

CONSPECTUS:
This Account highlights recent advances and discusses major challenges in investigations of cryptic (hidden) binding sites by molecular simulations. Cryptic binding sites are not visible in protein targets crystallized without a ligand and only become visible crystallographically upon binding events. These sites have been shown to be druggable and might provide a rare opportunity to target difficult proteins. However, due to their hidden nature, they are difficult to find through experimental screening. Computational methods based on atomistic molecular simulations remain one of the best approaches to identify and characterize cryptic binding sites. However, not all methods are equally efficient. Some are more apt at quickly probing protein dynamics, but do not provide thermodynamic or druggability information, while others that are able to provide such data are demanding in terms of time and resources. Here, we review the recent contributions of mixed-solvent simulations, metadynamics, Markov state models, and other enhanced sampling methods to the field of cryptic site identification and characterization. We discuss how these methods were able to provide precious information on the nature of the site opening mechanisms, to predict previously unknown sites which were used to design new ligands, and to compute the free energy landscapes and kinetics associated with the opening of the sites and the binding of the ligands. We highlight the potential and the importance of such predictions in drug discovery, especially for difficult (“undruggable”) targets. We also discuss the major challenges in the field and their possible solutions.
Graphical Abstract
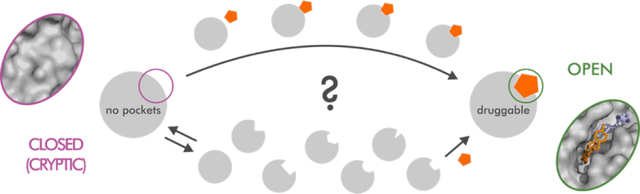
INTRODUCTION
In recent years, the number of biologically validated drug targets for complex diseases has considerably increased, especially due to large-scale genome sequencing.1,2 Unfortunately, many of these potential targets are considered “undruggable”, meaning they are unamenable to classic substrate-competitive drug discovery strategies.1 For this reason, significant efforts are put towards alternative strategies, including identifying allosteric ligands and characterizing hidden (cryptic) allosteric pockets. Cryptic binding pockets are absent in unliganded protein structures but open due to protein dynamics. They represent an attractive alternative to substrate-competitive sites that have been exploited in a number of high-profile targets, including K-Ras,3,4 an oncogene commonly found in human cancers. Despite their vast potential, the hidden nature of cryptic pockets makes it difficult to use rational drug discovery approaches based on structural experiments or computer modeling. Indeed, most known cryptic sites have been found serendipitously. Even the very mechanism of the cavity opening and whether it occurs through an induced fit, a conformational selection, or perhaps a combination of the two, is still debated in the literature. However, atomistic molecular dynamics (MD) simulations provide ways to both model the opening of unknown cryptic sites and describe possible binding mechanisms.
After many years of development, MD simulations are finally becoming useful in obtaining a detailed molecular description of protein (de)activation and ligand (un)binding. With the technological advancements in specialized computer architectures5 for MD simulations and distributed computing,6 as well as the parallelization of the MD simulation packages7 and their increasing use of GPUs,8 scientists are now able to reach unprecedented system sizes9 and timescales,10 enabling μs-long simulations on a routine basis. At the same time, both protein11–14 and ligand15 force fields are sufficiently accurate to capture the important features of target dynamics and ligand binding mechanisms. Prediction of previously unknown cryptic binding pockets that have successively been experimentally validated to design new drugs shows how promising the approach is, as was the case of HIV integrase where MD simulations predicted the opening of a new cleft.16 Still, despite the substantial progress, conventional approaches in MD simulations cannot adequately sample many biologically and pharmaceutically interesting processes. Among such processes is the opening of buried cryptic binding sites which is a prerequisite for their detection. Recent studies demonstrate how this challenge can be successfully tackled using MD-based approaches, which offer a more dynamic picture of the system (compared to X-ray crystallography) and provide high-resolution structural data for drug design.
In this Account, we review the recent contributions of MD simulations and modelling combined with experiments in understanding the nature of cryptic pockets and predicting their location.
NATURE OF CRYPTIC POCKETS AND MECHANISMS OF THEIR FORMATION
Similarly to all conformational changes of a target associated with ligand binding, the discussion on whether cryptic pockets emerge through conformational selection17,18 or induced fit19 is still ongoing. The former mechanism implies that the ligand’s role is to stabilize specific conformations that are also accessible in the unbound state. The latter mechanism proposes that the ligand causes the target to explore regions of the conformational space that are practically inaccessible to the unbound form. As discussed elsewhere,20 from the experimental point of view, only kinetic experiments might provide a clear distinction of the two mechanisms, as the binding rate in the case of conformational selection is dependent on the concentration of the ligand, while for the induced fit it is not. When the binding mechanism was ascertained experimentally, it emerged that, depending on the target, either induced fit or conformational selection are prevalent and in some cases both play a role.21,22 In the case of cryptic sites, an extensive analysis of multiple X-ray structures of proteins with validated cryptic sites performed by Beglov et al.23 showed that cryptic sites tend to be quite flexible which would hint towards conformational selection, but they are also almost always close to binding energy hot spots, often exploited by the bound ligands, suggesting that an induced fit component is also important for binding. This finding is in agreement with our simulations of the cryptic pocket found at the interface between α-helices H11 and H12 of TEM1 β-lactamase. Long timescale simulations of the system have detected this cryptic pocket in the absence of a ligand and suggested roles for both induced fit and conformational selection.24 The role of conformational selection was supported further by subsequent experiments that detected the cryptic pocket in the absence of ligand.25 Enhanced sampling simulations of small fragments binding to the pocket are also consistent with a picture in which large fluctuations lead to the opening of the pocket (conformational selection) which are then stabilized by small molecules wedging between the helices (induced fit).26
Analysis of ~90 available crystal structures harboring cryptic sites showed that the main conformational changes associated with their opening are linked to: lateral chain rotation, loop movements, secondary structure changes, and interdomain motions (Figure 1).23,27 Cryptic pockets have been also found to play a role at flexible protein-protein interfaces.28
Figure 1.
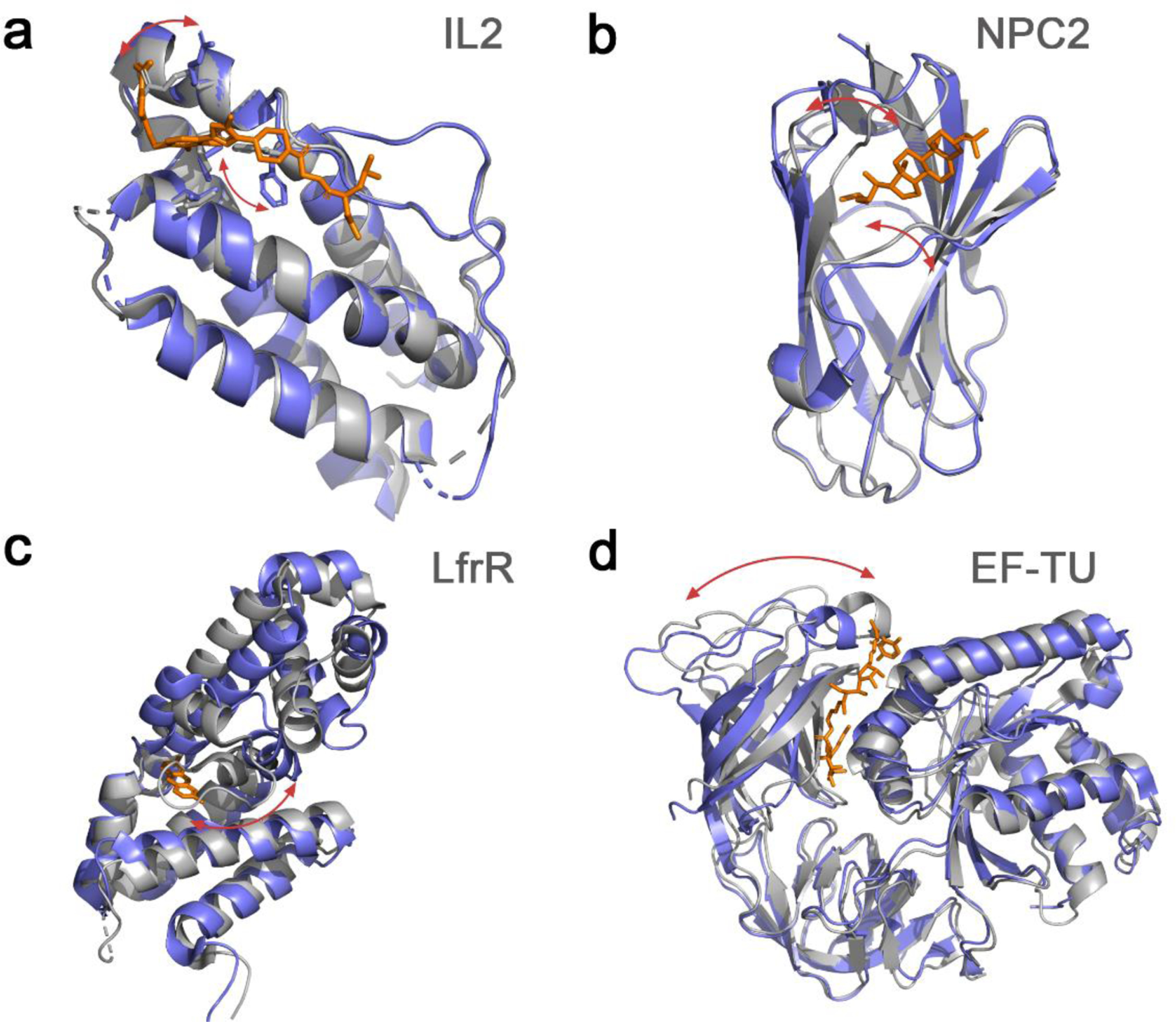
Illustration of common structural changes associated with cryptic binding site formation. a. Side chain rearrangements as shown in IL2 (apo: 1M47_A, holo: 1PY2_D), b. loop movements in NPC2 (apo: 1NEP_A, holo: 2HKA_C), c. secondary structure changes like the partial helix unfolding in LfrR (apo: 2WGB_A, holo: 2V57_A), and d. interdomain motions in EF-TU (apo: 1EXM_A, holo: 1HA3_B). Apo structures are show in gray and holo structures in slate blue, while red arrows highlight the main structural changes.
Thus, cryptic pocket opening is associated with conformational changes of the target and the extent of such change plays a role in the operative definition of crypticity (i.e., a site “being cryptic”). The operative definition of crypticity is based on whether the pocket is present or visible in the apo structure and is typically a continuous measure based on measures of pocket exposure or steric clash with the ligand. Thus, depending on the chosen threshold, a pocket can be considered cryptic or not, and even sites contiguous with the catalytic pocket can end up being labelled as cryptic.
Algorithms for the detection and analysis of pockets per se (including their transient formation), are thus instrumental to distinguish and identify the cryptic ones. To this aim, a number of pocket analysis tools have been developed by the community. Tools such as Fpocket,29 Epock,30 POVME,31 TRAPP,32 and Nanoshaper33 are able to detect and/or analyse both stable and transient pockets and exposons focus on transient pockets. When the pockets are not particularly buried and the opening mechanism is mainly conformational selection, these tools can successfully detect the presence of cryptic pockets in the trajectories of the unliganded target.
Finally, the mere detection of a cryptic pocket does not make a target tractable per se. The newly found pocket needs to be ligandable. Having a cryptic pocket that is close to a binding hotspot and can accommodate chemically diverse fragments increases the chances that they are ligandable.23 Some pocket analysis algorithms and mixed solvent simulations also provide an estimate of the pocket ligandability.29,32
If a cryptic pocket does not cdirectly coincide with a functional site, then an allosteric connection of the remote pocket to the catalytic site or other functionally important sites is also needed for its druggability. As allosteric connections are bidirectional, an MD-based analysis of allosteric signal propagation from and to the active site of a target can be used to discover previously unknown binding sites. For instance, in the case of Hsp90, such a method was used to characterize the allosteric “hot spots” involved in interdomain communication pathways. One of these binding hotspots is cryptic and was successively shown to be druggable.34,35
MIXED-SOLVENT MOLECULAR DYNAMICS SIMULATIONS
To facilitate the exploration of cryptic binding sites, one of the approaches based on MD simulations relies on the use of small probes mixed with water molecules that help open and stabilize cryptic pockets which are often hydrophobic. These simulations are known as mixed-solvent MD simulations.36–39 Probes that were successfully tested were either readily miscible with water (e.g., acetic acid, isopropanol, or resorcinol),36,40 or quite hydrophobic (e.g. benzene)26,41 and in such cases a repulsive potential is necessary to prevent the probes from clustering.37,42 Mixed-solvent MD simulations have been successfully used on a number of targets to characterize active and allosteric sites and are relatively simple to set up.28,36–38,43 Schmidt et al. tested the performance of various co-solvent compositions on seven diverse targets with known cryptic sites and reported that a composition of 90% water and 10% phenol was the most effective in opening the cavities without unfolding the proteins.44 The pocket occupancy and fragment residence time obtained from mixed-solvent MD simulations can also provide druggability estimates, which is important to assess the feasibility of targeting a pocket with small molecules.16,36 However, their application to cryptic pockets detection still requires substantial sampling of the system. For instance, we showed that, in the case of the cryptic pocket of TEM1 β-lactamase, 6 different hydrophobic ring probes were able to open the cryptic pocket only in a few 100-ns-long simulations out of 32 that we ran for each of the probes. Extending the mixed-solvent simulations with benzene to more than 1 μs resulted in the opening of the pocket in 1/3 of the simulations.26 Sampling problems also affect the ability of mixed-solvent MD simulations to identify deeply buried cryptic pockets as their exploration relies on the diffusion of probe molecules around the protein surface and/or overcoming high energy barriers. An additional problem with mixed solvent MD is the risk of unfolding the protein with hydrophobic probes44 which in some cases can be circumvented through a carefully selected set of position/distance restraints.41
COLLECTIVE-VARIABLE-DEPENDENT ENHANCED SAMPLING METHODS
When conventional MD simulations prove ineffective in sampling the event of interest, enhanced sampling methods can be used. Such techniques can be practically divided based on their dependence on collective variables (CV). In the case of CV-dependent methods, like metadynamics,45,46 umbrella sampling,47 and steered MD,48 the process of interest is approximated through CVs, i.e. the functions of system’s coordinates that can be as simple as a distance between two atoms or as complex as a contact map describing an active state of a protein. Unfortunately, choosing suitable CVs can be far from straightforward which is why these choices are often guided by experimental data. Recently, metadynamics was successfully applied to investigate the binding mechanism of SSR128129E (SSR), a newly developed inhibitor of fibroblast growth factor receptor 1 (FGFR1).49,50 This receptor tyrosine kinase arose as a potential anticancer drug target due to its involvement in numerous essential cellular processes, such as blood vessel formation. SSR was initially discovered through high-throughput screening and its binding location was narrowed down to the extracellular D3 Ig-like domain, but the exact binding site eluded both X-ray crystallography and NMR due to the disordered nature of the domain. Using a range of CVs that describe not only the binding process, but also the folding of the domain, the authors were able to capture inhibitor’s reversible binding to a hidden pocket formed through the elongation of a small α-helix in the D3 domain. The observed binding mechanism was further confirmed with a mutational analysis and subsequently used to design more potent inhibitors. Despite this success story, using CV methods based on geometric criteria to systematically predict cryptic binding sites is challenging as it is difficult to find a set of parameters that adequately captures the complexity of the cryptic binding pocket formation in the absence of previous information on the location and nature of the pocket.26
The JEDI methodology is a CV-based approach that overcomes these limitations and allows to bias sampling towards protein conformations of favorable druggability.51 It uses a 3D grid overlapped on a region of interest on a protein surface to determine the location of putative pockets, and calculates a druggability score, i.e., a quantification of how adequate those pockets are to accommodate a small organic molecule. This druggability scoring was inspired by structural bioinformatics methodologies trained on datasets of X-ray crystallography-derived binding sites,52,53 with the important difference that the druggability estimator was designed to be smooth and continuously differentiable with respect to protein atomic Cartesian coordinates. This enables the use of the JEDI score as a collective variable for biased MD simulations. Therefore, a typical workflow to seek cryptic pockets with JEDI involves working out the druggability score of a protein region covered by a manually placed 3D grid, and subsequently carrying out biased MD simulations to encourage the protein to adopt new conformations that increase (or decrease) the initial druggability score. Because the estimator scores favorably hydrophobic cavities suitably sized to accommodate a small molecule, the application of JEDI to a protein region that does not contain a pocket will encourage spontaneous sampling of a cavity without requiring a priori knowledge of which residues must undergo a conformational change to reveal a cryptic pocket. The process can be very rapid, requiring only a few nanoseconds of MD simulation (Figure 2). Current drawbacks of the first version of the algorithm include the relatively expensive cost of the CV, making long timescale simulations difficult, and support for implicit solvent simulations only. The JEDI CV can also suffer from degeneracy, leading to the biased simulations becoming stuck in an alternative conformational state that may not accurately describe the cryptic pocket owing to the approximate nature of the druggability estimator.
Figure 2.
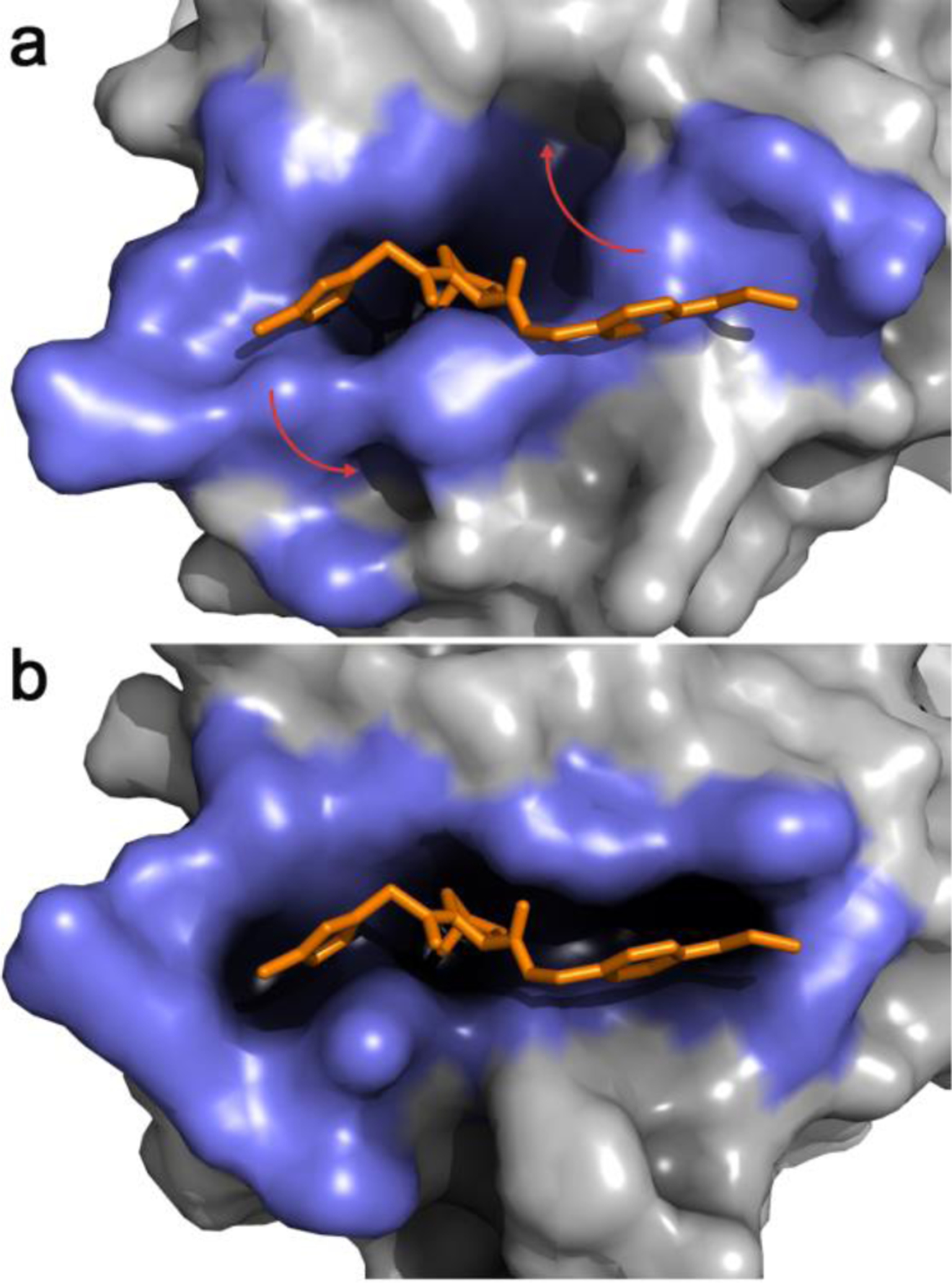
a. Representative snapshot from the most populated cluster of an equilibrium MD simulation of apo VHL. The binding-site lining residues (highlighted in slate blue) adopt a collapsed conformation with low druggability score b. A JEDI-biased MD simulation of apo VHL has rearranged the binding-site lining residues in a druggable conformation. In both panels an overlay of the crystallographic structure of a VHL ligand is displayed in orange sticks. Red arrows highlight the main structural changes.
COLLECTIVE-VARIABLE-INDEPENDENT ENHANCED SAMPLING METHODS
One of the most commonly used CV-independent enhanced sampling methods is parallel tempering (i.e., temperature replica exchange MD)54 where several replicas of the system are run at the same time, but at different temperatures. At regular intervals, an exchange between the neighboring replicas is attempted and either accepted or rejected based on the Metropolis criterion. The method is usually quite effective (albeit computationally expensive as it requires a large number of replicas to span a wide range of temperatures) in studying protein conformational landscapes. However when it came to studies of cryptic binding sites, it was unable to open the pockets in the targets that were tested, even at temperatures of 400 K.26 This is probably due to the entropic nature of the energy barriers involved in pocket opening.26 A different approach, based on non-equilibrium MD simulations (rotamerically induced perturbations or RIP) was proposed by Kokh et al. to initiate large-scale protein movements and sample cryptic sites.55 This approach proved useful for a rapid evaluation of pocket flexibility. However, RIP is not able to predict folding of protein segments, nor the kinetics or thermodynamics associated with the protein motions.56
Considering the hydrophobic nature of cryptic binding pockets, Oleinikovas et al. devised a method in which they sample water interactions through scaled Hamiltonians (SWISH).26 The method is based on a Hamiltonian replica exchange approach and, instead of altering the temperature of the replicas as in parallel tempering, it changes the interaction of apolar carbons and sulfurs with water oxygen. This renders the protein less hydrophobic at higher replicas and allows for more effective exploration of cryptic binding sites compared to conventional MD simulations. The method was tested on a range of pharmaceutically relevant targets, such as β-lactamase, IL2, and PLK1 in the initial study,26 and NPC2, LfrR, p38α, and hPNMT in the follow-up study.41 SWISH was able to successfully detect known cryptic binding sites in these proteins, typically within 1 μs of of sampling time with 6–8 replicas. When combined with probes which stabilized the open cryptic binding sites, it became apparent that the formation mechanism of cryptic binding pockets typically involves both conformational selection and induced fit. However, the approach combining SWISH with probes suffers from the same drawbacks as mixed-solvent simulations where the proteins can become denatured when the selected probes are hydrophobic in nature. Such behavior is even more pronounced in SWISH simulations where the protein is already pushed towards more hydrophilic states. A way to prevent the protein from unfolding is either through a more conservative set of scaling factors or by using a contact-map restraint41 which is applied on a small set of relevant contacts away from the site of interest that essentially keeps the secondary structure elements folded. Further iterations of the method involve a more targeted approach where only the interactions of hydrophobic residues with water are scaled which would hopefully also reduce the protein unfolding and rendering the contact-map restraint unnecessary. Another advantage of SWISH is that it can be easily combined with other enhanced sampling methods, such as metadynamics, if the cryptic binding pocket of interest forms through a more complex conformational change, as was the case of FGFR1.50
MARKOV STATE MODELS (MSMs)
MSMs are a powerful means to integrate many independent simulations into a map of protein’s conformational space that captures long-timescale events far beyond the reach of any individual simulation.57 These methods were originally developed to study protein folding mechanisms.58 However, they have since proved extremely effective in a wide variety of settings.59
Inspired by the conformational selection model, MSMs were first applied to the apo TEM1 β-lactamase enzyme introduced above.24 The resulting model successfully identified the cryptic pocket between helices 11 and 12, supporting a role for conformational selection in the opening of such pockets. A second model built in the presence of a known ligand for this cryptic site also demonstrated a role for induced fit, consistent with previous applications of MSMs to other protein-ligand binding processes60 that revealed an interplay between conformational selection and induced fit. In addition to recapitulating the known cryptic pocket, subsequent analysis of the β-lactamase MSM revealed at least two additional cryptic pockets with allosteric coupling to the active site.25 To test this prediction, an experimental thiol labeling assay was applied to detect the exposure of buried residues upon pocket opening. The results confirmed the two newly predicted pockets, a role for conformational selection in the opening of all three sites, and their ability to allosterically impact function.25 Since then, MSMs have been applied to hunt for cryptic pockets in a number of other proteins.61–63 New adaptive sampling algorithms have also been developed to reduce the computational cost of building such models.64 Strikingly, these approaches have revealed yet another cryptic allosteric site that is common to multiple β-lactamase families (Fig. 3).65 Experimental tests have confirmed the existence of this pocket and demonstrated that it exerts more potent allosteric control over enzymatic activity than the previously identified pockets.65,66 Applications to multiple related proteins are also beginning to reveal the sequence-dependence of the probabilities of potentially druggable sites.39,66,67
Figure 3.
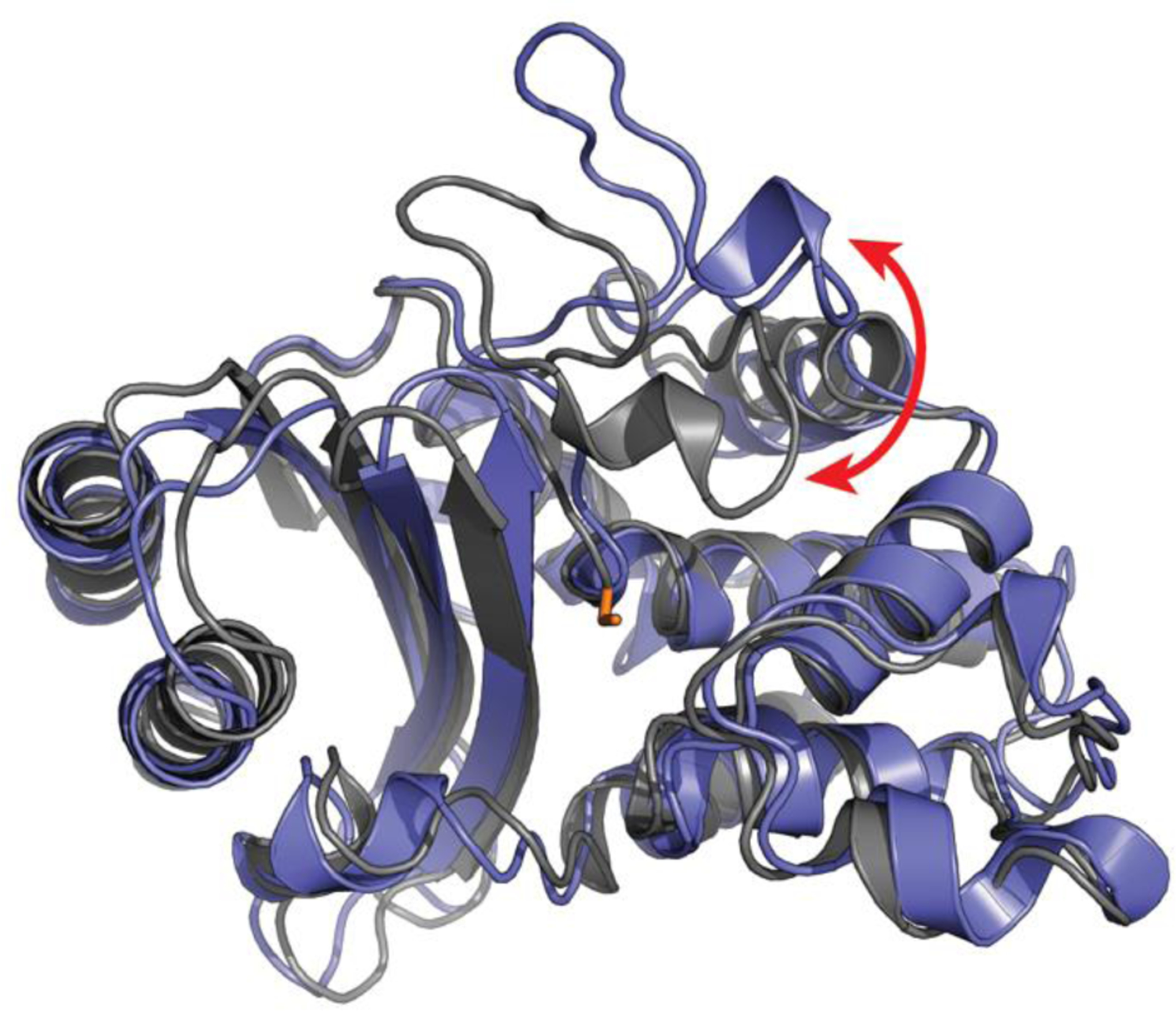
A new cryptic pocket in TEM1 β-lactamase that was discovered with MSMs and subsequently confirmed by experimental tests. An open structure (blue) is overlaid on the apo crystal structure (gray) with the key catalytic serine (S70) in sticks (orange). The red arrow highlights a large loop motion that creates a cryptic pocket adjacent to the active site.
Having succeeded in detecting cryptic pockets, MSMs are now actively being used to intentionally target these sites with small molecules. Hypothetically, this could be achieved by taking an individual structure with an open cryptic pocket and applying existing rational drug design tools. However, more effective approaches could systematically take advantage of all the conformational diversity an MSM captures. One proposal for achieving this end, called Boltzmann docking,67 works by docking a library of compounds against a representative structure from each state in an MSM and then, using the equilibrium probabilities of those states, to calculate a Boltzmann-weighted average docking score. Boltzmann docking is related to ensemble docking.68 The primary difference is that Boltzmann docking uses the equilibrium probabilities of each state instead of treating all conformations equally. In doing so, Boltzmann docking automatically favors compounds that bind to higher probability states over compounds that bind higher-energy (i.e. lower probability) states where a greater penalty has to be paid to stabilize the conformation. Boltzmann docking has been successfully applied to find new allosteric modulators of TEM1 β-lactamase, including both inhibitors and activators that have been confirmed experimentally.69 The discovery of activators is intriguing as it suggests the potential for enhancing desirable functions in other settings, e.g., to counteract the deleterious effects of a disease-causing mutation.
OTHER COMPUTATIONAL APPROACHES
Despite the aforementioned advances in computer software and hardware, MD simulations still require a significant amount of computational resources and time to adequately sample protein conformational landscapes. Thus, the appeal of inexpensive computational methods that rapidly predict cryptic binding sites is evident. Cimermancic et al. recently curated a data set of apo and holo protein pairs from the PDB harboring cryptic binding sites (composed of ~90 proteins) which they used to build a machine learning model (CryptoSite) to predict such sites in proteins considered undruggable.27 The authors initially used over 50 features they deemed potentially relevant for the cryptic binding site prediction, however, only three of them proved to be statistically significant - the average pocket score calculated for conformations obtained from MD simulations that rely on a simplified energy landscape, the sequence conservation of the site, and the likelihood of binding small-molecule fragments. While the approach is certainly fast and remarkably accurate for the testing set, it drew criticism due to the overall small size of the training and testing sets, as well as the use of a single apo structure in each pair which can lead to an overestimation of the site’s crypticity as the pocket could be quite apparent in other apo structures.23,70 Beglov et al. expanded the original CryptoSite data set by including these structures and analyzed the new set using their own tools (FTMap and FTFlex).23 The authors showed that cryptic binding sites in apo structures typically have a strong binding hot spot in their vicinity and that they exhibit an above-average flexibility. It should be, however, noted that the analysis of X-ray structures has its limitations due to their static nature and the presence of crystal contacts that can introduce a range of artifacts.71 Efforts are also underway to identify cryptic pockets using Rosetta-based Monte Carlo sampling of the conformational space.72
PERSPECTIVES
The past few years have seen the successful execution of a number of proof-of-concept studies that have demonstrated it is possible to computationally identify cryptic pockets and then target them with small molecules. Methods based on MD simulations combined with algorithms for the detection of pockets, mixed solvents, enhanced sampling, and Markov state models are increasingly effective in sampling and identifying cryptic pockets, providing high-quality structural models for the design of novel drug-like compounds. Some of these methodologies, namely CV-based enhanced sampling methods and MSMs, are also able to provide accurate estimates of the free energy penalty associated with the opening of the pockets, while simulations with mixed solvents and fragments provide precious information on the ligandability of the newly discovered sites. As many cryptic pockets are allosteric, i.e., remote from known functional sites, an important question is whether binding of a small molecule at a cryptic pocket has a functional effect on the protein. Even in this case, methods to characterize allosteric communication networks based on simulations can provide solid predictions. Looking ahead, as many new cryptic pockets are predicted by simulations, experimental validation becomes a crucial bottleneck in the search for cheaper solutions than the ones currently used. Machine learning approaches predict that a vast number of potential drug targets in the human genome harbor cryptic sites. It will be exciting to see how many of those, particularly in validated drug targets that small molecule drugs have yet to be discovered for, can be targeted by leveraging the cryptic sites predicted by simulation-based approaches.
Funding Sources
G.R.B. was funded by NIH R01GM124007 and NSF CAREER Award MCB-1552471, and holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund and a Packard Fellowship for Science and Engineering from the David & Lucile Packard Foundation.
F.L.G. and A.K. were funded by EPSRC (grant no. EP/P022138/1 and EP/P011306/1). F.L.G was also funded by the EC H2020 Human Brain Project SGA2 CDP6.
J.M. and J.J.J were funded by EPSRC (grant no EP/P011306/1).
Biographies
Gregory R. Bowman is an Associate Professor in the Department of Biochemistry & Molecular Biophysics at the Washington University School of Medicine. His research focuses on the mechanisms of allosteric coupling and the opportunities this insight provides for manipulating function with drugs and mutations. A major emphasis is on identifying and targeting cryptic pockets. Half the lab develops and applies novel computational tools, often based on Markov state model (MSM) methods, while the other half experimentally tests small molecule and protein designs.
Julien Michel is a Senior Lecturer at the School of Chemistry of the University of Edinburgh. His research interests cover methods and applications of molecular simulations and their integration with biophysical experiments for studies of protein dynamics, protein-ligand interactions, and for drug discovery. His group has developed the JEDI approach for cryptic site discovery, and characterized cryptic site formation mechanisms in important drug targets
Jordi Juarez-Jimenez is a Research Associate at the School of Chemistry of the University of Edinburgh. His research focuses on understanding how protein conformational changes can be exploited in rational drug design. He is currently working on enhancing the functionalities of the JEDI algorithm in combination with different computational tools.
Antonija Kuzmanic is a Research Associate at University College London. She splits her time between trying to understand what makes kinases active and cryptic pockets hidden using molecular dynamics simulations and enhanced sampling methods.
Francesco L. Gervasio is a full Professor in Chemistry and Structural and Molecular Biology at University College London and a full Professor in Biomolecular and Pharmaceutical Modelling at the University of Geneva. His research interests are in the field of computational chemistry, biophysics, structural biology, and drug discovery. He has co-developed a number of enhanced-sampling algorithms, including the SWISH approach for cryptic site discovery.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Hopkins AL; Groom CR The druggable genome. Nat. Rev. Drug Discov 2002, 1, 727–730. [DOI] [PubMed] [Google Scholar]
- (2).Oprea TI; Bologa CG; Brunak S; Campbell A; Gan GN; Gaulton A; Gomez SM; Guha R; Hersey A; Holmes J; Jadhav A; Jensen LJ; Johnson GL; Karlson A; Leach AR; Ma’ayan A; Malovannaya A; Mani S; Mathias SL; McManus MT; Meehan TF; von Mering C; Muthas D; Nguyen D-T; Overington JP; Papadatos G; Qin J; Reich C; Roth BL; Schürer SC; Simeonov A; Sklar LA; Southall N; Tomita S; Tudose I; Ursu O; Vidović D; Waller A; Westergaard D; Yang JJ; Zahoránszky-Köhalmi G Unexplored therapeutic opportunities in the human genome. Nat. Rev. Drug Discov 2018, 17, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Canon J; Rex K; Saiki AY; Mohr C; Cooke K; Bagal D; Gaida K; Holt T; Knutson CG; Koppada N; Lanman BA; Werner J; Rapaport AS; Miguel TS; Ortiz R; Osgood T; Sun J-R; Zhu X; McCarter JD; Volak LP; Houk BE; Fakih MG; O’Neil BH; Price TJ; Falchook GS; Desai J; Kuo J; Govindan R; Hong DS; Ouyang W; Henary H; Arvedson T; Cee VJ; Lipford JR The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [DOI] [PubMed] [Google Scholar]
- (5).Shaw DE; Grossman JP; Bank JA; Batson B; Butts JA; Chao JC; Deneroff MM; Dror RO; Even A; Fenton CH; Forte A; Gagliardo J; Gill G; Greskamp B; Ho CR; Ierardi DJ; Iserovich L; Kuskin JS; Larson RH; Layman T; Lee L-S; Lerer AK; Li C; Killebrew D; Mackenzie KM; Mok SY-H; Moraes MA; Mueller R; Nociolo LJ; Peticolas JL; Quan T; Ramot D; Salmon JK; Scarpazza DP; Schafer UB; Siddique N; Snyder CW; Spengler J; Tang PTP; Theobald M; Toma H; Towles B; Vitale B; Wang SC; Young C: Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer In Proceedings of the International Conference for High Performance Computing, Networking, Storage and Analysis; IEEE Press: New Orleans, Louisana, 2014; pp 41–53. [Google Scholar]
- (6).Shirts M; Pande VS COMPUTING: Screen savers of the world unite! Science 2000, 290, 1903–1904. [DOI] [PubMed] [Google Scholar]
- (7).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (8).Páll S; Abraham MJ; Kutzner C; Hess B; Lindahl E Tackling exascale software challenges in molecular dynamics simulations with GROMACS, In Solving Software Challenges for Exascale, Cham, 2015; Springer International Publishing. [Google Scholar]
- (9).von Bulow S; Siggel M; Linke M; Hummer G Dynamic cluster formation determines viscosity and diffusion in dense protein solutions. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 9843–9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Voelz VA; Bowman GR; Beauchamp K; Pande VS Molecular simulation of ab initio protein folding for a millisecond folder NTL9(1–39). J. Am. Chem. Soc 2010, 132, 1526–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmuller H; MacKerell AD Jr. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Robustelli P; Piana S; Shaw DE Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kuzmanic A; Pritchard RB; Hansen DF; Gervasio FL Importance of the force field choice in capturing functionally relevant dynamics in the von Willebrand factor. J. Phys. Chem. Lett 2019, 10, 1928–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Robertson MJ; Tirado-Rives J; Jorgensen WL Improved peptide and protein torsional energetics with the OPLS-AA force field. J. Chem. Theory Comput 2015, 11, 3499–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ghanakota P; Carlson HA Moving beyond active-site detection: MixMD applied to allosteric systems. J. Phys. Chem. B 2016, 120, 8685–8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Monod J; Wyman J; Changeux JP On the nature of allosteric transitions: A plausible model. J. Mol. Biol 1965, 12, 88–118. [DOI] [PubMed] [Google Scholar]
- (18).Tsai CJ; Ma B; Nussinov R Folding and binding cascades: shifts in energy landscapes. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9970–9972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Koshland DE Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. U. S. A 1958, 44, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gianni S; Dogan J; Jemth P Distinguishing induced fit from conformational selection. Biophys. Chem 2014, 189, 33–39. [DOI] [PubMed] [Google Scholar]
- (21).Hammes GG; Chang YC; Oas TG Conformational selection or induced fit: a flux description of reaction mechanism. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13737–13741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Morando MA; Saladino G; D’Amelio N; Pucheta-Martinez E; Lovera S; Lelli M; Lopez-Mendez B; Marenchino M; Campos-Olivas R; Gervasio FL Conformational selection and induced fit mechanisms in the binding of an anticancer drug to the c-Src kinase. Sci. Rep 2016, 6, 24439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Beglov D; Hall DR; Wakefield AE; Luo L; Allen KN; Kozakov D; Whitty A; Vajda S Exploring the structural origins of cryptic sites on proteins. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E3416–E3425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bowman GR; Geissler PL Equilibrium fluctuations of a single folded protein reveal a multitude of potential cryptic allosteric sites. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 11681–11686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bowman GR; Bolin ER; Hart KM; Maguire BC; Marqusee S Discovery of multiple hidden allosteric sites by combining Markov state models and experiments. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2734–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Oleinikovas V; Saladino G; Cossins BP; Gervasio FL Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J. Am. Chem. Soc 2016, 138, 14257–14263. [DOI] [PubMed] [Google Scholar]
- (27).Cimermancic P; Weinkam P; Rettenmaier TJ; Bichmann L; Keedy DA; Woldeyes RA; Schneidman-Duhovny D; Demerdash ON; Mitchell JC; Wells JA; Fraser JS; Sali A CryptoSite: Expanding the druggable proteome by characterization and prediction of cryptic binding sites. J. Mol. Biol 2016, 428, 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Schames JR; Henchman RH; Siegel JS; Sotriffer CA; Ni HH; McCammon JA Discovery of a novel binding trench in HIV integrase. J. Med. Chem 2004, 47, 1879–1881. [DOI] [PubMed] [Google Scholar]
- (29).Le Guilloux V; Schmidtke P; Tuffery P Fpocket: An open source platform for ligand pocket detection. Bmc Bioinformatics 2009, 10, 168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Laurent B; Chavent M; Cragnolini T; Dahl AC; Pasquali S; Derreumaux P; Sansom MS; Baaden M Epock: rapid analysis of protein pocket dynamics. Bioinformatics 2015, 31, 1478–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wagner JR; Sørensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: Software for mapping binding pocket flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kokh DB; Richter S; Henrich S; Czodrowski P; Rippmann F; Wade RC TRAPP: a tool for analysis of transient binding pockets in proteins. J. Chem. Inf. Model 2013, 53, 1235–1252. [DOI] [PubMed] [Google Scholar]
- (33).La Sala G; Decherchi S; De Vivo M; Rocchia W Allosteric communication networks in proteins revealed through pocket crosstalk analysis. ACS Cent. Sci 2017, 3, 949–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Morra G; Neves MA; Plescia CJ; Tsustsumi S; Neckers L; Verkhivker G; Altieri DC; Colombo G Dynamics-based discovery of allosteric inhibitors: selection of new ligands for the C-terminal domain of Hsp90. J. Chem. Theory Comput 2010, 6, 2978–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Sattin S; Tao J; Vettoretti G; Moroni E; Pennati M; Lopergolo A; Morelli L; Bugatti A; Zuehlke A; Moses M; Prince T; Kijima T; Beebe K; Rusnati M; Neckers L; Zaffaroni N; Agard DA; Bernardi A; Colombo G Activation of Hsp90 enzymatic activity and conformational dynamics through rationally designed allosteric ligands. Chemistry 2015, 21, 13598–13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Seco J; Luque FJ; Barril X Binding site detection and druggability index from first principles. J. Med. Chem 2009, 52, 2363–2371. [DOI] [PubMed] [Google Scholar]
- (37).Guvench O; MacKerell AD Jr. Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol 2009, 5, e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bakan A; Nevins N; Lakdawala AS; Bahar I Druggability assessment of allosteric proteins by dynamics simulations in thepresence of probe molecules. J. Chem. Theory Comput 2012, 8, 2435–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sultan MM; Kiss G; Pande VS Towards simple kinetic models of functional dynamics for a kinase subfamily. Nat. Chem 2018, 10, 903–909. [DOI] [PubMed] [Google Scholar]
- (40).Kimura SR; Hu HP; Ruvinsky AM; Sherman W; Favia AD Deciphering cryptic binding sites on proteins by mixed-solvent molecular dynamics. J. Chem. Inf. Model 2017, 57, 1388–1401. [DOI] [PubMed] [Google Scholar]
- (41).Comitani F; Gervasio FL Exploring cryptic pockets formation in targets of pharmaceutical interest with SWISH. J. Chem. Theory Comput 2018, 14, 3321–3331. [DOI] [PubMed] [Google Scholar]
- (42).Raman EP; Yu W; Guvench O; Mackerell AD Reproducing crystal binding modes of ligand functional groups using site-identification by ligand competitive saturation (SILCS) simulations. J. Chem. Inf. Model 2011, 51, 877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Eyrisch S; Helms V Transient pockets on protein surfaces involved in protein-protein interaction. J. Med. Chem 2007, 50, 3457–3464. [DOI] [PubMed] [Google Scholar]
- (44).Schmidt D; Boehm M; McClendon CL; Torella R; Gohlke H Cosolvent-enhanced sampling and unbiased identification of cryptic pockets suitable for structure-based drug design. J. Chem. Theory Comput 2019, 15, 3331–3343. [DOI] [PubMed] [Google Scholar]
- (45).Barducci A; Bonomi M; Parrinello M Metadynamics. Wiley Interdiscip. Rev. Comput. Mol. Sci 2011, 1, 826–843. [Google Scholar]
- (46).Laio A; Gervasio FL Metadynamics: a method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys 2008, 71, 126601. [Google Scholar]
- (47).Torrie GM; Valleau JP Nonphysical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comp. Phys 1977, 23, 187–199. [Google Scholar]
- (48).Gullingsrud JR; Braun R; Schulten K Reconstructing potentials of mean force through time series analysis of steered molecular dynamics simulations. J. Comp. Phys 1999, 151, 190–211. [Google Scholar]
- (49).Bono F; De Smet F; Herbert C; De Bock K; Georgiadou M; Fons P; Tjwa M; Alcouffe C; Ny A; Bianciotto M; Jonckx B; Murakami M; Lanahan AA; Michielsen C; Sibrac D; Dol-Gleizes F; Mazzone M; Zacchigna S; Herault JP; Fischer C; Rigon P; Ruiz de Almodovar C; Claes F; Blanc I; Poesen K; Zhang J; Segura I; Gueguen G; Bordes MF; Lambrechts D; Broussy R; van de Wouwer M; Michaux C; Shimada T; Jean I; Blacher S; Noel A; Motte P; Rom E; Rakic JM; Katsuma S; Schaeffer P; Yayon A; Van Schepdael A; Schwalbe H; Gervasio FL; Carmeliet G; Rozensky J; Dewerchin M; Simons M; Christopoulos A; Herbert JM; Carmeliet P Inhibition of tumor angiogenesis and growth by a small-molecule multi-FGF receptor blocker with allosteric properties. Cancer Cell 2013, 23, 477–488. [DOI] [PubMed] [Google Scholar]
- (50).Herbert C; Schieborr U; Saxena K; Juraszek J; De Smet F; Alcouffe C; Bianciotto M; Saladino G; Sibrac D; Kudlinzki D; Sreeramulu S; Brown A; Rigon P; Herault JP; Lassalle G; Blundell TL; Rousseau F; Gils A; Schymkowitz J; Tompa P; Herbert JM; Carmeliet P; Gervasio FL; Schwalbe H; Bono F Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 2013, 23, 489–501. [DOI] [PubMed] [Google Scholar]
- (51).Cuchillo R; Pinto-Gil K; Michel J A collective variable for the rapid exploration of protein druggability. J. Chem. Theory Comput 2015, 11, 1292–1307. [DOI] [PubMed] [Google Scholar]
- (52).Ghanakota P; DasGupta D; Carlson HA Free energies and entropies of binding sites identified by MixMD cosolvent simulations. J. Chem. Inf. Model 2019, 59, 2035–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Halgren T New method for fast and accurate binding-site identification and analysis. Chem. Biol. Drug Des 2007, 69, 146–148. [DOI] [PubMed] [Google Scholar]
- (54).Sugita Y; Okamoto Y Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett 1999, 314, 141–151. [Google Scholar]
- (55).Kokh DB; Czodrowski P; Rippmann F; Wade RC Perturbation approaches for exploring protein binding site flexibility to predict transient binding pockets. J. Chem. Theory Comput 2016. [DOI] [PubMed] [Google Scholar]
- (56).Le Guilloux V; Schmidtke P; Tuffery P Fpocket: An open source platform for ligand pocket detection. Bmc Bioinformatics 2009, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bowman G; Pande V; Noé F: An introduction to Markov state models and their application to long timescale molecular simulation, 2014; Vol. 797. [Google Scholar]
- (58).Bowman GR; Voelz VA; Pande VS Taming the complexity of protein folding. Curr. Opin. Struct. Biol 2011, 21, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Chodera JD; Noe F Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol 2014, 25, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Silva D-A; Bowman GR; Sosa-Peinado A; Huang X A role for both conformational selection and induced fit in ligand binding by the LAO protein. PLoS Comput. Biol 2011, 7, e1002054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Wagner JR; Lee CT; Durrant JD; Malmstrom RD; Feher VA; Amaro RE Emerging computational methods for the rational discovery of allosteric drugs. Chem. Rev 2016, 116, 6370–6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Knoverek CR; Amarasinghe GK; Bowman GR Advanced methods for accessing protein shape-shifting present new therapeutic opportunities. Trends Biochem. Sci 2019, 44, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Ferruz N; Doerr S; Vanase-Frawley MA; Zou Y; Chen X; Marr ES; Nelson RT; Kormos BL; Wager TT; Hou X; Villalobos A; Sciabola S; De Fabritiis G Dopamine D3 receptor antagonist reveals a cryptic pocket in aminergic GPCRs. Sci. Rep 2018, 8, 897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Zimmerman MI; Bowman GR FAST conformational searches by balancing exploration/exploitation trade-offs. J. Chem. Theory Comput 2015, 11, 5747–5757. [DOI] [PubMed] [Google Scholar]
- (65).Porter JR; Moeder KE; Sibbald CA; Zimmerman MI; Hart KM; Greenberg MJ; Bowman GR Cooperative changes in solvent exposure identify cryptic pockets, switches, and allosteric coupling. Biophys. J 2019, 116, 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chen S; Wiewiora RP; Meng F; Babault N; Ma A; Yu W; Qian K; Hu H; Zou H; Wang J; Fan S; Blum G; Pittella-Silva F; Beauchamp KA; Tempel W; Jiang H; Chen K; Skene RJ; Zheng YG; Brown PJ; Jin J; Luo C; Chodera JD; Luo M The dynamic conformational landscape of the protein methyltransferase SETD8. eLife 2019, 8, e45403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Hart KM; Ho CM; Dutta S; Gross ML; Bowman GR Modelling proteins’ hidden conformations to predict antibiotic resistance. Nat. Commun 2016, 7, 12965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Amaro RE; Baudry J; Chodera J; Demir O; McCammon JA; Miao Y; Smith JC Ensemble docking in drug discovery. Biophys. J 2018, 114, 2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hart KM; Moeder KE; Ho CMW; Zimmerman MI; Frederick TE; Bowman GR Designing small molecules to target cryptic pockets yields both positive and negative allosteric modulators. PLoS One 2017, 12, e0178678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Vajda S; Beglov D; Wakefield AE; Egbert M; Whitty A Cryptic binding sites on proteins: definition, detection, and druggability. Curr. Opin. Chem. Biol 2018, 44, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sondergaard CR; Garrett AE; Carstensen T; Pollastri G; Nielsen JE Structural artifacts in protein-ligand X-ray structures: implications for the development of docking scoring functions. J. Med. Chem 2009, 52, 5673–5684. [DOI] [PubMed] [Google Scholar]
- (72).Johnson DK; Karanicolas J Druggable protein interaction sites are more predisposed to surface pocket formation than the rest of the protein surface. PLoS Comput. Biol 2013, 9, e1002951. [DOI] [PMC free article] [PubMed] [Google Scholar]