

Abstract
Pancreatic cancer has poor survival rates and largely ineffective therapies. Aspirin is the prototypical anti-cancer agent but its long-term use is associated with significant side effects. NOSH-aspirin belongs to a new class of anti-inflammatory agents that were designed to be safer alternatives by releasing nitric oxide and hydrogen sulfide. In this study we evaluated the effects of NOSH-aspirin against pancreatic cancer using cell lines and a xenograft mouse model. NOSH-aspirin inhibited growth of MIA PaCa-2 and BxPC-3 pancreatic cancer cells with IC50s of 47 ± 5, and 57 ± 4 nM, respectively, while it did not inhibit growth of a normal pancreatic epithelial cell line at these concentrations. NOSH-aspirin inhibited cell proliferation, caused G0/G1 phase cycle arrest, leading to increased apoptosis. Treated cells displayed increases in reactive oxygen species (ROS) and caspase-3 activity. In MIA PaCa-2 cell xenografts, NOSH-aspirin significantly reduced tumor growth and tumor mass. Growth inhibition was due to reduced proliferation (decreased PCNA expression) and induction of apoptosis (increased TUNEL positive cells). Expressions of ROS, iNOS, and mutated p53 were increased; while that of NF-κB and FoxM1 that were high in vehicle-treated xenografts were significantly inhibited by NOSH-aspirin. Taken together, these molecular events and signaling pathways contribute to NOSH-aspirin mediated growth inhibition and apoptotic death of pancreatic cancer cells in vitro and in vivo.
Keywords: Hydrogen sulfide, Nitric oxide, NOSH-aspirin, pancreatic cancer, cell signaling, FoxM1, p53, reactive oxygen species, cell kinetics
Graphical Abstract
1. Introduction
Pancreatic cancer is one of the most aggressive human malignancies with poor survival rates of approximately 4% [1]. It is the fourth cause of death in cancer even though it constitutes 2–3% of all cancers [2]. Pancreatic cancer cells are infiltrating epithelium cells that are neoplastic and gland forming, and therefore, pancreatic cancers are ductal adenocarcinomas. Majority of pancreatic cancer patients present with advanced disease or distant metastatcies which is often non-operable. Monotherapies with gemcitabine, 5-fluorouracil (5-FU), cisplatin and capecitabine or combinations thereof have been used with modest improvements in survival, however, mutidrug resistance to chemotherapy is generally a significant problem. To control disease progression, it is believed that multiple modes of intervention are required. As more targeted investigational agents are developed, the major pathways of focus are those related to signaling pathways associated with induction of apoptosis or proliferation. Therefore, many of the current agents target growth factors mediated events, angiogenesis, and other activated signaling pathways in this malignant disease [3, 4]. Not surprizing therefore, newer therapies for pancreatic cancer are needed.
Inactivation of the TP53 gene is found in approximately 50%–70% of pancreatic cancers [5]. The p53 protein is needed for important functions in the cell, such as G1-S cell cycle checkpoint, arresting cells at G2-M, and inducing apoptosis of aberrant cells [5]. Mutated cells with loss of p53 function generally accumulate more genomic alterations and this allows abnormal pancreatic cells with damaged DNA to survive more and then continue to divide. The transcriptional regulator FoxM1 is also a player in the tumorigenesis process such as initiation, progression, and metastasis in many tumor types [6]. FoxM1 is activated in pancreatic cancer and other cancers such as lung cancer, glioblastomas, prostate cancer, basal cell and hepatocellular carcinoma [7, 8]. Considering the poor survival rates and burden of small window of time in pancreatic cancer, targeting FoxM1 constitutes a meaningful strategy. Pancreatic cancer chemotherapy is also associated with resistance. It is also reported that many primary pancreatic cancers and cell lines, but not normal pancreatic cells, show constitutive activity of NF-κB, which is an important transcription factor promoting survival of cells [9, 10].
Induction of reactive oxygen species (ROS) is also known to inhibit cell growth and induce apoptosis and cell death [11, 12]. In particular, in pancreatic adenocarcinoma cells with p53 deletions, cell growth is strongly inhibited by ROS-inducing compounds [13]. Further, it is reported that pancreatic cancer cells with lower levels of ROS are more resistant to chemotherapy [14, 15]. Collectively, there is an understanding that increases in ROS production presents a good strategy to induce apoptosis in those cancer cells with decreased antioxidant capacity) and also to improve upon chemotherapy-induced resistance during pancreatic cancer treatment [14]
Nonsteroidal anti-inflammatory drugs (NSAIDs) are prototypical anti-cancer agents whose long-term use is associated with adverse side effects mainly gastrointestinal, cardiovascular, and renal, reviewed in [16]. Nitric oxide (NO) and hydrogen sulfide (H2S) are endogenous gaseous mediators that are associated with improved mucosal defense mechanisms [17, 18]. For these reasons, we recently developed NOSH-aspirin, a hybrid entity capable of releasing both NO and H2S [19]. Our preliminary results indicated that NOSH-aspirin was a potent anti-inflammatory agent devoid of cellular toxicity, which was also active against a variety of cancer lines at very low concentrations [19]; was efficacious against established tumors in a xenograft mouse model of colon cancer [20]; was safe to the stomach yet it retained all the pharmacological hallmarks of its parent compound aspirin [21]. In this study we explored the effect of NOSH-aspirin on twopancreatic cancer cell lines kinetics and a xenograft mouse model evaluating redox sensitive proteins that are important in determining cellular mass and carcinogenesis.
2. Materials and Methods
2.1. Reagents and cell lines
NOSH-aspirin (NBS-1120) 4-(3-thioxo-3H-1,2-dithiol-5-yl)phenyl 2-((4-(nitrooxy)butanoyl)oxy), benzoate was synthesized as described previously [19] and was a gift from Avicenna Pharmaceuticals Inc, (New York, NY). The structural components of NOSH-aspirin are shown in Figure 1. Aspirin was obtained from Sigma (St Louis, MO). Stock solutions (10 mM or 100 mM) were made in DMSO; final DMSO concentration was adjusted in all media to 1%. The pancaspase inhibitor z-VAD-FMK (z-VAD) was obtained from Promega (Madison, WI) and used according to the manufacturer’s instructions. Human pancreatic cancer MIA PaCa-2 (CRL-1420) and BxPC-3 (CRL-1687) cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) with verification of authenticity indicating them to be mycoplasma-free; human normal pancreatic epithelial ACBRI 515 was purchased from the European Collection of Cell Culture (ECACC, Salisbury, UK) and certified as such by the supplier. Cells were maintained in RPMI-1640 media (Invitrogen, Carlsbad, CA) that was supplemented with 10% fetal bovine serum and 100 μg/mL penicillin, and 100 μg/mL streptomycin (Invitrogen, Carlsbad, CA) and maintained at 37°C and in an atmosphere of 5% CO2/95% air.
Figure 1.
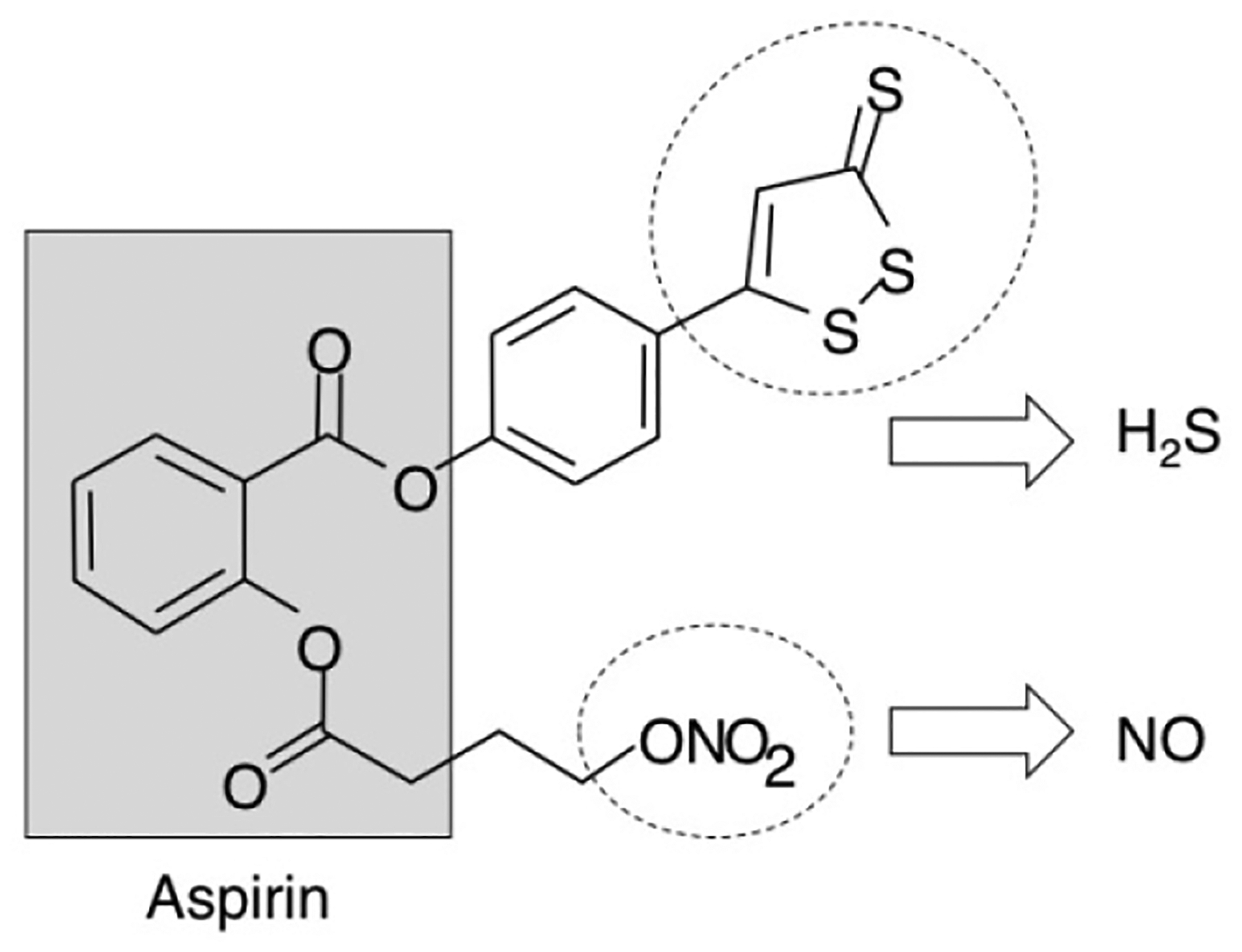
Structural components of NOSH-aspirin. The parent compound aspirin is shown in the shaded box. The parts of the molecule that releases NO and H2S are shown in the dotted ellipses.
2.2. Cell growth inhibition assay
The growth inhibitory effect of NOSH-aspirin was measured using a colorimetric MTT assay kit (Roche, Indianapolis, IN). Briefly, cells were plated in 96 well plates at a density of 104 cells per well and, following overnight incubation, were washed and treated with vehicle, NOSH-aspirin or ASA. At indicated time periods of treatment, viable cells were incubated with MTT substrate (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide, 5 mg/mL in phosphate buffered saline), further for 2 hrs, and formazan formed by viable cells was dissolved in 100 μL of the solubilization solution (10% SDS in 0.01 M HCl) and absorbance was measured on a spectrophotometric plate reader at a wavelength of 570 nm. Each experiment was performed in triplicate, and the entire experiment was repeated three times. In some experiments pan-caspase inhibitor z-VAD-FMK, pretreatment was performed prior to NOSH-aspirin addition to cells, followed by MTT assay.
2.3. Cell proliferation
Proliferating cell nuclear antigen (PCNA) was determined using an ELISA Kit (Calbiochem, La Jolla, CA), in accordance with the manufacturers protocol and read on spectrophotometric plate reader as previously described [22].
2.4. Flow cytometry for phase distribution in the cell cycle and detection of apoptosis
Cell proliferation and cell cycle phases:
Cell proliferation and cell cycle analysis were performed as previously described [22, 23]. Cell cycle phase distributions were obtained using a Coulter Profile XL equipped with a single argon ion laser. For each subset, >10,000 events were analyzed. All parameters were collected in list mode files. Data were analyzed on a Coulter XL Elite Work station using the Software programs Multigraph™ and Multicycle™. The percentage of cells in G0/G1, G2/M, and S phases was determined form DNA content histograms.
Apoptosis assay:
Cell death was determined by FITC-labeled Annexin V and propidium iodide (PI), using an Annexin V-FITC apoptosis detection kit, (BD Biosciences Pharmingen, San Jose, CA, USA), according to the manufacturer’s instructions, as reported previously [22]. Treated cells (floating and adherent cells) were collected, resuspended in an Annexin V binding buffer and transferred to test tubes containing FITC-labeled Annexin V and PI. The cells were then incubated for 15 minutes at room temperature in the dark, and analyzed by flow cytometry. Annexin V–FITC detects cells in early apoptosis by staining externalized phosphatidylserine, and the nuclear stain PI identifies cells that have lost their plasma membrane integrity such as necrotic or late apoptotic cells. Annexin V-FITC-positive/PI-negative cells were regarded as a good measure of early apoptosis. Percentage of apoptotic cells was obtained using a Becton Dickinson LSR II equipped with a single argon ion laser. For each subset, about 10,000 events were analyzed. All parameters were collected in list mode files. Data was analyzed by Flow Jo software.
2.5. Caspase-3 activity measurement
Cells were treated with NOSH-aspirin at its IC50 (50 nM) for cell growth inhibition and caspase-3 activity was assayed as a function of time (0, 3, 6, 12 hr) using a colorimetric kit from Abcam bcam (Cambridge, MA) following the manufacturer’s instructions. The assay is based on spectrophotometric detection of the chromophore p-nitroaniline (p-NA) after cleavage from the labeled substrate DEVD-p-NA. The p-NA light emission was quantified using a plate reader at 400 nm. In some experiments the pan-caspase inhibitor z-VAD-FMK was used following the manufacturer’s instructions.
2.6. Determination of reactive oxygen species (ROS)
Cells (0.3 × 106 cells/well) were plated in 6-well plates for 24 h after which they were treated with various concentrations of NOSH-aspirin for 6 h. Cells were then trypsinized, washed once in PBS resuspended and then incubated for 30 min at 37°C in the dark with the oxidation-sensitive fluorescent probes 2’,7’-dichlorodihydrofluorescein diacetate (H2DCFDA, 10 μM) or dihydroethidium (DHE, 5 μM) (Molecular Probes, Life Technologies, NY). Fluorescence intensity was then measured by flow cytometry using a FACS Calibur (BD Bioscience). H2DCFDA is a probe for H2O2, peroxyl radical, including both alkylperoxyl and hydroperoxyl, while DHE is an intracellular probe that preferentially measures superoxide anion [24, 25]. A minimum of 10,000 events were analyzed and expressed as fluorescent intensity versus events.
2.7. Mouse xenograft study
Our institutional animal care and research committee approved all experimental procedures described here. Male athymic SCID mice, age 5 weeks, were purchased from Charles River Laboratories, Inc., (Wilmington, MA) and were housed according to institutional and National Institute’s of Health (NIH) guidelines. In this post-initiation study, tumors were initiated and followed with NOSH-aspirin treatment. MIA PaCa-2 cells (2×106) were suspended in Matrigel (BD Biosciences, San Jose, CA) 50% v/v and were inoculated subcutaneously in the right flanks of each mouse (N=10) using a 1-mL syringe and 22-gauge needles. After 10 days (average tumor sizes of ~70 mm3) the mice were randomly divided into 2 groups (N=5/gp) and gavaged daily with NOSH-aspirin (100 mg/kg/d) or vehicle (1% methylcelloluse). The mice were closely monitored for signs of toxicity. Tumor volume and animal weight were recorded every 3 days. Tumor volumes were calculated using the following formula: length × width2/2. After 30 days of treatment, mice were sacrificed, tumors excised, weighed, photographed and fixed in 10% buffered formalin for immunohistochemistry studies.
2.8. Immunohistochemistry
Paraffin embedded sections were dewaxed, rehydrated and microwave heated for 15 min in 0.01 M citrate acid buffer (pH 6.0) for antigen retrieval. This was followed by 3% hydrogen peroxide for 15 min to block endogenous peroxidase activity and normal horse serum for 15 min as a blocking solution. The primary antibody or isotype IgG control were applied and incubated overnight at 4° C. Slides were washed 3 times with PBS, each for 5 min. The biotinylated secondary antibody and the streptavidin-biotin complex (Zymed Laboratories, San Francisco, CA) were applied, each for 30 min at room temperature with an interval washing. After rinsing with PBS, the slides were immersed for 5 min in the substrate 3,3’-diaminobenzidine (DAB, Sigma-Aldrich Corp. St. Louis, MO) 0.4 mg/mL with 0.003% hydrogen peroxide, then rinsed with distilled water, counterstained with hematoxylin, dehydrated and cover-slipped. Antibodies: PCNA, p53 and FoxM1 were from Santa Cruz Biotechnology (Santa Cruz, CA); NF-ĸB p65 (which recognizes activated NF-ĸB), was from Chemicon International (Temecula, CA).
2.9. TUNEL and ROS staining in tissues
TUNEL staining was performed using the In Situ Cell Death Detection kit (Roche Applied Science, Indianapolis, IN) following the manufacturer’s instructions. Briefly, 4 μm thick formalin-fixed, paraffin-embedded tissue sections were deparaffinized and rehydrated. Endogenous peroxidase activity was quenched by hydrogen peroxide and tissue protein was hydrolyzed with proteinase K (Roche Applied Science, Indianapolis, IN). Negative control: sections incubated with Label Solution (without TdT enzyme). All other sections were incubated with TUNEL reaction mixture (fluorescein-labeled nucleotides) at 37°C for 1 h in a humid chamber, incubated with Converter-POD solution (anti-fluorescein antibody conjugated with POD) for 30 min at 37°C, treated with DAB and counterstained with hematoxylin. For ROS detection by immunofluorescence in tumor tissues, paraffin embedded sections were stained with 5 μmol/L CM-H2DCFDA (Invitrogen, Carlsbad, CA) for 1.5 hours at 37°C. Images were taken using a fluorescent microscope (BX41, Olympus) controlled by IPlab (Scanalytics Inc., Fairfax, VA). Randomly selected areas were photographed with the same exposure time. The images were processed using the same fixed threshold in all samples by Imagej software; representative images are shown.
2.10. Scoring the expression of biomarkers
For each animal and treatment group, 5 slides were prepared and scored by a pathologist blind to the identity of the specimens (x400). For PCNA, and TUNEL staining, cells with a brown nucleus were considered labeled and those with a blue nucleus unlabeled. For each, the percentage of positive cells over the total cells counted was calculated. For NF-κB, FoxM1, and p53 the following semi-quantitative scoring system was used. The extent of staining was graded as follows: 0 = no staining; 1+ = ≤25% of cells positive; 2+ = 26% – 50% of cells positive; 3+ = ≥51% of cells positive. The intensity of staining was scored as follows: 0 = no staining; 1+ = faint; 2+ = moderate; 3+ = strong. 1+, 2+ and 3+ were recorded as 1, 2 and 3 points, respectively. To compare differences in staining, an Expression index (EI) was calculated by the following formula: EI = Extent of staining × Intensity of staining, as described previously [26].
2.11. Statistical analysis
In vitro data are presented as means ± SEM for at least 3 different sets of plates and treatment groups done in duplicate or triplicate. Xenograft data, tumor volume and mass are presented as means ± SEM for 5 animals in each group. Comparisons between groups were performed using a one-way analysis of variance followed by the Student-t test.
3. Results
3.1. NOSH-aspirin inhibits pancreatic cancer cell growth
The effects of NOSH-aspirin and ASA were examined on cell growth of two pancreatic cancer cell lines MIA PaCa-2 (Cox-1 and Cox-2 negative, p53 mutated, K-ras mutated, p16 deleted) and BxPC-3 (Cox-1 and Cox-2 positive, p53 mutated, K-ras wild type) [27], and a normal pancreatic ductile epithelial ACBRI 515 cell line. Cells were exposed to various concentrations of these compounds for 24 hrs and MTT assays were performed. From their growth inhibition curves, IC50 values were calculated (Table 1). NOSH-aspirin inhibited the growth of MIA PaCa-2 and BxPC-3 cells at nanomolar levels with calculated IC50 values 47 ± 5 nM and 57± 4 nM at 24h, respectively, while it did not inhibit growth of normal ACBRI 515 cells at these low concentrations. These results strongly suggest that NOSH-aspirin’s effect on cancer cell growth maybe COX independent, as MIA PaCa-2 cells are COX null while BxPC-3 cells express both COX-1 and COX-2. The IC50 for the normal cell line was determined to be 28,000 ± 2000 nM (Table 1), that is ~500-fold higher, demonstrating the differential behavior of this novel compound towards cancer cells. The IC50 of the parent compound aspirin was more than 5mM in these cells at 24 hr. Overall, NOSH-aspirin demonstrated an increase in potency as determined by the ratio of the IC50s (ASA / NOSH-aspirin) which was calculated to be ~80,000-fold more than ASA. Finally, as K-ras wild type and K-ras mutated cell lines responded to NOSH-aspirin treatment much the same way, it appears that NOSH-aspirin growth inhibition are independent of the RAS/RAF/MAPK pathway.
Table 1.
IC50s for cell growth inhibition at 24 hr.
| IC50 nM at 24 hr | |||
|---|---|---|---|
| MIA PaCa-2 | BxPc-3 | ACBRI 515 | |
| COX-1, −2 status | − | + | NK |
| NOSH-aspirin | 47± 5 | 57 ± 4 | 28,000 ± 2000 |
| ASA | > 5,000,000 | ND | |
| Enhanced potency | > 80,000* | ~500† | |
Pancreatic cancer cell lines MIA PaCa-2 and BxPC-3 and a normal pancreatic ductile epithelial cell line ACBRI 515 were treated with different concentrations of NOSH-aspirin or ASA for 24 hr and IC50s for cell growth inhibition determined. Results are mean ± SEM for at least 3–5 different experiments performed in triplicate.
ND = not determined. NK = not known
= Fold-increase for NOSH-ASA vs ASA determined as IC50 ASA / IC50 NOSH-aspirin.
= Fold-increase in potency towards cancer cells determined as IC50 normal / IC50 cancer cells.
3.2. NOSH-aspirin modulates cellular mechanisms
MIA PaCa-2 and BxPC-3 cells were analyzed for effects of NOSH-aspirin on their cell proliferation, cell cycle phase transitions and apoptosis by flow cytometry. The concentration of 50 nM was selected for these studies representing a dose very close to their respective IC50 values. Proliferation status, as measured by proportion of cells expressing PCNA when treated with the vehicle DMSO or 50 nM NOSH-aspirin for 0, 3, 6 and 12 hr showed a time-dependent anti-proliferative effect observed by reductions in PCNA expression for both cell types (Fig 2A, MIA PaCa-2 and 2D, BxPC-3). This was associated with block of cell cycle transitions by NOSH-aspirin at G0/G1 phase, based on the increase of population of cells in the G0/G1 phase from 42% to 59% for MIA PaCa-2 cells (Fig 2B), and from 39% to 58% for BxPC-3 cells (Fig 2E) at 12 hrs. Early apoptosis as quantified by Annexin V FITC-positive and PI-negative stained cell populations at these time points produced similar increases to approximately 65% to 70% early apoptotic cells for both cell types at 12 hrs (Figs 2C and 2F). Overall, it appears that NOSH-aspirin produces similar effects on these two cell lines and in general, inhibits cell growth via inhibition of proliferation, G0/G1 cell cycle arrest and apoptosis. We have reported earlier that effects of NOSH-aspirin are independent of cell type [19].
Figure 2.
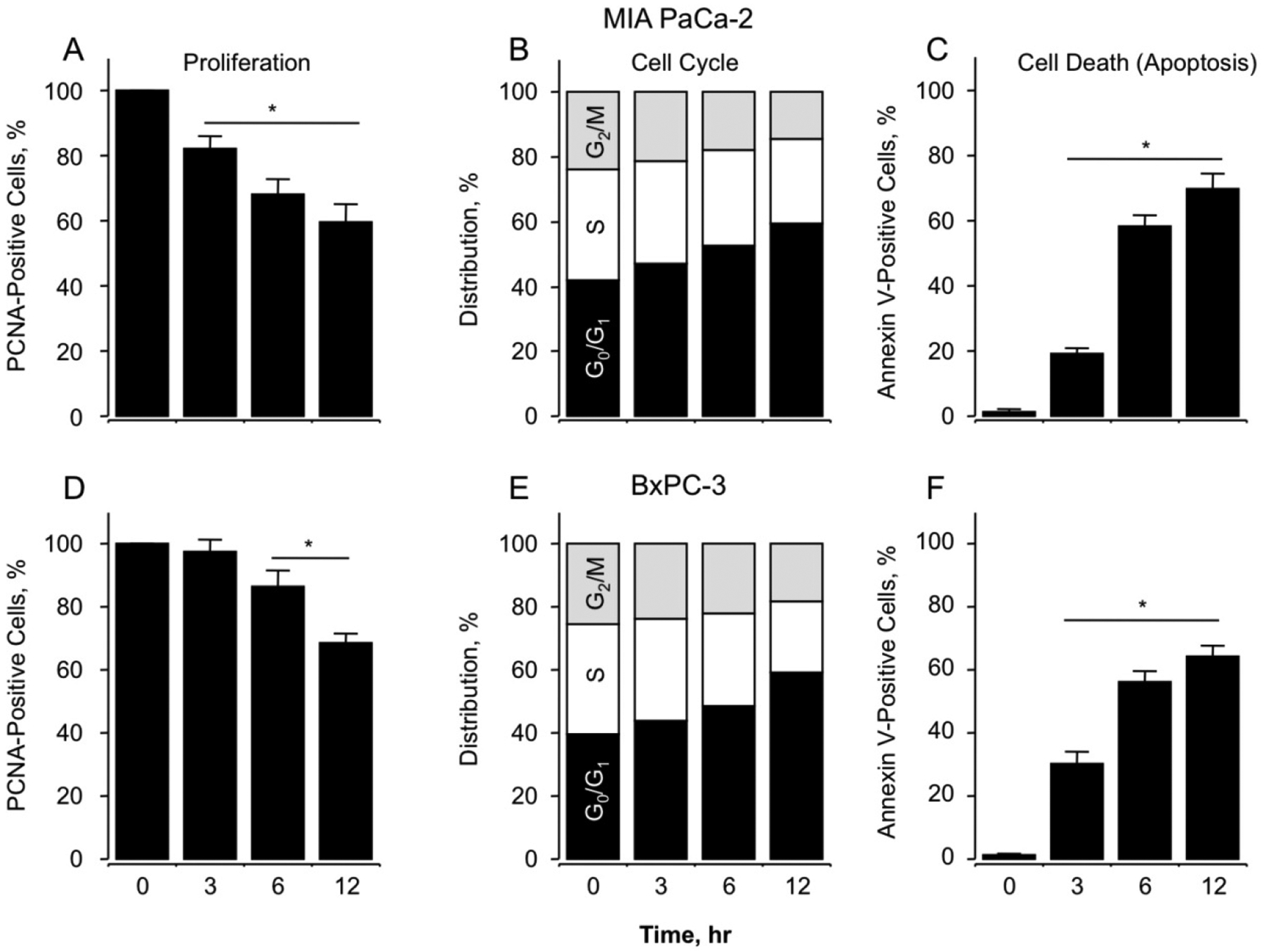
Effect of NOSH-aspirin on MIA PaCa-2 and BxPC-3 pancreatic cancer cell kinetics. NOSH-aspirin inhibits proliferation by altering cell cycle progression and inducing apoptosis. Cells were treated with vehicle or 50 nM NOSH-aspirin (close to IC50 for cell growth inhibition in both cell lines) and analyzed at the indicated times for proliferation by PCNA antigen expression (Panels A and D, MIA PaCa-2 and BxPC-3 cells respectively); cell cycle phases by PI staining and flow cytometry (Panels B and E, MIA PaCa-2 and BxPC-3 cells respectively); and apoptosis by Annexin V staining and flow cytometry (Panels C and F, MIA PaCa-2 and BxPC-3 cells respectively). In (A), (C), (D), and (F), results are mean ± SEM for 3 different experiments performed in duplicate, *P < 0.05 at the indicated time points. In (B) and (E), results are representative of two different experiments. This study was repeated twice generating results within 10% of those presented here.
3.3. NOSH-aspirin induces caspase-3 and reactive oxygen species
Caspase-3 activation, which is important for mediating the induction of apoptosis, was measured in both cell lines treated with 50 nM NOSH-aspirin at time intervals up to 12hrs. NOSH-aspirin increased the activity of caspase-3 in a time dependent manner to a statistically significant four- to five-fold increase above control levels at 12 hrs (P < 0.01) (Fig 3A, BxPC-3 and Fig 3B, MIA PaCa-2). Overall, both cell lines behaved similarly in their responses. Pretreatment for 1 hr with z-VAD-FMK, a cell permeable pan-caspase inhibitor abolished the increases in caspase-3 activity at all time intervals, demonstrating that NOSH-aspirin induces cell death through caspase activation in both cell lines.
Figure 3.
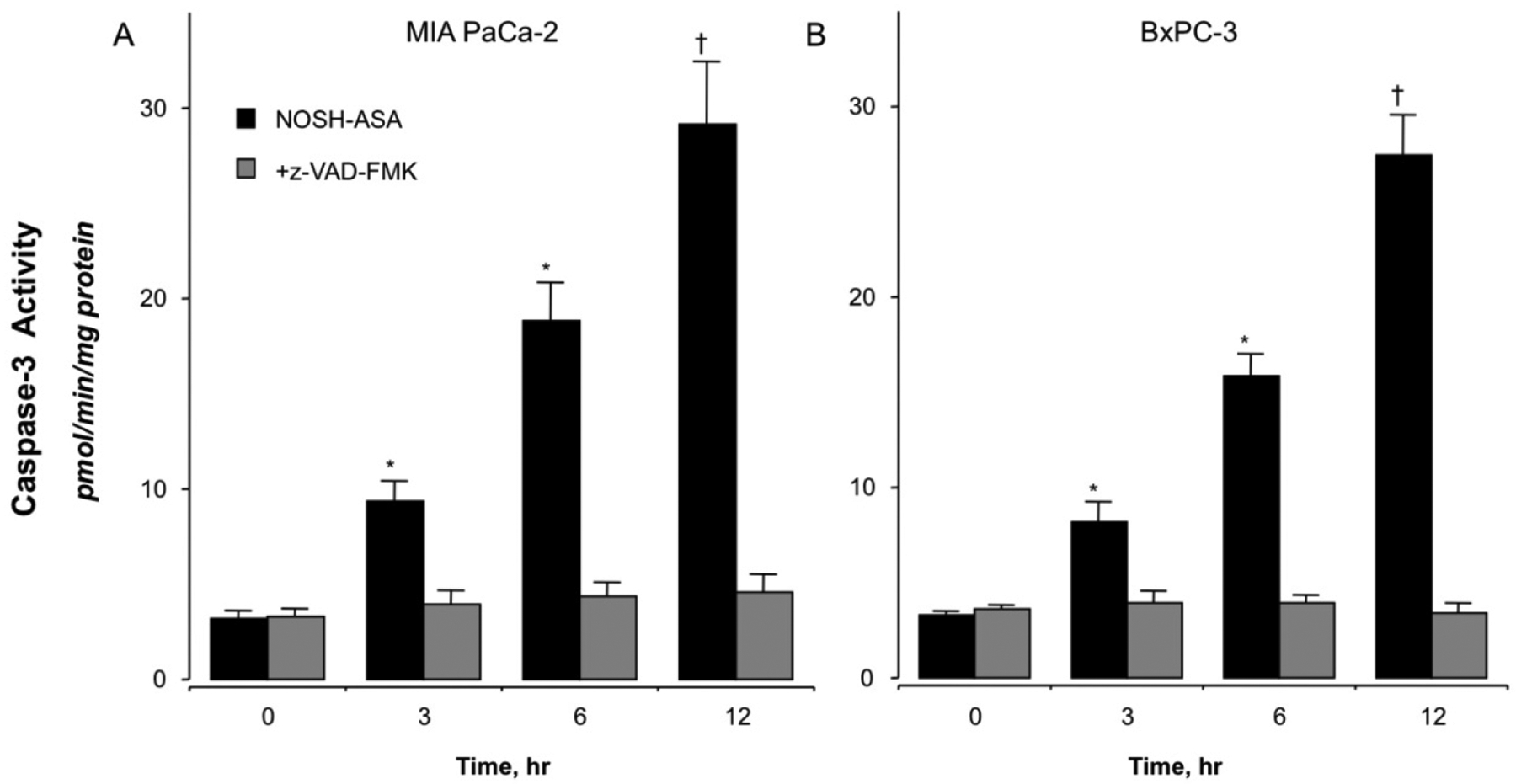
NOSH-aspirin treatment increases caspase-3 activity. MIA PaCa-2 (Pane A) and BxPC-3 (Panel B) pancreatic cancer cells were treated with vehicle or 50 nM NOSH-aspirin. Caspase-3 activity was increased in both cell lines at all time points, 3, 6, and 12 hr. Results are mean ± SEM for 4 different experiments performed in duplicates. *P < 0.05 and †P < 0.01 and the indicated times. The pan-caspase inhibitor z-VAD-FMK inhibited caspase-3 activity at all time points.
Very high ROS levels are known to push cells towards apoptosis [12]. We examined whether NOSH-aspirin induced ROS in these cells. A molecular probe that detects several individual reactive oxygen species including peroxides is H2DCFDA, also abbreviated as DCFDA [28, 29]. DHE is a selective probe for detection of superoxide anions (•O2-) [30]. Cells were treated with 50 nM NOSH-aspirin for 6 hr and analyzed for levels of intracellular peroxides by flow cytometry. At 6 hr, 50 nM NOSH-aspirin increased production of intracellular peroxide and •O2- levels in MIA PaCa2 cells by approximately two orders of magnitude (Fig 4A and 4B, respectively). Similar results were also obtained with the BxPC-3 cells (data not shown). We also determined the levels of ROS in MIA PaCa2 cells following treatment with z-VAD-FMK and NOSH-aspirin. ROS that was produced at 6 hr with NOSH-aspirin was blocked when caspase-3 was inhibited with z-VAD-FMK pretreatment in MIA PaCa2 (Fig. 4C) and BxPC-3 M cells (Fig. 4D). This strongly suggests that caspase-3 activation precedes and is necessary for ROS generation.
Figure 4.
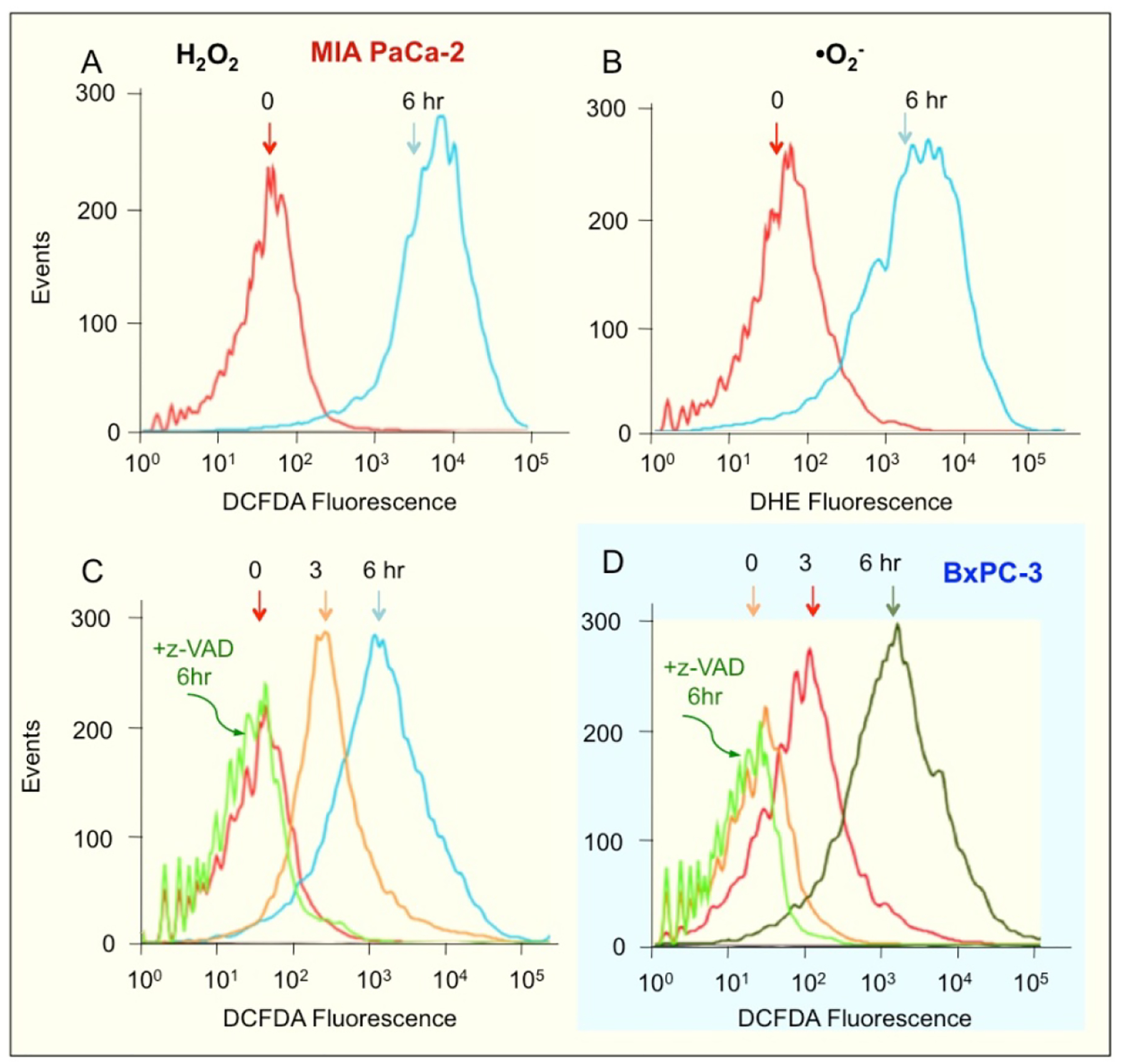
NOSH-aspirin induces ROS levels. MIA PaCA-2 cells were treated with 50 nM NOSH-aspirin for 6 h followed by staining with a general ROS probe DCFDA (A) or DHE which detects superoxide anions in cells (B). These experiments were repeated twice and representative histograms are shown. ROS in MIA PaCA-2 cells was also probed with DCFDA following treatment with z-VAD and NOSH-aspirin (C) and similarly examined in BxPC-3 cells (D). The coloring of the backgrounds in the histograms and cleaning of the axis was done by using Adobe Photoshop CS5.
3.4. Caspases are necessary for cell growth inhibition by NOSH-aspirin
Since caspase-3 activation and ROS production appear to be key features of NOSH-aspirin’s mechanism of action, we examined whether blocking caspase-3 activation could indeed block the inhibition of growth that we observed at the IC50 concentrations of NOSH-aspirin. We did not include ASA in these studies because ASA’s IC50 for cell growth inhibition at 24 hr was greater than 5mM. MIA PaCa-2 and BxPC-3 cells were pretreated with z-VAD (2 μM) for 4 hr followed by NOSH-aspirin (50 nM) or vehicle (control) for 24 h. Growth inhibition was measured by the MTT assay. Whereas cells were approximately 50% growth inhibited by NOSH-aspirin, this inhibition was markedly and partially reversed by z-VAD suggesting that caspases are necessary for the effects of NOSH-aspirin. (Fig 5).
Figure 5.
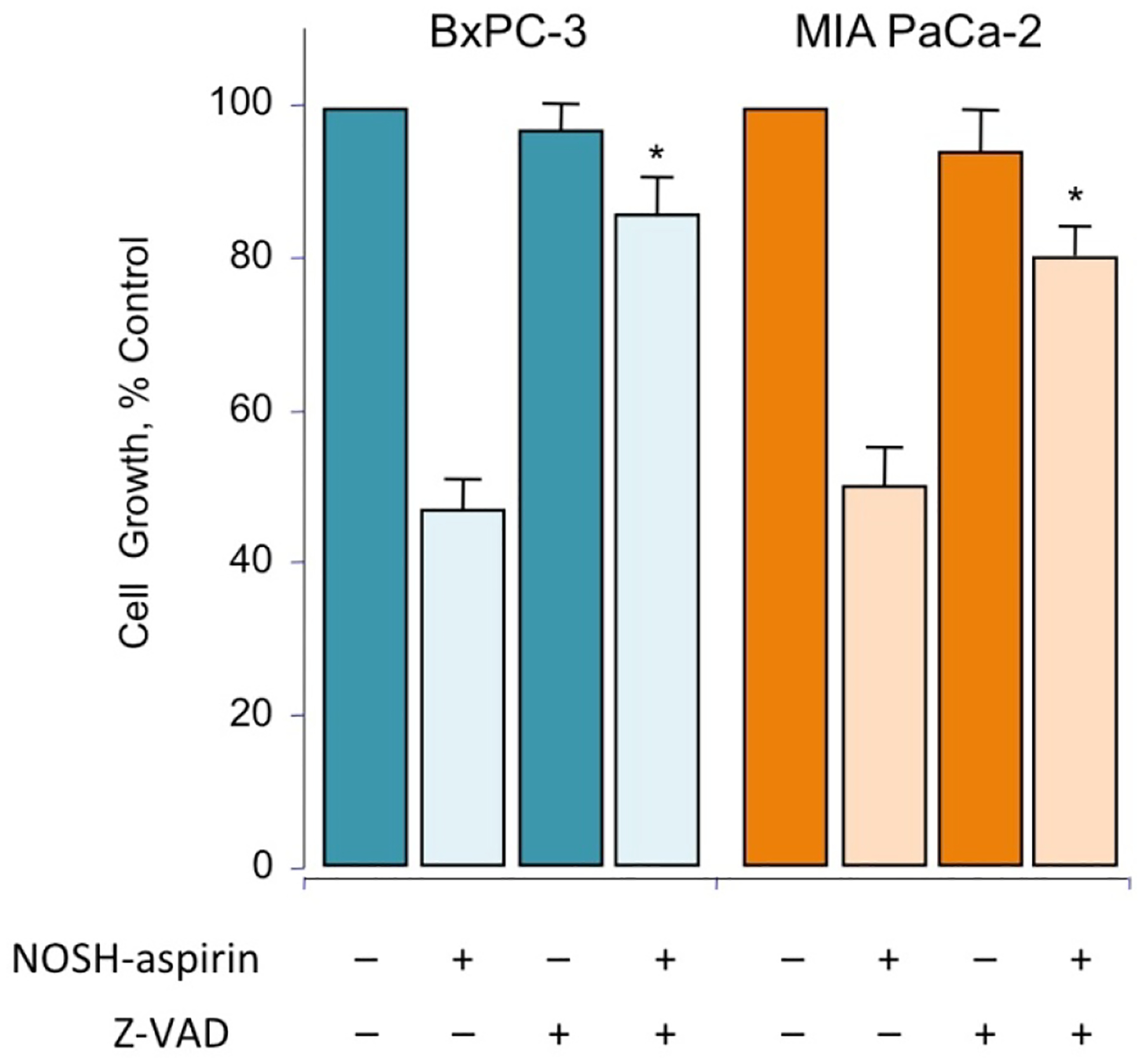
NOSH-aspirin mediated cell growth inhibition is reversed by zVAD. MIA PaCa-2 and BxPC-3 cells, pretreated with 2 μM z-VAD-FMK for 4 hr or untreated, were followed by 50 nM NOSH-aspirin (~IC50 concentration) and subsequently analyzed for growth as described in Section 2.2. Values represent means ± SEM of three representative experiments performed in triplicate. *P < 0.05 compared to absence of z-VAD.
3.5. NOSH-aspirin inhibits tumor growth in a xenograft model with no evidence of gross toxicity
In this treatment model, tumor xenografts were transplanted by human pancreatic cancer cells to evaluate the antitumor effects of NOSH-aspirin in vivo. Male athymic nude mice were used for subcutaneous implantation of MIA PaCa-2 cells (2 × 106) in the right flank. The mice were divided into two groups after 10 days when tumors were palpable and had a volume of about 70 mm3. The test group received daily administration of NOSH-aspirin (100 mg/kg, gavage), while the control group was gavaged with the vehicle (1% methylcellulose) daily till the end of the experiment, the protocol followed is shown in Fig 6A. During the 30 days of this study, mice did not die as a result of the received treatment; the body weights of the NOSH-aspirin-treated and vehicle-treated mice throughout the duration of the study did not vary (Fig 6B). After 30 days and post sacrifice, NOSH-aspirin-treated mice showed a significant reduction in tumor volume compared with the control mice (Fig 6C and inset). The control- and NOSH-aspirin- mean tumor volumes at the end of the study were 3265 ± 520 mm3 and 330 ± 95 mm3, respectively (Fig 6C), representing a mean reduction of about 90% (P=0.001). Tumor mass for control and NOSH-aspirin-treated mice at the end of the protocol were 2.47 ± 0.24 g and 0.62 ± 0.25 g respectively (Fig 6D), with 75% reduction (P=0.003). Figure 6E shows the vehicle and NOSH-aspirin-treated mice with the xenografts shown with arrows and Fig 6F shows the excised tumors in the two groups. After sacrifice, close inspection of the main organs (heart, liver, kidney, spleen) did not show any gross signs of toxicity (deformity, edema or bleeding) in the NOSH-aspirin treated mice.
Figure 6.
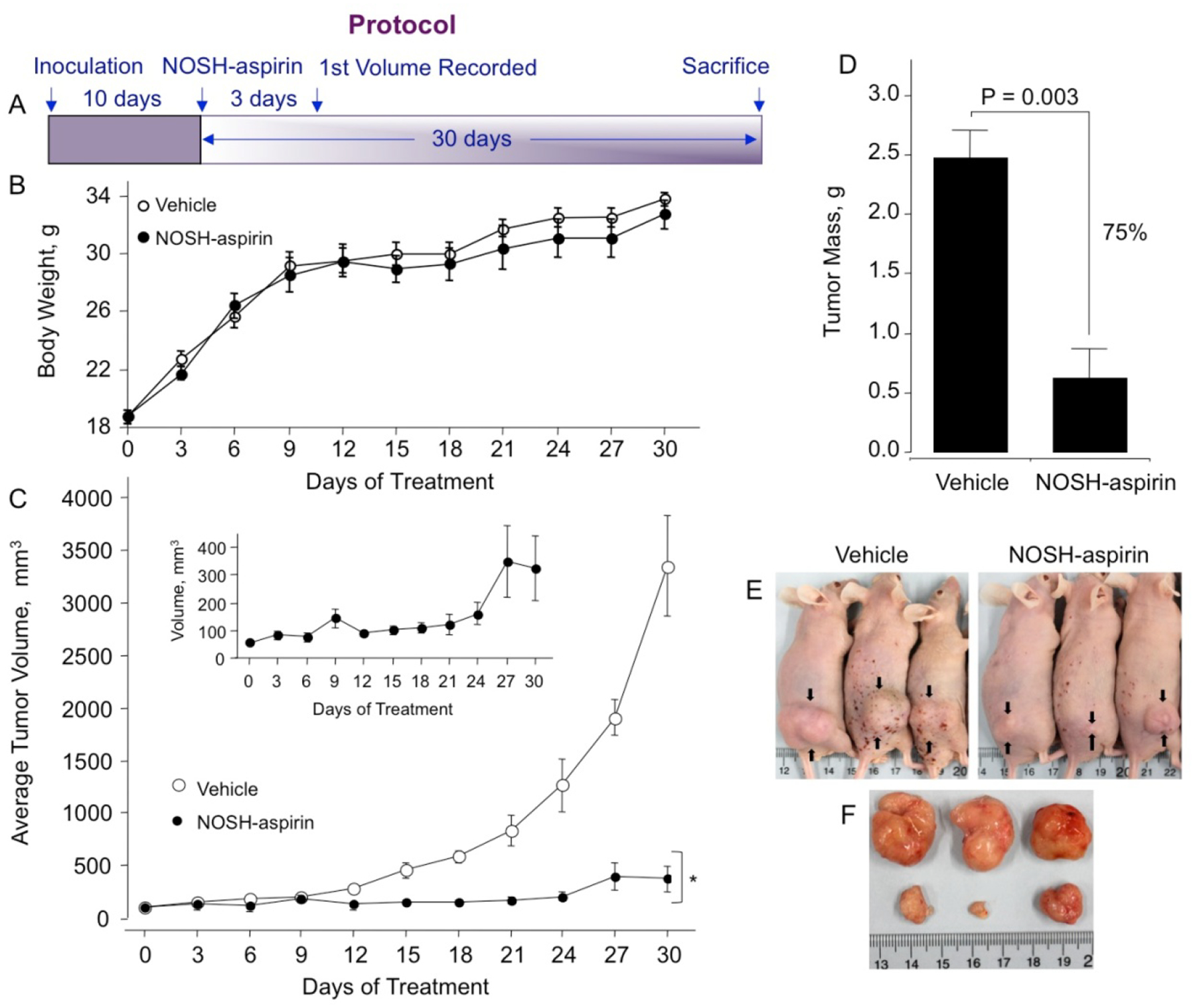
NOSH-aspirin inhibits growth of established tumor xenografts. Athymic SCID male mice were injected subcutaneously with MIA PaCa-2 cells for the development of subcutaneous tumors as described in Section 2.7. Following tumor formation, the mice were randomly divided into 2 groups (N = 5) and treated daily with vehicle or 100 mg/kg NOSH-aspirin for 30 days according to the protocol in panel A. Body weight of both groups over 30 days (panel B) and average tumor volume as function of time and tumor mass at sacrifice are shown in panels C and D. The inset shows an expanded scale for the NOSH-aspirin-treated mice. Representative mice and tumors for untreated and treated mice are shown in panels E and F, respectively. NOSH-aspirin significantly reduced tumor volume from day 15 of treatment to sacrifice, *P < 0.01. Tumor mass was also significantly reduced by NOSH-aspirin treatment, P = 0.003.
The tumor sections were examined for proliferating cells by the expression of PCNA stained cells, for apoptotic cells by TUNEL staining, and for promoters of cell survival by the expression by NF-ĸB p65. Compared to control tumors, the NOSH-aspirin group had reduced expression of PCNA (from 78.4 ± % to 25.2 ± 3 %, P < 0.01) (Fig 7A), increase in number of apoptotic cells (from 3.4 ± 0.3 % to 83.9 ± 3 %, P < 0.01) (Fig 7B), and reduced expression of NF-ĸB cells (from 63.6 ± 7 % to 14.7 ± 3 %, P < 0.01), (Fig 7C). These corroborated the in vitro observations from cell culture studies and demonstrate that in vivo tumor growth inhibition occurs by a combination of apoptosis induction and proliferation reduction, and reduced NF-ĸB expression. Since ROS induction was observed in vitro and may contribute to apoptosis, tumor sections were stained for ROS and observed by fluorescence. The NOSH-aspirin group showed marked increases in peroxide- (7 ± 1% control to 90 ± 2% treated) (Fig 8A) and superoxide (9 ± 1% control to 84 ± 3% treated) anion compared to vehicle treated controls (Fig 8B). Another target of interest that may be associated with induction of apoptosis is the inducible nitric oxide synthase (iNOS), which was visualized by staining in the NOSH-aspirin-treated group, and determined to be induced by NOSH-aspirin (1.6 ± 0.1% control versus 33 ± 2% treated), (Fig 8C).
Figure 7.
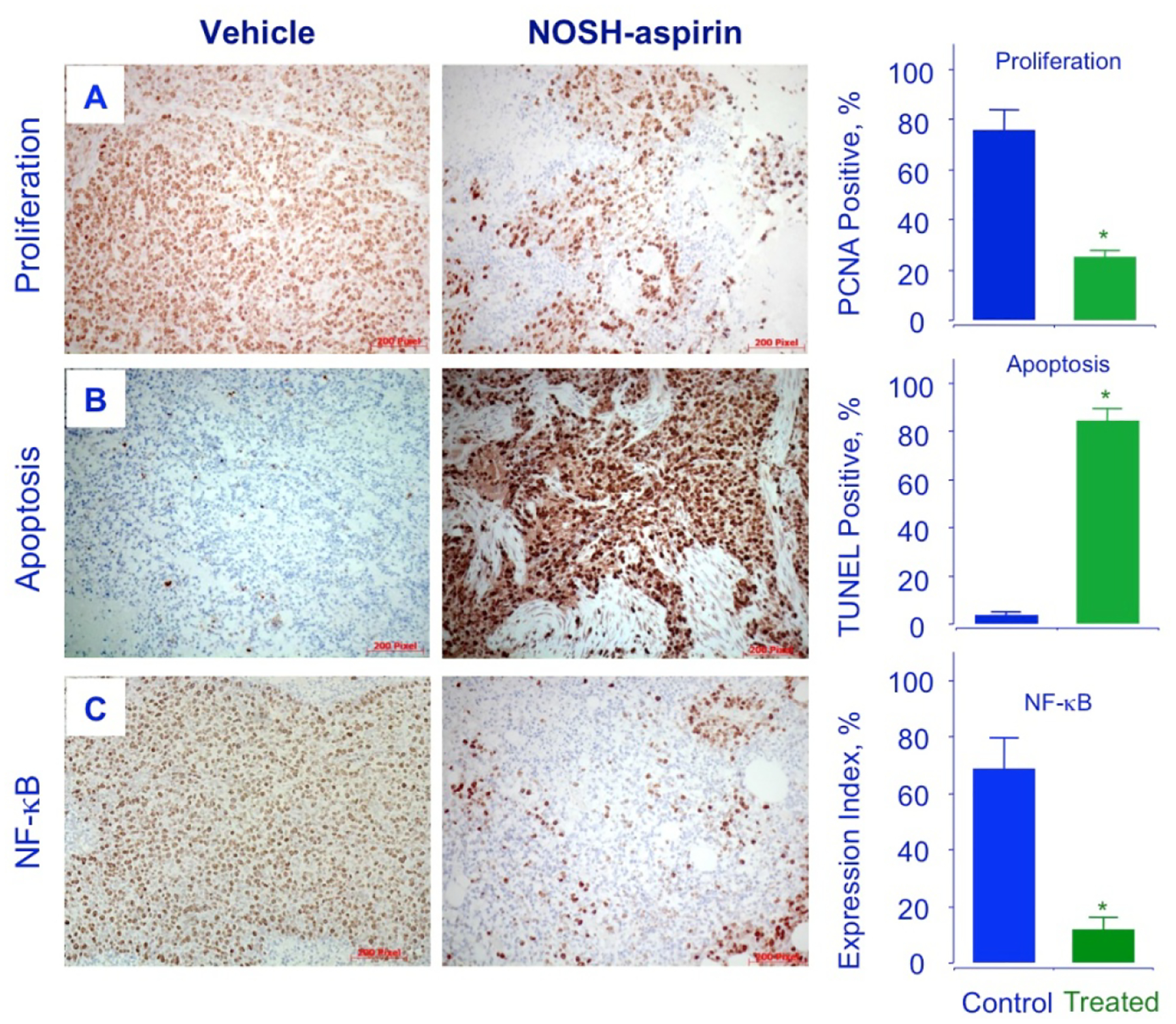
NOSH-aspirin inhibits proliferation, induces apoptosis and decreases NF-κB p65 in vivo. Stored tumors were sectioned, probed, and scored as described in Section 2.8–10. Average mitotic index at sacrifice was determined by PCNA staining (P < 0.05), TUNEL staining (P < 0.01) and p65 staining (P < 0.02). Representative fields used for quantification of the staining are shown. The scale bar represents 200 μm.
Figure 8.
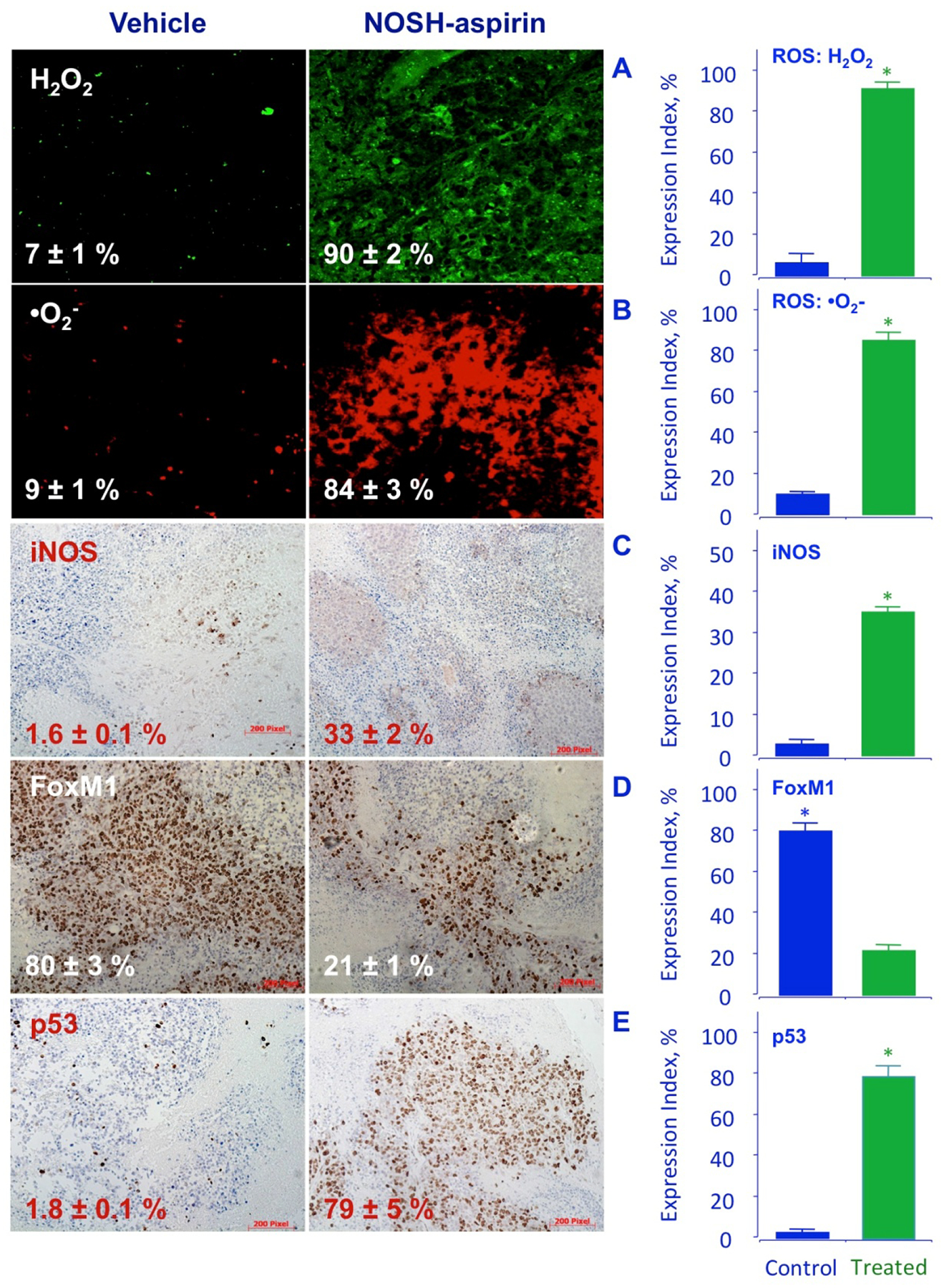
NOSH-aspirin induces ROS, iNOS, and p53 and inhibits FoxM1 in vivo. Stored tumors were sectioned, probed, and scored as described in Section 2.8–10. NOSH-aspirin-treatment increased peroxide and super oxide anions, as well as iNOS and p53 expression while it reduced FoxM1 levels. Five different representative fields were used for quantification from each tumor (N=5), in all cases P < 0.01 compared to vehicle treated mice. The scale bar represents 200 μm.
3.6. FoxM1 is modulated by NOSH-aspirin
FoxM1 (Forkhead box M1) is a transcription factor that regulates a network of proliferation-associated genes critical to mitosis [31, 32]. Its expression increases at G1 to S phase transition, and reaches maximal levels in G2 to M phase, promoting M phase entry [33] with depletion leading to cell cycle arrest. Importantly, aberrant upregulation of FoxM1 has been shown to be a key driver of cancer progression and has been proposed as an initiating factor of oncogenesis [34–36]. Since NOSH-aspirin accumulated cells in the G1 phase, we examined the expression of FoxM1 by immunohistochemical staining. The tumors from the NOSH-aspirin group had approximately 4-fold reduced levels of FoxM1 expression, compared to control (80 ± 3% control to 21 ± 1% treated, Fig 8D). It is known that p53 negatively regulates the expression of FoxM1 [8]. We therefore, evaluated the expression of p53 in our xenograft tumors. Staining demonstrated that in NOSH-aspirin-treated tumors, there was a large increase (about 40-fold) in p53 protein expression, 1.8 ± 0.1% to 79 ± 5% in control and treated tumors, respectively (Fig 8E).
4. Discussion
This study demonstrates that NOSH-aspirin inhibits tumor growth of pancreatic cell xenografts as well as pancreatic cancer cell lines with the advantages of strong potency and differential activity in cancer- versus normal- cell. Nanomolar concentrations inhibited pancreatic cancer cell growth, arrested cells in G0/G1 phase transition and induced apoptosis in vitro, while normal pancreatic epithelial cells were fairly unaffected. Mutations in the proto-oncogene Kras and tumor suppressor p53 are known to be major genetic alterations associated with cell cycle deregulation and apoptosis inhibition [37]. As MIA PaCa-2 cells have mutations in K-Ras and p53 [38], while BxPC-3 cells exhibit wild type K-Ras and mutated p53, it is promising that NOSH-aspirin inhibited both of these cell lines with low IC50 values. Apoptosis and cancer cell growth inhibition were also corroborated in the NOSH-aspirin xenograft model. Progression of MIA PaCa-2 xenograft tumors was strongly reduced and a combination of mutiple mechanisms may contribute to this process. Caspase-dependent apoptosis induction occurred in tumors and in cancer cells in vitro. This conclusion is based on our current observation that the pan-caspase inhibitor z-VAD-FMK inhibited caspase-3 activation and on our previous studies with NO-releasing aspirin [39] and H2S-releasing aspirin [40] where we had shown that the IC50 for cell growth inhibition in Jurkat T-leukemia cells increased by almost a factor of 5 in the presence z-VAD-FMK. Increases in ROS such as peroxide and superoxide anion production and an increase in iNOS expression by NOSH-aspirin were also associated with reduced tumor progression. Very high ROS levels are known to push cells to apoptosis by oxidative stress through both p53 and iNOS pathways [41].
As it is accepted that iNOS-mediated NO has the capacity to either promote or prevent tumorogenesis, therefore, the role of iNOS/NO in pancreatic cancer also remains controversial. For example, iNOS/NO promotes development and progression of pancreatic ductal adenocarcinoma [42] and increased iNOS expression is associated with poor survival in pancreatic ductal adenocarcinoma patients [43]. On the contrary, the aggressive pancreatic cancer cell line Panc02-H7 was associated with a low iNOS expression [44] and exogenous NO addition was able to reduce the proliferation and invasion [44]. Studies also note that iNOS affects the tumor microenvironment such that tumors escape the immune response [45], However, ectopic or orthotopic xenograft mouse models injected with iNOS transfectedpancreatic cancer cells did not form tumors or metastases likely due to NO-mediated apoptosis [46]. In our study, considering that the source of ROS production is not known, tumor microenvironment may be suspected to contribute to the tumor growth inhibition observed as iNOS expression is increased in the tumors. Desmoplastic stroma makes up 80% of tumor mass in pancreatic ductal adenocarcinoma [47], therefore, the interplay of tumor and its microenvironment components by NOSH-aspirin would be of interest for future investigation.
A molecular target that is relevant to inflammatory processes and is inhibited by NSAIDs is cyclo oxygenase (COX). When we compared the GI toxicities of NOSH-aspirin to that of aspirin [21], we showed that NOSH-aspirin was significantly less toxic than aspirin and that in the gastric mucosa, both aspirin and NOSH-aspirin reduced PGE2 content indicating significant COX inhibition. This is inline with another report from us indicating preferential dose-dependent inhibition of pure ovine COX-1 vs COX-2 enzymatic activity by NOSH-aspirin [20]. We also showed that NOSH-aspirin inhibits platelet aggregation, a COX-1 mediated effect; effectively reduces LPS-induced fever, a COX-2 mediated effect, and has excellent anti-inflammatory properties using the carrageenan paw oedema model representing a COX-2 mediated effect [21]. Using this model, we also showed that NOSH-aspirin dose dependently inhibited COX-1 and COX-2 expression [19]. These results underscore the importance of being cautious when applying in vitro data to any in vivo settings. In the present study the actions of NOSH-aspirin appear to be COX independent since the cell lines that we used, BxPc-3 that expresses both COX-1 and COX-2 and MIA PaCa-2, which does not express COX at all [48], responded similarly to the actions of NOSH-aspirin.
Pancreatic cancers are associated with activated FoxM1 signaling pathway and its downregulation produces anti-cancer effects in pancreatic cancer cell lines [49]. Our study demonstrated that NOSH-aspirin treatement lead to FoxM1 downregulation. FoxM1 has been implicated in regulation of proliferation [35] and in cell cycle control. For example it was found that G2/M arrest occurred if FoxM1 was repressed [50].
The current understanding is that p53 executes its pancreatic tumor-suppressive activity at least in part via downregulation of FoxM1 expression [51]. In tumors where p53 is inactivated, it is believed that this in part, may lead to FoxM1 overexpression [8, 50]. In other studies, although FoxM1 repression and its reduced protein levels has been associated with G2 arrest [50], however, here we found that NOSH-aspirin caused a large accumulation of the cells in the G1 phase and G2 arrest was not strongly evident.
Examining other pre-clinical studies on pancreatic cancer models and our current study, it is not possible to make a straight comparison. In this study, p53 defective xenograft pancreatic tumors showed strong reductions in tumor growth and mass. Yet it is important to note that in mice with p53-defective tumors, a targeted approach of EGFR downregulation did not inhibit tumor development [52]. NOSH-aspirin reduces tumors and inhibits cancer cells through mechanisms that converge on caspases and apoptosis, and which maybe in part ROS- mediated and NF-κB inhibition-mediated. ROS production appears to be caspase-dependent and a downstream event since inhibition of caspases completely blocked NOSH-aspirin-mediated ROS generation to basal levels of the cancer cells. It is known that caspase-induced ROS generation can contribute to the process of apoptosis [53]. In this regard, our overarching hypothesis, supported by in vitro and in vivo studies has been that NOSH-aspirin increases ROS within the cancer cells, shifting the balance towards apoptosis; of course these events may or may not be caspase-dependent. In our previous preliminary studies with breast cancer cells, we have shown that treatment of the cells with N-acetyl cysteine (NAC) partially reverses the effects of NOSH-aspirin on apoptosis [54].
Another aspect of our studies that leads to the conclusion that these effects/observations are COX-independent is the fact that our IC50s for cell growth inhibition in the two cell lines studied were very similar. MIA PaCa-2 cells are COX-null and BxPC-3 cells express both COX-1 and COX-2 [27]. COX-independent effects of NSAIDs are now well accepted [55].
Regarding the potency of NOSH-aspirin in terms of growth inhibition observed here, comparison of the IC50 values (aspirin/NOSH-aspirin) suggests that NOSH-aspirin was at least 80,000-fold more potent than aspirin. Such fold increases imply that the NO and H2S-related structural modifications of the aspirin molecule imparts a differential enhancement in potency. Currently we cannot explain the underlying mechanism(s) for the enhanced potency of NOSH-aspirin observed in these studies; however, our very preliminary data suggests that NO and H2S that are released from NOSH-aspirin react to form highly reactive polysulfides. Our earlier studies had indicated that both NO and H2S contribute towards the potency of the intact NOSH-aspirin molecule. We had shown that the biological activity of aspirin plus SNAP (S-Nitroso-N-acetylpenicillamine, which releases NO) plus ADT-OH (5-(4-hydroxyphenyl)-3H-1, 2-dithiole-3-thione, which releases H2S) was not the same as the biological activity of the intact NOSH-aspirin molecule [20]. Thus the sum of parts did not equal the whole. Other reports indicate that NO can react with H2S to produce HSNO (the smallest S-nitrosothiol) which is a highly reactive intermediate [56, 57]. Furthermore, NO and H2S signaling pathways appear to be intimately intertwined with mutual potentiation of biological responses [58].
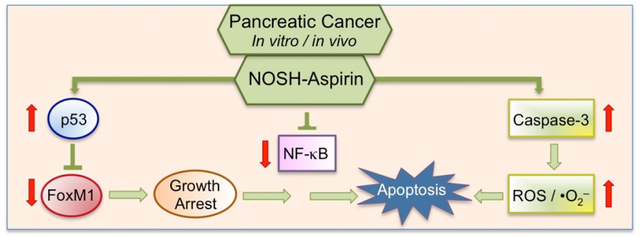
Overall, these studies demonstrate that NOSH-aspirin is inhibitory to human pancreatic tumor xenograft via combined effects of increases in ROS, iNOS, caspases, mutated p53 and downregulation of FoxM1. Collectively this study and our previous reports with H2S-releasing aspirin support our hypothesis and proposed working model that NOSH-aspirin induces ROS levels, induces p53, down-regulates FoxM1, which leads to growth arrest and apoptosis.
Acknowledment
This work was supported in part by NIH grant R24 DA018055. The funding agency had no role in the study design, collection, analysis and interpretation of data; in the writing of the manuscript; and in the decision to submit the manuscript for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors have nothing to disclose except for KK, who has an equity position in Avicenna Pharmaceuticals, Inc. the supplier of NOSH-aspirin used in these studies and to which NBS-1120 is licensed.
References
- [1].Siegel RL, Miller KD, Jemal A, Cancer Statistics, 2017, CA: a cancer journal for clinicians 67(1) (2017) 7–30. [DOI] [PubMed] [Google Scholar]
- [2].Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States, Cancer research 74(11) (2014) 2913–21. [DOI] [PubMed] [Google Scholar]
- [3].Von Hoff DD, What’s new in pancreatic cancer treatment pipeline?, Best practice & research. Clinical gastroenterology 20(2) (2006) 315–26. [DOI] [PubMed] [Google Scholar]
- [4].Garrido-Laguna I, Hidalgo M, Pancreatic cancer: from state-of-the-art treatments to promising novel therapies, Nature reviews. Clinical oncology 12(6) (2015) 319–34. [DOI] [PubMed] [Google Scholar]
- [5].Vogelstein B, Kinzler KW, Cancer genes and the pathways they control, Nature medicine 10(8) (2004) 789–99. [DOI] [PubMed] [Google Scholar]
- [6].Huang C, Du J, Xie K, FOXM1 and its oncogenic signaling in pancreatic cancer pathogenesis, Biochimica et biophysica acta 1845(2) (2014) 104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wierstra I, Alves J, FOXM1, a typical proliferation-associated transcription factor, Biological chemistry 388(12) (2007) 1257–74. [DOI] [PubMed] [Google Scholar]
- [8].Pandit B, Halasi M, Gartel AL, p53 negatively regulates expression of FoxM1, Cell cycle (Georgetown, Tex.) 8(20) (2009) 3425–7. [DOI] [PubMed] [Google Scholar]
- [9].Wang W, Abbruzzese JL, Evans DB, Larry L, Cleary KR, Chiao PJ, The nuclear factor-kappa B RelA transcription factor is constitutively activated in human pancreatic adenocarcinoma cells, Clinical cancer research : an official journal of the American Association for Cancer Research 5(1) (1999) 119–27. [PubMed] [Google Scholar]
- [10].Chandler NM, Canete JJ, Callery MP, Increased expression of NF-kappa B subunits in human pancreatic cancer cells, The Journal of surgical research 118(1) (2004) 9–14. [DOI] [PubMed] [Google Scholar]
- [11].Reczek CR, Chandel NS, ROS-dependent signal transduction, Current opinion in cell biology 33 (2015) 8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Reczek CR, Chandel NS, The Two Faces of Reactive Oxygen Species in Cancer, Annual Review of Cancer Biology 1 (2017) 79–98. [Google Scholar]
- [13].Donadelli M, Dalla Pozza E, Scupoli MT, Costanzo C, Scarpa A, Palmieri M, Intracellular zinc increase inhibits p53(−/−) pancreatic adenocarcinoma cell growth by ROS/AIF-mediated apoptosis, Biochimica et biophysica acta 1793(2) (2009) 273–80. [DOI] [PubMed] [Google Scholar]
- [14].Zhang L, Li J, Zong L, Chen X, Chen K, Jiang Z, Nan L, Li X, Li W, Shan T, Ma Q, Ma Z, Reactive Oxygen Species and Targeted Therapy for Pancreatic Cancer, Oxidative medicine and cellular longevity 2016 (2016) 1616781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Donadelli M, Dando I, Zaniboni T, Costanzo C, Dalla Pozza E, Scupoli MT, Scarpa A, Zappavigna S, Marra M, Abbruzzese A, Bifulco M, Caraglia M, Palmieri M, Gemcitabine/cannabinoid combination triggers autophagy in pancreatic cancer cells through a ROS-mediated mechanism, Cell death & disease 2 (2011) e152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kashfi K, Anti-inflammatory agents as cancer therapeutics, Advances in pharmacology (San Diego, Calif.) 57 (2009) 31–89. [DOI] [PubMed] [Google Scholar]
- [17].Wallace JL, Miller MJ, Nitric oxide in mucosal defense: a little goes a long way, Gastroenterology 119(2) (2000) 512–20. [DOI] [PubMed] [Google Scholar]
- [18].Fiorucci S, Prevention of nonsteroidal anti-inflammatory drug-induced ulcer: looking to the future, Gastroenterology clinics of North America 38(2) (2009) 315–32. [DOI] [PubMed] [Google Scholar]
- [19].Kodela R, Chattopadhyay M, Kashfi K, NOSH-Aspirin: A Novel Nitric Oxide-Hydrogen Sulfide-Releasing Hybrid: A New Class of Anti-inflammatory Pharmaceuticals, ACS medicinal chemistry letters 3(3) (2012) 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chattopadhyay M, Kodela R, Olson KR, Kashfi K, NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid is a potent inhibitor of colon cancer cell growth in vitro and in a xenograft mouse model, Biochem Biophys Res Commun 419(3) (2012) 523–8. [DOI] [PubMed] [Google Scholar]
- [21].Kodela R, Chattopadhyay M, Velazquez-Martinez CA, Kashfi K, NOSH-aspirin (NBS-1120), a novel nitric oxide- and hydrogen sulfide-releasing hybrid has enhanced chemo-preventive properties compared to aspirin, is gastrointestinal safe with all the classic therapeutic indications, Biochemical pharmacology 98(4) (2015) 564–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chattopadhyay M, Kodela R, Nath N, Dastagirzada YM, Velazquez-Martinez CA, Boring D, Kashfi K, Hydrogen sulfide-releasing NSAIDs inhibit the growth of human cancer cells: a general property and evidence of a tissue type-independent effect, Biochemical pharmacology 83(6) (2012) 715–22. [DOI] [PubMed] [Google Scholar]
- [23].Chattopadhyay M, Kodela R, Nath N, Barsegian A, Boring D, Kashfi K, Hydrogen sulfide-releasing aspirin suppresses NF-kappaB signaling in estrogen receptor negative breast cancer cells in vitro and in vivo, Biochemical pharmacology 83(6) (2012) 723–32. [DOI] [PubMed] [Google Scholar]
- [24].Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T, Requirement for generation of H2O2 for platelet-derived growth factor signal transduction, Science 270(5234) (1995) 296–9. [DOI] [PubMed] [Google Scholar]
- [25].Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS, Selective fluorescent imaging of superoxide in vivo using ethidium-based probes, Proc Natl Acad Sci U S A 103(41) (2006) 15038–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ouyang N, Williams JL, Tsioulias GJ, Gao J, Iatropoulos MJ, Kopelovich L, Kashfi K, Rigas B, Nitric oxide-donating aspirin prevents pancreatic cancer in a hamster tumor model, Cancer research 66(8) (2006) 4503–11. [DOI] [PubMed] [Google Scholar]
- [27].Kashfi K, Ryan Y, Qiao LL, Williams JL, Chen J, Del Soldato P, Traganos F, Rigas B, Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect, The Journal of pharmacology and experimental therapeutics 303(3) (2002) 1273–82. [DOI] [PubMed] [Google Scholar]
- [28].Bass DA, Parce JW, Dechatelet LR, Szejda P, Seeds MC, Thomas M, Flow cytometric studies of oxidative product formation by neutrophils: a graded response to membrane stimulation, J Immunol 130(4) (1983) 1910–7. [PubMed] [Google Scholar]
- [29].LeBel CP, Ischiropoulos H, Bondy SC, Evaluation of the probe 2’,7’-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress, Chem Res Toxicol 5(2) (1992) 227–31. [DOI] [PubMed] [Google Scholar]
- [30].Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT, Generation of superoxide in cardiomyocytes during ischemia before reperfusion, Am J Physiol 277(6 Pt 2) (1999) H2240–6. [DOI] [PubMed] [Google Scholar]
- [31].Gormally MV, Dexheimer TS, Marsico G, Sanders DA, Lowe C, Matak-Vinkovic D, Michael S, Jadhav A, Rai G, Maloney DJ, Simeonov A, Balasubramanian S, Suppression of the FOXM1 transcriptional programme via novel small molecule inhibition, Nature communications 5 (2014) 5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wonsey DR, Follettie MT, Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe, Cancer research 65(12) (2005) 5181–9. [DOI] [PubMed] [Google Scholar]
- [33].Costa RH, FoxM1 dances with mitosis, Nature cell biology 7(2) (2005) 108–10. [DOI] [PubMed] [Google Scholar]
- [34].Koo CY, Muir KW, Lam EW, FOXM1: From cancer initiation to progression and treatment, Biochimica et biophysica acta 1819(1) (2012) 28–37. [DOI] [PubMed] [Google Scholar]
- [35].Laoukili J, Kooistra MR, Bras A, Kauw J, Kerkhoven RM, Morrison A, Clevers H, Medema RH, FoxM1 is required for execution of the mitotic programme and chromosome stability, Nature cell biology 7(2) (2005) 126–36. [DOI] [PubMed] [Google Scholar]
- [36].Laoukili J, Stahl M, Medema RH, FoxM1: at the crossroads of ageing and cancer, Biochimica et biophysica acta 1775(1) (2007) 92–102. [DOI] [PubMed] [Google Scholar]
- [37].Wood LD, Hruban RH, Genomic landscapes of pancreatic neoplasia, J Pathol Transl Med 49(1) (2015) 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sipos B, Moser S, Kalthoff H, Torok V, Lohr M, Kloppel G, A comprehensive characterization of pancreatic ductal carcinoma cell lines: towards the establishment of an in vitro research platform, Virchows Archiv : an international journal of pathology 442(5) (2003) 444–52. [DOI] [PubMed] [Google Scholar]
- [39].Nath N, Labaze G, Rigas B, Kashfi K, NO-donating aspirin inhibits the growth of leukemic Jurkat cells and modulates beta-catenin expression, Biochem Biophys Res Commun 326(1) (2005) 93–9. [DOI] [PubMed] [Google Scholar]
- [40].Chattopadhyay M, Nath N, Kodela R, Sobocki T, Metkar S, Gan ZY, Kashfi K, Hydrogen sulfide-releasing aspirin inhibits the growth of leukemic Jurkat cells and modulates beta-catenin expression, Leukemia research 37(10) (2013) 1302–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Banerjee K, Ganguly A, Chakraborty P, Sarkar A, Singh S, Chatterjee M, Bhattacharya S, Choudhuri SK, ROS and RNS induced apoptosis through p53 and iNOS mediated pathway by a dibasic hydroxamic acid molecule in leukemia cells, European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences 52 (2014) 146–64. [DOI] [PubMed] [Google Scholar]
- [42].Wang B, Shi Q, Abbruzzese JL, Xiong Q, Le X, Xie K, A novel, clinically relevant animal model of metastatic pancreatic adenocarcinoma biology and therapy, International journal of pancreatology : official journal of the International Association of Pancreatology 29(1) (2001) 37–46. [DOI] [PubMed] [Google Scholar]
- [43].Wang J, He P, Gaida M, Yang S, Schetter AJ, Gaedcke J, Ghadimi BM, Ried T, Yfantis H, Lee D, Weiss JM, Stauffer J, Hanna N, Alexander HR, Hussain SP, Inducible nitric oxide synthase enhances disease aggressiveness in pancreatic cancer, Oncotarget 7(33) (2016) 52993–53004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sugita H, Kaneki M, Furuhashi S, Hirota M, Takamori H, Baba H, Nitric oxide inhibits the proliferation and invasion of pancreatic cancer cells through degradation of insulin receptor substrate-1 protein, Molecular cancer research : MCR 8(8) (2010) 1152–63. [DOI] [PubMed] [Google Scholar]
- [45].Wang L, Xie K, Nitric oxide and pancreatic cancer pathogenesis, prevention, and treatment, Current pharmaceutical design 16(4) (2010) 421–7. [DOI] [PubMed] [Google Scholar]
- [46].Vannini F, Kashfi K, Nath N, The dual role of iNOS in cancer, Redox biology 6 (2015) 334–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang Y, Lin J, Cui J, Han T, Jiao F, Meng Z, Wang L, Prognostic value and clinicopathological features of PD-1/PD-L1 expression with mismatch repair status and desmoplastic stroma in Chinese patients with pancreatic cancer, Oncotarget 8(6) (2017) 9354–9365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kashfi K, Rayyan Y, Qiao LL, Williams JL, Chen J, Del Soldato P, Traganos F, Rigas B, Ryann Y, Nitric oxide-donating nonsteroidal anti-inflammatory drugs inhibit the growth of various cultured human cancer cells: evidence of a tissue type-independent effect, The Journal of pharmacology and experimental therapeutics 303(3) (2002) 1273–82. [DOI] [PubMed] [Google Scholar]
- [49].Wang Z, Banerjee S, Kong D, Li Y, Sarkar FH, Down-regulation of Forkhead Box M1 transcription factor leads to the inhibition of invasion and angiogenesis of pancreatic cancer cells, Cancer research 67(17) (2007) 8293–300. [DOI] [PubMed] [Google Scholar]
- [50].Barsotti AM, Prives C, Pro-proliferative FoxM1 is a target of p53-mediated repression, Oncogene 28(48) (2009) 4295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Spurgers KB, Gold DL, Coombes KR, Bohnenstiehl NL, Mullins B, Meyn RE, Logothetis CJ, McDonnell TJ, Identification of cell cycle regulatory genes as principal targets of p53-mediated transcriptional repression, The Journal of biological chemistry 281(35) (2006) 25134–42. [DOI] [PubMed] [Google Scholar]
- [52].Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M, EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma, Cancer cell 22(3) (2012) 318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Tan S, Sagara Y, Liu Y, Maher P, Schubert D, The regulation of reactive oxygen species production during programmed cell death, J Cell Biol 141(6) (1998) 1423–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Nesbitt DE, Chattopadhyay M, Vannini F, Le T-T, Kodela C,R, Nath N, Kashfi K, NOSH-aspirin inhibits breast cancer cell growth: an effect modulated through reactive oxygen species and independent of the ER status., Proceedings of the 104th Annual Meeting of the American Association for Cancer Research; 2013 Apr 6–10; Washington, DC. Philadelphia (PA): AACR; Cancer Res 2013;73(8 Suppl):Abstract nr 4793. doi: 10.1158/1538-7445.AM2013-4793, Washington, DC, 2013, Apr 6–10. [DOI] [Google Scholar]
- [55].Kashfi K, Rigas B, Non-COX-2 targets and cancer: expanding the molecular target repertoire of chemoprevention, Biochemical pharmacology 70(7) (2005) 969–86. [DOI] [PubMed] [Google Scholar]
- [56].Cortese-Krott MM, Fernandez BO, Kelm M, Butler AR, Feelisch M, On the chemical biology of the nitrite/sulfide interaction, Nitric oxide : biology and chemistry 46 (2015) 14–24. [DOI] [PubMed] [Google Scholar]
- [57].Filipovic MR, Miljkovic J, Nauser T, Royzen M, Klos K, Shubina T, Koppenol WH, Lippard SJ, Ivanovic-Burmazovic I, Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols, Journal of the American Chemical Society 134(29) (2012) 12016–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Cortese-Krott MM, Kuhnle GG, Dyson A, Fernandez BO, Grman M, DuMond JF, Barrow MP, McLeod G, Nakagawa H, Ondrias K, Nagy P, King SB, Saavedra JE, Keefer LK, Singer M, Kelm M, Butler AR, Feelisch M, Key bioactive reaction products of the NO/H2S interaction are S/N-hybrid species, polysulfides, and nitroxyl, Proc Natl Acad Sci U S A 112(34) (2015) E4651–60. [DOI] [PMC free article] [PubMed] [Google Scholar]