

Abstract
Olanzapine is effective to treat for schizophrenia and other mood disorders, but limited by side effects such as weight gain, dyslipidemia, and liver injury. Obesity in the US is at epidemic levels, and is a significant risk factor for drug-induced liver injury. Obesity incidence in the psychiatric population is even higher than in the US population as a whole. The purpose of this study was to test the hypothesis that obesity worsens olanzapine-induced hepatic injury, and to investigate the potential protective effects of sulforaphane. 8-week old female C57BL/6 mice were fed either a high-fat or low-fat control diet (HFD and LFD). Mice also received either olanzapine (8 mg/kg/d) or vehicle by osmotic minipump for 4 weeks. A subset of mice in the HFD + olanzapine group was administered sulforaphane, a prototypical Nrf2 inducer (90 mg/kg/d). Olanzapine alone increased body weight, without a commensurate increase in food consumption. Olanzapine also caused hepatic steatosis and injury. Combining olanzapine and HFD caused further dysregulation of glucose and lipid metabolism. Liver damage from concurrent HFD and olanzapine was worse than liver damage from high-fat diet or olanzapine alone. Sulforaphane alleviated many metabolic side effects of olanzapine and HFD. Taken together, these data show that olanzapine dysregulates glucose and lipid metabolism and exacerbates hepatic changes caused by eating a HFD. Activation of the intrinsic antioxidant defense pathway with sulforaphane can partially prevent these effects of olanzapine and may represent a useful strategy to protect against liver injury.
Keywords: olanzapine, liver, NAFLD, HFD, oxidative stress, sulforaphane
1.0. Introduction
Schizophrenia is a severe chronic mental illness that affects between ~0.3 and ~0.6% of the US population [1]. Despite this relatively low prevalence, the annual total economic burden of schizophrenia is as high as $155.7 billion/yr in the US [2], a burden on par with societal costs of alcohol use disorders [AUD; prevalence 1:10; [3]]. Olanzapine (OLZ) is a lead second-generation (i.e., ‘atypical’) antipsychotic drug (APD) that is effective in treating schizophrenia and other mood disorders. Second-generation APDs are more commonly prescribed today because they limit the toxicities of first generation antipsychotics, such as tardive dyskinesia. Indeed, the lack of these severe side effects has expanded the off-label use of this class of drugs for indications such as dementia and treatment-resistant anxiety disorders [4]. In 2008, over 45 million prescriptions of APDs were written in the US alone, which emphasizes the increase in off-label use of this family of drugs in the US [5].
The prevalence of obesity in the United States is increasing at an alarming rate and will have significant impact on the US population in the years to come [6]. In this context, metabolic syndrome (abdominal obesity, dyslipidemia, high blood pressure, and insulin resistance) is considered the key factor impacting overall health [7]. Alarmingly, in the US National Health and Nutrition Examination Survey (NHANES), more than 1 in 3 adults have metabolic syndrome [8]. The major side effect of OLZ that impacts compliance and health is significant weight gain. The incidence rate is as high as 50% in patients taking OLZ for more than 4 weeks [9, 10]. Moreover, even within an increasingly overweight general population, obesity disproportionately affects individuals with severe mental illnesses [11]; life expectancy is shortened in this patient cohort by 15–25 years, largely owing to obesity and its sequelae [12]. The effectiveness of traditional drugs used to treat insulin resistance/metabolic syndrome [thiazolidinediones (TZD) and metformin] has been modest in patients taking OLZ [13, 14]. It is therefore necessary to develop strategies that prevent, minimize, or reverse the adverse metabolic effects that occur during OLZ treatment, especially for an already at risk population [15].
Work by this group and others has shown that OLZ administration to lean mice causes obesity, dyslipidemia and metabolic syndrome [16]. Although obesity rates are high in the general population and enriched in the psychiatric population, most studies to date have not investigated the potential of an interaction between obesity and OLZ exposure. This consideration is important, given that metabolic syndrome and its sequelae involve multiple hits, often with altered production of reactive oxygen species (ROS) at the center [17]. The purpose of the current study was to therefore investigate the potential interaction between an obesogenic high fat diet (HFD) and OLZ exposure in mice; the potential therapeutic effect of sulforaphane, an electrophilic activator of Nrf2-mediated antioxidant response, was also investigated.
Oxidative stress it is widely hypothesized to be a common mechanism in the initiation or progression of many diseases, including disorders of metabolism. Manifestations of oxidative stress such as lipid peroxidation are repeatedly observed in conjunction with metabolic diseases [18]. There is some evidence that oxidative stress may be a downstream effect of OLZ administration (e.g., [19]). In contrast to these findings, high-dose vitamin E did not improve insulin resistance in OLZ-medicated adults in a small clinical trial conducted [20]. However, direct antioxidants (e.g., vitamin E) have generally met with limited success in human patients [21]. In contrast, indirect agents such as sulforaphane (SFN) have neither reducing nor oxidizing capability, and work by inducing Nrf2, thereby enhancing the cell’s own antioxidant defense [18]. A secondary purpose of this study was to therefore test the effectiveness of SFN as an interventive strategy under these conditions.
2.0. Materials and Methods
2.1. Animals and treatments.
Female C57BL/6J mice (8 weeks old) were purchased from Jackson Laboratory (Bar Harbor, ME). In preliminary studies, no sexual dimorphism was observed in the response to OLZ in this strain of mice. Females were employed for continuity of the work from previous publications (e.g., [16]). Mice were housed in a pathogen-free barrier facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, and procedures were approved by the University of Louisville Institutional Animal Care and Use Committee. Food and tap water were allowed ad libitum. One week prior to the initiation of the experiment, animals were switched from standard chow to purified Western diet, containing 42% Kcal fat (milkfat), 42.7% Kcal carbohydrate (sucrose) and 15.2% Kcal protein (TD.88137; Envigo, Madison, WI) or purified low-fat control diet (TD.06416; Envigo, Madison, WI). Olanzapine (8 mg/kg/d; [22]) or vehicle (saline) was given s.c. via osmotic minipumps (Alzet, Cupertino, CA) for 28 days, as described previously [16]. To avoid concerns of OLZ degradation [23], pumps were replaced after two weeks. All surgeries were performed under isoflurane anesthesia, and all efforts were made to minimize suffering. Additional animals received sulforaphane (SFN; 90 mg/kg/d) by gavage [24], or tap water vehicle starting 1 week prior to OLZ administration. Animals were weighed on a weekly basis and food consumption was recorded twice per week. After 28 days, the mice were anesthetized with a mixture of ketamine and xylazine (100 and 15 mg/kg i.m.) and minipumps were removed. Body composition was assessed by dual-energy x-ray absorptiometry (DEXA) using a Lunar PIXImus densitometer (Lunar Corp., Madison, WI). Blood was collected from the vena cava just prior to sacrifice by exsanguination, and citrated plasma was stored at −80 °C for further analysis. Portions of liver tissue were frozen immediately in liquid nitrogen, fixed in 10% neutral buffered formalin, or frozen-fixed for subsequent sectioning and mounting on microscope slides. Fat pads were weighed and collected for later analysis.
2.2. Oral glucose tolerance test (OGTT)
Glucose tolerance was evaluated at 25 days of OLZ exposure using an OGTT method described by Andrikopoulos et al [25]. Mice were transferred to cages that had been cleared of food and bedding and fasted for 6 hours. Blood was sampled from the tail vein immediately after fasting, and then 15, 30, 60, 90 and 120 minutes after oral administration of 2 g/kg D-(+)-glucose (Sigma, St. Louis, MO) solution. Glucose concentrations were measured using an Accu-Chek® Aviva Plus glucometer and test strips (Roche Diagnostics Corp., Indianapolis, IN).
2.3. Glycogen determinations
To determine hepatic glycogen content, mouse livers (50–100mg) were weighed and placed immediately in 30% KOH (500 μl), digested at 100°C for 20 min, then cooled to room temperature. To the cooled digest, 95% EtOH (625 μl) was added and allowed to stand overnight at room temperature. The samples were then centrifuged at 16000 × g (4°C) for 15 min. The resultant pellet was resuspended in 1ml water. Hepatic glycogen content was then determined with anthrone reagent essentially as described by Seifter et al [26]. Glycogen content was calculated from glucose stand curve and reported as μg/g tissue.
2.4. Biochemical analyses and histology.
Plasma levels of aminotransferases (ALT and AST) were determined using standard kits (Thermo Scientific, Middleton, VA). Paraffin-embedded sections of liver were stained with hematoxylin and eosin (H&E) to assess overall hepatic structure, and periodic acid-Schiff reagent (PAS) to detect glycogen. Frozen sections of liver were stained with Oil Red O to detect neutral lipids, and counterstained with hematoxylin. For measurement of hepatic neutrophil infiltration, the naphthol AS-D chloroacetate esterase (CAE) kit (Sigma, St. Louis, MO) was used according to the manufacturer’s instructions. CAE-positive cells were quantitated by counting (positive cells per 1000 hepatocytes) in randomly selected fields [27]. Staining was quantitated by image analysis as described previously [28].
2.5. Lipid peroxidation
Adducts of 4-hydroxynonenal, an α,β-unsaturated hydroxyalkenal produced by lipid peroxidation, were detected with a modified version of the previously described immunohistochemical techniques [29, 30]. Specifically, tissue sections were stained using a rabbit polyclonal 4-HNE antibody (Alpha Diagnostic, San Antonio, Texas) and a kit based on a routine biotin-streptavidin-peroxidase staining technique (VECTASTAIN ABC kit, Vector Laboratories, Burlingame, CA). Once the antibody-biotin-peroxidase complex was formed, diaminobenzidine (ImmPACT™ DAB peroxidase substrate, Vector Laboratories) was added as the peroxidase substrate. After the immunostaining procedure, tissue sections were counterstained with hematoxylin and mounted. Staining was quantitated as the percentage of brown labeling (i.e., DAB) of the field area minus the area of the acellular spaces.
2.6. Lipid determinations.
Hepatic lipids [non-esterified fatty acids (NEFA) and triglycerides (TG)] were extracted by an aqueous solution of chloroform and methanol as described by Bligh and Dyer [31], with minor modifications [32]. Briefly, liver samples were pulverized in a mortar filled with liquid nitrogen. Tissue lipids were extracted with methanol:chloroform (1:2), dried under compressed nitrogen gas, and resuspended in 5% fat-free bovine serum albumin. NEFA and TG were determined colorimetrically using standard kits (Roche, Penzberg, Germany and Thermo Scientific, Middletown, VA). Values were normalized to protein in homogenate prior to extraction, as determined by the Bradford assay (Bio-Rad Laboratories, Hercules, CA).
2.7. Statistical analyses.
Quantitative data are reported as either means ± SEM (n=6–8), or as box and whisker plots, which show the median (thick line), interquartile range (box) and range (whiskers). Group sizes were based on previous studies and preliminary data, to get an 85% power for detecting a difference of at least 20% with a P<0.05 between the experimental groups for main effects (e.g., AST and ALT). 2-way ANOVA, repeated measures 2-way ANOVA (for OGTT) with Tukey’s post hoc test or the Mann–Whitney rank sum test was used for the determination of statistical significance among treatment groups, as appropriate. The effect of SFN on changes caused by HFD and OLZ were determined by 1-way ANOVA. A p value less than 0.05 was selected before the study as the level of significance.
3.0. Results
3.1. High-fat diet (HFD) increases food consumption and exacerbates weight gain caused by OLZ
Mice were switched to purified diets one week before OLZ administration, as described in Materials and Methods (see also Figure 1A). All animals gained weight over the course of the study and no mortality was observed in any group. Mice fed HFD consumed more food and gained more weight on average than mice fed LFD (Figure 1B). OLZ did not have any effect on food consumption, but contributed to body weight gain in the LFD-fed group (Figure 1B), as observed previously [16]. OLZ and HFD both also increased the ratio of gonadal fat pad mass to total body mass (Figure 1C). OLZ and HFD promoted overall body fat accumulation: DEXA scans revealed increases in soft matter after four weeks of HFD and/or OLZ (representative images, panel D).
Figure 1: OLZ exacerbates weight gain caused by high-fat diet (HFD).
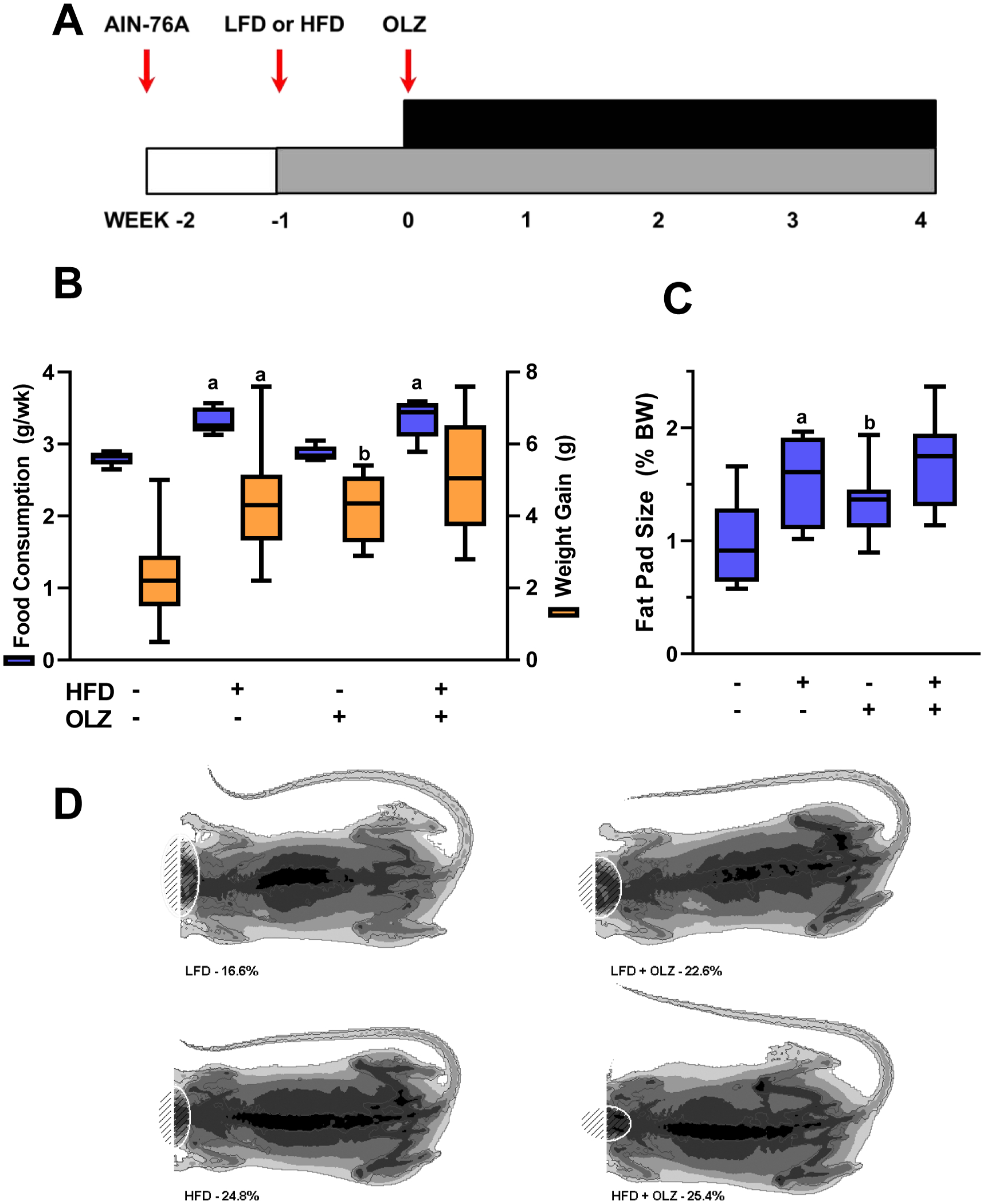
Mice were fed HFD or LFD and administered OLZ or vehicle by osmotic minipumps for four weeks (Timeline, panel A). Food consumption and weight gain over the course of the four week study is shown in panel B. Gonadal fat pads were removed at sacrifice and percentage of gonadal fat mass to total body mass was determined (panel C). Body composition was measured by dual energy x-ray absorptiometry; representative scans are depicted (panel D). aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
3.2. HFD aggravates OLZ-induced liver injury
Previous studies have shown that feeding mice a diet enriched in triglycerides and cholesterol (i.e., the “Western diet”) damages the liver and may contribute to the onset of NAFLD [33]. Studies from this laboratory have also linked OLZ administration to early indices of liver damage [16]. Liver injury was indeed observed in OLZ-treated animals, and this liver injury was aggravated by concurrent HFD feeding (Figure 2). As in previous work [16], four weeks of OLZ increased fat accumulation across all regions of the liver, as shown by H&E staining (Figure 2A, upper). Co-administration of HFD further increased the fat accumulation resulting from OLZ alone. Expression of key genes involved in hepatic inflammation (e.g., TNFα, IL-6 and iNOS) were not significantly altered by OLZ and/or HFD under these conditions (not shown). However, neutrophil infiltration in liver was increased by HFD or OLZ alone, and more so by HFD and OLZ together (Figure 2A, lower and Figure 2B). OLZ and HFD separately increased the liver weight to body weight ratio (Figure 2B), but the combination of OLZ and HFD produced no additional change. OLZ administration elevated circulating hepatic transaminases, a common indicator of hepatocellular injury (Figure 2B). HFD alone had no effect on transaminase levels; however, in conjunction with OLZ, HFD raised ALT and AST to levels greater than OLZ alone (Figure 2B).
Fig. 2: OLZ aggravates HFD-induced liver injury.
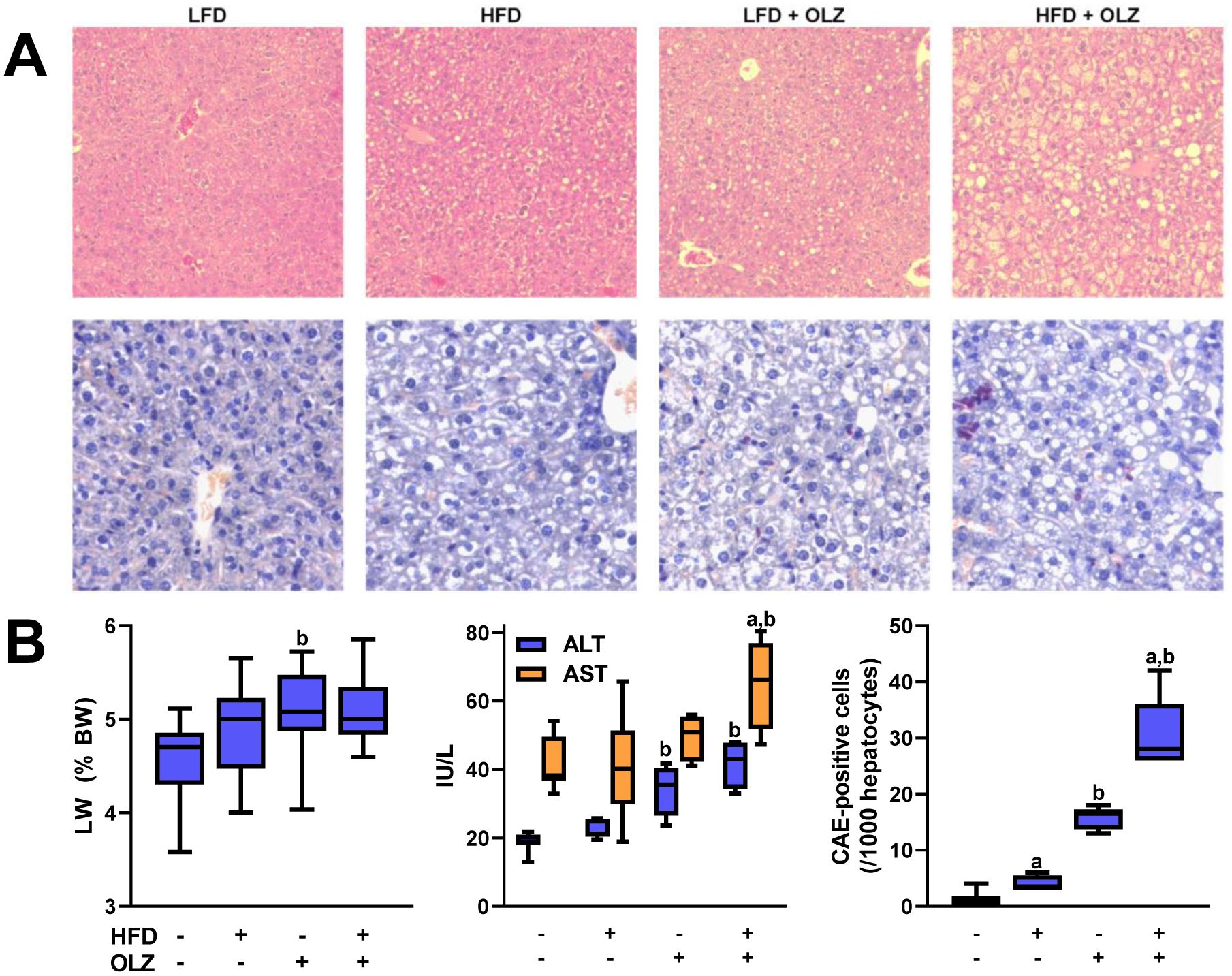
Livers were stained with hematoxylin and eosin to demonstrate general hepatocellular morphology (panel A; upper). Neutrophil infiltration is shown by staining with chloroacetate esterase (panel A, lower, representative pictures; panel B, quantitation). Liver weight is expressed as a percentage of total body weight (panel B, left). Transaminase levels were measured in plasma taken at sacrifice (panels B, middle). aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
3.3. OLZ and HFD promote hepatic lipid storage
Hematoxylin and eosin staining had demonstrated steatosis with OLZ and HFD, whether administered separately or in combination (see Figure 2A). Therefore, frozen liver sections were stained with Oil Red O (Figure 3A) and staining intensity (i.e., extent of lipid accumulation) was quantitated (Panel 3B, left). Minimal staining intensity was seen with LFD feeding, indicating very little hepatic fat accumulation. OLZ administration caused a 1.5-fold increase in micro-and macro-vesicular steatosis, as determined by image analysis. HFD by itself caused a 2-fold increase in steatosis. When OLZ and HFD were administered together, steatosis increased 2.5-fold compared to LFD alone. Hepatic lipid extraction elaborated upon these Oil Red O data: OLZ and HFD separately doubled triglyceride (TG) content in liver, and together increased TG 4-fold (Figure 3B, middle). Hepatic free fatty acids (NEFA) were elevated with HFD alone, but remained stable in all other treatment groups.
Fig. 3: OLZ and HFD promote hepatic lipid storage.
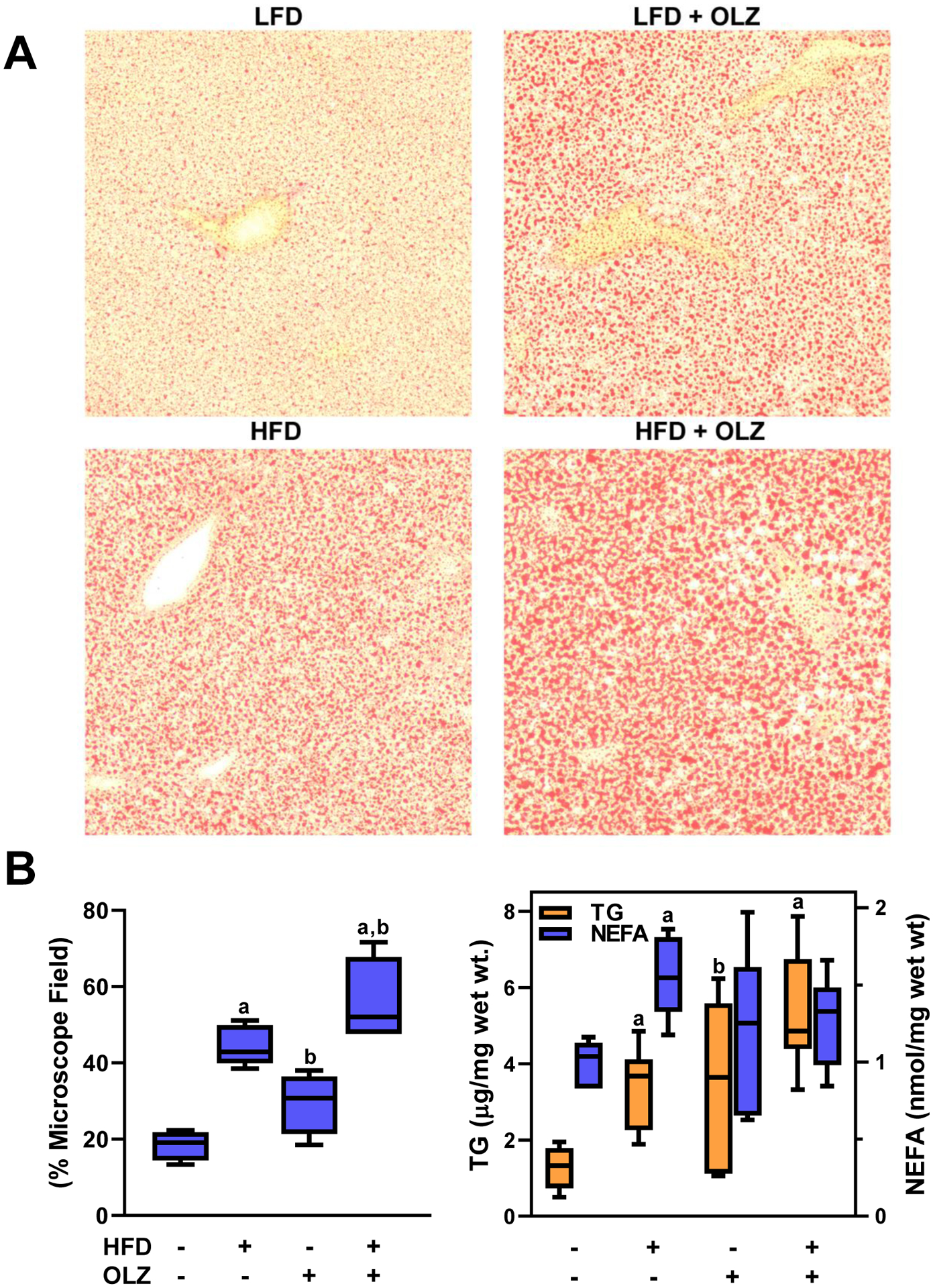
Frozen sections of liver were stained with Oil Red O as described in Materials and Methods (panel A, representative pictures) and staining was quantitated in all samples (panel B, left). Lipids (TG and NEFA) were extracted from pulverized liver samples and determined by colorimetric assay (panel B, right). Expression of key genes involved in de novo lipid genesis and catabolism were determined by real-time rtPCR (Panel B, right). aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
As observed previously [16], OLZ exposure impacted genes critical for the synthesis and catabolism of lipids. Carnitine palmitoyltransferase 1 (Cpt1), which encodes the rate-limiting enzyme in fatty acid β-oxidation, was unchanged (Figure 4A). In contrast, 4 weeks of OLZ administration significantly downregulated the expression of a number of genes involved in lipid biosynthesis, including fatty acid synthase (Fasn), sterol regulatory element-binding protein (Srebf), and ATP citrate lyase (Acly) (Figure 4A). HFD alone did not impact the expression of any of these genes. The interaction between HFD and OLZ only impacted expression of Srebf, which was significantly decreased compared to OLZ alone (Figure 4A).
Fig. 4: Effects of OLZ and HFD on expression of key genes involved in lipid and glucose metabolism.
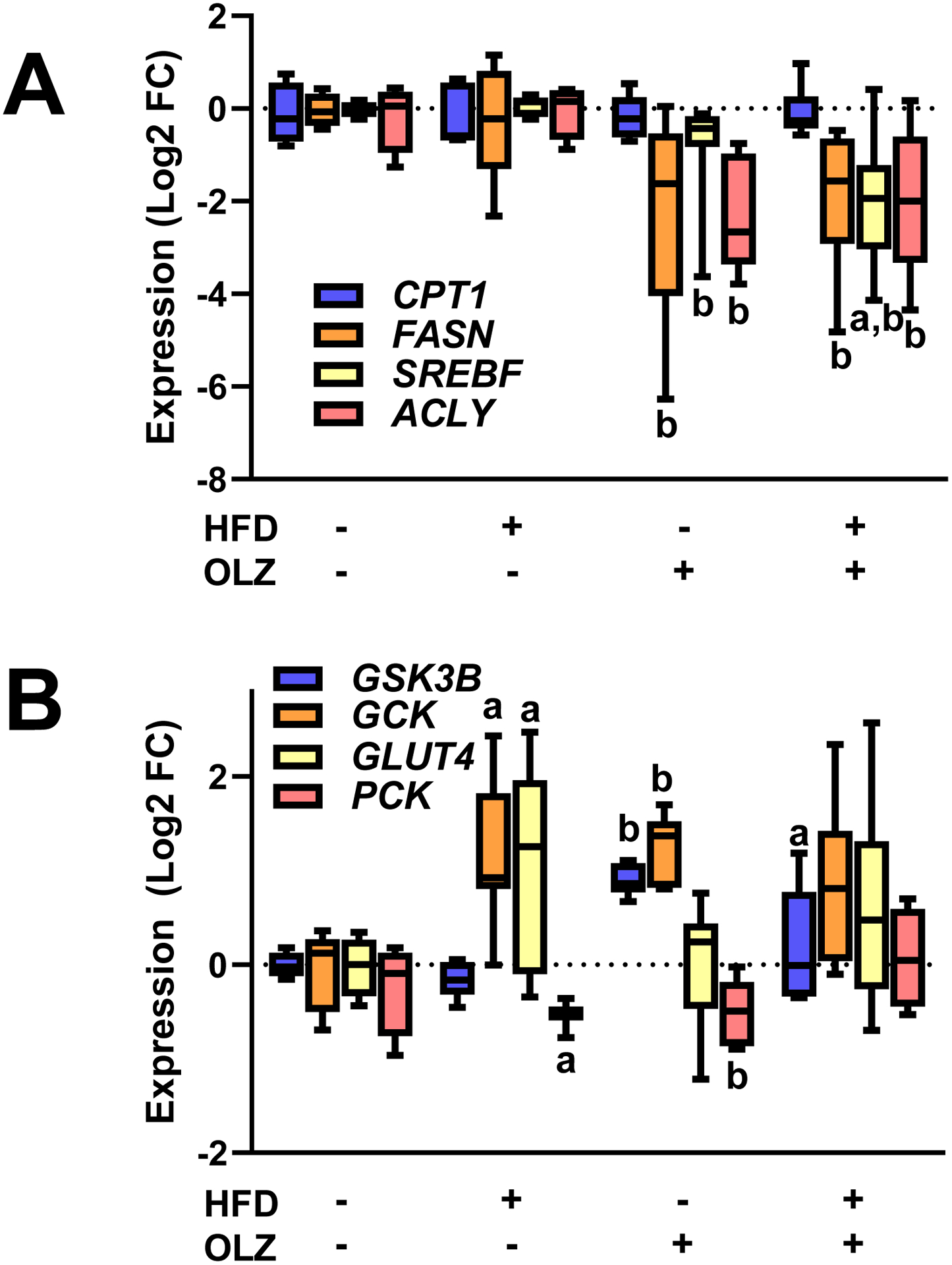
Expression of key genes involved in de novo lipid genesis and catabolism (Panel A) and glucose metabolism (Panel B) were determined by real-time rtPCR. aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
3.4. Effects of OLZ and HFD on glucose metabolism
In vivo glucose metabolism was measured by oral glucose tolerance test (OGTT) after 25 days of drug administration and LFD or HFD consumption (Figure 5A). Four weeks of OLZ and/or HFD did not significantly alter plasma glucose concentrations after a 6 hour fast. Following bolus glucose administration, however, HFD alone increased plasma glucose levels; the Area Under the Curve (AUC) was increased by 20% compared to LFD-fed controls in both HFD-and OLZ-exposed animals. Exposure to OLZ and/or HFD also influenced glucose kinetics. In mice given OLZ and HFD, either separately or in combination, peak plasma glucose was observed at 15 minutes in contrast to 30 minutes for LFD-fed controls. Interestingly, at the 30 minute time point, plasma glucose remained elevated in mice given HFD alone, but rapidly declined in mice given HFD and OLZ together.
Fig. 5: Effects of OLZ and HFD on glucose and glycogen metabolism.
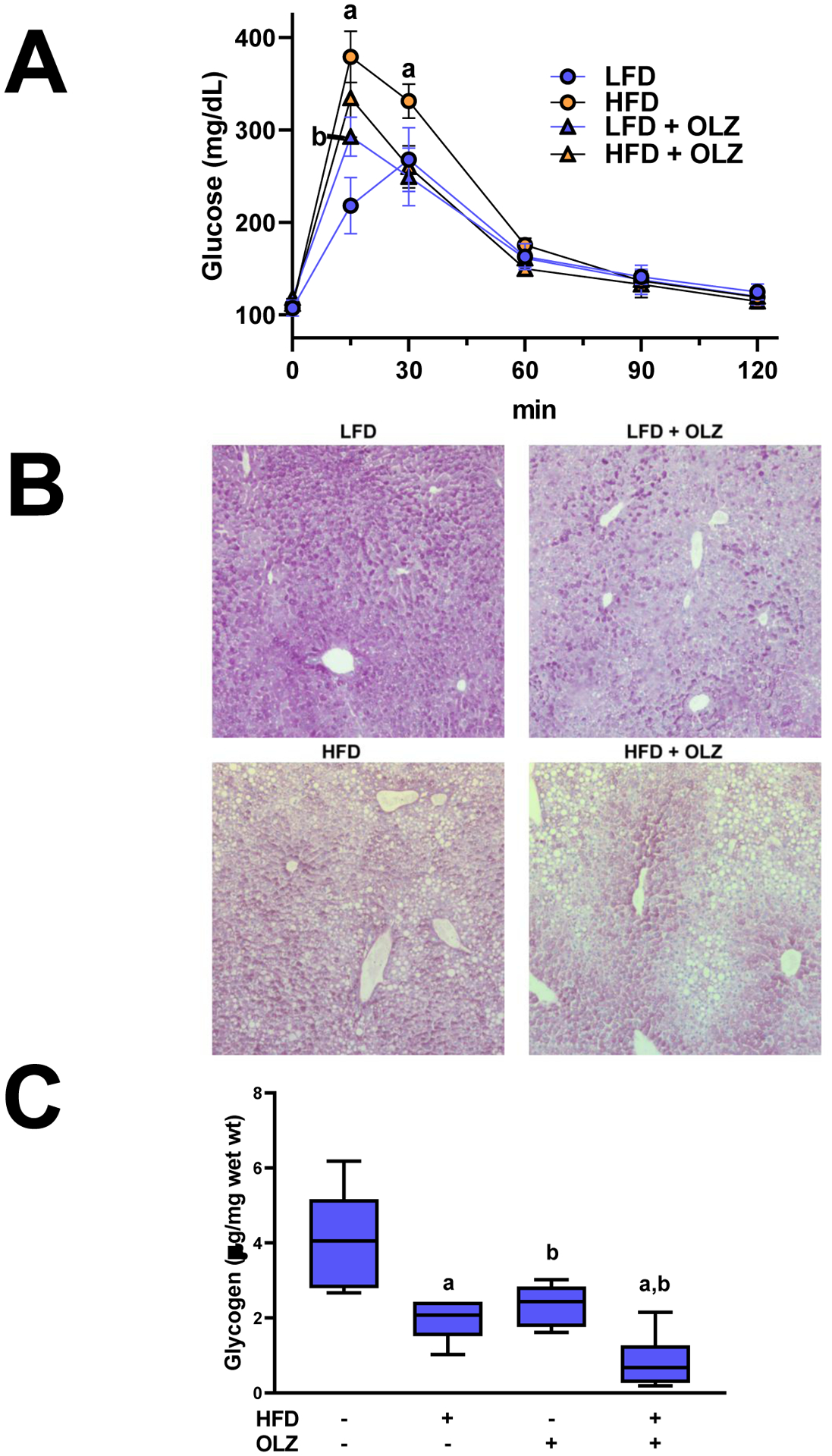
OGTT was performed after 25 days of HFD or LFD feeding and exposure to OLZ or vehicle (panel A). Liver samples fixed at sacrifice were stained with Periodic acid-Schiff reagent to identify glycogen (panel B). Glycogen was isolated from snap-frozen liver samples and concentrations were determined by colorimetric assay (panel B). aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
A previous study by this group showed that OLZ exposure attenuated glycogen storage in liver [16]. Here, the interaction between OLZ and HFD was investigated (Figure 5B and C). Periodic acid-Schiff (PAS) staining showed that OLZ alone and HFD alone decreased hepatic glycogen reserves (Figure 5B and C). Glycogen was further depleted by co-administration of OLZ and HFD. These effects correlated with an inhibition of the expression of the gluconeogenic gene, phosphoenolpyruvate carboxykinase 1 (Pck), and induction of glycogen synthase kinase-3β (Gsk3b), a protein that suppresses glycogen synthesis (Figure 4B).
3.5. Effects of HFD and OLZ on lipid peroxidation
To better determine the cause of aggravated liver injury with concurrent HFD and OLZ, liver sections were stained for 4-hydroxnonenal, a marker of lipid peroxidation (Figure 6A and B). Lipid peroxidation, as determined by 4-HNE staining, increased ~6-fold with OLZ alone and ~8-fold with HFD and OLZ in combination.
Fig. 6: Effects of OLZ on lipid peroxidation.

Formalin-fixed liver sections were used for immunohistochemical detection of 4-HNE adducts (panel A). Extent of staining was quantitated as described in Experimental Procedures (panel B). aP <0.05 compared to LFD; bP <0.05 compared to absence of OLZ by 2-way ANOVA with Tukey’s post-hoc analysis.
3.6. SFN protects against steatosis and liver injury caused by HFD and OLZ
The results indicate that HFD exacerbates metabolic and pathologic changes to the liver caused by OLZ. This effect correlated with an increase in lipid peroxidation, an index of oxidative stress (Figure 6). Much of the research regarding Nrf2 has focused on the pathway’s function in mitigating oxidative stress [34]. Furthermore, Nrf2 activation may also regulate lipid metabolism [35, 36]. Indices of pathology were assessed in the presence or absence of SFN, a compound that is known to induce Nrf2 signaling [37, 38]. SFN increased mRNA expression of the Nrf2 target gene, NQO1 (Figure 7B). In summary, SFN prevented much of the exacerbated liver injury caused by the interaction of HFD and OLZ (Figure 7). SFN treatment decreased fat accumulation in liver (Figure 7A) in addition to decreasing inflammation (Figure 7A, inset). ALT and AST decreased in parallel with the changes in histology (Figure 7B). SFN blunted the characteristic triglyceride accumulation caused by HFD and OLZ. SFN also rescued NEFA levels, which are paradoxically low during OLZ administration [39, 40], and increased hepatic glycogen storage, which is synergistically impaired by HFD and OLZ. Adducts of 4-HNE, which amassed in large quantity after HFD and OLZ exposure (Figure 6), appeared to be nearly absent from the tissue sections of SFN-treated mice; the visually apparent attenuation in staining was quantitated and determined to be a >20-fold decrease in 4-HNE binding (Figure 7B).
Fig. 7: SFN blunts liver injury caused by HFD and OLZ.
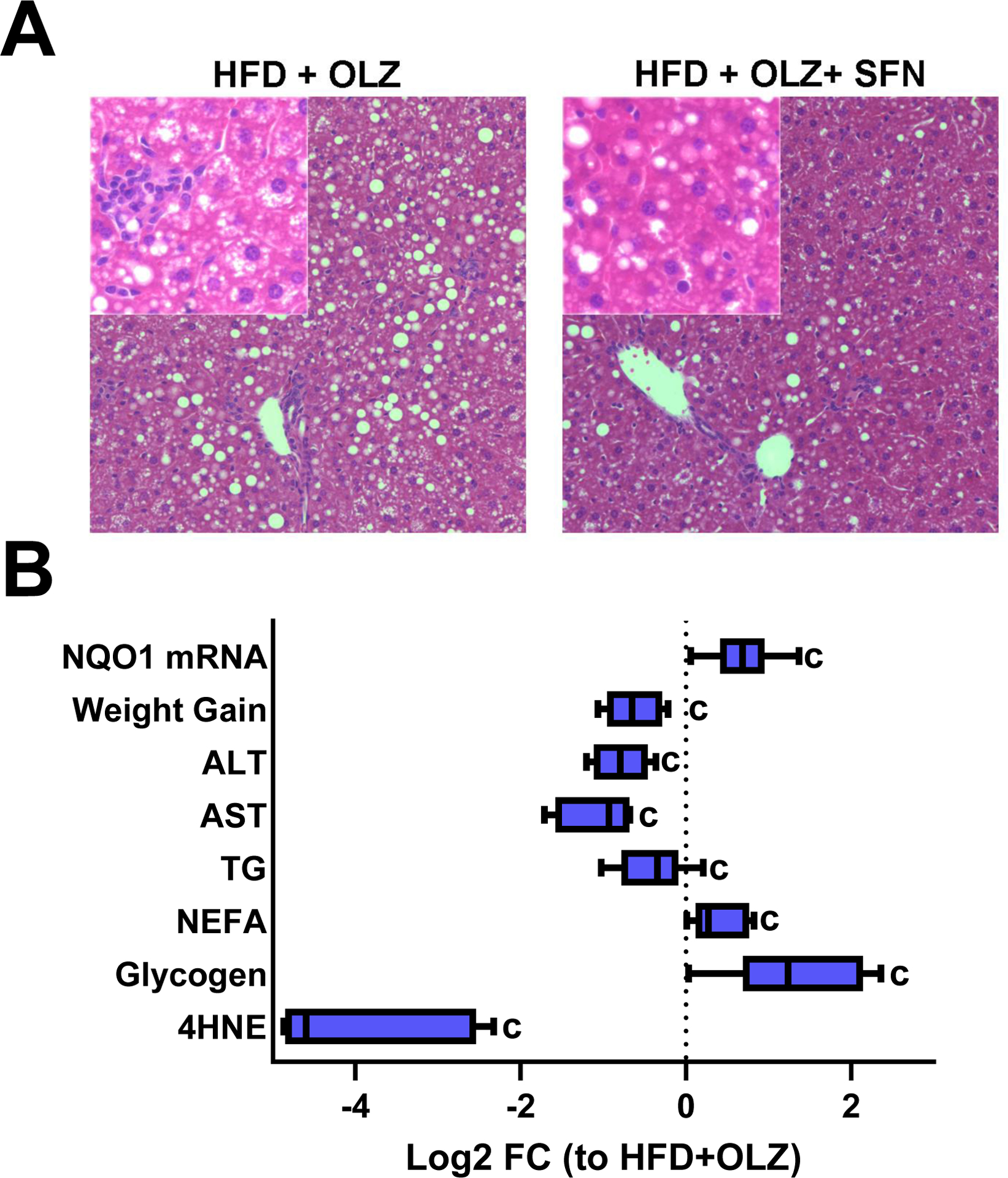
Panel A: H&E staining is shown at 100x and 400x (inset). Panel B: Weight and other variables were measured after HFD and OLZ treatment in the presence or absence of SFN. Values for the SFN treatment group are expressed as Log2 fold change vs. HFD and OLZ in the absence of SFN. cP <0.05 compared to HFD+OLZ; by 1-way ANOVA with Tukey’s post-hoc analysis.
4.0. DISCUSSION
Fatty liver disease is characterized by primary risks (e.g., obesity and metabolic syndrome) as well as other risk-modifying factors, be they environmental or genetic, that predict overall individual risk. For example, fatty livers are more sensitive to inflammatory liver injury [41]. For example, Yang et al. [42] demonstrated that liver injury in obese (fa/fa) Zucker rats is exacerbated by an inflammatory stimulus (LPS). In the study presented here, a different but complementary interaction was observed. Both OLZ and HFD alone produced toxic effects, but the combined effects of OLZ and HFD noticeably exacerbated toxicity, increasing fat accumulation and inflammatory response (Figures 2 and 3), among other observable injuries. Oxidative stress also markedly worsened during HFD + OLZ co-exposure (Figure 5).
Obesity rates in the US are at epidemic levels, with obesity a significant risk factor for drug-induced liver injury [43]. Given that the obesity incidence in the psychiatric population is even higher than in the US population as a whole [11], the effects of OLZ may exacerbate an underlying condition in these patients. Unfortunately, with few exceptions (e.g, [44]) most studies to date have employed lean experimental animals and/or healthy subjects in their studies with OLZ. The importance of the observed interaction between an obesogenic diet and OLZ exposure arises from its implications for two overlapping clinical populations. The expanding proportions of the national weight problem, coupled with the direct metabolic effects of OLZ, increase the likelihood of a hepatotoxic interaction (i.e., obesity and OLZ) for many patients taking this drug.
Consumption of OLZ and other antipsychotic drugs directly impacts peripheral organs and causes fatty liver [16]. Elevated plasma liver enzymes are common (~25%) in patients taking OLZ, as well as other antipsychotic drugs [45]. Although the reported incidence rate of clinically severe liver injury is relatively infrequent in patients taking OLZ (<0.5%) [46], even low subclinical (i.e., ‘asymptomatic’) increases in liver enzymes are independent risk factors for metabolic syndrome and cardiovascular disease [47]. NAFLD is not simply a consequence of obesity and metabolic syndrome, but also as a key player in these processes [48]. NAFLD is an independent risk factor for insulin resistance [48], hypertension [49], dyslipidemia [50], and even CVD [51]. Indeed, treating NAFLD is often sufficient to improve overall metabolic syndrome. The work summarized here indicates that OLZ exacerbates experimental NAFLD, which may then drive systemic dysmetabolism.
In addition to insulin resistance, mechanisms contributing to the initiation and progression of NAFLD can include lipotoxicity [52], inflammation [53] and oxidative stress [54]. Single-target drugs, although viewed to have better safety profiles by avoiding off-target reactions, are unlikely to treat complex disorders, such as NAFLD and metabolic syndrome [55]. A more contemporary view guiding this project is that the activation of pleiotropic pharmacologic responses by molecules designed to mimic endogenous mediators can represent a safe and effective drug strategy for diseases having a complex pathogenesis. This study identifies a new pharmacological adjunct to OLZ: the Nrf2 inducer sulforaphane counteracted several of the side effects of OLZ administration in this experimental model.
Although much of the research regarding SFN has focused on the pathway’s function in mitigating oxidative stress [34], it has also been shown to be antiinflammatory, antidiabetic and antilipogenic effects in mouse models of obesity-induced liver disease [56, 57]. SFN administration offset many of the side-effects caused by the interaction between HFD and OLZ without causing apparent injury to the mice (Figure 6). The most significant improvements seen were in weight, adiposity, and lipid peroxidation (Figure 6).
4.1. Summary and Conclusions
In summary, it was demonstrated that liver injury was exacerbated by the interaction between a drug (OLZ) and obesogenic HFD. Fatty liver disease is not simply a consequence of obesity, but also as a key player in driving negative effects of obesity [48]. Indeed, treating NAFLD is often sufficient to improve overall metabolic syndrome [58]. Although the reported incidence rate of clinically severe liver injury is relatively infrequent in patients taking OLZ (<0.5%) [46], elevated plasma liver enzymes is common (~25%) in patients taking OLZ [59]. The favorable results with SFN in this preclinical mouse model suggest that electrophilic activation of intrinsic defense mechanisms may be used to decrease the incidence of additional medical complaints that can be caused by OLZ. Consequently, SFN administration might also encourage patient adherence with psychiatric treatment regimens. Such future prospects would have to be clarified in human trials.
Highlights.
Olanzapine (OLZ) is effective for is effective in treating schizophrenia and other mood disorders.
Dysmetabolism and weight are serious side effects that impact OLZ therapy and patient health.
Obesity rates in individuals with schizophrenia iŝ2-fold higher than population as a whole (~60%)
We show that obesity and OLZ interact to exacerbate dysmetabolism and liver injury in mice
This drug/diet interaction can be prevented by the electrophilic antioxidant, sulforaphane.
Acknowledgements.
This work was supported by NIH grants R01AA021978, P50AA024337 and P30DKDK120531 (GEA), R01HL64937, R01HL132550 and P01-HL103455 (BAF), K01DK096042 (JIB) and University of Pittsburgh Medical Center Competitive Medical Research Fund Award (N.K.H.K.). RHI was supported by a predoctoral fellowship (T32EF011564).
Abbreviations.
- OLZ
olanzapine
- APD
antipsychotic drug
- NHANES
US National Health and Nutrition Examination Survey
- TZD
thiazolidinediones
- ROS
reactive oxygen species
- LFD
low-fat diet
- LPS
lipopolysaccharide
- HFD
high-fat diet
- DEXA
dual-energy x-ray absorptiometry
- OGTT
oral glucose tolerance test
- PAS
periodic acid-Schiff reagent
- CAE
naphthol AS-D chloroacetate esterase
- NEFA
non-esterified fatty acids
- 4HNE
4OH-nonenal
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- [1].Kessler RC, Birnbaum H, Demler O, Falloon IR, Gagnon E, Guyer M, et al. The prevalence and correlates of nonaffective psychosis in the National Comorbidity Survey Replication (NCS-R). Biol Psychiatry. 2005;58:668–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cloutier M, Aigbogun MS, Guerin A, Nitulescu R, Ramanakumar AV, Kamat SA, et al. The Economic Burden of Schizophrenia in the United States in 2013. J Clin Psychiatry. 2016;77:764–71. [DOI] [PubMed] [Google Scholar]
- [3].Sacks JJ, Gonzales KR, Bouchery EE, Tomedi LE, Brewer RD. 2010 National and State Costs of Excessive Alcohol Consumption. Am J Prev Med. 2015;49:e73–e9. [DOI] [PubMed] [Google Scholar]
- [4].Maher AR, Theodore G. Summary of the comparative effectiveness review on off-label use of atypical antipsychotics. J Manag Care Pharm. 2012;18:S1–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Alexander GC, Gallagher SA, Mascola A, Moloney RM, Stafford RS. Increasing off-label use of antipsychotic medications in the United States, 1995–2008. Pharmacoepidemiol Drug Saf. 2011;20:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999–2004. JAMA. 2006;295:1549–55. [DOI] [PubMed] [Google Scholar]
- [7].Grundy SM. Metabolic syndrome scientific statement by the American Heart Association and the National Heart, Lung, and Blood Institute. Arterioscler Thromb Vasc Biol. 2005;25:2243–4. [DOI] [PubMed] [Google Scholar]
- [8].Moore JX, Chaudhary N, Akinyemiju T. Metabolic Syndrome Prevalence by Race/Ethnicity and Sex in the United States, National Health and Nutrition Examination Survey, 1988–2012. Preventing chronic disease. 2017;14:E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Parsons B, Allison DB, Loebel A, Williams K, Giller E, Romano S, et al. Weight effects associated with antipsychotics: a comprehensive database analysis. Schizophr Res. 2009;110:103–10. [DOI] [PubMed] [Google Scholar]
- [10].Lieberman JA, Stroup TS, McEvoy JP, Swartz MS, Rosenheck RA, Perkins DO, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353:1209–23. [DOI] [PubMed] [Google Scholar]
- [11].Allison DB, Newcomer JW, Dunn AL, Blumenthal JA, Fabricatore AN, Daumit GL, et al. Obesity among those with mental disorders: a National Institute of Mental Health meeting report. Am J Prev Med. 2009;36:341–50. [DOI] [PubMed] [Google Scholar]
- [12].Colton CW, Manderscheid RW. Congruencies in increased mortality rates, years of potential life lost, and causes of death among public mental health clients in eight states. Preventing chronic disease. 2006;3:A42. [PMC free article] [PubMed] [Google Scholar]
- [13].Jarskog LF, Hamer RM, Catellier DJ, Stewart DD, Lavange L, Ray N, et al. Metformin for weight loss and metabolic control in overweight outpatients with schizophrenia and schizoaffective disorder. Am J Psychiatry. 2013;170:1032–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rado J, von Ammon Cavanaugh S. A Naturalistic Randomized Placebo-Controlled Trial of Extended-Release Metformin to Prevent Weight Gain Associated With Olanzapine in a US Community-Dwelling Population. J Clin Psychopharmacol. 2016;36:163–8. [DOI] [PubMed] [Google Scholar]
- [15].Heiskanen T, Niskanen L, Lyytikainen R, Saarinen PI, Hintikka J. Metabolic syndrome in patients with schizophrenia. J Clin Psychiatry. 2003;64:575–9. [DOI] [PubMed] [Google Scholar]
- [16].Schmidt RH, Jokinen JD, Massey VL, Falkner KC, Shi X, Yin X, et al. Olanzapine activates hepatic mammalian target of rapamycin: new mechanistic insight into metabolic dysregulation with atypical antipsychotic drugs. J Pharmacol Exp Ther. 2013;347:126–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vona R, Gambardella L, Cittadini C, Straface E, Pietraforte D. Biomarkers of Oxidative Stress in Metabolic Syndrome and Associated Diseases. Oxid Med Cell Longev. 2019;2019:8267234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Maher J, Yamamoto M. The rise of antioxidant signaling--the evolution and hormetic actions of Nrf2. Toxicol Appl Pharmacol. 2010;244:4–15. [DOI] [PubMed] [Google Scholar]
- [19].Padurariu M, Ciobica A, Dobrin I, Stefanescu C. Evaluation of antioxidant enzymes activities and lipid peroxidation in schizophrenic patients treated with typical and atypical antipsychotics. Neurosci Lett. 2010;479:317–20. [DOI] [PubMed] [Google Scholar]
- [20].Salmasi FB, Jazayeri M, Ghaeli P, Hashemian F, Akhondzadeh S, Raisi F, et al. Comparing the Effects of High-Dose Vitamin E With Those of Placebo on Insulin Resistance in Patients With Schizophrenia Treated With Olanzapine. Journal of Clinical Psychopharmacology. 2009;29:182–3. [DOI] [PubMed] [Google Scholar]
- [21].Miller ERr, Pastor-Barriuso R, Dalal D, Riemersma R, Appel LJ, Guallar E. Meta-analysis: high-dosage vitamin E supplementation may increase all-cause mortality. Annals of Internal Medicine. 2005;142:37–46. [DOI] [PubMed] [Google Scholar]
- [22].Kapur S, VanderSpek SC, Brownlee BA, Nobrega JN. Antipsychotic dosing in preclinical models is often unrepresentative of the clinical condition: a suggested solution based on in vivo occupancy. J Pharmacol Exp Ther. 2003;305:625–31. [DOI] [PubMed] [Google Scholar]
- [23].van der Zwaal EM, Luijendijk MC, Adan RA, la Fleur SE. Olanzapine-induced weight gain: chronic infusion using osmotic minipumps does not result in stable plasma levels due to degradation of olanzapine in solution. Eur J Pharmacol. 2008;585:130–6. [DOI] [PubMed] [Google Scholar]
- [24].Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, et al. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006;243:170–92. [DOI] [PubMed] [Google Scholar]
- [25].Andrikopoulos S, Blair AR, Deluca N, Fam BC, Proietto J. Evaluating the glucose tolerance test in mice. Am J Physiol Endocrinol Metab. 2008;295:E1323–32. [DOI] [PubMed] [Google Scholar]
- [26].Seifter S, Dayton S, et al. The estimation of glycogen with the anthrone reagent. Arch Biochem. 1950;25:191–200. [PubMed] [Google Scholar]
- [27].von Montfort C, Beier JI, Guo L, Kaiser JP, Arteel GE. Contribution of the sympathetic hormone epinephrine to the sensitizing effect of ethanol on LPS-induced liver damage in mice. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1227–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bergheim I, Guo L, Davis MA, Lambert JC, Beier JI, Duveau I, et al. Metformin prevents alcohol-induced liver injury in the mouse: Critical role of plasminogen activator inhibitor-1. Gastroenterology. 2006;130:2099–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arteel GE, Iimuro Y, Yin M, Raleigh JA, Thurman RG. Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology. 1997;25:920–6. [DOI] [PubMed] [Google Scholar]
- [30].McKim SE, Gabele E, Isayama F, Lambert JC, Tucker LM, Wheeler MD, et al. Inducible nitric oxide synthase is required in alcohol-induced liver injury: studies with knockout mice. Gastroenterology. 2003;125:1834–44. [DOI] [PubMed] [Google Scholar]
- [31].Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37:911–7. [DOI] [PubMed] [Google Scholar]
- [32].Kaiser JP, Beier JI, Zhang J, David Hoetker J, von Montfort C, Guo L, et al. PKCepsilon plays a causal role in acute ethanol-induced steatosis. Arch Biochem Biophys. 2009;482:104–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tan M, Schmidt RH, Beier JI, Watson WH, Zhong H, States JC, et al. Chronic subhepatotoxic exposure to arsenic enhances hepatic injury caused by high fat diet in mice. Toxicol Appl Pharmacol. 2011;257:356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Myint AM, Kim YK, Verkerk R, Park SH, Scharpe S, Steinbusch HW, et al. Tryptophan breakdown pathway in bipolar mania. J Affect Disord. 2007;102:65–72. [DOI] [PubMed] [Google Scholar]
- [35].Huang J, Tabbi-Anneni I, Gunda V, Wang L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kitteringham NR, Abdullah A, Walsh J, Randle L, Jenkins RE, Sison R, et al. Proteomic analysis of Nrf2 deficient transgenic mice reveals cellular defence and lipid metabolism as primary Nrf2-dependent pathways in the liver. J Proteomics. 2010;73:1612–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. 2004;310:263–71. [DOI] [PubMed] [Google Scholar]
- [38].Fimognari C, Lenzi M, Hrelia P. Interaction of the isothiocyanate sulforaphane with drug disposition and metabolism: pharmacological and toxicological implications. Curr Drug Metab. 2008;9:668–78. [DOI] [PubMed] [Google Scholar]
- [39].Albaugh VL, Vary TC, Ilkayeva O, Wenner BR, Maresca KP, Joyal JL, et al. Atypical antipsychotics rapidly and inappropriately switch peripheral fuel utilization to lipids, impairing metabolic flexibility in rodents. Schizophr Bull. 2012;38:153–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fahey JW, Talalay P. Antioxidant functions of sulforaphane: a potent inducer of Phase II detoxication enzymes. Food Chem Toxicol. 1999;37:973–9. [DOI] [PubMed] [Google Scholar]
- [41].You M, Arteel GE. Effect of ethanol on lipid metabolism. J Hepatol. 2019;70:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997;94:2557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Bessone F, Dirchwolf M, Rodil MA, Razori MV, Roma MG. Review article: drug-induced liver injury in the context of nonalcoholic fatty liver disease - a physiopathological and clinical integrated view. Aliment Pharmacol Ther. 2018;48:892–913. [DOI] [PubMed] [Google Scholar]
- [44].Townsend LK, Peppler WT, Bush ND, Wright DC. Obesity exacerbates the acute metabolic side effects of olanzapine. Psychoneuroendocrinology. 2018;88:121–8. [DOI] [PubMed] [Google Scholar]
- [45].Pae CU, Lim HK, Kim TS, Kim JJ, Lee CU, Lee SJ, et al. Naturalistic observation on the hepatic enzyme changes in patients treated with either risperidone or olanzapine alone. International clinical psychopharmacology. 2005;20:173–6. [DOI] [PubMed] [Google Scholar]
- [46].Bender S, Grohmann R, Engel RR, Degner D, Dittmann-Balcar A, Ruther E. Severe adverse drug reactions in psychiatric inpatients treated with neuroleptics. Pharmacopsychiatry. 2004;37 Suppl 1:S46–53. [DOI] [PubMed] [Google Scholar]
- [47].Afarideh M, Aryan Z, Ghajar A, Noshad S, Nakhjavani M, Baber U, et al. Complex association of serum alanine aminotransferase with the risk of future cardiovascular disease in type 2 diabetes. Atherosclerosis. 2016;254:42–51. [DOI] [PubMed] [Google Scholar]
- [48].Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–44. [DOI] [PubMed] [Google Scholar]
- [49].Sung KC, Wild SH, Byrne CD. Development of new fatty liver, or resolution of existing fatty liver, over five years of follow-up, and risk of incident hypertension. J Hepatol. 2014;60:1040–5. [DOI] [PubMed] [Google Scholar]
- [50].Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–36. [DOI] [PubMed] [Google Scholar]
- [51].Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non- alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011;43:617–49. [DOI] [PubMed] [Google Scholar]
- [52].Chang CY, Argo CK, Al-Osaimi AM, Caldwell SH. Therapy of NAFLD: antioxidants and cytoprotective agents. J Clin Gastroenterol. 2006;40 Suppl 1:S51–60. [DOI] [PubMed] [Google Scholar]
- [53].Choi S, Diehl AM. Role of inflammation in nonalcoholic steatohepatitis. Curr Opin Gastroenterol. 2005;21:702–7. [DOI] [PubMed] [Google Scholar]
- [54].McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40 Suppl 1:S17–29. [DOI] [PubMed] [Google Scholar]
- [55].Benedict M, Zhang X. Non-alcoholic fatty liver disease: An expanded review. World journal of hepatology. 2017;9:715–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Okada K, Warabi E, Sugimoto H, Horie M, Tokushige K, Ueda T, et al. Nrf2 inhibits hepatic iron accumulation and counteracts oxidative stress-induced liver injury in nutritional steatohepatitis. Journal of Gastroenterology. 2012;47:924–35. [DOI] [PubMed] [Google Scholar]
- [57].Shawky NM, Shehatou GSG, Suddek GM, Gameil NM. Comparison of the effects of sulforaphane and pioglitazone on insulin resistance and associated dyslipidemia, hepatosteatosis, and endothelial dysfunction in fructose-fed rats. Environ Toxicol Pharmacol. 2019;66:43–54. [DOI] [PubMed] [Google Scholar]
- [58].Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med. 2016;165:305–15. [DOI] [PubMed] [Google Scholar]
- [59].Atasoy N, Erdogan A, Yalug I, Ozturk U, Konuk N, Atik L, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1255–60. [DOI] [PubMed] [Google Scholar]