortium . Genetically determined height and coronary artery disease.N Engl J Med.2015;372(17):1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Paajanen TA,Oksala NK,Kuukasjärvi P,Karhunen PJ. Short stature is associated with coronary heart disease: a systematic review of the literature and a meta-analysis.Eur Heart J.2010;31(14):1802–1809. [DOI] [PubMed] [Google Scholar]
- 54. Emerging Risk Factors Collaboration Adult height and the risk of cause-specific death and vascular morbidity in 1 million people: individual participant meta-analysis.Int J Epidemiol.2012;41(5):1419–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bulik-Sullivan B,Finucane HK,Anttila V,Gusev A,Day FR,Loh P-R,Duncan L,Perry JRB,Patterson N,Robinson EB,Daly MJ,Price AL,Neale BM. An atlas of genetic correlations across human diseases and traits.Nat Genet.2015;47(11):1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chan Y,Salem RM,Hsu YHH,McMahon G,Pers TH,Vedantam S,Esko T,Guo MH,Lim ET,Franke L,Smith GD,Strachan DP,Hirschhorn JN;GIANT Consortium . Genome-wide analysis of body proportion classifies height-associated variants by mechanism of action and implicates genes important for skeletal development.Am J Hum Genet.2015;96(5):695–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Horikoshi M,Beaumont RN,Day FR,Warrington NM,Kooijman MN,Fernandez-Tajes J,Feenstra B,van Zuydam NR,Gaulton KJ,Grarup N,Bradfield JP,Strachan DP,Li-Gao R,Ahluwalia TS,Kreiner E,Rueedi R,Lyytikäinen LP,Cousminer DL,Wu Y,Thiering E,Wang CA,Have CT,Hottenga JJ,Vilor-Tejedor N,Joshi PK,Boh ETH,Ntalla I,Pitkänen N,Mahajan A,van Leeuwen EM,Joro R,Lagou V,Nodzenski M,Diver LA,Zondervan KT,Bustamante M,Marques-Vidal P,Mercader JM,Bennett AJ,Rahmioglu N,Nyholt DR,Ma RCW,Tam CHT,Tam WH,Ganesh SK,van Rooij FJ,Jones SE,Loh PR,Ruth KS,Tuke MA,Tyrrell J,Wood AR,Yaghootkar H,Scholtens DM,Paternoster L,Prokopenko I,Kovacs P,Atalay M,Willems SM,Panoutsopoulou K,Wang X,Carstensen L,Geller F,Schraut KE,Murcia M,van Beijsterveldt CE,Willemsen G,Appel EVR,Fonvig CE,Trier C,Tiesler CM,Standl M,Kutalik Z,Bonas-Guarch S,Hougaard DM,Sánchez F,Torrents D,Waage J,Hollegaard MV,de Haan HG,Rosendaal FR,Medina-Gomez C,Ring SM,Hemani G,McMahon G,Robertson NR,Groves CJ,Langenberg C,Luan J,Scott RA,Zhao JH,Mentch FD,MacKenzie SM,Reynolds RM,Lowe WL Jr,Tönjes A,Stumvoll M,Lindi V,Lakka TA,van Duijn CM,Kiess W,Körner A,Sørensen TI,Niinikoski H,Pahkala K,Raitakari OT,Zeggini E,Dedoussis GV,Teo YY,Saw SM,Melbye M,Campbell H,Wilson JF,Vrijheid M,de Geus EJ,Boomsma DI,Kadarmideen HN,Holm JC,Hansen T,Sebert S,Hattersley AT,Beilin LJ,Newnham JP,Pennell CE,Heinrich J,Adair LS,Borja JB,Mohlke KL,Eriksson JG,Widén EE,Kähönen M,Viikari JS,Lehtimäki T,Vollenweider P,Bønnelykke K,Bisgaard H,Mook-Kanamori DO,Hofman A,Rivadeneira F,Uitterlinden AG,Pisinger C,Pedersen O,Power C,Hyppönen E,Wareham NJ,Hakonarson H,Davies E,Walker BR,Jaddoe VW,Jarvelin MR,Grant SF,Vaag AA,Lawlor DA,Frayling TM,Davey Smith G,Morris AP,Ong KK,Felix JF,Timpson NJ,Perry JR,Evans DM,McCarthy MI,Freathy RM;CHARGE Consortium Hematology Working Group . Genome-wide associations for birth weight and correlations with adult disease.Nature.2016;538(7624):248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Silventoinen K,Sammalisto S,Perola M,Boomsma DI,Cornes BK,Davis C,Dunkel L,De Lange M,Harris JR,Hjelmborg JVBB,Luciano M,Martin NG,Mortensen J,Nisticò L,Pedersen NL,Skytthe A,Spector TD,Stazi MA,Willemsen G,Kaprio J. Heritability of adult body height: a comparative study of twin cohorts in eight countries.Twin Res.2003;6(5):399–408. [DOI] [PubMed] [Google Scholar]
- 59. Zaitlen N,Kraft P,Patterson N,Pasaniuc B,Bhatia G,Pollack S,Price AL. Using extended genealogy to estimate components of heritability for 23 quantitative and dichotomous traits.PLoS Genet.2013;9(5):e1003520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yang J,Bakshi A,Zhu Z,Hemani G,Vinkhuyzen AAE,Lee SH,Robinson MR,Perry JRB,Nolte IM,van Vliet-Ostaptchouk JV,Snieder H,Esko T,Milani L,Mägi R,Metspalu A,Hamsten A,Magnusson PKE,Pedersen NL,Ingelsson E,Soranzo N,Keller MC,Wray NR,Goddard ME,Visscher PM;LifeLines Cohort Study . Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index.Nat Genet.2015;47(10):1114–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chan Y,Holmen OL,Dauber A,Vatten L,Havulinna AS,Skorpen F,Kvaløy K,Silander K,Nguyen TT,Willer C,Boehnke M,Perola M,Palotie A,Salomaa V,Hveem K,Frayling TM,Hirschhorn JN,Weedon MN. Common variants show predicted polygenic effects on height in the tails of the distribution, except in extremely short individuals.PLoS Genet.2011;7(12):e1002439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Dauber A,Rosenfeld RG,Hirschhorn JN. Genetic evaluation of short stature.J Clin Endocrinol Metab.2014;99(9):3080–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Guo MH,Shen Y,Walvoord EC,Miller TC,Moon JE,Hirschhorn JN,Dauber A. Whole exome sequencing to identify genetic causes of short stature.Horm Res Paediatr.2014;82(1):44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hauer NN,Popp B,Schoeller E,Schuhmann S,Heath KE,Hisado-Oliva A,Klinger P,Kraus C,Trautmann U,Zenker M,Zweier C,Wiesener A,Abou Jamra R,Kunstmann E,Wieczorek D,Uebe S,Ferrazzi F,Büttner C,Ekici AB,Rauch A,Sticht H,Dörr H-G,Reis A,Thiel CT. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature.Genet Med.2018 Jun;20(6):630–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Abraham G,Havulinna AS,Bhalala OG,Byars SG,De Livera AM,Yetukuri L,Tikkanen E,Perola M,Schunkert H,Sijbrands EJ,Palotie A,Samani NJ,Salomaa V,Ripatti S,Inouye M. Genomic prediction of coronary heart disease.Eur Heart J.2016;37(43):3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Plenge RM,Scolnick EM,Altshuler D. Validating therapeutic targets through human genetics.Nat Rev Drug Discov.2013;12(8):581–594. [DOI] [PubMed] [Google Scholar]
- 67. Vinjamur DS,Bauer DE,Orkin SH. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies.Br J Haematol.2018;180(5):630–643. [DOI] [PubMed] [Google Scholar]
- 68. Shapiro MD,Tavori H,Fazio S. PCSK9: from basic science discoveries to clinical trials.Circ Res.2018;122(10):1420–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Capellini TD,Chen H,Cao J,Doxey AC,Kiapour AM,Schoor M,Kingsley DM. Ancient selection for derived alleles at a GDF5 enhancer influencing human growth and osteoarthritis risk.Nat Genet.2017;49(8):1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]