

Abstract
Nitric oxide (NO) is a gaseous molecule that plays a multifactorial role in several cellular processes. In the central nervous system, the NO dual nature in neuroprotection and neurotoxicity has been explored to unveil its involvement in Alzheimer’s disease (AD). A growing body of research shows that the activation of the NO signaling pathway leading to the phosphorylation of the transcription factor cyclic adenine monophosphate responsive element binding protein (CREB) (so-called NO/cGMP/PKG/CREB signaling pathway) ameliorates altered neuroplasticity and memory deficits in AD animal models. In addition to NO donors, several other pharmacological agents, such as phosphodiesterase 5 (PDE5) inhibitors have been used to activate the pathway and rescue memory disorders. PDE5 inhibitors, including sildenafil, tadalafil and vardenafil, are marketed for the treatment of erectile dysfunction and arterial pulmonary hypertension due to their vasodilatory properties. The ability of PDE5 inhibitors to interfere with the NO/cGMP/PKG/CREB signaling pathway by increasing the levels of cGMP has prompted the hypothesis that PDE5 inhibition might be used as an effective therapeutic strategy for the treatment of AD. To this end, newly designed PDE5 inhibitors belonging to different chemical classes with improved pharmacologic profile (e.g. higher potency, improved selectivity, and blood-brain barrier penetration) have been synthesized and evaluated in several animal models of AD. In addition, recent medicinal chemistry effort has led to the development of agents concurrently acting on the PDE5 enzyme and a second target involved in AD. Both marketed and investigational PDE5 inhibitors have shown to reverse cognitive defects in young and aged wild type mice as well as transgenic mouse models of AD and tauopathy using a variety of behavioral tasks. These studies confirmed the therapeutic potential of PDE5 inhibitors as cognitive enhancers. However, clinical studies assessing cognitive functions using marketed PDE5 inhibitors have not been conclusive. Drug discovery efforts by our group and others are currently directed towards the development of novel PDE5 inhibitors tailored to AD with improved pharmacodynamic and pharmacokinetic properties. In summary, the present perspective reports an overview of the correlation between the NO signaling and AD, as well as an outline of the PDE5 inhibitors used as an alternative approach in altering the NO pathway leading to an improvement of learning and memory. The last two sections describe the preclinical and clinical evaluation of PDE5 inhibitors for the treatment of AD, providing a comprehensive analysis of the current status of the AD drug discovery efforts involving PDE5 as a new therapeutic target.
1. Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder affecting millions of people worldwide. AD represents the most common and one of the most extensively studied forms of dementia. At the pathological level, it is characterized by intracellular amyloid plaques consisting of amyloid beta (Aβ) aggregates as well as extracellular neurofibrillary tangles (NFTs) formed by hyperphosphorylated tau fibrils that hamper proper neuronal functioning [1]. Currently used drugs, acetylcholinesterase (AChE) inhibitors or N-methyl-D-aspartate (NMDA) antagonists have very limited efficacy. Most of the efforts have been directed towards different therapeutic approaches, such us inhibiting neurofibrillary tangles (NFTs) formation, interfering with tau protein function, combatting inflammation and oxidative damage, decreasing Aβ load in the brain either by the use of agents that inhibit β-and γ-secretases or increase α-secretase (enzymes involved in the Aβ processing), inhibiting Aβ oligomerization [2, 3] or using treatments such as immunization with Aβ that appear to augment the removal of Aβ from the brain [4]. However, the role of tau, the amyloid-precursor protein (APP), Aβ, and the secretases in normal physiological processes [1, 5–19] might present a problem in providing effective and safe approaches to AD therapy. Developing agents that interact with tau and Aβ targets that lead to neuronal dysfunction and death is another approach that might became our sole resource in case all other strategies fail. Within this frame of thinking, the nitric oxide (NO) cascade offers unique possibilities that will be discussed in this review. Particularly, phosphodiesterase 5 (PDE5) looks very appealing due to the widespread use of its inhibitors with limited side effects. This review focuses on the drug discovery efforts to develop PDE5 inhibitors that cross the blood-brain barrier (BBB) and are suitable for chronic treatment of elderly people with comorbidity such as AD patients. Following a discussion of the involvement of the NO cascade in AD, medicinal chemistry efforts to develop novel selective PDE5 inhibitors that are also BBB permeant and efficacious in AD animal models are described. Finally, both preclinical and clinical studies relevant to AD and cognition using the inhibitors are reviewed.
2. Nitric Oxide Signaling and Alzheimer’s Disease
NO is a small gaseous molecule that is produced from the metabolism of L-arginine by the enzyme nitric oxide synthase (NOS). In the central nervous system (CNS), NO was characterized as an unconventional neurotransmitter since it is not stored in vesicles, but it might travel, upon production, from the post-synaptic to the pre-synaptic neuron, as a retrograde messenger [20, 21]. Due to its high lipophilicity, NO can easily diffuse through cell membranes, participating in a wide range of physiological functions, including vasodilation, inflammation, neuroprotection, neurotoxicity and synaptic transmission [22].
The action of NO is mediated either after binding to its biological receptor soluble guanylate cyclase (sGC) or via S-nitrosylation, a posttranslational modification that regulates the activity of substrate proteins and involves the formation of S-nitrosothiol (SNO) [23]. Binding of NO to sGC stimulates the production of the second messenger cyclic guanosine monophosphate (cGMP) from guanosine triphosphate (GTP) (Fig 1). Subsequently, cGMP activates its downstream effector protein kinase G (PKG) that activates, among other substrates [24], the transcription factor cyclic adenosine monophosphate (cAMP) response element-binding element (CREB), promoting neurotransmission, synaptic plasticity and memory formation [25–27]. PKG also activates the phosphatidylinositol 3-kinase (PI3k)/Akt signaling pathway [28] that mediates neuroprotection via the inhibition of apoptosis [29]. Additionally, NO could act at the presynaptic cell and promote the release of neurotransmitters, like glutamate, via activation of the sGC/cGMP/PKG pathway [30] (Fig. 1). Another mechanism by which NO can facilitate CREB signaling is via S-nitrosylation of nuclear proteins associated with CREB target genes and thereafter promote CREB-DNA binding [31, 32]. Other proteins that exhibit enhanced activity after nitrosylation include cyclic nucleotide-gated ion channels (CNGC) and ryanodine receptor Ca2+ release channels (RyR). Importantly, nitrosylation reduces the activity of apoptotic caspase enzymes and N-methyl-D-aspartate receptor (NMDAR) [33, 34]. The inhibition of NMDAR is suggested to be part of the neuroprotective action of NO, since excessive NMDAR activity leads to abnormal elevation in intracellular Ca2+ levels resulting in excitotoxicity [35].
Figure 1.

Nitric oxide signaling pathway in the brain. NO, produced by NOS, binds to and activates sGC, which catalyzes the production of cGMP, a second messenger activator of PKG. Levels of cGMP are regulated by the cGMP-degrading enzyme PDE5. Finally, PKG phosphorylates CREB protein, an important transcription factor responsible for the expression of memory-related genes. An alternative route shows the activation of the PI3k/Akt signaling pathway by PKG, leading to the promotion of cell survival. S-nitrosylation of NP associated with CREB-binding genes facilitates the activation of CREB, while S-nitrosylation of NMDAR inhibits the receptor activity. At the presynaptic neurons, NO is involved in the retrograde signaling and promotes the release of neurotransmitters, like glutamate, after activation of the sGC/cGMP/PKG pathway. CaMKII: Ca2+/calmodulin-dependent protein kinase II; cGMP: cyclic guanosine monophosphate; CREB: cAMP response element-binding element; NMDAR: N-methyl-D-aspartate receptor; NO: nitric oxide; NOS: nitric oxide synthase; NP: nuclear proteins; PI3k: phosphatidylinositol 3-kinase; PDE: phosphodiesterase; PKG: protein kinase G; SNO: S-nitrosothiol; sGC: soluble guanylate cyclase.
Apart from the important role in neurotransmission and synaptic plasticity, unbalanced production of NO induces neurotoxicity. The concentration of NO in the brain is determined by the activity of the 3 isoenzymes that constitute the NOS family: i) the neuronal form (nNOS or NOS1) that is widely expressed in the brain and mainly located in the striatum, nucleus accumbens, hippocampus and amygdala, ii) the inducible form (iNOS or NOS2) that is present in neurons, astrocytes, microglial cells, smooth cells, and macrophages, and is produced in response to inflammation or trauma, and iii) the endothelial form (eNOS or NOS3) expressed in the endothelial cells. nNOS and eNOS are classified as constitutive forms of NO (cNOS) that produce low amounts of NO transiently and their activation requires Ca2+ and more precisely phosphorylation by Ca2+/calmodulin-dependent protein kinase II (CaMKII) [36]. On the contrary, activation of iNOS is Ca2+-independent and could lead to the production of high levels of NO that last even for days [37]. At high concentrations, NO reacts with oxygen anion superoxide to form peroxynitrite, a highly reactive species that could cause cellular damage and it is responsible for the so-called nitrosative stress [22, 38].
Due to the multifactorial role of NO during several cellular processes, and especially its biphasic nature leading to both neuroprotection and neurotoxicity, several research lines have focused on the involvement of NO signaling in Alzheimer’s disease (AD).
In addition to the well-recognized Aβ and tau pathological hallmarks, AD is characterized by abnormal NO/sGC/cGMP/PKG/CREB signaling in the brain [38–41], an alteration in the expression of the different NOS isoforms [42, 43], and aberrant S-nitrosylation of target proteins [44], providing a strong link between AD and NO signaling. Nevertheless, the role of NO in AD remains controversial, since there are studies suggesting a neuroprotective function, while others support a neurotoxic action.
In agreement with the involvement of iNOS in the production of neurotoxic NO, iNOS expression is significantly increased in postmortem brain tissue from AD patients [45–47]. Of note, augmented immunoreactivity for iNOS was found in astrocytes surrounding amyloid plaques [45, 46]. The study from Lüth et al. has further shown that in addition to iNOS, the expression of eNOS was also increased in astrocytes in both postmortem AD brains and APP23 transgenic AD mice. Importantly, the authors suggested that the presence of iNOS-and eNOS-expressing astrocytes close to early, but not late, stage amyloid plaques indicates that elevation of NOS isoforms is a byproduct of the disease and does not participate in its etiopathogenesis [46]. In this regard, it is possible that increased expression of iNOS and eNOS represents an inflammatory response triggered by Aβ deposition. This notion is supported by an in vitro study showing that synthetic Aβ1–42 peptide increased the expression of iNOS and, in turn, NO production in neuronal-glial cultures from rats [47]. Noticeably, a recent study suggested that the elevation of NOS activity could be a compensatory mechanism to restore the subtle synaptic changes occurring in pre-symptomatic AD, as shown with the 3xTg-AD transgenic mouse model [48]. In these mice, blockage of NO resulted in synaptic depression, suggesting that increased NO signaling is required for maintaining synaptic plasticity that is essential for memory formation. The latter observation could explain the increased levels of nNOS expression in AD brain [49, 50]. However, during the progression of the disease, excessive production of NO could lead to neurotoxicity through formation of peroxynitrate and S-nitrosylated proteins, that will eventually accelerate the pathology of AD [44, 48, 51].
The importance of NO signaling to maintain intact cellular functions in AD was also outlined by studies showing that eNOS deficiency in aged wild-type mice (14–15 months old) led to increased production of Aβ, promoted inflammatory processes, and impaired hippocampus-dependent memory formation [52, 53]. In addition, genetic depletion of iNOS in AD mice overexpressing the amyloid precursor protein Swedish mutation (APPSW), elevated Aβ levels, tau hyperphosphorylation, and neuronal degeneration [54]. Noteworthy, NO signaling appears to have a direct effect on Aβ and tau pathology (Fig. 2). In particular, it was shown that both low and high levels of NO prevent the cleavage of APP by beta-secretase 1 (BACE1), a process involved in the production of Aβ. Lower concentration of NO blocked BACE1 expression, whereas higher concentration prevented its enzymatic activity via S-nitrosylation. The latter finding comes in agreement with the observed decrease in S-nitrosylation in AD brains [55]. With regard to the involvement of NO in tau pathology, a study from Reynolds and coworkers showed that peroxynitrate-mediated nitrosylation in specific tyrosine residues of the tau protein inhibits its assembly and oligomerization, preventing formation of NFTs. [56]. In addition, NO can regulate tau aggregation via activation of the P13K/Akt pathway. Specifically, Akt inhibits glycogen synthase kinase 3β (GSK-3β) that mediates tau phosphorylation, and therefore the reduction of NO could enhance tau hyperphosphorylation [54, 57, 58].
Figure 2.

Effects of the NO signaling on Aβ and tau pathology. Low and high levels of NO prevent the cleavage of amyloid precursor protein (APP) by beta-secretase 1 (BACE1), a process involved in the production of Aβ. Lower concentration of NO block BACE1 expression, whereas higher concentration prevent its enzymatic activity. At the same time, NO inhibits tau assembly and oligomerization, preventing formation of neurofibrillary tangles (NFTs).
Considering the neuroprotective role of NO signaling, at least at the early phase of AD, several studies have shown that activation of NO per se or other molecules in the NO/sGC/cGMP/PKG/CREB pathway can ameliorate altered neuroplasticity and memory deficits in animal models exhibiting AD-like pathology. Consistent with a plethora of studies reporting decreased phosphorylated CREB (pCREB) in AD brains [59–67], it has been shown that Aβ or tau oligomers lead to a reduction in pCREB during neuronal plasticity and memory formation. To this extent, two studies have provided electrophysiological, behavioral and biochemical evidence showing that the activation of the NO/sGC/cGMP/PKG/CREB pathway could rescue Aβ or tau pathology and restore pCREB levels [39, 40]. This has been shown for several pharmacological agents, including NO donors, sGC stimulators, cGMP analogs, phosphodiesterase 5 (PDE5) inhibitors, and PKG activators.
3. Drug Development of PDE5 Inhibitors against Alzheimer’s disease
Among the various components of the NO cascade, PDE5 is currently used as a target for the therapy of male erectile dysfunction (ED) and pulmonary hypertension, strongly supporting the possibility of exploiting the enzyme for the cure of other conditions in which the cascade is involved, including AD. PDE5 is one of the eleven subfamilies of PDE enzymes that are responsible for the degradation of cGMP and/or cAMP. While PDE1, 2, 3, 10, and 11 are dual-substrate PDEs, having affinity for both cyclic nucleotides, PDE5, 6, and 9 are cGMP-specific enzymes, and PDE4, 7, and 8 are cAMP-specific enzymes [68]. PDE5 inhibitors have been historically developed for ED treatment due to their vasodilation and smooth muscle relaxant effects. A challenge in developing PDE5 inhibitors is to attain high selectivity especially versus the PDE6 isozyme, which shares high structural similarity with PDE5 [69]. In addition, the inhibition of PDE isozymes such as PDE1, 6 and PDE11 has been associated with the onset of adverse effects, including tachycardia, vision disturbances and back pain [70]. The first synthesized PDE5 inhibitor was zaprinast (IC50 values is 0.76μM). It was an effective bronchodilator in exercise-associated asthma and was able to produce smooth muscles relaxation and a NO/cGMP-dependent relaxation of the corpus cavernosum. Zaprinast was a precursor of the chemically related PDE5 inhibitor cGMP-based derivative, sildenafil (Viagra) [71]. Sildenafil was originally developed as an anti-hypertensive drug, but ultimately was approved by FDA for ED in 1998 [72]. Subsequently, new PDE5 inhibitors were approved by FDA for ED treatment, namely vardenafil, tadalafil (both introduced in 2003), and avanafil (2012). Sildenafil exhibits a PDE5 and PDE6 IC50 of 2.2 and 9.5 nM, respectively. Tadalafil (Cialis), developed by Eli Lilly, is a PDE5 as well as PDE11 inhibitor, with IC50 values of 1.2 and 11nM, respectively, while vardenafil (Levitra) shows an inhibitory profile similar to that one of sildenafil [73]. A new generation of PDE5 inhibitors lodenafil (Helleva), udenafil (Zydena), and mirodenafil (Mvix) are also available in Brazil and Korea for ED treatment, but none of them have been approved by the FDA, yet (Fig. 3) [74].
Figure 3.

Chemical structures of commercial PDE5 inhibitors.
The involvement of the NO cascade in memory mechanisms led to the hypothesis that PDE5 inhibition might be used in AD therapy, a disorder characterized by subtle mnesic problems since the disease onset [75]. Given that AD is a chronic condition, affecting an elderly population with likely comorbidity, it became evident that should PDE5 inhibitors be used in AD, they should have minimal side effects. Quinoline-based, naphthyridine-based and 1H-pyrroloquinolinone-based PDE5 inhibitors were thought to have potential for chronic treatment of AD patients (Fig. 4 and Table 1). Compounds 1 and 2 bearing the quinoline scaffold showed excellent selectivity against all eleven PDEs [73], thus suggesting that they might have less side effects than classic PDE5 inhibitors. Importantly, both compounds exhibited an improved PDE5/PDE6 potency compared to sildenafil, vardenafil and tadalafil (PDE5/PDE6 IC50 of 1 is 0.27/339 nM, PDE5/PDE6 IC50 of 2 is 0.4/5100 nM) and no inhibitory activity versus the other PDE isozymes [73]. Compound 1 crossed the BBB and increased the level of cGMP in the APP/PS1 mouse model of AD, thus counteracting the synaptic plasticity and memory damage caused by the elevation of Aβ. However, compound 1 showed a low water solubility, which unfortunately prevented the advancement of the compound into preclinical studies. Naphthyridine-based and 1H-pyrroloquinolinone-based PDE5 inhibitors, compounds 3 and 4 (Fig. 4 and Table 1), were designed by locking the rotatable bonds of the hydroxymethyl group of the quinoline-base compounds into a ring [76]. Compound 3 showed improved water solubility with respect to 1 together with excellent PDE5 potency and selectivity (IC50 (PDE5)=0.056 nM, IC50 (PDE6)=30.1 nM). Compound 3 was tested in the APP/PS1 mouse model of AD and showed to ameliorate learning and memory deficits. These in vivo positive pharmacologic effects were correlated to an increased cGMP levels in the mouse hippocampus. 1H-pyrroloquinolinone-based PDE5 inhibitor, compound 4, showed a potency in picomolar range (IC50 (PDE5)=0.059 nM, IC50 (PDE6)=6.6 nM) and led to a selectivity for PDE5 of ∼100-fold compared to PDE6. An in silico docking study was performed with both compounds 3 and 4, showing their interaction with residues in the cGMP pocket of PDE5A1. The targeted pocket was chosen based on the fact that multiple complex crystal structures show that diverse PDE5 small molecule inhibitors consistently bind in it, showing that it has highly druggable characteristics. However both compounds, 3 and 4, lacked in vitro metabolic stability, which suggests the need of further effort toward the optimization of these drugs property physicochemical and pharmacological properties [76].
Figure 4.

Chemical structures of quinoline-based (1 and 2), naphthyridine-based (3) and 1H-pyrroloquinolinone-based (4) PDE5 inhibitors.
Table 1.
Current most relevant PDE5 inhibitors, PDE5/HDAC inhibitors and PDE5/AChE inhibitors.
| COMPOUNDS | TARGET | INDICATION | EFFECT | REFERENCE | LIMITATIONS |
|---|---|---|---|---|---|
| 1 | PDE5 | Alzheimer’s disease | Increased cGMP levels in the hippocampus of mice and improved learning and memory deficits in transgenic mouse model of AD. | Fiorito et al. 2013 [57] | Low water solubility. |
| 3 | PDE5 | Alzheimer’s disease | Increased cGMP levels in the hippocampus of mice and improved learning and memory deficits in transgenic mouse model of AD. | Fiorito et al. 2017 [58] | Lack of in vitro metabolic stability. |
| 5 | PDE5-HDAC | Alzheimer’s disease | Reduced Aβ levels and tau phosphorylation levels | Cuadrado-Tejedor et al. 2017 [62] | Moderate HDAC class I inhibitor and HDAC6 and PDE5 PDE9 inhibitor. not every isoform-selective HDACis target each class I isoform (HDAC1, HDAC2, and HDAC3) is associated with cytotoxicity. |
| 6 | PDE5-HDAC | Alzheimer’s disease | Reduced tau phosphorylation levels | Rabal et al. 2018 [63] | Poor BBB penetration; Lack of in vitro metabolic stability. |
| 7 | PDE5-AChE | Alzheimer’s disease | Inhibition of cortical AChE; improved CREB phosphorylation | Mao et al. 2018 [67] | Nearly insoluble in water. More selective for AChE |
| 8 | PDE5-AChE | Alzheimer’s disease | Inhibition of cortical AChE; improved CREB phosphorylation | Mao et al. 2018 [67] | Nearly insoluble in water. More selective for AChE |
| 9 | PDE5-AChE | Alzheimer’s disease | Inhibition of cortical AChE; improved CREB phosphorylation | Ni et al. 2018 [68] | More selective for AChE and BuChE |
An alternative therapeutic strategy that has recently drawn much attention concerns the development of multi target-directed ligands (MTDLs) that are able to act on different molecular targets involved in the pathological processes in AD [77]. To this end, dual inhibitors targeting PDE5 and histone deacetylases (HDACs) have been tested as a potentially novel therapeutic approach against AD. Similar to PDE5 inhibitors, HDAC inhibitors are potential modulators of cognitive impairment in AD [78, 79]. A series of cGMP-based derivatives and β-carboline derivatives were designed and synthesized with the aim to validate this multi target approach (Fig. 5 and Table 1) [80, 81]. Compound 5 shares the 1H-pyrazolo[4,3-d]pyrimidine scaffold of sildenafil, with an additional hydroxamic acid moiety that typically confers HDAC inhibitory activity. Compound 5 showed a PDE5 IC50 value of 60 nM and moderate inhibition activity against HDAC class I (HDAC1: 310 nM, HDAC2: 490 nM, HDAC3: 322 nM, HDAC4: 91 nM). A follow-up study showed that compound 5 was able to cross the BBB and increase histone acetylation and CREB phosphorylation in the hippocampus, leading to the rescue of LTP in APP/PS1 mice [80]. The next goal in developing dual target ligands with PDE5/HDAC activity focused on reducing the toxicity deriving from the inhibition of other HDAC class I enzymes by improving the HDAC6 selectivity. Compound 6 demonstrated potent activities against PDE5 (IC50=11 nM) and HDAC6 (IC50=15 nM), retaining excellent HDAC6 selectivity over HDAC1 (1.3 log units). Although compound 6 reduced the levels of the AD-related markers APP and phosphorylated tau protein in Tg2576 neurons, however in vivo studies did not produce a significant memory improvement [81].
Figure 5.
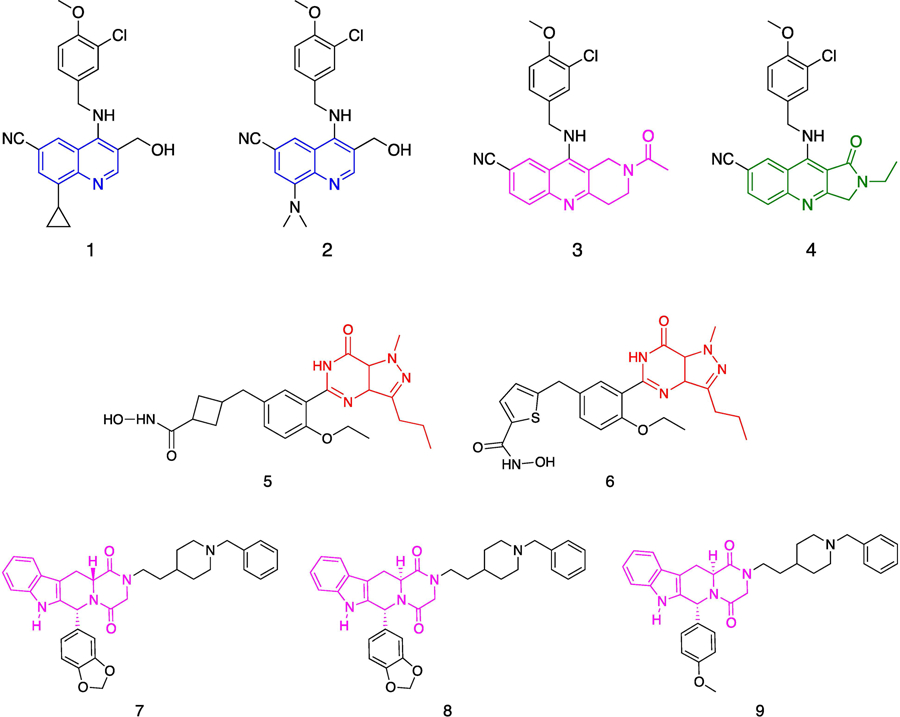
Chemical structures of PDE5/AChE inhibitors.
In the last decades, AChE/PDE5 dual target inhibitors have been investigated for the treatment of AD. Such approach is based on the rationale that the currently available AD drugs are AChE inhibitors. Two generations of dual-target AChE/PDE5 inhibitors with β-carboline scaffold were designed. The first generation includes compounds 7 and 8 (Fig. 5 and Table 1), which are derived from modifying the substituent at the nitrogen atom of piperazine-2,5-dione (compound 7) and the substituent at the phenyl ring (compound 8) of tadalafil. According to this study, the substituent ethyl-(1-benzylpiperidin-4-yl) can represent the AChE inhibition pharmacophore, which modulates liposolubility and improved BBB permeability [82]. As a result, these two candidate compounds exhibited the expected properties. Both compounds showed good AChE inhibition (IC50 7: 36 nM; IC50 8: 32 nM) as well as moderate PDE5 inhibitory activities (IC50 7: 153 nM; IC50 8: 1.53 μM). Importantly, in in vivo studies compound 7 reversed the cognitive dysfunction of scopolamine-induced AD mice and enhanced CREB phosphorylation. Unfortunately, this compound possessed poor water solubility, which represents a limitation to its further development [82]. To overcome this limitation, a second generation of dual target AChE/PDE5 inhibitors was synthesized with improved water solubility [83]. The structure-activity relationship studies revealed that the stereo-configuration at position 6 of the tadalafil core was vital to AChE inhibitory activity with the favored configuration was 6R, while the stereo-configuration at position 12 of the tadalafil nucleus had little influence on AChE inhibitory activity, but it was important for exerting PDE5A1 inhibitory activity. Compound 9 exhibited a dual-target AChE/PDE5 inhibitory activity of 15 nM and 3.23 μM, respectively, and good BBB permeability. Compound 9 was characterized by improved water solubility and good therapeutic effects against scopolamine-induced cognitive impairment, with an inhibition of cortical AChE activities and an enhancement of CREB phosphorylation ex vivo [83].
When proposing a new class of drugs as therapeutic agents it is imperative to consider their possible side effects that might be dependent upon interference with other targets or with the activity of the NO/cGMP/CREB axis and the role that it plays in many physiologic processes in the brain and elsewhere in the body. An advantage of using PDE5 inhibitors is that their side effects are known as they have already been utilized for many years, such that FDA has authorized the daily use of PDE5 inhibitors in arterial pulmonary hypertension. Recently, a number of clinical studies have explored the association between PDE5 inhibitors and the risk for melanoma and, interestingly, both positive and negative correlations have been reported [84–88]. However, those studies showing an increased risk of incident melanoma upon PDE5 inhibitors exposure did not account for confounding risk factors, such as sun exposure, or adjusted their results for different factors (e.g. age, comorbidities, smoking). Thus, it has been concluded that there is no causality between the use of PDE5 inhibitors and melanoma and further evidence to show the association is needed [89, 90]. A few cases of priapism have also been reported following the intake of PDE5 inhibitors. However, it is possible that this side effect is due to a dysregulation of PDE5 function following NO cascade down-regulation [91] - a phenomenon that is also caused by Aβ increase [39] - such that, paradoxically, PDE5 inhibitors have been proposed as therapeutic agents against priapism [92, 93]. Additional adverse events of the PDE5 inhibitors include mild vasodilatory events such as headache, flushing, dyspepsia, and nasal congestion or rhinitis, hypotension (if nitrates are given concurrently), transient visual impairment. However, although Aβ is primarily accumulating in the CNS, Aβ is also present in the blood of patients with AD and related dementia [94, 95]. Interestingly, Aβ potentiates vasoconstriction both in cerebral vasculature and in other districts of the vascular system [96–102]. Moreover, hypertension has been often associated with AD [97–99]. Thus, it is very attractive to hypothesize that PDE5 inhibitors might counteract not only CNS symptoms, but also vascular symptoms that often affect AD patients.
4. Preclinical Studies of PDE5 Inhibitors in Alzheimer’s Disease
At the preclinical level, there is a growing body of evidence showing that PDE5 inhibition can improve memory function. The first PDE5 inhibitor exhibiting cognitive enhancing properties in animals was zaprinast, a phosphodiesterase inhibitor that targets PDE5, PDE6, PDE9 and PDE11 [103]. During the training in the object recognition test, zaprinast and 7-nitroindazole, a putative selective inhibitor of nNOS, were administered i.p. to rats, immediately after the exposure to two identical objects [103]. After 1hr, control rats spent more time exploring the new object, indicating that they recognized the previously seen object. However, animals injected with 7-nitroindazole could not discriminate between the two objects. At 4hrs, control rats did not discriminate between the objects, whereas zaprinast facilitated object recognition and reversed the recognition memory deficit induced by 7-nitroindazole [103]. In addition to this original study performed in young rats, it was later confirmed that this effect on improving memory consolidation could be extended to older animals [104]. Moreover, the memory enhancing effect of zaprinast was extended to memory acquisition and consolidation in rats administered with the compound and tested with an object recognition task [104].
Following these initial studies with zaprinast, considering that the compound inhibits several PDEs [105], other studies were performed using inhibitors that are selective towards PDE5 in view of a potential therapeutic use of a strategy including enhancement of cGMP levels. Sildenafil was found to improve acquisition and consolidation of object recognition memory in both mice and rats [106–108], spatial memory in the elevated plus maze in mice [109], and performance in the active avoidance task in mice [110]. Additionally, it was shown that treatment with both sildenafil or zaprinast improves memory in young and age-impaired mice in an adapted version of the elevated plus maze and passive avoidance task [111]. As a matter of fact, the memory improving effect was more pronounced in aged mice than in young mice. Nevertheless, a later study reported that sildenafil had no effect on the memory performance of young and aged rats tested in the passive avoidance task [112]. Recently, aged mice were also tested after chronic sildenafil treatment [113]. It was found that sildenafil improved age-impaired object and spatial memory performance in the object recognition task and Morris water maze task, respectively. Sildenafil also restored phosphorylation of hippocampal CREB in these aged mice. Altogether, these data suggest the possibility of using PDE5 inhibitors to enhance normal memory and age-related memory decline.
The possibility of using both genetically modified models of AD and pharmacologic models displaying cognitive dysfunction led to experiments assessing the efficacy of PDE5 inhibitors against memory loss associated with the disease [75, 114, 115]. Puzzo et al. showed that chronic administration of sildenafil in a 3-month-old transgenic APP/PS1 mouse model could restore AD related cognitive deficits, synaptic dysfunction, decreased activity of the memory related transcription factor CREB, and, interestingly, diminished hippocampal Aβ levels. In addition, administration of sildenafil to hippocampal slices reversed the impairment of LTP in these APP/PS1 mice [75]. Zhang and coworkers also showed that sildenafil reversed memory deficits in APP/PS1 mice in the object recognition task and led to improved cGMP/PKG/pCREB signaling and a decrease in soluble Aβ levels. These findings were in part consistent with a study conducted by Cuadrado-Tejedor et al. demonstrating that chronic administration of sildenafil reverses memory deficits in Tg2576 transgenic mice without changing Aβ load [114]. Moreover these studies were in agreement with the observation that pre-training administration of sildenafil reverses the spatial learning impairment induced by the nonspecific NOS inhibitor Nω-L-nitro-arginine methyl ester (L-NAME) [116, 117]. Finally, consistent with these studies, Rutten et al. extended their earlier findings from rodents to monkeys by showing that sildenafil dose-dependently improved cognitive function in a prefrontal task, i.e., the object retrieval task, in cynomolgus macaques [118].
Later studies have replicated the sildenafil data in the object recognition task in rats using other PDE5 inhibitor, such as vardenafil or tadalafil [75, 119, 120]. For instance, acute treatment with vardenafil improved spatial memory tested with the object location test [121]. Additionally, in a study from Devan et al., repeated daily injections of vardenafil for one week improved long-term memory performance [119]. Tadalafil, in turn, was able to reverse the reduction of LTP when directly administered to slices of the APP/PS1 mouse model of AD but failed to achieve behavioral benefits similar to sildenafil when administered to adult APP/PS1 mice in vivo. The lack of in vivo efficacy was attributed to the inability of tadalafil to cross the BBB that had been reported by the producer of the compound [75]. Conversely, Garcia-Barroso et al. showed data indicating that tadalafil improves the Morris water maze performance in the J20 mouse model of AD, and reduced tau phosphorylation without affecting the Aβ burden [120]. They also reported that the compound crosses the BBB [120]. Nevertheless, in 2017 Fei Mao et al. prepared a series of tadalafil analogues with improved BBB penetrability and confirmed that tadalafil possesses poor BBB penetration [82]. Thus, a plausible explanation for the beneficial effect of the compound in the Garcia-Barroso et al. manuscript [120] might be related to a possible alteration in the BBB of the AD mice, which facilitated tadalafil access to the CNS. Alternatively, one cannot exclude that the inhibitor might act through vascular mechanisms, as PDE5 inhibitors were initial developed for their antihypertensive effects due to the resulting vasodilation effects deriving from an increase of cGMP in the endothelial tissue.
Most recently, other PDE5 inhibitors such as icariin, yonkenafil, compound 7a (1 in Fig. 4) and compound 6c (3 in Fig. 4) were developed specifically for exploring their therapeutic potential in learning and memory. Jin et al. showed a beneficial effect of icariin against memory loss in APP/PS1 transgenic mice [122]. They found that icariin treatment significantly improves the memory defect in the Y-maze task. Moreover, levels of amyloid precursor protein (APP), amyloid-beta (Aβ1–40/42) and both PDE5 mRNA and protein were increased in the mouse hippocampus and cortex.
The effects of yonkenafil were investigated on cognitive behaviors as well as the pathological hallmarks of AD using transgenic models of the disease. Daily treatment of seven-month-old APP/PS1 mice with yonkenafil for 3 months improved nesting-building ability, working memory deficits in the Y-maze task, and Morris water maze performance. In addition, yonkenafil reduced Aβ plaque area, and inhibited over-activation of microglia and astrocytes. Furthermore, yonkenafil increased neurogenesis in the dentate gyrus. These results are consistent with the hypothesis that PDE5 inhibition attenuates cognitive deficits in AD by reducing the amyloid burden, and inhibiting over-activation of microglia and astrocytes as well as restoring neurogenesis [123].
The pharmacological efficacy of compound 7a (1 in Fig. 4) was investigated in several transgenic mouse models of AD [40, 73]. Compound 7a was able to improve contextual memory in both mice pre-treated with Aβ or tau oligomers, and APP/PS1mice. Additionally, an amelioration of spatial memory in tau oligomer treated and APP/PS1 mice was observed. These behavioral findings were corroborated by the biochemical analysis of the hippocampus of mice that revealed an increase of cGMP and pCREB levels. Thus, compound 7a confirmed the therapeutic potential of using PDE5 inhibitors in AD and extended their usefulness to tauopathies [40, 73].
The drug optimization of compound 7a led to the discovery of compound 6c (3 in Fig. 4), which was profiled for its ability to rescue learning and memory in the APP/PS1 transgenic mouse model [76]. LTP studies with compound 6c showed strengthening of synaptic plasticity in hippocampal slices of APP/PS1 mice. In vivo studies also demonstrated positive behavioral outcomes when compound 6c was administered to young mice that were assessed with a radial arm water maze task. Remarkably, the beneficial effects of the PDE5 inhibitor were still observed three months after the administration of the therapy, suggesting that the inhibition of PDE5, and therefore the elevation of cGMP, is inducing persisting biochemical changes [76].
The study of PDE5 inhibition as potential cognition enhancement strategy gained particular interest, since the PDE5 protein is highly expressed in the cytoplasm of neuronal cells in human brains [124]. Specifically, in cortex and hippocampus, PDE5 is expressed primarily in large, pyramidal-type neurons, whereas in cerebellum, it is prominent in Purkinje neurons [124]. However, PDE5 inhibition is currently used as a therapeutic approach for treating erectile dysfunction and pulmonary hypertension due to the resulting vasodilation effects deriving from an increase of cGMP in the endothelial tissue. These cardiovascular effects of PDE5 inhibitors caused a controversy with regard to their mechanism of action as cognitive enhancers. Vasodilation might lead to an increase in blood flow and glucose metabolism in the brain and, as such, could induce improved brain functions without directly affecting neuroplasticity mechanisms. However, it has been preclinically shown that the effective dose of PDE5 inhibitors required for improving cognition is below the dose that induces vascular and metabolic effects. For example, a study has shown that sildenafil could increase the mean arterial blood pressure in rats following 10 mg/kg oral administration [125]. Yet, the memory improvement effect of sildenafil has been observed at a dose of 1–3 mg/kg, as demonstrated by Puzzo et al. [75]. Also, sildenafil had no effect in humans on blood flow in the middle cerebral artery, just as there were no changes in radial and temporal artery diameters [126–128]. Another study showed that the cognitive enhancement observed in rats treated with vardenafil was not related to any main effects on blood flow and glucose utilization in the brain [129]. Finally, a recent report by Akkerman et al. showed results in support of the hypothesis that PDE5 inhibitors act through central mechanisms in a study in which they administered vardenafil via the intracerebroventricular route [130]. Nevertheless, one cannot exclude that in some circumstances (see for instance the beneficial effect of tadalafil in Garcia-Barroso et al. [120]), PDE5 inhibitors might enhance cognition through vascular mechanisms.
5. Clinical Studies of PDE5 Inhibitors as Cognitive Enhancers in Normal Conditions and Disease Status
Despite the robust memory improving action of PDE5 inhibitors in animal studies, these effects have not yet been translated into clinical efficacy to enhance cognition. Several clinical studies have involved health volunteers as well as different populations of patients. However, none of the investigated drugs has reached the market as a cognitive enhancer/anti-AD drug so far.
Data obtained from administration in humans have shown a very wide variety of results ranging from no effect to beneficial effect on normal memory. For instance, in a study sildenafil was tested on various cognitive functions in six male healthy volunteers [131]. A single oral dose of 100 mg sildenafil enhanced performance in a simple reaction time test, but showed no significant effects on short-term memory, divided attention and other psychomotor tasks. Another study conducted on young healthy male volunteers confirmed that the same dose of sildenafil did not significantly improve memory performance in healthy subjects [132]. In this placebo-controlled trial, sildenafil significantly affected patterns of auditory event-related brain potentials consistent with enhanced ability to focus attention and to select relevant target stimuli, although induced no direct cognition enhancing effect on auditory attention and word recognition [132]. Moreover, sildenafil showed an effect on information processing since a reduced negativity in the electroencephalogram was found in the word recognition experiment [132].
The cognition enhancing potential of sildenafil was also investigated in a trial for schizophrenia and another one for Parkinson’s disease. In the first study, pairing of sildenafil with antipsychotic treatment did not affect cognitive performance [133, 134]. It is possible that the doses of 50 and 100 mg used in this study were not optimal or repeated dosing may be necessary to achieve therapeutic effects. In the latter study, sildenafil was tested for its efficacy in reducing dyskinesia in patients with Parkinson’s disease (ClinicalTrials.gov Identifier: NCT02162979). However, this study was terminated due to insufficient participant inclusion. Sildenafil has also been evaluated in a Phase I study for its neuroprotective properties in the treatment of stroke (ClinicalTrials.gov Identifier: NCT00452582). However, in 2011 this study was terminated because of a failure to recruit in the expected time period. Also, a recent study on AD patients demonstrated that a single, 50 mg dose of sildenafil could improve significantly cerebral hemodynamic function and increase cerebral oxygen metabolism [135]. These results together suggest that PDE5 inhibitors might enhance memory via a cerebrovascular mechanism.
Tadalafil is considered to be less efficacious than sildenafil or vardenafil for cognition enhancement because of its poor brain penetration [75]. This inhibitor is currently evaluated in a phase II clinical trial initiated at St George’s (University of London) in elderly patients with cerebral small vessel disease. Cognitive performance was measured after the treatment of a single oral dose of 20 mg tadalafil. The overall idea of the clinical trial is that the vasodilatory properties of tadalafil should improve blood flow to these brain areas thus preventing vascular cognitive impairment (ClinicalTrials.gov Identifier: NCT02450253).
Two studies from Shim et al. reported that chronic treatment with the PDE5 inhibitor udenafil (100 mg) for 2 months in patients with erectile dysfunction improved cognitive and frontal executive function [136]. Moreover, a follow-up study from the same group showed that even a low dose of udenafil (50 mg) could exert cognitive enhancing action after daily administration for 2 months [137]. The latter findings probably indicate that chronic treatment of PDE5 inhibitors is necessary in order to exert their cognitive enhancing action in humans. Importantly, no severe adverse events were observed in both studies, and no patient discontinued the treatment. The only adverse events were mild dizziness in 2 out of 27 patients in the first study [136] and hot flushing or nasal congestion in 4 out of 24 patients in the second study [137].
6. Conclusions
Altogether, while PDE5 inhibitors have shown clear cognition enhancing potential in preclinical studies, these effects have not yet resulted in a clear demonstration of clinical efficacy. Although PDE5 inhibitors can induce vasodilatory effects when administered at high doses, the cognition enhancing properties observed in the current studies using lower doses were not related to any main effect on blood flow and glucose utilization in the brain. Interestingly, due to the FDA-approval of PDE5 inhibitors for the treatment of non-neurological conditions, their side effects have been extensively documented and consist of mild adverse effects that are generally well-tolerated by patients like headache, flushing, runny nose, stomach pain, back pain, and indigestion [138]. Nevertheless, it is acknowledged that currently available PDE5 inhibitors are characterized by suboptimal specificity, which could limit their clinical application. For instance, sildenafil also inhibits PDE6 that is expressed in the retina and therefore causes visual disturbances after chronic administration. As a result, development of novel PDE5 inhibitors is focusing on improving specificity of the inhibitor for the enzyme, optimizing pharmacokinetic properties in humans, and increasing BBB permeability. Medicinal chemistry data that have been obtained so far together with a growing body of preclinical and clinical evidence here described strongly support the possibility that PDE5 inhibitors represent a novel class of compounds that might effectively counteract AD progression.
Table 2.
PDE5 inhibitors in cognition. EEG, electroencephalogram, i.m. intramuscular, i.p. intraperitoneal, ORT object recognition test, p.o. per os, s.c. subcutaneously.
| PDE5 inhibitor | Doses | Route | Test | Animals | Behavioral Results | Reference |
|---|---|---|---|---|---|---|
| Zaprinast | 3 or 10 mg/kg | i.p. | ORT (4h interval) | 3 months old Tryon–Maze–Bright rats | 10 mg/kg improved memory consolidation | Prickaerts et al. 1997 [69] |
| Zaprinast | 0.5, 1 or 2 mg/kg | i.p. | Elevate plus-maze and passive avoidance task | 3 and 20–22 months old Swiss mice | All doses improved memory in aged animals; 1 and 2 mg/kg improved memory in young animals | Patil et al. 2004 [77] |
| Zaprinast | 0.3 mg/kg | s.c. | ORT (2h interval) | 3 and 12 months old Wistar rats | Pro-cognitive action only for the young animals. No effect in older animals. | Domek-Łopacińska and Strosznajder 2008 [70] |
| Sildenafil | 1, 3, 10 or 30 mg/kg | i.p. | Active avoidance learning | 2 months old Swiss mice | 3 mg/kg improved performance | Baratti and Boccia 1999 [76] |
| Sildenafil | 0.25, 0.5 or 1 mg/kg | i.p. | Elevate plus-maze and passive avoidance task | 3 and 20–22 months old Swiss mice | All doses improved memory in aged animals-0.5 and 1 mg/kg improved memory in young animals | Patil et al. 2004 [77] |
| Sildenafil | 1, 3 or 10 mg/kg | p.o. | ORT (24h interval) | 4 months old Wistar rats | 3 and 10 mg/kg improved memory consolidation | Prickaerts et al. 2005 [73] |
| Sildenafil | 0.3, 1 or 3 mg/kg | p.o. | ORT (24h interval) | 6 months old Swiss mice | 1 mg/kg improved memory consolidation | Rutten et al. 2005 [74] |
| Sildenafil | 1, 3, 10 or 20 mg/kg | i.p. | Passive avoidance task | 2 and 12-month old Wistar rats | No memory enhancing effect | Shafiei et al. 2006 [78] |
| Sildenafil | 0.3, 1 or 3 mg/kg | i.m. | Object retrieval task | Cynomolgus macaque | 1 and 3 mg/kg improved memory performance | Rutten et al. 2008 [84] |
| Sildenafil | 3mg/kg daily for 3 weeks | i.p. | ORT and MWM | 26–30 months old C57Bl/6J mice | Improved performance in both tests | Palmeri et al. 2013 [79] |
| Vardenafil | 0.5 mg/kg | p.o. | 14-unit T-maze | 3,17, and 24-month-old rats | Improved spatial memory retention | Devan et al. 2014 [85] |
| Vardenafil | 0.3 mg/kg | i.p. | Object location test (24h interval) | 4–5 months old mice | Improved spatial memory acquisition and early consolidation | Argyrousi et al. 2019 [87] |
| Tadalafil | 15 mg/kg | p.o. | MWM | 3-month-old J20 transgenic mice | Improved memory performance | Garcia-Barroso et al. 2013 [86] |
Table 3.
PDE5 inhibitors oral single dose in clinical trials.
| Drugs | Doses | Field | Phase | Patients | References and ID clinical number |
|---|---|---|---|---|---|
| Sildenafil | 100 mg | cognition | Completed Phase VI | Healthy males | Grass et al. 2001 [97] |
| Sildenafil | 100 mg | cognition | Completed Phase VI | Healthy males | Schultheiss et al. 2001 [98] |
| Sildenafil | 50 and 100 mg | schizophrenia | Completed Phase VI | Schizophrenic patients | Goff et al. 2009 [99] |
| Sildenafil | 50 mg | Parkinson’s disease | Phase II (insufficient participants) | Parkinson’s disease patients | Identifier: NCT02162979 |
| Sildenafil | 50 mg | hemodynamic function | N/A | AD patients | Sheg et al. 2017 [101] |
| Sildenafil | ischemic stroke | Phase I (terminated due to non-recruitment of patients within the scheduled time period) | Ischemic stroke patients | Identifier: NCT00452582 | |
| Tadalafil | 20 mg | cognition | Phase II | Elderly with small vessel disease | Identifier: NCT02450253 |
| Vardenafil | 10, 20 mg | sensory gating | Completed phase II | Healthy young adults | Reneerkens et al 2013 [105] |
| Vardenafil | 10, 20 mg | cognition | Completed phase II | Healthy young adults | Reneerkens et al 2013 [106] |
| Udenafil | 100 mg | cognition | N/A | Patients with ED | Shim et al 2011 [102] |
| Udenafil | 50 mg | cognition | N/A | Patients with ED | Shim et al. 2014 [103] |
Acknowledgments
Financial Support
This work was supported by the NIH grants R01NS110024 and R01AG049402 to OA, and grant AARF-17-504483 from the Alzheimer’s Association to JF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Maccioni RB, Muñoz JP, Barbeito L, The molecular bases of Alzheimer’s disease and other neurodegenerative disorders, Archives of medical research 32(5) (2001) 367–381. [DOI] [PubMed] [Google Scholar]
- [2].Nakagami Y, Nishimura S, Murasugi T, Kaneko I, Meguro M, Marumoto S, Kogen H, Koyama K, Oda T, A novel beta-sheet breaker, RS-0406, reverses amyloid beta-induced cytotoxicity and impairment of long-term potentiation in vitro, Br J Pharmacol 137(5) (2002) 676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Walsh DM, Townsend M, Podlisny MB, Shankar GM, Fadeeva JV, El Agnaf O, Hartley DM, Selkoe DJ, Certain inhibitors of synthetic amyloid beta-peptide (Abeta) fibrillogenesis block oligomerization of natural Abeta and thereby rescue long-term potentiation, J Neurosci 25(10) (2005) 2455–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P, Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse, Nature 400(6740) (1999) 173–7. [DOI] [PubMed] [Google Scholar]
- [5].Wu J, Anwyl R, Rowan MJ, beta-Amyloid-(1–40) increases long-term potentiation in rat hippocampus in vitro, Eur J Pharmacol 284(3) (1995) R1–3. [DOI] [PubMed] [Google Scholar]
- [6].Kowalska MA, Badellino K, beta-Amyloid protein induces platelet aggregation and supports platelet adhesion, Biochemical and biophysical research communications 205(3) (1994) 1829–35. [DOI] [PubMed] [Google Scholar]
- [7].Mattson MP, Guo ZH, Geiger JD, Secreted form of amyloid precursor protein enhances basal glucose and glutamate transport and protects against oxidative impairment of glucose and glutamate transport in synaptosomes by a cyclic GMP-mediated mechanism, J Neurochem 73(2) (1999) 532–7. [DOI] [PubMed] [Google Scholar]
- [8].Binder LI, Frankfurter A, Rebhun LI, The distribution of tau in the mammalian central nervous system, J Cell Biol 101(4) (1985) 1371–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Guzman-Martinez L, Farias GA, Maccioni RB, Emerging noninvasive biomarkers for early detection of Alzheimer’s disease, Arch Med Res 43(8) (2012) 663–6. [DOI] [PubMed] [Google Scholar]
- [10].Maccioni RB, Cambiazo V, Role of microtubule-associated proteins in the control of microtubule assembly, Physiological reviews 75(4) (1995) 835–64. [DOI] [PubMed] [Google Scholar]
- [11].Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP, Molecular motors implicated in the axonal transport of tau and alpha-synuclein, J Cell Sci 118(Pt 20) (2005) 4645–54. [DOI] [PubMed] [Google Scholar]
- [12].Cuchillo-Ibanez I, Seereeram A, Byers HL, Leung KY, Ward MA, Anderton BH, Hanger DP, Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin, FASEB J 22(9) (2008) 3186–95. [DOI] [PubMed] [Google Scholar]
- [13].Reddy PH, Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease, Brain Res 1415 (2011) 136–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, Misgeld T, Brion JP, Anderton B, Hanger DP, Coleman MP, Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse, Neurobiol Aging 33(3) (2012) 621 e1–621 e15. [DOI] [PubMed] [Google Scholar]
- [15].Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, Wolfing H, Chieng BC, Christie MJ, Napier IA, Eckert A, Staufenbiel M, Hardeman E, Gotz J, Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models, Cell 142(3) (2010) 387–97. [DOI] [PubMed] [Google Scholar]
- [16].Mondragon-Rodriguez S, Trillaud-Doppia E, Dudilot A, Bourgeois C, Lauzon M, Leclerc N, Boehm J, Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation, The Journal of biological chemistry 287(38) (2012) 32040–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Trepanier CH, Jackson MF, MacDonald JF, Regulation of NMDA receptors by the tyrosine kinase Fyn, FEBS J 279(1) (2012) 12–9. [DOI] [PubMed] [Google Scholar]
- [18].Moreno H, Choi S, Yu E, Brusco J, Avila J, Moreira JE, Sugimori M, Llinas RR, Blocking Effects of Human Tau on Squid Giant Synapse Transmission and Its Prevention by T-817 MA, Front Synaptic Neurosci 3 (2011) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH, Motile axonal mitochondria contribute to the variability of presynaptic strength, Cell Rep 4(3) (2013) 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Arancio O, Kiebler M, Lee CJ, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD, Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiationin cultured hippocampal neurons, Cell 87(6) (1996) 1025–1035. [DOI] [PubMed] [Google Scholar]
- [21].Paakkari I, Lindsberg P, Nitric oxide in the central nervous system, Annals of medicine 27(3) (1995) 369–377. [DOI] [PubMed] [Google Scholar]
- [22].Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AMG, Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity, Nature Reviews Neuroscience 8(10) (2007) 766. [DOI] [PubMed] [Google Scholar]
- [23].Nakamura T, Tu S, Akhtar MW, Sunico CR, S.-i. Okamoto, S.A. Lipton, Aberrant protein s-nitrosylation in neurodegenerative diseases, Neuron 78(4) (2013) 596–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Garthwaite J, Boulton C, Nitric oxide signaling in the central nervous system, Annual review of physiology 57(1) (1995) 683–706. [DOI] [PubMed] [Google Scholar]
- [25].Lu Y-F, Kandel ER, Hawkins RD, Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus, Journal of Neuroscience 19(23) (1999) 10250–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Silva AJ, Kogan JH, Frankland PW, Kida S, CREB and memory, Annual review of neuroscience 21(1) (1998) 127–148. [DOI] [PubMed] [Google Scholar]
- [27].Yin JC, Tully T, CREB and the formation of long-term memory, Current opinion in neurobiology 6(2) (1996) 264–268. [DOI] [PubMed] [Google Scholar]
- [28].Kawasaki K, Smith RS, Hsieh C-M, Sun J, Chao J, Liao JK, Activation of the phosphatidylinositol 3-kinase/protein kinase Akt pathway mediates nitric oxide-induced endothelial cell migration and angiogenesis, Molecular and cellular biology 23(16) (2003) 5726–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Keynes RG, Garthwaite J, Nitric oxide and its role in ischaemic brain injury, Current molecular medicine 4(2) (2004) 179–191. [DOI] [PubMed] [Google Scholar]
- [30].Arancio O, Kandel ER, Hawkins RD, Activity-dependent long-term enhancement of transmitter release by presynaptic 3’,5’-cyclic GMP in cultured hippocampal neurons, Nature 376(6535) (1995) 74–80. [DOI] [PubMed] [Google Scholar]
- [31].Contestabile A, Ciani E, Role of nitric oxide in the regulation of neuronal proliferation, survival and differentiation, Neurochemistry international 45(6) (2004) 903–914. [DOI] [PubMed] [Google Scholar]
- [32].Riccio A, Alvania RS, Lonze BE, Ramanan N, Kim T, Huang Y, Dawson TM, Snyder SH, Ginty DD, A nitric oxide signaling pathway controls CREB-mediated gene expression in neurons, Molecular cell 21(2) (2006) 283–294. [DOI] [PubMed] [Google Scholar]
- [33].Edwards T, Rickard N, New perspectives on the mechanisms through which nitric oxide may affect learning and memory processes, Neuroscience & Biobehavioral Reviews 31(3) (2007) 413–425. [DOI] [PubMed] [Google Scholar]
- [34].Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH, Protein S-nitrosylation: a physiological signal for neuronal nitric oxide, Nature cell biology 3(2) (2001) 193–197. [DOI] [PubMed] [Google Scholar]
- [35].Choi Y-B, Tenneti L, Le DA, Ortiz J, Bai G, Chen H-SV, Lipton SA, Molecular basis of NMDA receptor-coupled ion channel modulation by S-nitrosylation, Nature neuroscience 3(1) (2000) 15–21. [DOI] [PubMed] [Google Scholar]
- [36].Alderton WK, Cooper CE, Knowles RG, Nitric oxide synthases: structure, function and inhibition, Biochemical journal 357(3) (2001) 593–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Guix F, Uribesalgo I, Coma M, Munoz F, The physiology and pathophysiology of nitric oxide in the brain, Progress in neurobiology 76(2) (2005) 126–152. [DOI] [PubMed] [Google Scholar]
- [38].Malinski T, Nitric oxide and nitroxidative stress in Alzheimer’s disease, Journal of Alzheimer’s disease 11(2) (2007) 207–218. [DOI] [PubMed] [Google Scholar]
- [39].Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O, Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity, Journal of Neuroscience 25(29) (2005) 6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Acquarone E, Argyrousi EK, van den Berg M, Gulisano W, Fà M, Staniszewski A, Calcagno E, Zuccarello E, D’Adamio L, Deng S-X, Synaptic and memory dysfunction induced by tau oligomers is rescued by up-regulation of the nitric oxide cascade, Molecular Neurodegeneration 14(1) (2019) 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Law A, Gauthier S, Quirion R, Say NO to Alzheimer’s disease: the putative links between nitric oxide and dementia of the Alzheimer’s type, Brain research reviews 35(1) (2001) 73–96. [DOI] [PubMed] [Google Scholar]
- [42].Suzanne M, Bloch KD, Aberrant expression of the constitutive endothelial nitric oxide synthase gene in Alzheimer disease, Molecular and chemical neuropathology 30(1–2) (1997) 139–159. [DOI] [PubMed] [Google Scholar]
- [43].De la Monte S, Lu B, Sohn Y, Etienne D, Kraft J, Ganju N, Wands J, Aberrant expression of nitric oxide synthase III in Alzheimer’s disease: relevance to cerebral vasculopathy and neurodegeneration, Neurobiology of aging 21(2) (2000) 309–319. [DOI] [PubMed] [Google Scholar]
- [44].Zhao Q-F, Yu J-T, Tan L, S-Nitrosylation in Alzheimer’s disease, Molecular neurobiology 51(1) (2015) 268–280. [DOI] [PubMed] [Google Scholar]
- [45].Wallace MN, Geddes JG, Farquhar DA, Masson MR, Nitric oxide synthase in reactive astrocytes adjacent to β-amyloid plaques, Experimental neurology 144(2) (1997) 266–272. [DOI] [PubMed] [Google Scholar]
- [46].Lüth H-J, Holzer M, Gärtner U, Staufenbiel M, Arendt T, Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: evidence for an induction by amyloid pathology, Brain research 913(1) (2001) 57–67. [DOI] [PubMed] [Google Scholar]
- [47].Haas J, Storch-Hagenlocher B, Biessmann A, Wildemann B, Inducible nitric oxide synthase and argininosuccinate synthetase: co-induction in brain tissue of patients with Alzheimer’s dementia and following stimulation with β-amyloid 1–42 in vitro, Neuroscience letters 322(2) (2002) 121–125. [DOI] [PubMed] [Google Scholar]
- [48].Chakroborty S, Kim J, Schneider C, West AR, Stutzmann GE, Nitric oxide signaling is recruited as a compensatory mechanism for sustaining synaptic plasticity in Alzheimer’s disease mice, Journal of Neuroscience 35(17) (2015) 6893–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Law A, O’donnell J, Gauthier S, Quirion R, Neuronal and inducible nitric oxide synthase expressions and activities in the hippocampi and cortices of young adult, aged cognitively unimpaired, and impaired Long–Evans rats, Neuroscience 112(2) (2002) 267–275. [DOI] [PubMed] [Google Scholar]
- [50].Lüth H-J, Münch G, Arendt T, Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation, Brain research 953(1–2) (2002) 135–143. [DOI] [PubMed] [Google Scholar]
- [51].Koppal T, Drake J, Yatin S, Jordan B, Varadarajan S, Bettenhausen L, Butterfield DA, Peroxynitrite-induced alterations in synaptosomal membrane proteins: insight into oxidative stress in Alzheimer’s disease, Journal of neurochemistry 72(1) (1999) 310–317. [DOI] [PubMed] [Google Scholar]
- [52].Austin SA, Santhanam AV, Hinton DJ, Choi DS, Katusic ZS, Endothelial nitric oxide deficiency promotes Alzheimer’s disease pathology, Journal of neurochemistry 127(5) (2013) 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Austin SA, Santhanam AV, Katusic ZS, Endothelial nitric oxide modulates expression and processing of amyloid precursor protein, Circulation research 107(12) (2010) 1498–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Colton C, Vitek M, Wink D, Xu Q, Cantillana V, Previti M, Van Nostrand W, Weinberg J, Dawson H, NO synthase 2 (NOS2) deletion promotes multiple pathologies in a mouse model of Alzheimer’s disease, Proceedings of the National Academy of Sciences 103(34) (2006) 12867–12872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kwak Y-D, Wang R, Li JJ, Zhang Y-W, Xu H, Liao F-F, Differential regulation of BACE1 expression by oxidative and nitrosative signals, Molecular neurodegeneration 6(1) (2011) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Reynolds MR, Berry RW, Binder LI, Site-specific nitration and oxidative dityrosine bridging of the τ protein by peroxynitrite: implications for Alzheimer’s disease, Biochemistry 44(5) (2005) 1690–1700. [DOI] [PubMed] [Google Scholar]
- [57].Chen F, David D, Ferrari A, Gotz J, Posttranslational modifications of tau-Role in human tauopathies and modeling in transgenic animals, Current drug targets 5(6) (2004) 503–515. [DOI] [PubMed] [Google Scholar]
- [58].Iqbal K, Alonso A.d.C., Chen S, Chohan MO, El-Akkad E, Gong C-X, Khatoon S, Li B, Liu F, Rahman A, Tau pathology in Alzheimer disease and other tauopathies, Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1739(2–3) (2005) 198–210. [DOI] [PubMed] [Google Scholar]
- [59].Vitolo OV, Sant’Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M, Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling, Proceedings of the National Academy of Sciences 99(20) (2002) 13217–13221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Müller M, Cárdenas C, Mei L, Cheung K-H, Foskett JK, Constitutive cAMP response element binding protein (CREB) activation by Alzheimer’s disease presenilin-driven inositol trisphosphate receptor (InsP3R) Ca2+ signaling, Proceedings of the National Academy of Sciences 108(32) (2011) 13293–13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Teich AF, Nicholls RE, Puzzo D, Fiorito J, Purgatorio R, Arancio O, Synaptic therapy in Alzheimer’s disease: a CREB-centric approach, Neurotherapeutics 12(1) (2015) 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Bartolotti N, Segura L, Lazarov O, Diminished CRE-induced plasticity is linked to memory deficits in familial Alzheimer’s disease mice, Journal of Alzheimer’s Disease 50(2) (2016) 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Bartolotti N, Bennett D, Lazarov O, Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells, Molecular psychiatry 21(9) (2016) 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Yamamoto M, Ozawa H, Saito T, Hatta S, Riederer P, Takahata N, Ca 2+/CaM-sensitive adenylyl cyclase activity is decreased in the Alzheimer’s brain: possible relation to type I adenylyl cyclase, Journal of neural transmission 104(6–7) (1997) 721–732. [DOI] [PubMed] [Google Scholar]
- [65].Yamamoto M, Ozawa H, Saito T, Frölich L, Riederer P, Takahata N, Reduced immunoreactivity of adenylyl cyclase in dementia of the Alzheimer type, Neuroreport 7(18) (1996) 2965–2970. [DOI] [PubMed] [Google Scholar]
- [66].Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P, Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type, Brain research 824(2) (1999) 300–303. [DOI] [PubMed] [Google Scholar]
- [67].Pugazhenthi S, Wang M, Pham S, Sze C-I, Eckman CB, Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons, Molecular neurodegeneration 6(1) (2011) 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Beavo JA, Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms, Physiological reviews 75(4) (1995) 725–48. [DOI] [PubMed] [Google Scholar]
- [69].Pissarnitski D, Phosphodiesterase 5 (PDE 5) inhibitors for the treatment of male erectile disorder: attaining selectivity versus PDE6, Med Res Rev 26(3) (2006) 369–95. [DOI] [PubMed] [Google Scholar]
- [70].Bischoff E, Potency, selectivity, and consequences of nonselectivity of PDE inhibition, Int J Impot Res 16 Suppl 1 (2004) S11–4. [DOI] [PubMed] [Google Scholar]
- [71].Gibson A, Phosphodiesterase 5 inhibitors and nitrergic transmission-from zaprinast to sildenafil, Eur J Pharmacol 411(1–2) (2001) 1–10. [DOI] [PubMed] [Google Scholar]
- [72].Goldstein I, Lue TF, Padma-Nathan H, Rosen RC, Steers WD, Wicker PA, Oral sildenafil in the treatment of erectile dysfunction. Sildenafil Study Group, N Engl J Med 338(20) (1998) 1397–404. [DOI] [PubMed] [Google Scholar]
- [73].Fiorito J, Saeed F, Zhang H, Staniszewski A, Feng Y, Francis YI, Rao S, Thakkar DM, Deng SX, Landry DW, Arancio O, Synthesis of quinoline derivatives: discovery of a potent and selective phosphodiesterase 5 inhibitor for the treatment of Alzheimer’s disease, Eur J Med Chem 60 (2013) 285–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Hong JH, Kwon YS, Kim IY, Pharmacodynamics, pharmacokinetics and clinical efficacy of phosphodiesterase-5 inhibitors, Expert Opin Drug Metab Toxicol 13(2) (2017) 183–192. [DOI] [PubMed] [Google Scholar]
- [75].Puzzo D, Staniszewski A, Deng SX, Privitera L, Leznik E, Liu S, Zhang H, Feng Y, Palmeri A, Landry DW, Arancio O, Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-beta load in an Alzheimer’s disease mouse model, J Neurosci 29(25) (2009) 8075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Fiorito J, Vendome J, Saeed F, Staniszewski A, Zhang H, Yan S, Deng SX, Arancio O, Landry DW, Identification of a Novel 1,2,3,4-Tetrahydrobenzo[b][1,6]naphthyridine Analogue as a Potent Phosphodiesterase 5 Inhibitor with Improved Aqueous Solubility for the Treatment of Alzheimer’s Disease, J Med Chem 60(21) (2017) 8858–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sheng R, Tang L, Jiang L, Hong L, Shi Y, Zhou N, Hu Y, Novel 1-Phenyl-3-hydroxy-4-pyridinone Derivatives as Multifunctional Agents for the Therapy of Alzheimer’s Disease, ACS Chem Neurosci 7(1) (2016) 69–81. [DOI] [PubMed] [Google Scholar]
- [78].Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, Jain G, Capece V, Burkhardt S, Navarro-Sala M, Nagarajan S, Schutz AL, Johnsen SA, Bonn S, Luhrmann R, Dean C, Fischer A, HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models, The Journal of clinical investigation 125(9) (2015) 3572–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Graff J, Tsai LH, The potential of HDAC inhibitors as cognitive enhancers, Annu Rev Pharmacol Toxicol 53 (2013) 311–30. [DOI] [PubMed] [Google Scholar]
- [80].Cuadrado-Tejedor M, Garcia-Barroso C, Sanchez-Arias JA, Rabal O, Perez-Gonzalez M, Mederos S, Ugarte A, Franco R, Segura V, Perea G, Oyarzabal J, Garcia-Osta A, A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer’s Disease Mice, Neuropsychopharmacology 42(2) (2017) 524–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Rabal O, Sanchez-Arias JA, Cuadrado-Tejedor M, de Miguel I, Perez-Gonzalez M, Garcia-Barroso C, Ugarte A, Estella-Hermoso de Mendoza A, Saez E, Espelosin M, Ursua S, Haizhong T, Wei W, Musheng X, Garcia-Osta A, Oyarzabal J, Design, synthesis, biological evaluation and in vivo testing of dual phosphodiesterase 5 (PDE5) and histone deacetylase 6 (HDAC6)-selective inhibitors for the treatment of Alzheimer’s disease, Eur J Med Chem 150 (2018) 506–524. [DOI] [PubMed] [Google Scholar]
- [82].Mao F, Wang H, Ni W, Zheng X, Wang M, Bao K, Ling D, Li X, Xu Y, Zhang H, Li J, Design, Synthesis, and Biological Evaluation of Orally Available First-Generation Dual-Target Selective Inhibitors of Acetylcholinesterase (AChE) and Phosphodiesterase 5 (PDE5) for the Treatment of Alzheimer’s Disease, ACS Chem Neurosci 9(2) (2018) 328–345. [DOI] [PubMed] [Google Scholar]
- [83].Ni W, Wang H, Li X, Zheng X, Wang M, Zhang J, Gong Q, Ling D, Mao F, Zhang H, Li J, Novel Tadalafil Derivatives Ameliorates Scopolamine-Induced Cognitive Impairment in Mice via Inhibition of Acetylcholinesterase (AChE) and Phosphodiesterase 5 (PDE5), ACS Chem Neurosci 9(7) (2018) 1625–1636. [DOI] [PubMed] [Google Scholar]
- [84].Li WQ, Qureshi AA, Robinson KC, Han J, Sildenafil use and increased risk of incident melanoma in US men: a prospective cohort study, JAMA Intern Med 174(6) (2014) 964–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Loeb S, Folkvaljon Y, Lambe M, Robinson D, Garmo H, Ingvar C, Stattin P, Use of Phosphodiesterase Type 5 Inhibitors for Erectile Dysfunction and Risk of Malignant Melanoma, JAMA : the journal of the American Medical Association 313(24) (2015) 2449–55. [DOI] [PubMed] [Google Scholar]
- [86].Lian Y, Yin H, Pollak MN, Carrier S, Platt RW, Suissa S, Azoulay L, Phosphodiesterase Type 5 Inhibitors and the Risk of Melanoma Skin Cancer, European urology 70(5) (2016) 808–815. [DOI] [PubMed] [Google Scholar]
- [87].Matthews A, Langan SM, Douglas IJ, Smeeth L, Bhaskaran K, Phosphodiesterase Type 5 Inhibitors and Risk of Malignant Melanoma: Matched Cohort Study Using Primary Care Data from the UK Clinical Practice Research Datalink, PLoS Med 13(6) (2016) e1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pottegard A, Schmidt SA, Olesen AB, Achacoso N, Van Den Eeden SK, Hallas J, Sorensen HT, Friis S, Habel LA, Use of sildenafil or other phosphodiesterase inhibitors and risk of melanoma, Br J Cancer 115(7) (2016) 895–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Wang JZ, Le S, Alexanian C, Boddu S, Merleev A, Marusina A, Maverakis E, No Causal Link between Phosphodiesterase Type 5 Inhibition and Melanoma, World J Mens Health 37(3) (2019) 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Feng S, Zhou L, Liu Q, He Q, Liao B, Wei X, Li H, Wang K, Zhu Y, Are phosphodiesterase type 5 inhibitors associated with increased risk of melanoma?: A systematic review and meta-analysis, Medicine (Baltimore) 97(3) (2018) e9601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Champion HC, Bivalacqua TJ, Takimoto E, Kass DA, Burnett AL, Phosphodiesterase-5A dysregulation in penile erectile tissue is a mechanism of priapism, Proceedings of the National Academy of Sciences of the United States of America 102(5) (2005) 1661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Burnett AL, Bivalacqua TJ, Champion HC, Musicki B, Long-term oral phosphodiesterase 5 inhibitor therapy alleviates recurrent priapism, Urology 67(5) (2006) 1043–8. [DOI] [PubMed] [Google Scholar]
- [93].Rajfer J, Gore JL, Kaufman J, Gonzalez-Cadavid N, Case report: Avoidance of palpable corporal fibrosis due to priapism with upregulators of nitric oxide, J Sex Med 3(1) (2006) 173–6. [DOI] [PubMed] [Google Scholar]
- [94].Basun H, Nilsberth C, Eckman C, Lannfelt L, Younkin S, Plasma levels of Abeta42 and Abeta40 in Alzheimer patients during treatment with the acetylcholinesterase inhibitor tacrine, Dementia and geriatric cognitive disorders 14(3) (2002) 156–60. [DOI] [PubMed] [Google Scholar]
- [95].Andreasen N, Sjogren M, Blennow K, CSF markers for Alzheimer’s disease: total tau, phospho-tau and Abeta42, World J Biol Psychiatry 4(4) (2003) 147–55. [DOI] [PubMed] [Google Scholar]
- [96].Suhara T, Magrane J, Rosen K, Christensen R, Kim HS, Zheng B, McPhie DL, Walsh K, Querfurth H, Abeta42 generation is toxic to endothelial cells and inhibits eNOS function through an Akt/GSK-3beta signaling-dependent mechanism, Neurobiol Aging 24(3) (2003) 437–51. [DOI] [PubMed] [Google Scholar]
- [97].Gentile MT, Vecchione C, Maffei A, Aretini A, Marino G, Poulet R, Capobianco L, Selvetella G, Lembo G, Mechanisms of soluble beta-amyloid impairment of endothelial function, The Journal of biological chemistry 279(46) (2004) 48135–42. [DOI] [PubMed] [Google Scholar]
- [98].Price JM, Hellermann A, Hellermann G, Sutton ET, Aging enhances vascular dysfunction induced by the Alzheimer’s peptide beta-amyloid, Neurol Res 26(3) (2004) 305–11. [DOI] [PubMed] [Google Scholar]
- [99].Pasquier F, Leys D, [Blood pressure and Alzheimer’s disease], Rev Neurol (Paris) 154(11) (1998) 743–51. [PubMed] [Google Scholar]
- [100].Khalil Z, Poliviou H, Maynard CJ, Beyreuther K, Masters CL, Li QX, Mechanisms of peripheral microvascular dysfunction in transgenic mice overexpressing the Alzheimer’s disease amyloid Abeta protein, J Alzheimers Dis 4(6) (2002) 467–78. [DOI] [PubMed] [Google Scholar]
- [101].Smith CC, Stanyer L, Betteridge DJ, Soluble beta-amyloid (A beta) 40 causes attenuation or potentiation of noradrenaline-induced vasoconstriction in rats depending upon the concentration employed, Neurosci Lett 367(1) (2004) 129–32. [DOI] [PubMed] [Google Scholar]
- [102].Kalaria RN, Vascular factors in Alzheimer’s disease, Int Psychogeriatr 15 Suppl 1 (2003) 47–52. [DOI] [PubMed] [Google Scholar]
- [103].Prickaerts J, Steinbusch HW, Smits JF, de Vente J, Possible role of nitric oxide-cyclic GMP pathway in object recognition memory: effects of 7-nitroindazole and zaprinast, European journal of pharmacology 337(2–3) (1997) 125–136. [DOI] [PubMed] [Google Scholar]
- [104].Domek-Łopacińska K, Strosznajder J, The effect of selective inhibition of cyclic GMP hydrolyzing phosphodiesterases 2 and 5 on learning and memory processes and nitric oxide synthase activity in brain during aging, Brain research 1216 (2008) 68–77. [DOI] [PubMed] [Google Scholar]
- [105].Bender AT, Beavo JA, Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use, Pharmacological reviews 58(3) (2006) 488–520. [DOI] [PubMed] [Google Scholar]
- [106].Deumens R, Blokland A, Prickaerts J, Modeling Parkinson’s disease in rats: an evaluation of 6-OHDA lesions of the nigrostriatal pathway, Exp Neurol 175(2) (2002) 303–17. [DOI] [PubMed] [Google Scholar]
- [107].Prickaerts J, Sik A, van der Staay FJ, de Vente J, Blokland A, Dissociable effects of acetylcholinesterase inhibitors and phosphodiesterase type 5 inhibitors on object recognition memory: acquisition versus consolidation, Psychopharmacology (Berl) 177(4) (2005) 381–90. [DOI] [PubMed] [Google Scholar]
- [108].Rutten K, De Vente J, Şik A, Markerink-Van Ittersum M, Prickaerts J, Blokland A, The selective PDE5 inhibitor, sildenafil, improves object memory in Swiss mice and increases cGMP levels in hippocampal slices, Behavioural brain research 164(1) (2005) 11–16. [DOI] [PubMed] [Google Scholar]
- [109].Singh N, Parle M, Sildenafil improves acquisition and retention of memory in mice, Indian J Physiol Pharmacol 47(3) (2003) 318–24. [PubMed] [Google Scholar]
- [110].Baratti C, Boccia M, Effects of sildenafil on long-term retention of an inhibitory avoidance response in mice, Behavioural pharmacology 10(8) (1999) 731–737. [DOI] [PubMed] [Google Scholar]
- [111].Patil CS, Jain NK, Singh VP, Kulkarni SK, Differential effect of the PDE5 inhibitors, sildenafil and zaprinast, in aging-and lipopolysaccharide-induced cognitive dysfunction in mice, Drug development research 63(2) (2004) 66–75. [Google Scholar]
- [112].Shafiei M, Mahmoudian M, Rostami P, Nemati F, Effect of sildenafil (Viagra) on memory retention of a passive avoidance response in rats, Acta Physiologica Hungarica 93(1) (2006) 53–59. [DOI] [PubMed] [Google Scholar]
- [113].Palmeri A, Privitera L, Giunta S, Loreto C, Puzzo D, Inhibition of phosphodiesterase-5 rescues age-related impairment of synaptic plasticity and memory, Behav Brain Res 240 (2013) 11–20. [DOI] [PubMed] [Google Scholar]
- [114].Cuadrado-Tejedor M, Hervias I, Ricobaraza A, Puerta E, Perez-Roldan JM, Garcia-Barroso C, Franco R, Aguirre N, Garcia-Osta A, Sildenafil restores cognitive function without affecting beta-amyloid burden in a mouse model of Alzheimer’s disease, Br J Pharmacol 164(8) (2011) 2029–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Zhang J, Guo J, Zhao X, Chen Z, Wang G, Liu A, Wang Q, Zhou W, Xu Y, Wang C, Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice, Behav Brain Res 250 (2013) 230–7. [DOI] [PubMed] [Google Scholar]
- [116].Devan BD, Bowker JL, Duffy KB, Bharati IS, Jimenez M, Sierra-Mercado D Jr., Nelson CM, Spangler EL, Ingram DK, Phosphodiesterase inhibition by sildenafil citrate attenuates a maze learning impairment in rats induced by nitric oxide synthase inhibition, Psychopharmacology (Berl) 183(4) (2006) 439–45. [DOI] [PubMed] [Google Scholar]
- [117].Devan BD, Pistell PJ, Daffin LW Jr., Nelson CM, Duffy KB, Bowker JL, Bharati IS, Sierra-Mercado D, Spangler EL, Ingram DK, Sildenafil citrate attenuates a complex maze impairment induced by intracerebroventricular infusion of the NOS inhibitor Nomega-nitro-L-arginine methyl ester, Eur J Pharmacol 563(1–3) (2007) 134–40. [DOI] [PubMed] [Google Scholar]
- [118].Rutten K, Basile J, Prickaerts J, Blokland A, Vivian J, Selective PDE inhibitors rolipram and sildenafil improve object retrieval performance in adult cynomolgus macaques, Psychopharmacology 196(4) (2008) 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Devan BD, Pistell PJ, Duffy KB, Kelley-Bell B, Spangler EL, Ingram DK, Phosphodiesterase inhibition facilitates cognitive restoration in rodent models of age-related memory decline, NeuroRehabilitation 34(1) (2014) 101–11. [DOI] [PubMed] [Google Scholar]
- [120].Garcia-Barroso C, Ricobaraza A, Pascual-Lucas M, Unceta N, Rico AJ, Goicolea MA, Salles J, Lanciego JL, Oyarzabal J, Franco R, Cuadrado-Tejedor M, Garcia-Osta A, Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD, Neuropharmacology 64 (2013) 114–23. [DOI] [PubMed] [Google Scholar]
- [121].Argyrousi EK, Heckman PR, van Hagen BT, Muysers H, van Goethem NP, Prickaerts J, Pro-cognitive effect of upregulating cyclic guanosine monophosphate signalling during memory acquisition or early consolidation is mediated by increased AMPA receptor trafficking, J Psychopharmacol (2019) 269881119885262. [DOI] [PMC free article] [PubMed]
- [122].Jin F, Gong QH, Xu YS, Wang LN, Jin H, Li F, Li LS, Ma YM, Shi JS, Icariin, a phosphodiesterase-5 inhibitor, improves learning and memory in APP/PS1 transgenic mice by stimulation of NO/cGMP signalling, The international journal of neuropsychopharmacology / official scientific journal of the Collegium Internationale Neuropsychopharmacologicum (CINP) 17(6) (2014) 871–81. [DOI] [PubMed] [Google Scholar]
- [123].Zhu L, Yang JY, Xue X, Dong YX, Liu Y, Miao FR, Wang YF, Xue H, Wu CF, A novel phosphodiesterase-5 Inhibitor: Yonkenafil modulates neurogenesis, gliosis to improve cognitive function and ameliorates amyloid burden in an APP/PS1 transgenic mice model, Mech Ageing Dev 150 (2015) 34–45. [DOI] [PubMed] [Google Scholar]
- [124].Teich AF, Sakurai M, Patel M, Holman C, Saeed F, Fiorito J, Arancio O, PDE5 Exists in Human Neurons and is a Viable Therapeutic Target for Neurologic Disease, J Alzheimers Dis (2016). [DOI] [PMC free article] [PubMed]
- [125].Rehse K, Scheffler H, Reitner N, Interaction of Viagra with the NO donors molsidomine and RE 2047 with regard to antithrombotic and blood pressure lowering activities, Archiv der Pharmazie: An International Journal Pharmaceutical and Medicinal Chemistry 332(5) (1999) 182–184. [DOI] [PubMed] [Google Scholar]
- [126].Arnavaz A, Aurich A, Weißenborn K, Hartmann U, Emrich HM, Schneider U, Effect of sildenafil (Viagra®) on cerebral blood flow velocity: a pilot study, Psychiatry Research: Neuroimaging 122(3) (2003) 207–209. [DOI] [PubMed] [Google Scholar]
- [127].Kruuse C, Hansen AE, Larsson HB, Lauritzen M, Rostrup E, Cerebral haemodynamic response or excitability is not affected by sildenafil, Journal of Cerebral Blood Flow & Metabolism 29(4) (2009) 830–839. [DOI] [PubMed] [Google Scholar]
- [128].Kruuse C, Thomsen LL, Birk S, Olesen J, Migraine can be induced by sildenafil without changes in middle cerebral artery diameter, Brain 126(1) (2003) 241–247. [DOI] [PubMed] [Google Scholar]
- [129].Reneerkens OA, Rutten K, Steinbusch HW, Blokland A, Prickaerts J, Selective phosphodiesterase inhibitors: a promising target for cognition enhancement, Psychopharmacology (Berl) 202(1–3) (2009) 419–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Akkerman S, Blokland A, van Goethem NP, Cremers P, Shaffer CL, Osgood SM, Steinbusch HW, Prickaerts J, PDE5 inhibition improves acquisition processes after learning via a central mechanism, Neuropharmacology 97 (2015) 233–9. [DOI] [PubMed] [Google Scholar]
- [131].Grass H, Klotz T, Fathian-Sabet B, Berghaus G, Engelmann U, Kaferstein H, Sildenafil (Viagra): is there an influence on psychological performance?, Int Urol Nephrol 32(3) (2001) 409–12. [DOI] [PubMed] [Google Scholar]
- [132].Schultheiss D, Muller SV, Nager W, Stief CG, Schlote N, Jonas U, Asvestis C, Johannes S, Munte TF, Central effects of sildenafil (Viagra) on auditory selective attention and verbal recognition memory in humans: a study with event-related brain potentials, World J Urol 19(1) (2001) 46–50. [DOI] [PubMed] [Google Scholar]
- [133].Goff DC, Cather C, Freudenreich O, Henderson DC, Evins AE, Culhane MA, Walsh JP, A placebo-controlled study of sildenafil effects on cognition in schizophrenia, Psychopharmacology 202(1–3) (2009) 411–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Safarinejad MR, Taghva A, Shekarchi B, Safarinejad S, Safety and efficacy of sildenafil citrate in the treatment of Parkinson-emergent erectile dysfunction: a double-blind, placebo-controlled, randomized study, Int J Impot Res 22(5) (2010) 325–35. [DOI] [PubMed] [Google Scholar]
- [135].Sheng M, Lu H, Liu P, Li Y, Ravi H, Peng SL, Diaz-Arrastia R, Devous MD, Womack KB, Sildenafil Improves Vascular and Metabolic Function in Patients with Alzheimer’s Disease, J Alzheimers Dis 60(4) (2017) 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Shim YS, Pae CU, Kim SW, Kim HW, Kim JC, Koh JS, Effects of repeated dosing with Udenafil (Zydena) on cognition, somatization and erection in patients with erectile dysfunction: a pilot study, Int J Impot Res 23(3) (2011) 109–14. [DOI] [PubMed] [Google Scholar]
- [137].Shim YS, Pae CU, Cho KJ, Kim SW, Kim JC, Koh JS, Effects of daily low-dose treatment with phosphodiesterase type 5 inhibitor on cognition, depression, somatization and erectile function in patients with erectile dysfunction: a double-blind, placebo-controlled study, Int J Impot Res 26(2) (2014) 76–80. [DOI] [PubMed] [Google Scholar]
- [138].Goldstein I, Tseng L-J, Creanga D, Stecher V, Kaminetsky JC, Efficacy and safety of sildenafil by age in men with erectile dysfunction, The journal of sexual medicine 13(5) (2016) 852–859. [DOI] [PubMed] [Google Scholar]