

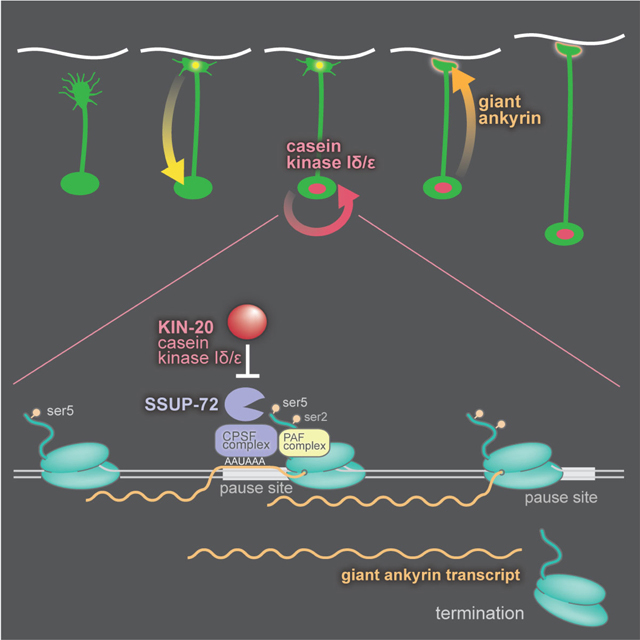
Summary
After axon outgrowth and synapse formation, the nervous system transitions to a stable architecture. In C. elegans, this transition is marked by the appearance of casein kinase 1δ (CK1δ) in the nucleus. In CK1δ mutants, neurons continue to sprout growth cones into adulthood leading to a highly ramified nervous system. Nervous system architecture in these mutants is completely restored by suppressor mutations in 10 genes involved in transcription termination. CK1δ prevents termination by phosphorylating and inhibiting SSUP-72. SSUP-72 would normally remodel the C-terminal domain of RNA polymerase in anticipation of termination. The antitermination activity of CK1δ establishes the mature state of a neuron by promoting the expression of the long isoform of a single gene, the cytoskeleton protein Ankyrin.
Keywords: Nervous system maturation, growth cone, casein kinase 1δ, alternative polyA sites, gene loops, transcription termination, kin-20, unc-44, Ssu72, Paf1C, CPSF, Ankyrin
eTOC paragraph
In C. elegans, Casein Kinase 1 delta accumulates in the nucleus after synapse formation, suppressing growth cones and stabilizing synapses and axons. LaBella et al. show that CK1δ phosphorylates and inhibits transcription termination at a short alternative polyadenylation site within ankyrin and promoting expression of giant Ankyrin, stabilizing axonal growth.
Graphical Abstract
Introduction
Axon guidance during nervous system development is mediated by growth cones that pioneer a pathway from the cell body to the target. When a growth cone reaches its goal, the growth cone collapses, a synapse is formed, the axon is stabilized, and the neuron acquires its mature morphology. This transition from an immature to a mature neuron must require reprograming of the nucleus, presumably in response to interactions of adhesion molecules and the formation of a synapse. Thereafter, neuronal architecture and synaptic connectivity is maintained for the lifetime of the animal in spite of mechanical stress, growth and aging of the animal. By contrast, neurons that fail to mature are marked by retraction of axons and continued sprouting of growth cones throughout the course of the animal’s lifetime.
Mutants with defects in axon outgrowth superficially resemble those with defects in maturation: axons are misplaced or truncated when imaged in adult animals. However, mutants with defects in maturation will exhibit normal axon guidance and synapse formation during development. Even demonstrating that axon outgrowth is normal during development can be inadequate. For example, mutants lacking β-spectrin exhibit normal outgrowth during development followed by continued emergence of growth cones in mature neurons in C. elegans (Hammarlund et al., 2000). However, ectopic growth cones arise due to axon breaks during movement, not defects in maturation. If movement is blocked in β-spectrin mutants, axon breaks are suppressed, and the architecture of the nervous system is stable (Hammarlund et al., 2007). Thus, neuron maturation is normal, and growth cone sprouting is due to normal regenerative processes after axon damage in spectrin mutants.
To date relatively few genes have been identified that play a specific role in the establishment of the mature state. Mutations in unc-119 in C. elegans display normal axon outgrowth in time-lapse imaging and form synapses, yet continue sprouting growth cones into adulthood (Knobel et al., 2001). UNC-119 is a conserved protein that binds acyl groups of G-proteins (Zhang et al., 2011) but its specific binding target required to establish the mature state of the nervous system remains unknown. The cytoskeleton protein Ankyrin has also been implicated in maintenance of axon morphology in invertebrates and vertebrates. In addition, to the ubiquitous short isoforms, Ankyrins encode giant isoforms that are specifically expressed in neurons (Bennett et al., 1982; Jegla et al., 2016; Kordeli et al., 1995; Kunimoto et al., 1991). In Drosophila, mutants lacking the giant isoform of Ankyrin form synapses but then retract them, suggesting that there is a defect in the establishment of the mature state (Koch et al., 2008; Pielage et al., 2008). In C. elegans, Ankyrin mutants exhibit axon defects (Hedgecock et al., 1985; McIntire et al., 1992; Otsuka et al., 1995, 2002), and the expression of giant ankyrin in neurons is regulated by termination at an alternative polyadenylation site (Chen et al., 2015, 2017). These data suggest that UNC-119 and giant Ankyrin are structural components that establish the mature state of an axon. However, ‘maturation’ implies that there must be a signaling pathway. Specifically, the neuron must ‘sense’ that it has formed synapses to the correct target and reprogram its nucleus to repress outgrowth and to maintain a stable architecture.
To identify new genes required for neuron maturation, we screened for mutants which continue to sprout growth cones after nervous system differentiation in C. elegans. We identified mutations in casein kinase 1δ (CK1δ / kin-20) which result in massive disruption of axon morphology in the adult animal. Mutations in kin-20 do not disrupt axon outgrowth or synapse formation during development, instead neurons continue to sprout growth cones after development is complete, which eventually leads to a highly branched nervous system. To identify the signaling pathway downstream of CK1δ, we performed a suppressor screen and found that CK1δ functions to promote expression of the giant isoform of Ankyrin. The screen identified 39 mutations in 13 genes that when mutated restore nervous system stability in the CK1δ mutant. Second-site mutations in 10 components of the RNA polymerase-II termination complex restore expression of Ankyrin with remarkable specificity. These results suggest that casein kinase 1δ might function as a switch to stabilize mature nervous system architecture.
Results
CK1δ is required to establish a mature nervous system architecture
To identify genes required for the maintenance of nervous system architecture, we screened for mutants exhibiting highly branched axons in C. elegans. The mutation ox423 causes animals to be dumpy and exhibit progressive paralysis, similar to β-spectrin / unc-70 mutants. Mapping and genome sequencing demonstrated that ox423 is a mutation in kin-20, which encodes the kinase CK1δ. The CK1δ ortholog in Drosophila was isolated as doubletime, a mutant with defects in circadian rhythms (Kloss et al., 1998; Price et al., 1998). A conserved pathway comprised of doubletime, period, and timeless regulate circadian rhythms in most animals (Young and Kay, 2001), and regulate heterochronic gene expression in nematodes (Rhodehouse et al., 2018; Temmerman et al., 2011; Tennessen et al., 2006, 2010). In nematodes, knockdown of kin-20 by RNA interference promoted formation of precocious, stage-specific cuticle structures called alae (Banerjee et al., 2005; Rhodehouse et al., 2018).
In C. elegans, the CK1 family includes kin-19 (CK1α), csnk-1 (CK1γ), and kin-20 (CK1δ); in vertebrates, a fourth homolog, called CK1ε, has arisen from a duplication of CK1δ (Fish et al., 1995). kin-20(ox423) contains a nonsense mutation (Q344stop) in the kinase domain (Figure 1A). A deletion allele ok505, obtained from the knock-out consortium (C. elegans Deletion Mutant Consortium, 2012), exhibits an identical phenotype and fails to complement ox423; we conclude that they both represent null mutations. To confirm that kin-20 is the relevant gene we rescued the mutant with a single copy transgene (Figure S1A). kin-20 mutations exhibit a maternal effect, uncoordinated phenotype. Specifically, heterozygous mothers (kin-20(ox423)/+) produce homozygous offspring that are healthy and coordinated, but slightly constipated. By contrast, offspring from homozygous kin-20(ox423) mothers are paralyzed, dumpy and egg-laying defective. These phenotypes are not due to a strict maternal effect since they exhibit paternal rescue; kin-20/+ offspring generated by crossing a wild-type male to a kin-20(−) mutant mother are grossly wild-type. Thus, either a paternal chromosome or gene products from the maternal germline are sufficient for normal development.
Figure 1. CK1δ is required to stabilize neuronal architecture.

(A) The C. elegans kin-20 gene locus. The ox423 allele is a nonsense mutation changing glutamine to a premature stop codon (KIN-20a Q344X) in the middle of the conserved kinase domain (KHQHIKHXHI). The large 5’ exon is only found in Caenorhabditids.
(B) Wild-type L4 larva expressing GFP in GABA DD and VD neurons (strain EG1285). Fluorescence in all images is inverted to improve the visibility of processes.
(C) The nervous system in a mature kin-20 mutant is highly disorganized. A kin-20(ox423) L4 larva expressing GFP in all GABA neurons (EG5202). Boxes show examples of structures quantified in C–E.
(D) Ectopic growth cones, (E) branches, and (F) spindly axons (mean ±SEM per animal) in larval stages L2, L4 and young adults (YA) (N≥5 animals, n≥80 axons, Welch’s t-test: *p<0.05, **p<0.01, ***p<0.001, ns p>0.05).
The nervous system in kin-20 mutants is highly disorganized in late larval stage animals compared to wild-type animals (Figure 1B, C). During normal development, the DD GABA motor neurons extend axons from the ventral nerve cord to the dorsal nerve cord during embryogenesis, and the VD GABA motor neurons extend axons during the late L1 stage; thereafter the architecture of the nervous system remains stable. In kin-20 mutants, the motor neurons exhibit extra commissures, misrouted or branched axons, persistent growth cones, and spindly axons. To determine whether these defects arise during axon elongation or in mature neurons, we characterized the development of DD and VD motor neurons. We quantified the presence of supernumerary branches, ectopic growth cones, and thin, spindly axons during all larval stages and adults (Figure 1D–F). Axon defects accumulate in kin-20(ox423) mutants as the animal ages: ectopic growth cones sprouting from the axon or cell body increased from 0.2 per animal in the L2 larval stage to 6.4 in the adult (Figure 1D). The increase in ectopic growth cones gave rise to a concomitant increase in ectopic branches, which increased from 3 branched axons per animal in the L2 stage to 24 branched axons in the adult (Figure 1E).
These data suggest that the disorganized nervous system in kin-20 mutants is caused by axon sprouting in larvae and adults.
CK1 is not required for axon guidance or synapse formation
To determine if CK1δ is required for axon outgrowth during the initial formation of the nervous system, we performed time-lapse confocal microscopy during the late L1 to L2 transition (Figure 2A–B, and Movie S1–S2). We monitored outgrowth by imaging VD motor neurons. VD motor neurons are born in the late L1 larval stage on the ventral side and extend growth cones to the dorsal cord during the L1 to L2 molt (Knobel et al., 1999). In kin-20 early L2 larvae, growth cones from the VD neurons extend from the ventral to dorsal nerve cord with no signs of ectopic branching. These data suggest that axon outgrowth is normal and that branching occurs after the growth cones have reached their targets.
Figure 2. CK1δ is required for neuronal maturation, not axon guidance.

(A) Wild-type VD growth cones (arrows) extend during early L2 larval stage (EG1285). The DD axon has already extended and formed neuromuscular junctions during embryogenesis.
(B) Primary VD growth cones extend normally in kin-20 larvae (EG5202).
(C) Mature neurons continue to extend growth cones in kin-20 mutants. An interstitial growth cone extends within a differentiated DD axon shaft and terminalizes in the dorsal nerve cord.
(D) The dorsal cord retracts (panel 2), and the secondary growth cone extends into the synaptic region in a kin-20 mutant.
(E) Growth cones sprout continuously from commissures in older kin-20 larvae and adults, in this example the growth cone fuses with its own axon and collapses.
(F) CK1δ inhibits regeneration. Left, most cut axons (green arrows) in the wild type remain as stalled growth cones or stumps 24 hours after axotomy. Middle, in kin-20 mutants growth cones rapidly sprout from cut axons immediately after laser axotomy (red arrows). Right, 24 hours later, these same axons have reached the dorsal cord (red arrows).
(G) Following laser axotomy, growth cones reach the dorsal cord more frequently in kin-20 mutants than in the wild type (WT 16%; kin-20(ox423) 78%; Fisher’s exact test **p<0.01, error bars represent 95% CI, n>100).
To monitor axon stability, we imaged DD motor neurons. The DD motor neurons are born on the ventral side and differentiate during embryogenesis; by the L1 stage, fully extended commissures are present (White et al., 1978, 1986). In kin-20 mutants, the DD commissures are largely normal in L1 larvae: 78% of DD commissures are composed of a single unbranched axon. However, 17% of DD axons generate an interstitial growth cone, which migrates within the existing axon shaft, and terminalizes in the dorsal nerve cord (Figure 2C and Movie S3). The formation of interstitial growth cones is occasionally accompanied by retraction of the distal portion of the axon and re-extension along the dorsal cord by the new growth cone (Figure 2D and Movie S4). In the early L2 larvae, ectopic growth cones are observed sprouting from the side of the axon shaft of DD neurons and extending toward the dorsal nerve cord (Figure 2B and Movie S2). We conclude that kin-20 mutant animals exhibit normal axon outgrowth but fail to repress the emergence of new growth cones after reaching their target.
CK1δ is required for neuronal maturation
Growth cones continue to sprout from existing axon shafts even in late larvae and adults in CK1δ mutants (Figure 2E and Movie S5). Ectopic growth cone formation in late larvae and adult stages is also observed in β-spectrin mutants (Hammarlund et al., 2007). In β-spectrin mutants, ectopic growth cone formation is caused by axon breakage during movement; these ectopic growth cones can be fully suppressed by paralyzing the animal. To determine if ectopic growth cones were caused by movement-induced axon breaks in CK1δ mutants, we paralyzed animals by knocking down the muscle gene Titin-related / unc-22 using RNA interference. Chronic paralysis rescued nervous system architecture in spectrin mutants but failed to eliminate ectopic growth cone formation and axonal branching in CK1δ mutants (Figure S1B). These data suggest that the defects in CK1δ mutants are not due to mechanical stress to the axon but rather appear to be a defect in maturation of the axon to a stable differentiated state.
Another explanation for the CK1δ outgrowth phenotype is that the neurons never mature. To determine whether neurons are chronically immature in kin-20 mutants, we assayed the regenerative capacity of neurons. In C. elegans, young neurons regenerate better than older neurons after laser axotomy (Byrne et al., 2014; Gabel et al., 2008; Hammarlund et al., 2009; Nix et al., 2011; Wu et al., 2007; Zou et al., 2013). We cut axons in the L4 stage and assayed regeneration 24 hours later. Most axons retracted or remained as persistent stumps both in the wild type and in kin-20 mutants (Figure S1C WT 53%; kin-20(ox423) 66%, Fisher exact test p=0.05). In the remaining axons, growth cones sprouted, but in wild-type animals only 16% reach the dorsal cord, whereas, in kin-20 mutants, 78% of the growth cones reach the dorsal nerve cord (Figure 2F, G; Figure S1C; regeneration to DNC Fisher exact test ***p=0.0004). This result indicates that kin-20 mutants, like young animals, can regenerate cut axons better than in the wild type.
Axons may be unstable in CK1δ mutants because they fail to recognize their targets and form synapses, and the nucleus is not reprogrammed to become a fully differentiated neuron. To determine if synapse formation was normal, we tagged three synaptic proteins, liprin-α (SYD-2), neurexin (NRX-1), and RIM-binding protein (RIMB-1) with Skylan-S using CRISPR. Synapses in the ventral nerve cord were imaged using super-resolution microscopy during the L2 larval stage before the nervous system sprouted supernumerary growth cones (Figure 3A). The number, volume, and density of the presynaptic puncta for these proteins were normal in kin-20 mutants (Figure 3B, Figure S2A–F). Thus, persistent axon outgrowth in CK1δ mutants is not due to a failure to form synapses.
Figure 3. CK1δ localizes to neuron cell bodies during synapse formation.

(A) Synapse formation is normal in kin-20 mutants. Super-resolution imaging of liprin-α/ SYD-2 tagged with Skylan-S along the ventral cord of wild-type and kin-20(−) L2 larvae (EG9812 and EG9811, respectively). Colors distinguish individual clusters.
(B) Synapses are normal for the number of SYD-2 labeled synapses (p=0.85, unpaired t-test), volume of SYD-2 dense projections (p=0.36, unpaired t-test), and SYD-2 protein density (p=0.12, unpaired t-test). (See Figure S2 for RIMB-1 and NRX-1 data, N>5, n>120). (C) CK1δ (tagRFP) is observed in cell bodies of mature DD neurons in late L1 animals, but not in cell bodies of immature VD neurons (top). Tagged CK1δ also appears in cell bodies of VD neurons after synapse formation in the late L2 stage (bottom). (EG9581). (D) Tagged CK1δ fluorescence in VD cell bodies is significantly lower than in DD cell bodies in L1s (***p=<0.001, unpaired t-test). CK1δ expression level is not significantly different in VD neurons after axon extension and synapse formation (ns p=0.8, unpaired t-test)(bottom panel).
CK1δ acts cell autonomously after axon outgrowth and synapse formation
To determine in which tissue kin-20 acts to stabilize neurons, we selectively expressed KIN-20 in the epidermis, muscle, intestine, and nervous system in kin-20(ox423) mutants (Table S1). Only expression of KIN-20 in neurons rescued the uncoordinated and axon branching phenotypes (Figure S3A), demonstrating that CK1δ acts cell autonomously to stabilize neuronal architecture.
To determine the expression pattern and localization of CK1δ, we fluorescently tagged the protein with tagRFP (Figure 1A). During embryogenesis, expression of KIN-20 is absent in early embryos, expression increases at three-fold stage, and is observed as a few dim speckles in the nuclei of developing DD motor neurons (Figure S3D, E). In L1 hatchlings, expression is dim throughout the animal but KIN-20 can be observed in the nuclei of neurons (Figure S3F). The VD neurons send out growth cones postembryonically during the L1 / L2 molt (Knobel et al., 1999), and during axon extension expression in the VDs is lower than in DD neurons (Figure 3C, D & S3G). At the mid-L2 stage the VD neurons form synapses (Hallam and Jin, 1998; Kurup and Jin, 2015; Sulston, 1976) and KIN-20 expression is identical in DD and VD cell bodies (Figure 3C, D). At the L2 stage, KIN-20 is also highly expressed in the nuclei of epidermal cells, and is visible in the nuclei of motor neurons (Figure S3H). Expression tends to peak at the end of each larval stage, consistent with expression of other heterochronic genes (Rhodehouse et al., 2018). In the L4 stage, kin-20 is localized to the nucleus of all cell types (Figure S3B) and at adherens junctions in the spermatheca (Figure S3C). Ubiquitous expression is consistent with CK1δ expression in other organisms (Knippschild et al., 2005). Together, these data suggest that appearance of KIN-20 in the nuclei of neurons coincides with axon outgrowth and the formation of synapses just preceding the maturation of the nervous system.
CK1δ suppressors rescue axonal defects
To characterize the genetic pathway downstream of kin-20, we performed a suppressor screen using the chemical mutagen ENU (N-ethyl-N-nitrosourea). To identify strong suppressors, we performed a multigenerational fitness screen (Hollopeter et al., 2014). We isolated 44 independent suppressor strains that were healthy and exhibited normal locomotion (Figure 4A, D & S4A). Analysis of the GABA motor neurons revealed rescue of the kin-20 neuronal branching defect in all suppressors (Figure 4E). We were able to characterize suppressor mutations in 39 of the strains comprising mutations in 13 genes (Tables 1 & S2).
Figure 4. CK1δ acts on transcription termination.

(A) Suppressors restore locomotion to kin-20(ox423) mutants. Worm tracks for 5 worms were imaged for 2 minutes and traced. (For other suppressors see Figure S4).
(B) Above, CK1δ phosphorylates SSUP-72 in an in-vitro phosphorylation assay. Rat CK1δ1 was added to recombinant SSUP-72. Left, phosphorylated SSUP-72 (predicted MW 22.8 kDa); middle, phosphorylation control of phospho-primed casein; right, CK1δ autophosphorylation. Below, a consensus CK1δ phosphorylation site is conserved from yeast to humans in Ssu72 orthologs. The target serine is mutated in the suppressor ssup-72(ox542).
(C) The phosphomimetic mutation of SSUP-72 S39 is sufficient to suppress kin-20 locomotory defects. Specific mutations in ssup-72 were generated by CRISPR.
(D) Quantification of distances traveled in 2 minutes (±SEM, n=5). SSUP-72 S39E rescues kin-20(ox423) motility (***p<0.001 Welch’s t-test), SSUP-72 S39A does not rescue (ns, p>0.05), (EG9899 and EG9914). SSUP-72 S39E and S39A mutants alone are behaviorally wild type (EG9900 and EG9913). Because SSUP-72 S39A does not phenocopy the kin-20 null suggests that there are other targets of CK1δ. SSUP-72 S39A exhibits slightly increased motility compared to the wild type (*p<0.05, EG9913).
(E) kin-20(ox423) suppressors rescue axon defects. Some suppressors, for example zfp-3(ox653), exhibit occasional, remaining defects.
Table 1.
CK1δ suppressors are in transcription termination components and unc-44 / Ankyrin.
| gene | # of alleles | cellular process | mutation |
|---|---|---|---|
| kin-20 | 4 | Casein kinase 1 delta | Missense, revertants |
| kin-19 | 1 | Casein kinase 1 alpha | Missense |
| ssup-72 | 5 | Phosphatase for RNAP II CTD | Missense |
| pinn-1 | 3 | Prolyl isomerase for RNAP II CTD | Missense and null |
| pcf-11 | 1 | 3’ pre-mRNA processing | Missense |
| cpsf-4 | 3 | Cleavage polyadenylation factor complex | Missense |
| pfs-2 | 1 | Cleavage polyadenylation factor complex | Missense |
| ctr-9 | 3 | Elongation/termination, Paf1 complex | Missense |
| cdc-73 | 1 | Elongation/termination, Paf1 complex | Splice acceptor, null |
| zfp-3 | 7 | Likely RNA pol II complex | Missense and nulls |
| rpb-2 | 5 | RNAP II subunit B | Missense |
| cdk-8 | 1 | Mediator and RNA pol II complex | Missense |
| unc-44 | 4 | Ankyrin, cytoskeletal | Missense |
| Unconfirmed hits | |||
| zfp-3 | 1 | Likely RNA pol II complex | Intronic mutation |
| swd-2.2 | 1 | Set1/COMPASS complex | Missense |
| pinn-1 | 1 | Prolyl isomerase for RNAP II CTD | Intronic mutation |
The genes identified in the kin-20 suppressor screen. Four kin-20(ox423) revertants and pseudo-revertants were identified, indicating that the screen was approaching saturation. kin-19(ox689) is a predicted hypermorphic mutation of casein kinase 1α, a CK1 isoform closely related to CK1δ. unc-44 alleles are found in the polyadenylation site (ox685, ox832, ox833) and within a conserved intron upstream of the polyadenylation site (ox686). All other mutations are in components of the RNA polymerase II complex, most with functions in transcription termination. The same mutation, pfs-2(R158Q), was identified previously in a neuronal branching suppressor screen (Van Epps et al., 2010).
Suppressors prevent premature termination of Ankyrin transcripts
We identified CK1δ suppressor mutations by mapping them against a set of integrated fluorescence markers (Frøkjær-Jensen et al., 2014), whole genome resequencing, and in silico complementation tests (Minevich et al., 2012). Most single hit genes were validated by CRISPR; other genes were validated by transgene rescue (Table S3). Among the 39 characterized suppressors were 4 intracodon revertants of the nonsense mutation, suggesting the screen was approaching saturation. We also recovered a kin-20 suppressor mutation in the related kin-19 gene, which encodes casein kinase 1 alpha (Tables 1 & S2). The suppressor KIN-19(L300F) is a missense mutation in a residue at the C-terminus that is conserved in all CK1 isoforms. Casein kinase isoforms are known to function redundantly in some pathways (Knippschild et al., 2005); therefore, it is likely that this mutation leads to a promiscuous or constitutively active CK1α that compensates for the loss of CK1δ.
Among the remaining suppressors were 30 missense mutations in 10 RNA Polymerase II subunits: rpb-2, pinn-1, ssup-72, cpsf-4, pfs-2, cdc-73, ctr-9, zfp-3, pcf-11, and cdk-8 (Tables 1 & S1). The suppressor mutations can be divided into five classes: RNA polymerase B catalytic subunit, RNA polymerase C-terminal domain modifiers, CPSF complex, Paf1 complex (Paf1C), and Mediator complex. Each of these proteins and their associated complexes is implicated in alternative polyA site selection and transcription termination, with the exception of cdk-8 which functions in transcription initiation via Mediator (Allen and Taatjes, 2015; Shilatifard, 2012). These results suggest that CK1δ acts to inhibit transcription termination at loci important for maintaining the nervous system; in the absence of CK1δ premature termination occurs. Premature termination at these loci can be suppressed by secondary defects in termination.
CK1δ phosphorylates SSUP-72
Since CK1δ is upstream of the RNA polymerase II termination complex, it is possible that one or more proteins in the complex are phosphorylation targets of CK1δ. SSUP-72 is a particularly interesting potential target. SSUP-72 removes the phosphate from Ser5 of the C-terminal domain of RNA polymerase 1 subunit, and this dephosphorylation marks RNA polymerases that are approaching a termination site (Hsin and Manley, 2012; Kuehner et al., 2011). SSUP-72 was also identified previously as a regulator of alternative polyadenylation during neuron development (Chen et al., 2015). The SSUP-72 S39F suppressor mutation we identified is at a conserved serine/threonine from yeast to humans (Figure 4B, S4B), which is in a consensus CK1 phosphorylation site (p[S/T]xx[S/T]). Phosphorylation of the first Ser/Thr residue primes CK1δ phosphorylation of the second Ser/Thr (Knippschild et al., 2005).
To determine if SSUP-72 can be phosphorylated by CK1δ, recombinant C. elegans SSUP-72 was used as a target in an in vitro phosphorylation assay. We found that truncated rat CK1δ is able to phosphorylate SSUP-72 directly (Figure 4B). If SSUP-72 S39 is phosphorylated by CK1 in vivo to inhibit transcription termination, we would expect an SSUP-72 S39E phosphomimetic mutation would suppress the kin-20 null phenotype. The SSUP-72(S39E) allele was generated by CRISPR, and was crossed into the kin-20 null. ssup-72(S39E) suppressed the kin-20 null phenotype, restoring motility to these animals (Figure 4C, D). The phosphorylation-defective allele ssup-72(S39A) did not rescue kin-20 null animals (Figure 4C, D). These data demonstrate that SSUP-72 S39 is likely a key CK1δ phosphorylation target in this axon stabilization pathway. If SSUP-72 were the sole target of CK1δ, the ssup-72(S39A) mutant animals should phenocopy kin-20 null animals. However, the ssup-27(S39A) animals look and behave like the wild type (Figure 4C, D). Thus, SSUP-72 phosphorylation by CK1δ is sufficient, but not necessary for the stabilization of axons, suggesting that SSUP-72 is not the sole target of CK1δ.
CK1δ promotes expression of the giant Ankyrin isoform
In additison to mutations in RNA polymerase II termination subunits, we identified four suppressor mutations that disrupt termination at an early polyadenylation site in the unc-44 gene. unc-44 encodes Ankyrin, a component of the spectrin cytoskeleton (Otsuka et al., 1995). unc-44 generates three isoforms, one short, one medium and one very long isoform, each of which uses a different polyadenylation site (Figure 5A and S5A). The long form of UNC-44 is related to the giant isoforms of Ankyrin expressed in the neurons of all bilaterians (Jegla et al., 2016). The long isoform of UNC-44 is repressed in other tissues; for example the kinase DAPK-1 inhibits expression of the long form of Ankyrin in the epidermis of C. elegans (Chen et al., 2017), but it is unclear how long Ankyrin is activated in neurons. Three of our suppressors are mutations in the AATAAA polyadenylation signal that generates the transcript for one of the short isoforms (Figure 5A). A fourth variant, unc-44(ox686), is in the intron immediately upstream of this polyadenylation site in an adenine-rich sequence conserved in nematodes (Figure 5A and S5A). This mutation caused skipping of the downstream exon, and constitutive splicing in-frame to the giant exon of the long isoform. The medium isoform controlled by polyA site #2 is not detectable by qPCR in the unc-44(ox686) suppressor (Figure S6B). These mutations in unc-44 suggest that CK1δ suppressors promote the specific expression of the long form of Ankyrin.
Figure 5. CK1δ stabilizes mature axons by upregulating giant ankyrin transcription.

(A) Ankyrin gene model. C. elegans has three alternative polyA sites. Expression of the giant isoform requires read-through at polyA site #2. Yellow, kin-20 suppressors unc-44(ox685), unc-44(ox832), and unc-44 (ox833). These mutations disrupt the polyA motif and likely disrupt termination. Orange, kin-20 suppressor unc-44(ox686) (EG9097). The mutation is upstream of polyA site #2 in the preceding intron, which causes exon skipping and only giant ankyrin is transcribed.
(B) kin-20(ox423) lacks mRNA for the giant isoform of ankyrin. qPCR of the giant isoform of unc-44 mRNA in kin-20(ox423) and suppressed animals. mRNA levels are normalized to the wild type. In kin-20 mutants, the giant isoform of unc-44 is decreased ~16-fold compared to wild-type levels, ***p<0.001. All suppressors exhibited increased levels of unc-44 mRNA compared to kin-20(ox423) alone, with the exception of pcf-11(ox688) (Dunnett’s test * p<0.05, ** p<0.01, *** p < 0.001).
(C) Quantification of axon morphology of CK1δ mutant rescue by expression of giant Ankyrin. Single-copy expression of the wild-type kin-20 allele, oxSi1087, in kin-20 null animals fully rescues axon stability. Axon defects of kin-20(ox423) animals are rescued by expression of a chimeric copy of unc-44 in which the polyadenylation site #2 has been replaced by cDNA on an extrachromosomal array (EG9248, *** p<0.001). Both the unc-44(ox685) and unc-44(ox686) suppressor mutation rescues CK1(−) (***p<0.001). Mean ectopic growth cones, branches, and spindly axons were scored per adult animal. N ≥5 animals, n≥80 commissures. For the wild type and kin-20(ox423), data are repeated from Figure 1. All comparisons were made with Dunnett’s multiple comparison test.
(D) Expression of giant Ankyrin rescues axon morphology of the CK1δ mutant. Compared to kin-20(ox423), axon morphology of a kin-20(ox423) strain expressing the giant isoform of Ankyrin is largely rescued; occasionally an axon is branched or spindly (EG9248).
In the simplest model, CK1δ inhibits transcription termination at polyA site #2 of unc-44 and promotes expression of giant Ankyrin; in the absence of CK1δ, the giant Ankyrin isoform is not expressed. In kin-20 suppressors, transcription termination at polyA site#2 is reduced in neurons, and giant Ankyrin is restored. If this model is correct, (1) giant Ankyrin should be absent in the CK1δ null mutant, (2) giant Ankyrin should be restored in the suppressor mutants, and (3) expression of the giant Ankyrin should rescue CK1δ mutant animals.
To determine whether giant Ankyrin is absent in the CK1δ mutant we performed quantitative RT-PCR. Compared to wild-type embryos, the unc-44 long isoform was decreased approximately 16-fold in kin-20 mutant embryos (Figure 5B). The short and medium unc-44 isoforms were detected at wild-type levels in kin-20 mutant animals, indicating that the long isoform contributes little to the total level of unc-44 mRNAs (Figures S5A for primer locations, S6A, B). We conclude that giant Ankyrin is specifically depleted in CK1δ mutants.
To determine if the suppressors restore giant Ankyrin transcripts, we performed qPCR on ten kin-20 suppressor strains (Figure 5B). In all strains tested, expression of the giant Ankyrin isoform was significantly increased relative to kin-20(ox423). The expression of both short isoforms of Ankyrin remained constant in all strains except the exon-skipping mutation unc-44(ox686), which lacked the short isoform controlled by polyA site #2 (Figure S6A, B). Because the giant Ankyrin transcript is restored in all suppressors, it is likely that the function of CK1δ is to drive expression of the long isoform of Ankyrin.
If CK1δ mutants have unstable axons because they lack giant Ankyrin, then expressing giant Ankyrin in the CK1δ mutants should bypass the requirement for the kinase. We overexpressed the giant isoform of unc-44 from an extrachromosomal array in kin-20(ox423) animals (Figure S5B). The axon morphology of VD and DD motor neurons in the kin-20 mutant was rescued by unc-44-long overexpression (Figure 5C, D). The numbers of supernumerary branches, ectopic growth cones and spindly axons in kin-20 mutants were greatly reduced, although not completely rescued (Figure 5C). The suppressor mutation in the AAUAAA polyadenylation signal (unc-44(ox685)) rescued branching and spindly axon defects, but ectopic growth cones persisted (Figure 5C). The exon-skipping suppressor (unc-44(ox686)) fully rescued the kin-20 axon phenotype (Figure 5C). This suppressor expresses the short and long isoforms, but not the medium isoform (Figures 5C & S6A,B). These data demonstrate that the major defect in CK1δ mutant neurons is the lack of giant Ankyrin.
It is surprising that the most critical target for CK1δ is termination at a single polyA site in the Ankyrin gene. To more fully survey the transcriptome, we performed RNAseq on the wild type, kin-20(ox423) null animals, and kin-20 strains with suppressor mutations (ssup-72(ox542) and cpsf-4(ox649)). The only transcript with significantly reduced expression in the kin-20 mutant is the long form of Ankyrin (Table S4; adjusted P-value<0.1), and levels were restored in the suppressed strains. In addition, transcripts from the non-coding RNA tts-1 (transcribed telomeric sequence) (Essers et al., 2015) were significantly increased in kin-20 mutants, but transcript levels of tts-1 were not restored in the suppressor mutants, suggesting that changes in tts-1 may be due to other targets of CK1. To verify that unc-44 is the sole target and that our RNAseq data is correct, candidate genes were selected based on the presence of alternative polyA sites, as well as genes with known roles in axon and synapse stability (Table S5). None of these candidates are differentially expressed in kin-20(ox423) compared to the wild type (Figure S6C). Taken together, these data demonstrate that the interaction between CK1δ and giant Ankyrin is remarkably specific.
Discussion
Mutations in kin-20, which encodes CK1δ, result in a destabilized nervous system. CK1δ null axons grow out normally and form synapses, but continue to sprout growth cones and send new axons to synaptic targets. By adulthood, essentially all axons are highly branched, with many extending active growth cones, and the animals are paralyzed. To determine the pathway downstream of CK1δ we performed a genetic screen for suppressors. The mutations fall into two major classes: Most are in genes implicated in transcription termination and polyadenylation; the remainder disrupt the alternative polyadenylation site that precedes the long exon of giant Ankyrin. Overexpressing giant Ankyrin rescues CK1δ null animals, demonstrating that expression of giant Ankyrin is the primary target of CK1δ for stabilizing axons in mature neurons.
Giant Ankyrin mediates axon stability
Typically, there are two types of Ankyrins expressed in bilaterians: a short and a long isoform (Jegla et al., 2016). In C. elegans these isoforms are generated by alternative polyadenylation sites from a single gene unc-44. In Drosophila, there are two Ankyrin genes Ank and Ank2; Ank only expresses a short isoform, whereas Ank2 expresses short, medium, long and extra-long isoforms. Vertebrate genomes contain three Ankyrin genes, ANK1, ANK2, and ANK3 (which encode the proteins AnkR, AnkB, and AnkG). AnkR only expresses a short isoform; AnkB and AnkG express both short and long isoforms.
Short isoforms of Ankyrin are expressed ubiquitously, and are composed of 24 Ankyrin repeats, two ZU5 domains, a UPA domain, and a DEATH domain (Bennett and Lorenzo, 2013). The Ankyrin repeat domain binds transmembrane proteins including ion channels, transporters and adhesion molecules such as L1CAM (Koenig and Mohler, 2017). The Ankyrin ZU5-UPA domain links transmembrane proteins to the spectrin cytoskeleton and transport vesicles to dynactin (Bennett and Lorenzo, 2016). The function of the DEATH domain is unknown but the C-terminal domain that follows the DEATH domain has autoregulatory functions (Hall and Bennett, 1987; Tsytlonok et al., 2015).
The long forms or ‘giant Ankyrins’ are specifically expressed in neurons, and are localized to synapses and axons. In axons, Ankyrin and Spectrin are periodically spaced every 190nm (Otsuka et al., 2002; Pielage et al., 2008; Xu et al., 2013). Giant Ankyrins are so named because they are unusually large proteins; in C. elegans, the long form is 6994 amino acids, whereas the shorter isoform is 1867 amino acids. Giant Ankyrins contain the conserved domains found in short isoforms, but also include a long insertion at the C-terminus lacking recognizable domains. The insertion is encoded in large part by a single exon composed of unconserved DNA repeats in worms, flies and mice (Jenkins et al., 2015; Koch et al., 2008; Otsuka et al., 2002; Pielage et al., 2008; Stephan et al., 2015).
CK1δ is required to switch expression from the short isoform to the long isoform of Ankyrin and the long isoform stabilizes the axon architecture of the nervous system. What is the giant isoform doing that the short isoform cannot? The mechanism is not understood but where it has been tested giant ankyrin appears to inhibit axon branching. Specific loss of giant Ankyrin leads to axon defects in C. elegans and cultured mouse neurons (Hedgecock et al., 1985; McIntire et al., 1992; Otsuka et al., 1995, 2002; Yang et al., 2019). Giant Ankyrin also plays a role in synapse stability: loss of Ank2-L in Drosophila causes the disassembly and retraction of neuromuscular junctions (Koch et al., 2008; Pielage et al., 2008). Giant Ankyrin is also required to maintain the spacing and localization of microtubules in Drosophila axons and cultured mouse neurons (Ank2-XL and AnkB, respectively) (Fréal et al., 2016, 2019; He et al., 2019; Stephan et al., 2015; Yang et al., 2019). Which of these represents the primary defect is not known.
Molecular mechanism of termination at the proximal Ankyrin polyadenylation site
The giant form of Ankyrin in C. elegans is generated when RNA polymerase reads through alternative polyadenylation sites in the unc-44 locus and incorporates the giant exon into the transcript. In the absence of CK1δ, transcription is terminated at the proximal polyA site, and giant Ankyrin is not made. The suppressors of kin-20 prevent termination at the proximal site and giant Ankyrin is restored. Thus, these mutations tell us what processes are required for termination at alternative polyadenylation sites. In overview, termination is controlled by the phosphorylation pattern of the C-terminal domain of RNA polymerase, and by specific sequence motifs in the nascent mRNA. After the nascent transcript is cut, polyadenylated and released, the RNA polymerase must release the DNA and terminate transcription. Our genetic analysis suggests a model for how the RNA polymerase machinery directs selection of the proximal alternative polyadenylation site in the unc-44 gene, and how CK1δ blocks termination in neurons.
The first step in termination is likely to be the dephosphorylation of the C-terminal domain of RNA polymerase by the phosphatase SSUP-72. SSUP-72 was previously implicated in stimulating pausing and termination at the unc-44 polyA site #2 (Chen et al., 2015). SSUP-72 functions by dephosphorylating the C-terminal domain (CTD) of RNA polymerase subunit 1 (RPB1) (Krishnamurthy et al., 2004). The CTD is comprised of heptad amino acid repeats: 26 in S. cerevisiae, 42 in C. elegans, and 52 in mammals, encompassing the amino acid sequence YSPTSPS. Phosphorylation of the CTD domain controls the state of the RNA polymerase complex by recruiting elongation or termination factors. Phosphorylation of Ser5 in each repeat signals active transcription and processivity of the polymerase. During termination, the phosphorylation state of the CTD is rewritten. This requires the prolyl-isomerase PINN-1 to reshape the CTD, which allows the phosphatase SSUP-72 to erase the phosphate code (Krishnamurthy et al., 2004; Xiang et al., 2010). Specifically, SSUP-72 de-phosphorylates Serine 5 (Ser5) of the CTD, a key step in the transition from transcription elongation to termination (Kuehner et al., 2011).
In neurons, CK1δ prevents termination at the proximal polyadenylation site by inhibiting SSUP-72. Ser39 of SSUP-72 is a conserved consensus phosphorylation site for casein kinases (p[S/T]xx[S/T]). Phosphorylation of the first Ser/Thr residue primes phosphorylation of the second Ser/Thr by CK1δ (Knippschild et al., 2005). Mutations in SSUP-72 were previously found to restore a subtle loss of giant Ankyrin in sydn-1 mutant animals, SYDN-1 is a worm-specific nuclear protein thought to promote read-through during polyA site regulation (Chen et al., 2015). We found that CK1δ phosphorylates SSUP-72 in vitro, and the phosphomimetic mutation S39E bypasses the loss of CK1δ and restores motility in kin-20 nulls. Most of the other kin-20 suppressor alleles that we isolated in SSUP-72 are predicted to eliminate phosphatase activity (Rosado-Lugo and Hampsey, 2014; Xiang et al., 2010).
In the simplest model, CK1δ phosphorylates Ser39 of SSUP-72, which disrupts phosphatase activity. The phosphorylated CTD of the polymerase maintains the processive state; the polymerase reads through the polyadenylation signal to generate the long form of unc-44. CK1δ is known to bind the CTD of RNA polymerase: The yeast homolog of casein kinase 1 delta, Hrr25, directly binds the CTD in a phosphorylation dependent manner (Phatnani and Greenleaf, 2006). Hrr25 requires Ser2 and Ser5 to be phosphorylated – the state of the CTD during elongation – placing the kinase at the correct transcriptional time and place to phosphorylate SSUP-72.
An alternative but not mutually exclusive model is that SSUP-72 maintains a gene loop within the Ankyrin gene. Gene loops form when the termination site is associated with the gene’s promoter and thereby couples transcription termination to transcription reinitiation. In addition to a role in transcription termination, SSUP-72 is required for gene loop formation (Ansari and Hampsey, 2005; Perkins et al., 2008; Singh and Hampsey, 2007; Tan-Wong et al., 2012). Thus SSUP-72 may form a gene loop that promotes expression of the short form of Ankyrin. Loss of SSUP-72 function would disrupt loop formation, and the polymerase would proceed through the pause site and transcribe the giant isoform. In fact, Ank2 in mouse neurons forms multiple gene loops both including and excluding the giant exon (Bertolini et al., 2019). CK1δ phosphorylation of SSUP-72 may constitute the first example of gene loop regulation.
In the absence of CK1δ, dephosphorylation of Ser5 on the CTD allows the binding of the Cleavage Factor II complex (CF II). The CF II complex is comprised of PCF11 and CLP1 (Zhang and Gilmour, 2006; Zhang et al., 2005). PCF11 is recruited to the CTD by Ser2-P, binds the nascent RNA and disrupts the elongation complex (Licatalosi et al., 2002; Meinhart and Cramer, 2004; Zhang and Gilmour, 2006). The PCF-11(F83Y) mutation we recovered is in the CTD-binding domain (Meinhart and Cramer, 2004) and therefore is likely to interfere with PCF-11 recruitment to the CTD, and thereby leads to an increase in giant Ankyrin mRNA. These results are consistent with an observed increase in transcription read-through in yeast, flies and human cells depleted of PCF-11 (Baejen et al., 2017; West and Proudfoot, 2008; Zhang and Gilmour, 2006).Interestingly, PCF-11 is specifically involved in selecting proximal polyA sites in alternative gene isoforms in mammalian cells (Wang et al., 2019).
Selection of a site for cleavage and polyadenylation is under the control of the cleavage and polyadenylation specificity factor (CPSF) complex. There are two nucleotide motifs that coordinate transcription termination and polyadenylation (Figure S4). The canonical polyadenylation site AAUAAA is recognized by CPSF and pauses elongation; the U-rich downstream elements (DSE) are recognized and bound by the cleavage stimulation factor complex (CstF) (Porrua and Libri, 2015). The transcript is cleaved by CPSF and polyadenylated at a CA dinucleotide 18–30 nucleotides after the AAUAAA motif (Porrua and Libri, 2015). Transcription is terminated no more than 150 nucleotides downstream of the cleavage site, probably coincident and mechanistically linked to cleavage and polyadenylation of the mRNA (Bentley, 2014).
Four of our suppressor mutations are in residues of the CPSF complex (Figure 6), which is responsible for recognizing the AAUAAA polyadenylation signaling motif. One mutation is in PFS-2 in the AAUAAA binding pocket at an interface with CPSF-4 (Clerici et al., 2018); the identical mutation was isolated in a suppression screen of an RNA polymerase subunit in C. elegans (Van Epps et al., 2010). Three other mutated residues are on the surface of CPSF-4 directly adjacent to the binding pocket. (Casañal et al., 2017; Clerici et al., 2018). These residues are positioned to potentially convey AAUAAA recognition, directly to the RNA polymerase or possibly via the Paf1 complex.
Figure 6. CK1δ suppressors describe a pathway in transcription termination.

(A) The kin-20(ox423) suppressor screen generated mutations in the RNA polymerase II complex involved in termination (See Table 2.2 for full list). The genetics and structure together describe a pathway for transcription termination at the unc-44 second polyadenylation site.
(1) CK1 is associated with the C-terminal domain (CTD) of RNA polymerase subunit RPB-1 (Phatnani and Greenleaf, 2006). In differentiated neurons CK1δ phosphorylates SSUP-72, and thereby inhibits remodeling of the CTD. This step is likely the key regulatory step in the switch from expression of short to long Ankyrin.
(2) The phosphatase SSUP-72 requires the prolyl isomerase PINN-1 to isomerize proline-6 in the heptad repeat so that SSUP-72 can hydrolyze Ser5-P (Werner-Allen et al., 2011; Xu et al., 2003).
(3) PCF-11 binds the CTD after dephosphorylation of Ser5-P. PCF-11 interacts with the transcribed RNA and the CPSF complex.
(4) The CPSF complex recognizes the AAUAAA motif and signals for cleavage and polyadenylation. Mutations in the binding pocket formed by the PFS-2 and CPSF-4 likely interfere with recognition of the polyA signal motif. Mutations of CPSF-4 surface residues may interfere with transduction of that signal.
(5) The elongation Paf1 complex is also required for termination at the unc-44 polyadenylation site. The Paf1 complex recruits CPSF to RNA polymerase and possibly SSUP-72 to the CTD.
(6) Alternatively, the Paf1 complex could be acting as a transduction pathway to the RNA polymerase. Suppressor mutations in the Paf1 complex were obtained in CTR-9, CDC-73, and potential interaction sites in the RPB-2 polymerase subunit .
(B) Model. Top, wild type. When the elongating RNA polymerase II (Pol II) complex approaches PolyA #2, KIN-20 / CK1δ phosphorylates SSUP-72. Phosphorylation inhibits association with the CPSF complex, promoting Pol II processivity.
Middle, kin-20 null. Without CK1δ, CPSF is recruited to the RNA pol II complex and pauses at the polyA site. The transcript is cleaved at downstream CA elements terminating transcription at the middle isoform of unc-44.
Bottom, kin-20 suppressor. In the kin-20 null and suppressor double mutants, KIN-20/CK1δ is no longer able to inhibit pausing, resulting in production of unc-44 long.
The Paf1 complex binds and recruits the CPSF complex to the polymerase via multiple interactions (Nordick et al., 2008; Rozenblatt-Rosen et al., 2009). Thus, it is possible that the surface residues that we identified in the CPSF complex are communicating with the RNA polymerase via the Paf1 complex. Specifically, we recovered mutations on surface residues of the CTR-9 protein (Figure 6) (Vos et al., 2018). These mutations are within the main domain composed of 19 pairs of α-helices (Vos et al., 2018). We also recovered a mutation in CDC-73, which is the main subunit that recruits the CPSF complex to the polymerase (Nordick et al., 2008; Rozenblatt-Rosen et al., 2009). CDC-73 also links the Paf1 complex to the chromatin of actively transcribed genes (Amrich et al., 2012; Rozenblatt-Rosen et al., 2009). The most abundant target was ZFP-3 / ZC3H4 (7 mutations), which has been shown to bind the Paf1 protein and components of the CstF cleavage stimulation complex in high-throughput screens (Li et al., 2004; Stark et al., 2006). ZFP-3 was previously identified in screens for termination defects in the genes lin-15 and unc-44 (Chen et al., 2015; Cui et al., 2008).
The mutations in RNA polymerase recovered in our screen also suggest a role for the Paf1 complex in termination. RPB-2(S101L) disrupts an interaction site with the Paf1C member LEO-1 (Mueller and Jaehning, 2002; Vos et al., 2018). Although direct contacts between LEO-1 and the RPB-2 residues M600 and I603 could not be resolved, electron density from LEO-1was observed at this position (pers. comm. S. Vos). The RPB-2 residues Y537 and V540 are deep within a pocket that does not interact with LEO-1 directly, but may nevertheless destabilize LEO-1 interactions.
The Paf1 complex is normally associated with the initiation and elongation phases of transcription (Jaehning, 2010); however, our genetic data suggest that it is intimately involved in transcription termination in unc-44. In addition, mutations in Paf1 complex in yeast suggest that loss of the complex affects 3’ end formation and polyadenylation, again, indicating that the complex also functions during termination (Jaehning, 2010). How the Paf1 complex contributes to termination is not clear. The complex may simply recruit cleavage and polyadenylation factors to the polymerase; our mutations which destabilize the Paf1 complex could lead to a failure to recruit termination machinery. Alternatively, the complex might be involved in transducing cleavage and polyadenylation signals to the RNA polymerase to terminate transcription and dismount from the DNA helix. The mutations we identified may be specifically involved in transducing that allosteric signal.
It is curious that unc-44 was the only gene exhibiting premature termination in kin-20 mutants. One might expect that CK1δ could potentially act on the termination complex of all genes expressed in neurons. It is likely that specificity factors recruit KIN-20 to particular transcription units. For example, the SSUP-72 binding protein SYDN-1 selects among alternative polyadenylation sites in different tissues in the nematode (Chen et al., 2015; Van Epps et al., 2010). SYDN-1 biases transcription toward the long isoform of the unc-44 gene but the short isoform of the dlk-1 gene. Thus, SYDN-1 is likely to be a polyadenylation site selector protein rather than a part of the termination complex, whereas KIN-20 apparently only acts as an anti-termination factor on the unc-44 gene.
Is this mechanism for generation of giant ankyrin likely to be conserved in other organisms? The ankyrin genes of other invertebrates such as Drosophila have ankyrin gene models that are consistent with giant exon selection by alternative polyadenylation sites. Vertebrate ankyrins generate the giant isoform by alternatively splicing rather than alternative termination (Bennett and Lorenzo, 2016), suggesting that this mechanism of isoform selection is not conserved. On the other hand, it is possible that gene loop formation is the mechanism of isoform selection in both invertebrates and vertebrates. Polyadenylation sites are required for gene loop formation (Perkins et al., 2008), and the alternative exons in vertebrate Ankyrins are preceded and followed by canonical polyadenylation and transcriptional pause sites. It is possible that the machinery for polyadenylation site recognition, such as SSUP-72 and the CPSF complex, may be involved in alternative splicing as well as termination since transcriptional pausing impacts splice site availability (Herzel et al., 2017). In fact, splicing itself may drive gene loop formation, a cause and effect conundrum that is being actively investigated (Herzel et al., 2017; Moabbi et al., 2012).
How does CK1δ function as a maturation signal?
It is possible that CK1δ is active in all neurons at their birth to make the axon-specific, long isoform of Ankyrin. However, it is intriguing that axon outgrowth and synapse formation is normal in CK1δ mutants, that is, a single growth cone maintains apical dominance at the tip of the axon, and synapses are assembled normally on muscles. Only after the axons reach their targets and build synapses, do growth cones sprout from the cell body or shaft of mature axons in the CK1δ mutant. Presumably in the wild type, expression of the long isoform of Ankyrin coincides with synapse formation, and further growth cone initiation is blocked. An intriguing possibility is that the synapse itself launches the signaling cascade. Such a retrograde signal must be upstream of CK1δ, since the kinase is localized and functions in the nucleus. CK1δ then instructs the nucleus to adopt a mature profile. By contrast, in the CK1δ mutant the neuron remains forever young, and in a juvenile state.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Erik M. Jorgensen Jorgensen@biology.utah.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
C. elegans strains were grown and maintained using standard methods (Brenner, 1974). The wild type is N2 Bristol. For a full list of strains and mutations used in this work, see Key resource table.
KEY RESOURCES TABLE
kin-20(ox423) and wild-type axon quantification
C. elegans hermaphrodites were immobilized for microscopy on a 2% agarose pad in M9 + 25mM sodium azide solution on a glass slide. Animals were allowed to equilibrate for 5 minutes. The cover slip was sealed with Vaseline. Animals were imaged on a Zeiss LSM 510 META with a 60X oil immersion lens (Carl Zeiss, Jena, Germany). Right side was imaged because GABA motor neurons in the ventral cord send commissures along the right side of the body wall to the dorsal nerve cord. Spectral unmixing was performed to remove auto-fluorescence in gut granules, allowing better visualization of the motor neuron commissures to be scored.
kin-20 gene locus
Six isoforms are generated by alternative splicing of 5’ and 3’ exons. The large 5’ exon is only found in the genus Caenorhabditids. All isoforms contain a conserved casein kinase 1 domain. The ok505 allele is a complete deletion of the kinase domain and an out-of-frame insertion. KIN-20 was fluorescently tagged at the start of the second exon, after the alternative start-site methionine, with tagRFP (oxSi1087). The second exon is the start of the conserved kinase domain found in all isoforms. Inserting the fluorophore at this exon tags all isoforms.
Generation of super-resolution tags by CRISPR/Cas9
nrx-1, rimb-1, and syd-2 were endogenously tagged by CRISPR-mediated insertion of DNA encoding Skylan-s using the SapTrap toolkit as previously described (Schwartz and Jorgensen, 2016). A single plasmid containing the repair template and guide RNA was built using the SapTrap plasmid assembly. The repair template was composed of 57bp homology arms and SKYLAN-S (Zhang et al., 2015) containing a loxP::unc-119(+)::loxP. The SapTrap assembled plasmid was mixed with plasmids to express Cas9 in the germline, a histamine-gated chloride channel in neurons (Pokala et al., 2014), and fluorescent array markers. This mix was injected into the gonads of young adult EG9823 animals. After selecting for unc-119(+) and selecting against extrachromosomal arrays by histamine application, and loss of the fluorescent array markers, animals were injected with pDD104[Peft-3::CRE], selected for excision of loxP::unc-119::loxP, and 1x outcrossed before super-resolution microscopy.
Super-resolution imaging
Methods are similar to those described (Kurshan et al., 2018; Li et al., 2016). 3D super-resolution images were recorded with a Vutara SR 350 biplane microscope (Bruker, Nanosurfaces, Inc., Madison, WI) (Juette et al., 2008; Mlodzianoski et al., 2009). Live intact C. elegans L2 larva were immobilized for microscopy on a 2% agarose pad in M9 + 50mM sodium azide solution on a glass slide and the cover slip was sealed with Vaseline. The ventral cord in the region of the developing germline was positioned directly against the cover slip and imaged to avoid the intestinal auto-fluorescent granules. SKYLAN-S was excited by 488nm light at 1kW/cm2 and photoactivated by 405nm light. Images were recorded using a 60x/1.2 NA Olympus water immersion objective and Hamamatsu Flash4 V1 sCMOS camera with the gain set at 50 and frame rate at 50 Hz. Vutara SRX software (version 6.02) was used in data analysis. The background was removed, and single molecule localizations were identified based on brightness in each frame. Three dimensional localizations were obtained by fitting the raw data in a 12×12-pixel region of interest centered around each particle in each plane with a 3D model function built from recorded bead data sets. A density-based denoising algorithm to remove isolated particles was used for filtering. The remaining localizations were classified into clusters by density-based spatial clustering of applications with noise (DBSCAN), and a minimum of 10 localizations were connected around a 100nm search radius.
Time-lapse imaging
Methods are similar to those described (Knobel et al., 1999) with the following changes. Worms were immobilized for microscopy by placing them on a mixture of 1ul of 2.65% polystyrene 0.1um diameter beads in water and 1ul 20mM muscimol on an agarose pad under a cover slip. Slides were sealed with Vaseline to prevent evaporation. Time-lapse images were collected (Nikon Eclipse 90i) every 3 minutes over a Z range of 10–15 μm at 0.1 μm/pixel resolution. Figure 2A VD growth cones in a wild-type larva expressing GFP in GABA neurons (EG1285), Figure 2B VD growth cones in a kin-20(ox423) early L2 larva, Figure 2C L1 larva, Figure 2D kin-20(ox423) L1 larva.
KIN-20 localization
Preparation of worms for microscopy was done as previously described above. Animals for Figure S3B,C were imaged on an Opterra swept-field confocal microscope (Bruker) using a 60× 1.2NA water objective. Images were captured on an EM-CCD camera (Hamamatsu ImagEMX2). Animals for Figures 3F,G & S3D–H were imaged on the Zeiss LSM 880 confocal microscope using a 63x oil objective.
kin-20(ox423) motor neuron confocal microscopy
Preparation of worms for microscopy was done as previously described. Animals were imaged on the Pascal LSM5 (Zeiss) with a 63× 1.4NA oil objective. Z-stacks were acquired through the entire animal then a Z-projection from maximum intensity was made using Image J. Z-projections were stitched together with the MosaicJ plugin in Image J. The oxIs12 allele was used for expressing soluble GFP in GABA neurons.
Tissue-specific rescue of kin-20 mutants
kin-20(ox423) animals expressing soluble GFP in GABA neurons were injected with a kin-20 cDNA rescue gene under different promoters expressing in different tissues as seen in (construct details in Table S1). Minigenes were assembled with Gateway cloning. kin-20 cDNA was amplified from C. elegans cDNA library constructed from Superscript-III kits from Invitrogen. The kin-20 cDNA was amplified based on isoform ‘B’, which is simply the kinase domain and a small exon C-terminally. This isoform excludes the alternate first exon as seen in Figure 1F. The let-858 3’UTR was used for each construct.
kin-20 null animals have a low frequency of transgenic array formation for an unknown reason. The only constructs that were able to form stable transgenic arrays were those that could also rescue the null phenotype, ubiquitous expression, and pan-neuronal expression. This result suggests that array formation in kin-20 nulls is so low, a rescue construct is necessary to stabilize the array. Note, germline expression from an array is highly unlikely. The two constructs designed to express in the gametes would need to be integrated to assess rescue potential. This approach was abandoned since the genetics of the kin-20 maternal effect rescue indicated that germline contribution of KIN-20 gene products are sufficient to rescue offspring neuron defects in kin-20 mutant animals.
Uncoordination screen, generating the kin-20(ox423) allele
The kin-20(ox423) premature stop allele was generated in a screen looking for uncoordinated mutants. Adult hermaphrodites were mutagenized using 50mM ethyl methanesulfonate (EMS) (Brenner 1974), and 3,544 haploid genomes were screened for constipated phenotypes. These were then rescreened in the F3 for behavioral defects. ox423 was dumpy and paralyzed. Outcrossing demonstrated that ox423 mutation is maternal effect uncoordinated (Mau); F2 exhibited a constipated phenotype, whereas F3 animals were paralyzed and egg-laying defective. The kin-20(ox423) allele failed to complement a kin-20(ok505) full deletion allele. The kin-20(ox423) mutation was mapped to the X chromosome, then whole-genome sequenced and SNPs were identified to determine the molecular identity. A kin-20 rescue construct was built using the kin-20 promoter, gene isoform ‘a’ and the native 3’UTR, tagging the first common exon with tagRFP (Figure 1F). This construct was inserted as a single copy gene insertion using the MosSCI (Frøkjaer-Jensen et al., 2008; Frøkjær-Jensen et al., 2012), and fully rescued kin-20(ox423) phenotypes.
kin-20(ox423) suppressor screen
Methods are similar to those described (Hollopeter et al., 2014). Late L4 Caenorhabditis elegans larvae were mutagenized with 0.5 mM N-ethyl-N-nitrosourea (ENU) for 4hrs at 22C. Animals were washed with M9 buffer, then ~1000 L4 to young adults were pipetted onto a dense lawn of NA22 E. coli grown on 10-cm nematode growth medium (NGM) enriched peptone agar plates, on 66 plates in all. Animals were grown to starvation, then a ~2.5 × 2.5 cm piece of agar was cut from each plate and transferred to new NA22 bacteria on NGM enriched agar plates. In this screen, multiple independent mutagenized populations were generated, each containing enough genetic diversity to give rise to at least one suppressor. The populations were propagated for ~ ten generations, and then one mobile animal was selected from each population.
kin-20 suppressor identification
To identify the kin-20 suppressor mutations, we performed whole-genome resequencing. We first resequenced five suppressor strains to test our method of SNP identification. We did phenotypic mapping experiments with multicolor mapping strains, in which expression constructs were arrayed on different chromosomes (Frøkjær-Jensen et al., 2014). In the first mapping experiment, we found the causative SNP mapped to chromosome II in strain EG8338. There was only a mutation in one conserved gene in our list of candidates on chromosome II in this strain, SSUP-72 S39F, and cloned the mutant gene. We then overexpressed the mutant version of ssup-72 in a kin-20 null mutant and were able to rescue the neuronal defect in these animals (data not shown). We Sanger sequenced the remaining suppressor strains at the ssup-72 locus and found four additional alleles.
We leveraged the known binding partners of SSUP-72 as a candidate gene list and discovered mutations in RPB-2 and PINN-1 among our 5 whole-genome sequenced strains. We discovered four additional alleles of rpb-2 and three additional alleles of pinn-1 in our suppressor collection using Sanger sequencing.
To identify the remaining suppressor alleles, 31 strains were resequenced with an average depth of 22-fold. Four components of the RNA polymerase (RNAP) II complex and unc-44 were identified as suppressors by in silico complementation testing, we found multiple alleles in each gene: zfp-3, cpsf-4, ctr-9, and unc-44 (Minevich et al., 2012). Single mutations in the following suppressors were identified from the sequencing data for their known interactions with the RNAP II complex; cdc-73, pfs-2, pcf-11, and cdk-8.
Generating endogenous point mutations.
Methods are similar to those described using the SapTrap toolkit (Schwartz and Jorgensen, 2016). One critical update is the use of the EG9881 strain. This strain constitutively expresses Peft-3::Cas9 and Phsp::Cre from an endogenous insertion in chromosome III, as well as a fluorescent maker for easy tracking of the allele. SapTrapv31k software was used to design the potential suppressor point mutations in pfs-2, nrd-1, pcf-11, cdk-8, cdc-73, and ssup-72. Plasmids pMLS415 and pMLS256, as well as the 57 bp homology arm and sgRNA annealed oligos, were annealed into a single plasmid that was injected into the EG9881 strain along with the three array markers (Prab-3:mCherry:unc-54 UTR, Pmyo-2:mCherry:unc-54 UTR, Pmyo-3:mCherry:unc-54 UTR) used in MosSCI (Frøkjær-Jensen et al., 2012). Animals were selected for unc-119(−) rescue and no array marker expression. Strains were genotyped by PCR, then heat-shocked to express Cre and selected for unc-119(+) loss. Each CRISPR-generated allele was then Sanger sequenced and outcrossed.
Axotomy
Animals were mounted and axotomized as previously described (Hammarlund et al., 2009). Scoring kin-20 mutants for regeneration after axotomy was more difficult than wild type due to the severity of the branching phenotype. Images were taken immediately after axotomy and then again 24 hours later. These were compared to aid in scoring. If the original axotomized axon was unable to be identified, due to extensive branching, this was still included in the total axons cut and likely underestimated the extent of regeneration. 95% confidence intervals were calculated using the modified Wald method and P values were calculated using the Fisher exact test as previously described (Hammarlund et al., 2009).
Mapping strains
Fluorescent balancer strains, generated by random gene insertion using miniMos, were crossed together to generate two mapping strains (Frøkjær-Jensen et al., 2014). EG8040 is used to map chromosomes I, II, and III. EG8041 is used to map chromosomes IV, V, and X. These strains also contain a null allele of him-8 and him-5 respectively, to spontaneously generate males for crossing. The overexpression of Peft-3::H2B::GFP and Peft-3::H2B::tdTomato in these strains seems to make the males a bit temperature sensitive. Therefore, the first mapping crosses were done between 15–20C.
qPCR
C. elegans hermaphrodites were grown on NA22 E. coli to adulthood and were dissolved in hypochlorite solution. Embryos were washed, and total RNA was extracted using a Qiagen RNeasy mini kit + Qiagen on-column DNase treatment. Total RNA was then reverse transcribed to cDNA using an Invitrogen Superscript III First-Strand Synthesis System using the supplied oligo(dT) option and using 300ng of total RNA as template. Relative mRNA quantity was measured by qPCR on the Masterplex ep realplex instrument (Eppendorf, Hauppauge, NY) with SYBR green fluorescent dye. cDNA libraries were tested for contaminated gDNA by measuring a reverse transcriptase (−) control.
unc-44 long overexpression in kin-20(ox423)
kin-20(ox423) mutant animals were injected with overlapping PCR products of the entire unc-44 gene locus. The PCR products span from the beginning of the 5’UTR to the end of the 3’UTR of the native unc-44f locus. However cDNA was used as a template for the region that spanned the polyA site #2, thus amplifying only the long form of unc-44. C. elegans can assemble a functional gene in an extrachromosomal array by recombination of the overlapping PCR fragments (Hobert, 2002).
unc-44(ox686) molecular analysis
The mutation in unc-44(ox686) is a three-nucleotide substitution, conserved in nematodes, in the intron preceding exon 15 and the alternative polyA site #2 (Figure 5A & S6B). We synthesized a cDNA library (as previously described) from the unc-44(ox686) strain and sequenced from flanking exons across exon 15 using primers oML520 and oML596 (Key resource table). We found that exon 15 is skipped in this suppressor and bypasses polyA site #2 resulting in only the long form of Ankyrin. We see no obvious phenotype in these animals which no longer have short form #2 of Ankyrin. However, the short and long isoforms determined by polyA site #1 and #3 are still present in this strain (Figures S6A & 5B).
RNA seq
C. elegans embryos were prepared using standard methods (Stiernagle, 2006). Three biological replicates were used for each genome. Total RNA was extracted using a Qiagen RNeasy kit. The University of Utah sequencing core did library prep by poly(A) capture and Illumina RNA sequencing.
SSUP-72 protein purification
C. elegans ssup-72 and kin-20 cDNAs were amplified from a C. elegans cDNA library with primers oML929/oML930 and oML933/oML934 respectively (Key resource table). PCR products were gel purified and cloned by Gibson assembly into the pGEX-6P1 bacterial expression vector and transformed into BL21 chemically competent expression E. coli. This vector fuses the GST purification tag to the cloned sequence in the ORF via a PreScission protease linker sequence. A test expression was performed and SSUP-72 and KIN-20 protein were expressed at the correct size (KIN-20 37kD + GST 26kDa = ~63kDa; SSUP-72 22.8kDa + GST 26kDA = ~49kDa). Samples were run on a 10% polyacrylamide gel and stained with Coomassie G-250 (data not shown).
SSUP-72 was purified in a similar manner to other GST fusion RNA pol II complex components (Gross and Moore, 2001). Starter cultures were first grown O/N in LB with carbenicillin selection. 500ml of terrific broth + carb was inoculated to an OD 0.1nm and cells were grown at 37C in baffled flasks shaken at 180rpms to an optical density of 0.5–0.6nm at 37C. Protein expression was then induced with 0.4mM IPTG and cells were allowed to express for 24hrs at 20C. Cells were harvested, spun at 7K for 10min at 4C, washed with wash buffer (1xPBS, 1mM DTT, 1mM EDTA) and bacterial pellets were frozen at −80C. All purification steps were done at 4C. Cells were thawed in lysis buffer (50mM Tris/pH7.5, 250mM KCl/1mM EDTA/0.5mM DTT/10% glycerol/1mg/ml lysozyme/Roche protein inhibitor cocktail) and sonicated on a Branson sonicator until lysates clarified slightly and cells were disrupted. Cells debris was removed by centrifugation at 11,000 x g for 30min. The supernatant was diluted with 1:2 volumes of buffer D (20mM Tris HCl, pH8.0/50mM KCl/0.2mM EDTA/ 0.5mM DTT/ 10% Glycerol/Roche protease inhibitor cocktail) and bound to 1ml of glutathione sepharose 4B resin (that had been equilibrated to buffer D) O/N at 4C on a nutator (GE Cat#17075601). The beads were washed 4× 15ml PBS and GST was cleaved from the fusion protein using PreScission protease in cleavage buffer (50mM Tris-HCL pH7.5/15mM NaCl/1mM EDTA/1mM DTT/0.01% Triton) as per the GE protocol. Cleaved purified protein was collected through a column, dialyzed to buffer D, and stored at −80C.
KIN-20 was unfortunately insoluble and even after thorough testing of 10 different buffers and many purification conditions the protein remained insoluble in the pellet (data not shown). Since C. elegans CK1δ proved to be insoluble in our hands and because casein kinases are conserved, we purchased recombinant rat CK1δ protein from NEB (Cat# P06030). This protein is truncated, removing its self-phosphorylated inhibitory domain. Casein kinases self-inhibit so we used the truncated protein as opposed to the full-length purified protein, which has orders of magnitude lower activity (NEB CK1 ~1000pmol/min/ul, Abcam purified full-length CK1δ ~8pmol/min/ul).
Radioactive in vitro kinase assay
The reaction conditions for the in vitro kinase assay were based on the NEB protocol available online. 5ul of purified SSUP-72 substrate was incubated with 2ul of 100ng/ul CK1δ in NEB protein kinase buffer (50mM Tris-HCl pH7.5/10mM MgCl2/0.1mM EDTA/2mM DTT/0.01% Brij 35 + 200uM ATP + [γ−32P]ATP) in a reaction volume of 25ul at 30C for 2 hours. The commercially available protein Casein was used as a positive control. Samples (+/− CK1) and controls were run on a Bio-Rad pre-cast gel, washed with 7% acetic acid for 20min, then dried on Whatman 3.0 paper, exposed to a phosphor plate for 1 hour and imaged on a GE-Typhoon.
Protein structure visualization
Structure references for the human CPSF complex (cleavage-polyadenylation specificity factor), S. cerevisiae PCF-11, murine CK1δ, human PIN-1, human RNA polymerase II, and human SSUP-72 respectively: (Clerici et al., 2018; Meinhart and Cramer, 2004; Shinohara et al., 2017; Verdecia et al., 2000; Vos et al., 2018; Xiang et al., 2010).
Molecular graphics and analyses performed with UCSF Chimera (Pettersen et al., 2004), developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311.
QUANTIFICATION AND STATISTICAL ANALYSIS
KIN-20 quantification
Quantification of KIN-20 expression for Figure 3C was performed using an optical section at the center of the neuron cell body. The region of interest comprising the cell body was selected using Image J software. The fluorescence was measured and normalized to area in Image J. Cell bodies were excluded from analysis if the proper cell assignment, either DD or VD, could not be determined, or if an auto-fluorescent gut granule obstructed the image.
Worm track quantification
A single C. elegans young adult hermaphrodite was placed on a 5cm NGM plate seeded with OP50. The animal behaved freely for 2 minutes, and a photo was taken of the worm track with an Allied Vision Technologies model Stingray camera, using a Nikon AF Micro Nikkor 60mm lens, a MicroBrightField biosciences LED Illuminator, and MicroBrightField biosciences WormLab (4.1.0) software. Worm tracks were then traced on a WAACOM touchscreen monitor, x,y coordinates and length measurements were taken with in-house ImageJ macros. ImageJ x,y coordinates were transformed into a scalable vector graphics file (svg) with an in-house Tcl script.
Axons
Axon defect counts data were graphed in GraphPad Prism software and analyzed by Welch’s unpaired t-tests in GraphPad in Figure 1. Dunnett’s multiple comparison test was used for data in Figure 5C.
RNA seq
Reads were aligned with STAR (Dobin et al., 2013). DEXseq analysis was used for the discovery of mRNA differential expression at the isoform level (Anders et al., 2012). DESeq2 analysis was used for mRNA differential expression at the whole gene level (Love et al., 2014). Data analysis and graphing was done on GraphPad Prism software.
qPCR
Samples were measured in triplicate, and quantified using a standard curve of pooled, serially diluted samples. Three biological replicates were measured per sample. mRNA quantity was normalized to the quantity of ribosomal protein 18 (rpl-18) mRNA. rpl-18 was selected for its consistent expression level across biological replicates as well as developmental stages, making it an ideal normalization control. rpl-19, eif-3.d, ebg-1, and clp-3 were also tested by qPCR as potential normalization controls, but in our hands, rpl-18 was best. Primer design for qPCR reactions was done using Primer3Plus online software. All comparisons were done by Dunnett’s multiple comparison test.
DATA AND CODE AVAILABILITY
R-script for mutation filtering
The programming language R was used to write a SNP filter with the option to exclude mutations that fall under the following categories: parental, synonymous, intergenic, heterozygous, intronic, and pseudogenes. An in-silico complementation test was also written in R. Scripts can be accessed at https://github.com/jorgensenlab/WGS-variant-filtering.
Supplementary Material
Wild-type VD motor neuron commissures, extending in the late L1 stage. Movie related to Figure 2A.
kin-20(ox423) VD motor neuron commissures, extending in the late L1 stage. Movie related to Figure 2B.
kin-20(ox423) DD motor neuron commissures, interstitial growth cone. Movie related to Figure 2C.
kin-20(ox423) DD motor neuron retraction in the dorsal nerve cord followed by re-extension along the dorsal cord. Movie related to Figure 2D.
kin-20(ox423) sprouting ectopic growth cones in the adult GABA motor-neuron axons. Movie related to Figure 2E.
Differential gene expression in embryos: wild type, kin-20(ox423), and kin-20(ox423) suppressors related to Figures 5 & S6.
Differential gene expression analysis of wild-type embryos vs kin-20(ox423) embryos: giant unc-44 is the top differentially expressed gene when RNA sequencing data is analyzed by either DEXseq (adjusted P value=0.077, false discovery cutoff 10% or Padj<0.1) or DESeq2 (adjusted P value=0.0001, false discovery cutoff 10% or Padj<0.1). Genotypes: EG1285 lin-15(n765ts) X; oxIs12 X, EG5202 oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X, EG8338 ssup-72(ox542) II ; oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X, EG9090 zfp-3(ox649) X oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X
Highlights.
Casein kinase 1 δ (CK1δ) stabilizes nervous system architecture after axon outgrowth.
CK1δ phosphorylates and inhibits SSUP-72, an RNA polymerase II CTD phosphatase.
CK1δ inhibits transcription termination to promote giant Ankyrin expression.
Expression of giant Ankyrin in CK1δ mutants rescues axon maturation defects.
Acknowledgments
We thank the students of Biol531 Genetics Lab class for isolating the kin-20(ox423) mutation. Nels Jorgensen for kin-20 strain construction. Craig Kaplan for conversations about RNA pol II structure and termination and connecting us with ideas and people within the field. Seychelle Vos, Lucas Farnung and Patrick Cramer for sharing unpublished results. Rob Hobson and Nathan Okerlund for assistance imaging EG9581. Wayne Davis, Matt Schwartz and Matt Rich for suggestions to improve the manuscript. Matt LaBella was supported by a predoctoral training fellowship from the National Institute of Health under award number 5T32GM007464. This work was supported by NINDS R01 NS034307 to (EMJ) and NIGMS R35 GM119540 (JH). EMJ is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Declaration of Interests
No declared interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen BL, and Taatjes DJ (2015). The Mediator complex: a central integrator of transcription. Nat. Rev. Mol. Cell Biol 16, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrich CG, Davis CP, Rogal WP, Shirra MK, Heroux A, Gardner RG, Arndt KM, and VanDemark AP (2012). Cdc73 Subunit of Paf1 Complex Contains C-terminal Ras-like Domain That Promotes Association of Paf1 Complex with Chromatin. J. Biol. Chem 287, 10863–10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, and Huber W. (2012). Detecting differential usage of exons from RNA-seq data. Genome Res. 22, 2008–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari A, and Hampsey M. (2005). A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 19, 2969–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baejen C, Andreani J, Torkler P, Battaglia S, Schwalb B, Lidschreiber M, Maier KC, Boltendahl A, Rus P, Esslinger S, et al. (2017). Genome-wide Analysis of RNA Polymerase II Termination at Protein-Coding Genes. Mol. Cell 66, 38–49.e6. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Kwok A, Lin S-Y, and Slack FJ (2005). Developmental timing in C. elegans is regulated by kin-20 and tim-1, homologs of core circadian clock genes. Dev. Cell 8, 287–295. [DOI] [PubMed] [Google Scholar]
- Bennett V, and Lorenzo DN (2013). Chapter One - Spectrin- and Ankyrin-Based Membrane Domains and the Evolution of Vertebrates In Current Topics in Membranes, Bennett V, ed. (Academic Press; ), pp. 1–37. [DOI] [PubMed] [Google Scholar]
- Bennett V, and Lorenzo DN (2016). Chapter Five - An Adaptable Spectrin/Ankyrin-Based Mechanism for Long-Range Organization of Plasma Membranes in Vertebrate Tissues In Current Topics in Membranes, Bennett V, ed. (Academic Press; ), pp. 143–184. [DOI] [PubMed] [Google Scholar]
- Bennett V, Davis J, and Fowler WE (1982). Brain spectrin, a membrane-associated protein related in structure and function to erythrocyte spectrin. Nature 299, 126–131. [DOI] [PubMed] [Google Scholar]
- Bentley DL (2014). Coupling mRNA processing with transcription in time and space. Nat. Rev. Genet 15, 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolini JA, Favaro R, Zhu Y, Pagin M, Ngan CY, Wong CH, Tjong H, Vermunt MW, Martynoga B, Barone C, et al. (2019). Mapping the Global Chromatin Connectivity Network for Sox2 Function in Neural Stem Cell Maintenance. Cell Stem Cell 24, 462–476.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne AB, Walradt T, Gardner KE, Hubbert A, Reinke V, and Hammarlund M. (2014). Insulin/IGF1 Signaling Inhibits Age-Dependent Axon Regeneration. Neuron 81, 561–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- elegans C. Deletion Mutant Consortium (2012). large-scale screening for targeted knockouts in the Caenorhabditis elegans genome. G3 Bethesda Md 2, 1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casañal A, Kumar A, Hill CH, Easter AD, Emsley P, Degliesposti G, Gordiyenko Y, Santhanam B, Wolf J, Wiederhold K, et al. (2017). Architecture of eukaryotic mRNA 3′- end processing machinery. Science 358, 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhou Y, Qi YB, Khivansara V, Li H, Chun SY, Kim JK, Fu X-D, and Jin Y. (2015). Context-dependent modulation of Pol II CTD phosphatase SSUP-72 regulates alternative polyadenylation in neuronal development. Genes Dev. 29, 2377–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Chisholm AD, and Jin Y. (2017). Tissue-specific regulation of alternative polyadenylation represses expression of neuronal ankyrin isoform in C. elegans epidermal development. Development dev.146001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici M, Faini M, Muckenfuss LM, Aebersold R, and Jinek M. (2018). Structural basis of AAUAAA polyadenylation signal recognition by the human CPSF complex. Nat. Struct. Mol. Biol 25, 135–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Allen MA, Larsen A, Macmorris M, Han M, and Blumenthal T. (2008). Genes involved in pre-mRNA 3’-end formation and transcription termination revealed by a lin-15 operon Muv suppressor screen. Proc. Natl. Acad. Sci. U. S. A 105, 16665–16670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers PB, Nonnekens J, Goos YJ, Betist MC, Viester MD, Mossink B, Lansu N, Korswagen HC, Jelier R, Brenkman AB, et al. (2015). A Long Noncoding RNA on the Ribosome Is Required for Lifespan Extension. Cell Rep. 10, 339–345. [DOI] [PubMed] [Google Scholar]
- Fish KJ, Cegielska A, Getman ME, Landes GM, and Virshup DM (1995). Isolation and Characterization of Human Casein Kinase Iϵ (CKI), a Novel Member of the CKI Gene Family. J. Biol. Chem 270, 14875–14883. [DOI] [PubMed] [Google Scholar]
- Fréal A, Fassier C, Le Bras B, Bullier E, De Gois S, Hazan J, Hoogenraad CC, and Couraud F. (2016). Cooperative Interactions between 480 kDa Ankyrin-G and EB Proteins Assemble the Axon Initial Segment. J. Neurosci. Off. J. Soc. Neurosci 36, 4421–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fréal A, Rai D, Tas RP, Pan X, Katrukha EA, van de Willige D, Stucchi R, Aher A, Yang C, Altelaar AFM, et al. (2019). Feedback-Driven Assembly of the Axon Initial Segment. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjaer-Jensen C, Davis MW, Hopkins CE, Newman BJ, Thummel JM, Olesen S-P, Grunnet M, and Jorgensen EM (2008). Single-copy insertion of transgenes in Caenorhabditis elegans. Nat. Genet 40, 1375–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Ailion M, and Jorgensen EM (2012). Improved Mos1-mediated transgenesis in C. elegans. Nat. Methods 9, 117–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Sarov M, Taylor J, Flibotte S, LaBella M, Pozniakovsky A, Moerman DG, and Jorgensen EM (2014). Random and targeted transgene insertion in Caenorhabditis elegans using a modified Mos1 transposon. Nat. Methods 11, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel CV, Antoine F, Chuang C-F, Samuel ADT, and Chang C. (2008). Distinct cellular and molecular mechanisms mediate initial axon development and adult-stage axon regeneration in C. elegans. Development 135, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Gross S, and Moore C. (2001). Five subunits are required for reconstitution of the cleavage and polyadenylation activities of Saccharomyces cerevisiae cleavage factor I. Proc. Natl. Acad. Sci 98, 6080–6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall TG, and Bennett V. (1987). Regulatory domains of erythrocyte ankyrin. J. Biol. Chem 262, 10537–10545. [PubMed] [Google Scholar]
- Hallam SJ, and Jin Y. (1998). lin-14 regulates the timing of synaptic remodelling in Caenorhabditis elegans. Nature 395, 78–82. [DOI] [PubMed] [Google Scholar]
- Hammarlund M, Davis WS, and Jorgensen EM (2000). Mutations in beta-spectrin disrupt axon outgrowth and sarcomere structure. J. Cell Biol 149, 931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Jorgensen EM, and Bastiani MJ (2007). Axons break in animals lacking beta-spectrin. J. Cell Biol 176, 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Nix P, Hauth L, Jorgensen EM, and Bastiani M. (2009). Axon regeneration requires a conserved MAP kinase pathway. Science 323, 802–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Kooistra R, Das R, Oudejans E, Leen E.V. van, Ziegler J, Portegies S, Haan B. de, Altena A.S. van R, Stucchi R, et al. (2019). Cortical anchoring of the microtubule cytoskeleton is essential for neuron polarity and functioning. BioRxiv 783837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedgecock EM, Culotti JG, Thomson JN, and Perkins LA (1985). Axonal guidance mutants of Caenorhabditis elegans identified by filling sensory neurons with fluorescein dyes. Dev. Biol 111, 158–170. [DOI] [PubMed] [Google Scholar]
- Herzel L, Ottoz DSM, Alpert T, and Neugebauer KM (2017). Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat. Rev. Mol. Cell Biol 18, 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobert O. (2002). PCR Fusion-Based Approach to Create Reporter Gene Constructs for Expression Analysis in Transgenic C. elegans. BioTechniques 32, 728–730. [DOI] [PubMed] [Google Scholar]
- Hollopeter G, Lange JJ, Zhang Y, Vu TN, Gu M, Ailion M, Lambie EJ, Slaughter BD, Unruh JR, Florens L, et al. (2014). The membrane-associated proteins FCHo and SGIP are allosteric activators of the AP2 clathrin adaptor complex. ELife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J-P, and Manley JL (2012). The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26, 2119–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehning JA (2010). The Paf1 complex: Platform or player in RNA polymerase II transcription? Biochim. Biophys. Acta BBA - Gene Regul. Mech 1799, 379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jegla T, Nguyen MM, Feng C, Goetschius DJ, Luna E, van Rossum DB, Kamel B, Pisupati A, Milner ES, and Rolls MM (2016). Bilaterian Giant Ankyrins Have a Common Evolutionary Origin and Play a Conserved Role in Patterning the Axon Initial Segment. PLoS Genet. 12, e1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins PM, Kim N, Jones SL, Tseng WC, Svitkina TM, Yin HH, and Bennett V. (2015). Giant ankyrin-G: A critical innovation in vertebrate evolution of fast and integrated neuronal signaling. Proc. Natl. Acad. Sci 112, 957–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juette MF, Gould TJ, Lessard MD, Mlodzianoski MJ, Nagpure BS, Bennett BT, Hess ST, and Bewersdorf J. (2008). Three-dimensional sub–100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 5, 527–529. [DOI] [PubMed] [Google Scholar]
- Kloss B, Price JL, Saez L, Blau J, Rothenfluh A, Wesley CS, and Young MW (1998). The Drosophila Clock Gene double-time Encodes a Protein Closely Related to Human Casein Kinase Iε. Cell 94, 97–107. [DOI] [PubMed] [Google Scholar]
- Knippschild U, Gocht A, Wolff S, Huber N, Löhler J, and Stöter M. (2005). The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell. Signal 17, 675–689. [DOI] [PubMed] [Google Scholar]
- Knobel KM, Jorgensen EM, and Bastiani MJ (1999). Growth cones stall and collapse during axon outgrowth in Caenorhabditis elegans. Dev. Camb. Engl 126, 4489–4498. [DOI] [PubMed] [Google Scholar]
- Knobel KM, Davis WS, Jorgensen EM, and Bastiani MJ (2001). UNC-119 suppresses axon branching in C. elegans. Dev. Camb. Engl 128, 4079–4092. [DOI] [PubMed] [Google Scholar]
- Koch I, Schwarz H, Beuchle D, Goellner B, Langegger M, and Aberle H. (2008). Drosophila ankyrin 2 is required for synaptic stability. Neuron 58, 210–222. [DOI] [PubMed] [Google Scholar]
- Koenig SN, and Mohler PJ (2017). The evolving role of ankyrin-B in cardiovascular disease. Heart Rhythm 14, 1884–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordeli E, Lambert S, and Bennett V. (1995). AnkyrinG. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. - PubMed - NCBI. J. Biol. Chem 270, 2352–2359. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy S, He X, Reyes-Reyes M, Moore C, and Hampsey M. (2004). Ssu72 Is an RNA polymerase II CTD phosphatase. Mol. Cell 14, 387–394. [DOI] [PubMed] [Google Scholar]
- Kuehner JN, Pearson EL, and Moore C. (2011). Unravelling the means to an end: RNA polymerase II transcription termination. Nat. Rev. Mol. Cell Biol 12, 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunimoto M, Otto E, and Bennett V. (1991). A new 440-kD isoform is the major ankyrin in neonatal rat brain. J. Cell Biol 115, 1319–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan PT, Merrill SA, Dong Y, Ding C, Hammarlund M, Bai J, Jorgensen EM, and Shen K. (2018). γ-Neurexin and Frizzled Mediate Parallel Synapse Assembly Pathways Antagonized by Receptor Endocytosis. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurup N, and Jin Y. (2015). Neural circuit rewiring: insights from DD synapse remodeling. Worm 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Merrill SA, Jorgensen EM, and Shen K. (2016). Two Clathrin Adaptor Protein Complexes Instruct Axon-Dendrite Polarity. Neuron 90, 564–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain P-O, Han J-DJ, Chesneau A, Hao T, et al. (2004). A Map of the Interactome Network of the Metazoan C. elegans. Science 303, 540–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, Geiger G, Minet M, Schroeder S, Cilli K, McNeil JB, and Bentley DL (2002). Functional Interaction of Yeast Pre-mRNA 3′ End Processing Factors with RNA Polymerase II. Mol. Cell 9, 1101–1111. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Garriga G, White J, Jacobson D, and Horvitz HR (1992). Genes necessary for directed axonal elongation or fasciculation in C. elegans. Neuron 8, 307–322. [DOI] [PubMed] [Google Scholar]
- Meinhart A, and Cramer P. (2004). Recognition of RNA polymerase II carboxy-terminal domain by 3′-RNA-processing factors. Nature 430, 223–226. [DOI] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O. (2012). CloudMap: A Cloud-Based Pipeline for Analysis of Mutant Genome Sequences. Genetics 192, 1249–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mlodzianoski MJ, Juette MF, Beane GL, and Bewersdorf J. (2009). Experimental characterization of 3D localization techniques for particle-tracking and super-resolution microscopy. Opt. Express 17, 8264–8277. [DOI] [PubMed] [Google Scholar]
- Moabbi AM, Agarwal N, Kaderi BE, and Ansari A. (2012). Role for gene looping in intron-mediated enhancement of transcription. Proc. Natl. Acad. Sci 109, 8505–8510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller CL, and Jaehning JA (2002). Ctr9, Rtf1, and Leo1 Are Components of the Paf1/RNA Polymerase II Complex. Mol. Cell. Biol 22, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix P, Hisamoto N, Matsumoto K, and Bastiani M. (2011). Axon regeneration requires coordinate activation of p38 and JNK MAPK pathways. Proc. Natl. Acad. Sci 108, 10738–10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordick K, Hoffman MG, Betz JL, and Jaehning JA (2008). Direct Interactions between the Paf1 Complex and a Cleavage and Polyadenylation Factor Are Revealed by Dissociation of Paf1 from RNA Polymerase II. Eukaryot. Cell 7, 1158–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka AJ, Franco R, Yang B, Shim KH, Tang LZ, Zhang YY, Boontrakulpoontawee P, Jeyaprakash A, Hedgecock E, and Wheaton VI (1995). An ankyrin-related gene (unc-44) is necessary for proper axonal guidance in Caenorhabditis elegans. J. Cell Biol 129, 1081–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuka AJ, Boontrakulpoontawee P, Rebeiz N, Domanus M, Otsuka D, Velamparampil N, Chan S, Vande Wyngaerde M, Campagna S, and Cox A. (2002). Novel UNC-44 AO13 ankyrin is required for axonal guidance in C. elegans, contains six highly repetitive STEP blocks separated by seven potential transmembrane domains, and is localized to neuronal processes and the periphery of neural cell bodies. J. Neurobiol 50, 333–349. [DOI] [PubMed] [Google Scholar]
- Perkins KJ, Lusic M, Mitar I, Giacca M, and Proudfoot NJ (2008). Transcription-Dependent Gene Looping of the HIV-1 Provirus Is Dictated by Recognition of Pre-mRNA Processing Signals. Mol. Cell 29, 56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Phatnani HP, and Greenleaf AL (2006). Phosphorylation and functions of the RNA polymerase II CTD. Genes Dev. 20, 2922–2936. [DOI] [PubMed] [Google Scholar]
- Pielage J, Cheng L, Fetter RD, Carlton PM, Sedat JW, and Davis GW (2008). A presynaptic giant ankyrin stabilizes the NMJ through regulation of presynaptic microtubules and transsynaptic cell adhesion. Neuron 58, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokala N, Liu Q, Gordus A, and Bargmann CI (2014). Inducible and titratable silencing of Caenorhabditis elegans neurons in vivo with histamine-gated chloride channels. Proc. Natl. Acad. Sci 111, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, and Libri D. (2015). Transcription termination and the control of the transcriptome: why, where and how to stop. Nat. Rev. Mol. Cell Biol 16, 190–202. [DOI] [PubMed] [Google Scholar]
- Price JL, Blau J, Rothenfluh A, Abodeely M, Kloss B, and Young MW (1998). double-time is a novel Drosophila clock gene that regulates PERIOD protein accumulation. Cell 94, 83–95. [DOI] [PubMed] [Google Scholar]
- Rhodehouse K, Cascino K, Aseltine L, Padula A, Weinstein R, Spina JS, Olivero CE, and Van Wynsberghe PM (2018). The Doubletime Homolog KIN-20 Mainly Regulates let-7 Independently of Its Effects on the Period Homolog LIN-42 in Caenorhabditis elegans. G3 Bethesda Md 8, 2617–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, and Manley JL (2009). Transcription termination by nuclear RNA polymerases. Genes Dev. 23, 1247–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosado-Lugo JD, and Hampsey M. (2014). The Ssu72 phosphatase mediates the RNA polymerase II initiation-elongation transition. J. Biol. Chem 289, 33916–33926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Nagaike T, Francis JM, Kaneko S, Glatt KA, Hughes CM, LaFramboise T, Manley JL, and Meyerson M. (2009). The tumor suppressor Cdc73 functionally associates with CPSF and CstF 3’ mRNA processing factors. Proc. Natl. Acad. Sci 106, 755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ML, and Jorgensen EM (2016). SapTrap, a Toolkit for High-Throughput CRISPR/Cas9 Gene Modification in Caenorhabditis elegans. Genetics 202, 1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. (2012). The COMPASS Family of Histone H3K4 Methylases: Mechanisms of Regulation in Development and Disease Pathogenesis. Annu. Rev. Biochem 81, 65–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara Y, Koyama YM, Ukai-Tadenuma M, Hirokawa T, Kikuchi M, Yamada RG, Ukai H, Fujishima H, Umehara T, Tainaka K, et al. (2017). Temperature-Sensitive Substrate and Product Binding Underlie Temperature-Compensated Phosphorylation in the Clock. Mol. Cell 67, 783–798.e20. [DOI] [PubMed] [Google Scholar]
- Sidorenkov I, Komissarova N, and Kashlev M. (1998). Crucial Role of the RNA:DNA Hybrid in the Processivity of Transcription: Molecular Cell. Mol. Cell 2, 55–64. [DOI] [PubMed] [Google Scholar]
- Singh BN, and Hampsey M. (2007). A Transcription-Independent Role for TFIIB in Gene Looping. Mol. Cell 27, 806–816. [DOI] [PubMed] [Google Scholar]
- Stark C, Breitkreutz B-J, Reguly T, Boucher L, Breitkreutz A, and Tyers M. (2006). BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan R, Goellner B, Moreno E, Frank CA, Hugenschmidt T, Genoud C, Aberle H, and Pielage J. (2015). Hierarchical microtubule organization controls axon caliber and transport and determines synaptic structure and stability. Dev. Cell 33, 5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiernagle T. (2006). Maintenance of C. elegans (WormBook; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulston JE (1976). Post-embryonic development in the ventral cord of Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci 275. [DOI] [PubMed] [Google Scholar]
- Tan-Wong SM, Zaugg JB, Camblong J, Xu Z, Zhang DW, Mischo HE, Ansari AZ, Luscombe NM, Steinmetz LM, and Proudfoot NJ (2012). Gene loops enhance transcriptional directionality. Science 338, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temmerman L, Meelkop E, Janssen T, Bogaerts A, Lindemans M, Husson SJ, Beets I, and Schoofs L. (2011). C. elegans homologs of insect clock proteins: a tale of many stories. Ann. N. Y. Acad. Sci 1220, 137–148. [DOI] [PubMed] [Google Scholar]
- Tennessen JM, Gardner HF, Volk ML, and Rougvie AE (2006). Novel heterochronic functions of the Caenorhabditis elegans period-related protein LIN-42. Dev. Biol 289, 30–43. [DOI] [PubMed] [Google Scholar]
- Tennessen JM, Opperman KJ, and Rougvie AE (2010). The C. elegans developmental timing protein LIN-42 regulates diapause in response to environmental cues. Development 137, 3501–3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsytlonok M, Ibrahim SM, Rowling PJE, Xu W, Ruedas-Rama MJ, Orte A, Klenerman D, and Itzhaki LS (2015). Single-Molecule FRET Reveals Hidden Complexity in a Protein Energy Landscape. Struct. England1993 23, 190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Epps H, Dai Y, Qi Y, Goncharov A, and Jin Y. (2010). Nuclear pre-mRNA 3’-end processing regulates synapse and axon development in C. elegans. Dev. Camb. Engl 137, 2237–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdecia MA, Bowman ME, Lu KP, Hunter T, and Noel JP (2000). Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat. Struct. Biol 7, 639–643. [DOI] [PubMed] [Google Scholar]
- Vos SM, Farnung L, Boehning M, Wigge C, Linden A, Urlaub H, and Cramer P. (2018). Structure of activated transcription complex Pol II–DSIF–PAF–SPT6. Nature 560, 607. [DOI] [PubMed] [Google Scholar]
- Wang R, Zheng D, Wei L, Ding Q, and Tian B. (2019). Regulation of Intronic Polyadenylation by PCF11 Impacts mRNA Expression of Long Genes. Cell Rep. 26, 2766–2778.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Allen JW, Lee C-J, Liu P, Nicely NI, Wang S, Greenleaf AL, and Zhou P. (2011). cis-Proline-mediated Ser(P)5 Dephosphorylation by the RNA Polymerase II C-terminal Domain Phosphatase Ssu72. J. Biol. Chem 286, 5717–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West S, and Proudfoot NJ (2008). Human Pcf11 enhances degradation of RNA polymerase II-associated nascent RNA and transcriptional termination. Nucleic Acids Res. 36, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Albertson DG, and Anness MA (1978). Connectivity changes in a class of motoneurone during the development of a nematode. Nature 271, 764–766. [DOI] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, and Brenner S. (1986). The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci 314. [DOI] [PubMed] [Google Scholar]
- Wu Z, Ghosh-Roy A, Yanik MF, Zhang JZ, Jin Y, and Chisholm AD (2007). Caenorhabditis elegans neuronal regeneration is influenced by life stage, ephrin signaling, and synaptic branching. Proc. Natl. Acad. Sci. U. S. A 104, 15132–15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Nagaike T, Xiang S, Kilic T, Beh MM, Manley JL, and Tong L. (2010). Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature 467, 729–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang K, Tong L, and Manley JL (2014). Delineating the Structural Blueprint of the Pre-mRNA 3′-End Processing Machinery. Mol. Cell. Biol 34, 1894–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhong G, and Zhuang X. (2013). Actin, spectrin, and associated proteins form a periodic cytoskeletal structure in axons. Science 339, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y-X, Hirose Y, Zhou XZ, Lu KP, and Manley JL (2003). Pin1 modulates the structure and function of human RNA polymerase II. Genes Dev. 17, 2765–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Walder-Christensen KK, Kim N, Wu D, Lorenzo DN, Badea A, Jiang Y-H, Yin HH, Wetsel WC, and Bennett V. (2019). ANK2 autism mutation targeting giant ankyrin-B promotes axon branching and ectopic connectivity. Proc. Natl. Acad. Sci 116, 15262–15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MW, and Kay SA (2001). Time zones: a comparative genetics of circadian clocks. Nat. Rev. Genet 2, 702. [DOI] [PubMed] [Google Scholar]
- Zhang Z, and Gilmour DS (2006). Pcf11 Is a Termination Factor in Drosophila that Dismantles the Elongation Complex by Bridging the CTD of RNA Polymerase II to the Nascent Transcript. Mol. Cell 21, 65–74. [DOI] [PubMed] [Google Scholar]
- Zhang H, Constantine R, Vorobiev S, Chen Y, Seetharaman J, Huang YJ, Xiao R, Montelione GT, Gerstner CD, Davis MW, et al. (2011). UNC119 is required for G protein trafficking in sensory neurons. Nat. Neurosci 14, 874–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Chen X, Zeng Z, Zhang M, Sun Y, Xi P, Peng J, and Xu P. (2015). Development of a Reversibly Switchable Fluorescent Protein for Super-Resolution Optical Fluctuation Imaging (SOFI). ACS Nano 9, 2659–2667. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Fu J, and Gilmour DS (2005). CTD-dependent dismantling of the RNA polymerase II elongation complex by the pre-mRNA 3’-end processing factor, Pcf11. Genes Dev. 19, 1572–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Chiu H, Zinovyeva A, Ambros V, Chuang C-F, and Chang C. (2013). Developmental decline in neuronal regeneration by the progressive change of two intrinsic timers in C. elegans. Science 340, 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Wild-type VD motor neuron commissures, extending in the late L1 stage. Movie related to Figure 2A.
kin-20(ox423) VD motor neuron commissures, extending in the late L1 stage. Movie related to Figure 2B.
kin-20(ox423) DD motor neuron commissures, interstitial growth cone. Movie related to Figure 2C.
kin-20(ox423) DD motor neuron retraction in the dorsal nerve cord followed by re-extension along the dorsal cord. Movie related to Figure 2D.
kin-20(ox423) sprouting ectopic growth cones in the adult GABA motor-neuron axons. Movie related to Figure 2E.
Differential gene expression in embryos: wild type, kin-20(ox423), and kin-20(ox423) suppressors related to Figures 5 & S6.
Differential gene expression analysis of wild-type embryos vs kin-20(ox423) embryos: giant unc-44 is the top differentially expressed gene when RNA sequencing data is analyzed by either DEXseq (adjusted P value=0.077, false discovery cutoff 10% or Padj<0.1) or DESeq2 (adjusted P value=0.0001, false discovery cutoff 10% or Padj<0.1). Genotypes: EG1285 lin-15(n765ts) X; oxIs12 X, EG5202 oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X, EG8338 ssup-72(ox542) II ; oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X, EG9090 zfp-3(ox649) X oxIs12[Punc-47:GFP, lin-15+] X kin-20(ox423) X
Data Availability Statement
R-script for mutation filtering
The programming language R was used to write a SNP filter with the option to exclude mutations that fall under the following categories: parental, synonymous, intergenic, heterozygous, intronic, and pseudogenes. An in-silico complementation test was also written in R. Scripts can be accessed at https://github.com/jorgensenlab/WGS-variant-filtering.