

Abstract
Many malignant tumors use endogenous nitric oxide (NO) to promote survival, growth, and metastatic migration. This NO, which is typically generated by inducible nitric oxide synthase (iNOS), can also antagonize various anti-cancer therapies and its source is most often assumed to be constitutive or pre-existing iNOS. In this paper, we provide evidence (i) that many different cancer cells exhibit resistance to oxidative killing by photodynamic therapy (PDT), and (ii) that cells surviving the challenge grow, migrate and invade more aggressively, as do non-targeted bystander cells. Accompanying these effects are activation or upregulation of pro-survival/progression effector proteins such as NF-κB, Akt, and Survivin. Observed in the author’s laboratory, these responses were not attributed to basal iNOS/NO in most cases, but rather to NO from enzyme that was strongly upregulated by photodynamic stress. Each of these effects and how they can be mitigated by inhibitors of iNOS activity or transcription, or by NO scavengers will be discussed. When approved for clinical use, such pharmacologic agents could improve PDT efficacy as well as reduce potentially negative side-effects of this therapy.
Keywords: Photodynamic Therapy, Cancer, Nitric Oxide, iNOS, iNOS Inhibitors
1. Introduction
Photodynamic therapy (PDT) was introduced in the mid-1970s as an innovative new approach for selectively eradicating solid tumors in cancer patients with minimal off-target cytotoxicity [1–3]. PDT consists of three basic components: (i) a light-absorbing photosensitizing agent (PS), PS-exciting light in the visible-to-near infrared range, and molecular oxygen [2,3]. All three components (PS, light, and O2) must be engaged concurrently for PDT to be effective, and light delivery via fiber optic channels localizes treatment to the tumor site [3]. Thus, most PS are innocuous until photo-activated. Consequently, unlike many chemotherapies or radiotherapies, PDT has few negative side effects on normal tissues. A typical photodynamic reaction involves energy transfer from PS in the triplet excited state to ground state O2, giving singlet molecular oxygen (1O2), a reactive oxygen species (ROS) [3,4]. 1O2 can kill cancer cells by (i) irreversibly oxidizing vital proteins, lipids or nucleic acids, or (ii) initiating death signaling cascades, many of which are reversible [3,4]. The hematoporphyrin oligomer Photofrin® was the first PS to receive FDA approval for selected clinical use in 1996, and since then has been applied for a variety of solid tumors, including bladder, breast, prostate, and brain malignancies [3]. In addition to preexisting sensitizers like Photofrin®, which are administered as such, pro-sensitizers have been developed. A prominent example is 5-aminolevulinic acid (ALA) or an ester thereof, which is metabolized to protoporphyrin IX (PpIX), the active PS, via the heme biosynthetic pathway [5,6]. Because of pro-growth requirements, this pathway is more active in tumor cells than in normal counterparts. PpIX accumulates initially in mitochondria, making these organelles prominent targets of photodamage, which promotes apoptotic cell death [6].
Many cancers exhibit an innate or acquired resistance to various types of chemotherapy or radiotherapy [7]. It is now clear that resistance mechanisms also exist for PDT, and some of them are like those for other therapeutic modalities. For example, there is evidence that ROS-scavenging enzymes such as superoxide dismutase (SOD1) and glutathione peroxidase-type 1 (GPx1) are upregulated in cancer cells after a PDT-like oxidative challenge [8,9]. Type 4 glutathione peroxidase (GPx4) has also been shown to play a role in cell resistance [10]. A different mode of PDT resistance is exemplified by the ability of many cancer cells to export PpIX and other photosensitizers via the ABCG2 transporter, which has been shown to be upregulated after several rounds of PDT [11]. Another PDT resistance mechanism, which was discovered in the authors’ laboratory, involves nitric oxide generated by inducible nitric oxide synthase (iNOS/NOS2), particularly enzyme that is overexpressed in response to PDT stress [12].
2. Anti-PDT effects of exogenous NO: early evidence
Nitric oxide (NO) is a short-lived bioactive free radical molecule (τ <2 sec) that diffuses freely on its own and tends to partition into hydrophobic environments such as cell membranes [13–15]. NO is produced naturally by three nitric oxide synthase (NOS) enzymes: nNOS (neuronal), iNOS (inducible), and eNOS (endothelial), iNOS being of special interest vis-à-vis cancer. During an immune response, NO produced at relatively high steady state concentrations (0.5–1 μM) by iNOS in activated macrophages is cytotoxic and potentially oncogenic when it reacts with superoxide (O2−) to give peroxynitrite (ONOO−), a strong oxidant [14]. However, at lower levels (0.1–0.5 μM range) NO can play a key role in tumor survival and progression as well as resistance to various therapeutic interventions [14,15].
In early studies carried out in the authors’ laboratory, we asked whether NO might antagonize PDT by inhibiting membrane-damaging lipid peroxidation. Studies by others had already shown that NO could suppress free radical-mediated lipid peroxidation in liposomes and lipoproteins by scavenging oxyl (LO•) and peroxyl (LOO•) radicals arising from 1-electron reduction of lipid hydroperoxides (LOOHs) [16,17]. Rate constants for NO interception of these radicals were reported to be extremely high, viz. 1–3 × 109 M−1s−1 [16]. Using high performance liquid chromatography with mercury cathode electrochemical detection [HPLC-EC(Hg)] to analyze membrane LOOHs, Niziolek et al. [18,19] found that 5α-OOH, a 1O2-generated cholesterol hydroperoxide (ChOOH), accumulated linearly during irradiation of large unilamellar liposomes (LUVs) composed of an unsaturated phospholipid (POPC), cholesterol (Ch), and protoporphyrin (PpIX), the latter serving as a photosensitizing agent (Fig. 1A). The LUV lipid proportions mimicked those found in a typical mammalian cell plasma membrane. At the concentrations used, a lipophilic iron chelate (ferric-8-hydroxyquinoline [Fe(HQ)3]) and the reductant ascorbate (AH−) had no significant effect on 5α-OOH buildup over the fluence (light dose) range used (Fig. 1A). During 5α-OOH formation, another positional hydroperoxide of Ch (7α/7β-OOH) accumulated, but exponentially in this case, and in Fe/AH-dependent manner (Fig. 1B). In accord with previous findings [16,17], these results indicated that 1O2-derived primary hydroperoxides of POPC and Ch (e.g. 5α-OOH) were undergoing light-independent, Fe/AH-induced 1-electron reduction to free radical intermediates which triggered chain lipid peroxidation, as informed by 7α/7β-OOH appearance [18,19]. There was no effect of Fe/AH on 5α-OOH buildup, suggesting that photogeneration was much faster than reductive turnover; however, POPC-OOH competition for Fe/AH could also have been a factor. The chemical NO donor, spermine NONOate (SPNO; t1/2 ~39 min at pH 7.4, 37 °C), had no effect on 5α-OOH formation (Fig. 1A), but dose-dependently inhibited 7α/7β-OOH formation such that at 0.2 mM SPNO and above, none of this epimeric hydroperoxide was observed (Fig. 1B). When fully decomposed SPNO was used (from a stock SPNO solution kept at pH 7.4 overnight), no inhibitory effect was seen, confirming that liberated NO was the active agent. The non-effect of SPNO on 5α-OOH buildup ruled out any possible effect of the donor or NO itself on primary photochemistry in this system, e.g. quenching of photoexcited PpIX or of 1O2 generated by energy transfer from this PpIX [18,19]. The results shown in Fig. 1B are consistent with NO acting as a chain-breaking antioxidant, i.e. intercepting LO• and LOO• intermediates arising from 1-electron reduction of 5α-OOH and other primary LOOHs arising from PpIX-sensitized photooxidation of membrane lipids. In a subsequent study, Niziolek et al. [20] found that at light fluences >80 J/cm2 (cf. Fig. 1A/B), 5α-OOH accumulation slowed progressively with increasing fluence, and also with increasing unsaturation of liposomal phospholipid. SPNO (0.4 mM) prevented this slowdown, allowing undiminished linear accumulation of 5α-OOH throughout irradiation while abolishing 7α/7β-OOH accumulation. In this case, the NO effect was found to be mainly due to protection of PpIX from degradation by free radical peroxidation; thus, its photosensitization lifetime was prolonged [20].
Figure 1.
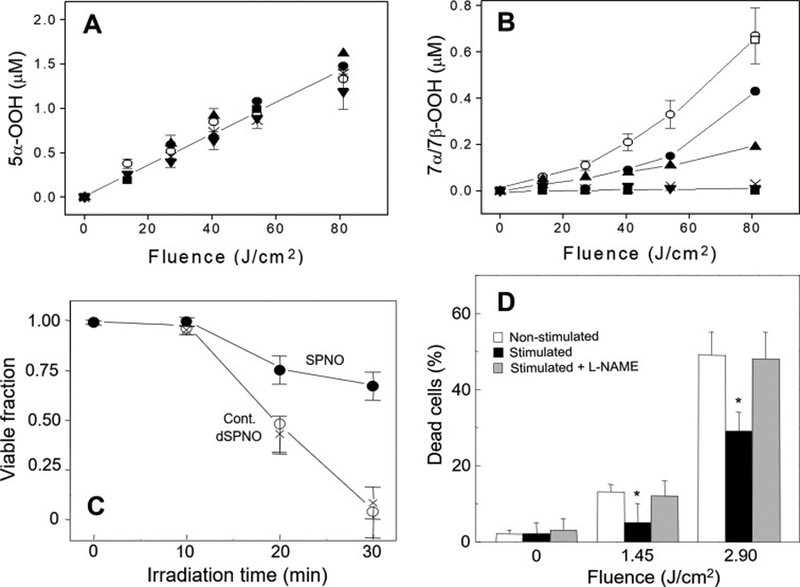
Effect of NO on photosensitized ChOOH formation in liposomes and on necrotic killing of cancer cells. (A) Accumulation of 5α-OOH and (B) 7α/7β-OOH in POPC/Ch/PpIX (100:80:0.2 by mol) liposomes irradiated in the absence vs. presence of 1 mM ascorbate (AH), 0.5 μM ferric-8-hydroxyquinoline (Fe) and spermine-NONOate (SPNO): −Fe/AH (x); +Fe/AH (○); + Fe/AH and 20 μM SPNO (●), 60 μM SPNO (▲), 200 μM SPNO (▼), 400 μM SPNO (■), or decomposed 400 μM SPNO (□). ChOOHs in lipid extracts were determined by HPLC-EC(Hg). (C) Photokilling of disseminated PpIX-sensitized COH-BR1 cells in the absence (○) vs. presence of 400 μM SPNO (●) or decomposed SPNO (x). Light fluence at 30 min irradiation: 2.2 J/cm2. (D) Photokilling of COH-BR1 cells during exposure to non-stimulated or stimulated RAW 264.7 macrophages (15 h after LPS added) in absence vs. presence of 4 mM L-NAME. All values are means ± SEM (n=3). *P<0.005 vs. non-stimulated ± L-NAME. (Reproduced from Refs. 18 and 19, with permission.)
Chain-breaking antioxidant activity of NO could also be demonstrated at the cellular level [18,19]. A breast cancer subline, COH-BR1, was metabolically sensitized with PpIX by incubating with the precursor ALA and allowing most of the PpIX generated to diffuse from mitochondria, where it originates, to the plasma membrane. When these cells were irradiated with broad band visible light, they died mainly via propidium iodide-assessed necrosis after a short lag period (Fig. 1C). Irradiation in the presence of active SPNO resulted in a dramatic reduction in necrotic cell death, e.g. ~32% decrease at 30 min (Fig. 1C). Decomposed SPNO had no protective effect, confirming that NO was the cytoprotective agent. Evidence from accompanying experiments indicated that the NO protection was predominantly due to suppression of post-irradiation chain lipid peroxidation in the plasma membrane of these cells [18,19]. A natural source of NO, viz. iNOS-derived NO from lipopolysaccharide (LPS)-stimulated murine RAW 264.7 macrophages, was also shown to protect cancer cells against photodynamic membrane damage [21]. The RAW cells on microporous inserts were activated with LPS and 15 h later (when superoxide production had subsided) were placed onto wells containing COH-BR1 cells sensitized with ALA-induced PpIX as described for Fig. 1C. After a 1 h interval, the cancer cells were irradiated in the absence vs. presence of L-NAME, non-stimulated RAW cells serving as controls. As shown in Fig. 1D, COH-BR1 cells exposed to stimulated RAW cells were much more resistant to photokilling than controls. This resistance was lost when L-NAME was present, (Fig. 1D), consistent with iNOS-derived NO being the predominant mediator of the resistance. Evidence for NO-inhibitable chain lipid peroxidation was obtained in this system [21], suggesting again that this was the underlying mechanism of increased resistance to photokilling in the presence of NO. The action of NO as a chain-breaking antioxidant in the experiments of Fig. 1 A–D is illustrated in Scheme 1.
Scheme 1.
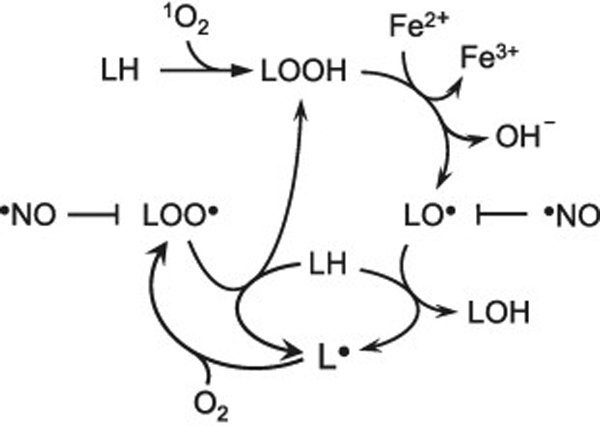
Lipid peroxidation induced by photogenerated 1O2 and amplified by iron-catalyzed reduction of lipid hydroperoxide (LOOH) intermediates. Free radical intermediates that are susceptible to interception/inactivation by NO are indicated.
3. Anti-PDT effects of endogenous NO
The first studies on how endogenous NO might affect PDT efficacy in vivo were carried out about 20 years ago by two separate groups using mouse syngeneic tumors and Photofrin®-sensitized PDT. Focusing on radiation-induced fibrosarcoma (RIF) tumors, Henderson et al. [22] found that PDT outcome was improved when L-NNA (a non-specific inhibitor of NOS activity) was administered before and after irradiation. Shortly thereafter, Korbelik et al. [23] found that PDT suppression of RIF and SCCVII tumors, but not EMT6 counterparts, was greatly improved when L-NNA or L-NAME (another NOS inhibitor) was injected immediately after irradiation. Importantly, the NO output of RIF and SCCVII tumors was much greater than that of EMT6, explaining the greater responsiveness of the former two to the inhibitors [23]. Similar findings were reported more recently by other investigators using mouse syngeneic tumors sensitized with ALA-induced PpIX [24]. The findings in each of these studies were attributed to competing effects of NO and PDT on the tumor microvasculature, i.e. NO-induced vasodilation acting in opposition to tumor ablation via PDT-induced vasoconstriction. Although this early work [22,23] was groundbreaking in identifying NO resistance to PDT in vivo, it left the following important issues unsettled: (i) the cellular source(s) of NO, e.g. tumor cells per se, proximal endothelial cells or macrophages, or possibly contributions from each, (ii) which NOS isoform was the principal source of resistance NO, (iii) whether the NOS/NO acted at constitutive levels or was upregulated by PDT stress, and (iv) the underlying mechanism(s) of NO’s anti-PDT activity.
In addressing these issues, the authors’ group discovered that several types of cancer cells can provide their own cytoprotective NO in response to photodynamic stress. For example, when breast COH-BR1 cells sensitized with ALA-induced PpIX were irradiated (fluence ~1 J/cm2), qRT-PCR analysis revealed a 75% increase in iNOS mRNA relative to dark controls 4 h after irradiation, and this persisted for at least another 16 h [25,26]. Accompanying immunoblot analyses revealed a 2–3-fold upregulation of iNOS protein over the same time period. The other NOS isoforms, nNOS and eNOS, did not change from very low basal levels. In these experiments, the PpIX prior to irradiation was localized mainly in mitochondria. Relatively modest photodynamic challenges were used, resulting in only ~25% cell death via intrinsic apoptosis after 20 h of post-irradiation incubation [25]. When an iNOS activity inhibitor (1400W) or a NO scavenger (cPTIO) was present throughout, or when COH-BR1 cells with shRNA-induced iNOS knockdown were used, a large increase in photosensitized apoptosis was observed [25,26]. Taken together, these findings indicated that NO from basal and/or stress-induced iNOS was eliciting greater stress resistance in these cancer cells.
More recent studies in the authors’ laboratory demonstrated that human prostate carcinoma cells could also exploit iNOS-generated NO for greater resistance to PDT cytotoxicity [27]. Pre-incubation of subconfluent PC3 cells with ALA for 30 min resulted in PpIX accumulation in mitochondria, as shown by the overlap of Mito-Green and PpIX fluorescence (Fig. 2A). When these cells were exposed to a broad band visible light fluence (~1 J/cm2), there was a progressive increase in immunodetectable iNOS during post-hν incubation, its level reaching ~8-times that of the dark control after 20 h (Fig. 2A). This dramatic upregulation of iNOS was reflected in a large increase in NO yield, as detected with the cellular NO fluorescent probe DAF-2DA. At 22 h post-hν, there was a 50-fold increase in fluorescence intensity over that of the dark control, and 1400W strongly inhibited the increase, consistent with iNOS being the major NO source (Fig. 2B). One of the earliest consequences of photostress-induced iNOS in PC3 cells was elevated resistance to photokilling. As shown in Fig. 2C, the viable fraction of ALA/light-challenged cells decreased progressively during post-hν dark incubation, reaching ~0.6 relative to time-0 after 24 h. When 1400W or cPTIO was present, viability loss was significantly greater, indicating that NO was acting cytoprotectively [28], most likely by inhibiting apoptosis, as observed with COH-BR1 cells. Since pre-existing iNOS in PC3 cells accounted for only ~10% of total iNOS at 20 h, the observed cytoprotective effect must have been mainly due to photostress-upregulated iNOS/NO. When viable PC3 cell count was tracked beyond the 24 h post-hν point, a striking effect was observed [28], viz. that surviving cells were proliferating faster than controls over at least two additional days, and in 1400W- and cPTIO-inhibitable fashion (Fig. 2C). This growth spurt was validated when photostressed and control cells were “normalized” to the same starting cell count [28]. To further validate endogenous iNOS/NO’s pro-growth effects on PC3 cells, we showed that an exogenous donor, DETA/NO at a starting concentration of 10 μM and half-life ~20 h at 37 °C [29] also stimulated cell growth significantly [28]. In addition to proliferating more rapidly, PC3 cells that could withstand an ALA/light challenge migrated and invaded more aggressively than controls, as demonstrated by gap-closure (“wound-healing”) and trans-well (Boyden-type) assays, respectively [28]. These responses also exhibited iNOS/NO-dependency. Accelerated invasion (Fig. 2D), was preceded by changes in proteins known to facilitate this process, e.g. activation of matrix metalloproteinase-9 (MMP-9), downregulation of MMP-9 inhibitor TIMP-1, and upregulation of integrins α6 and β1 [28]. Most of the above findings on PC3 cells could be replicated with another prostate cancer line, DU145, suggesting general applicability for PDT-treated prostate cancer [28].
Figure 2.
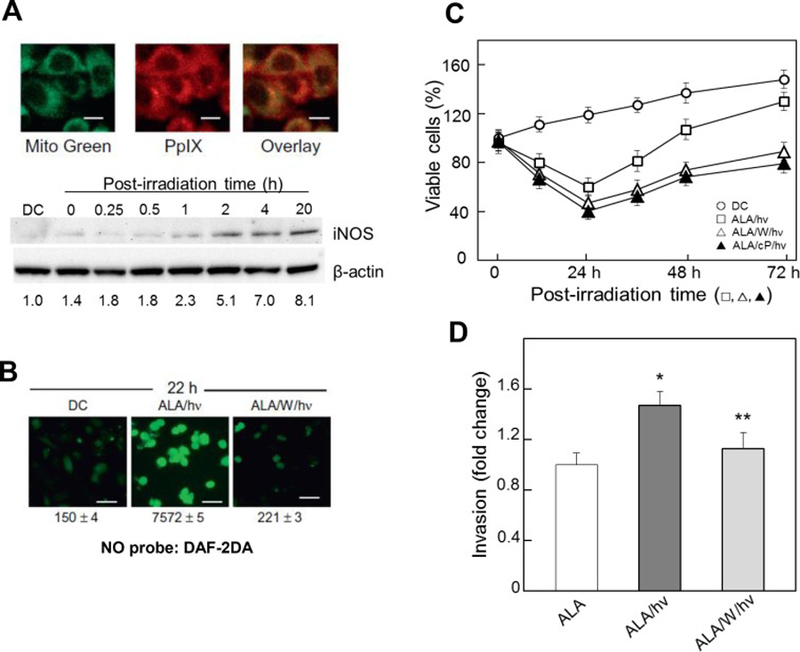
Upregulated iNOS/NO in ALA/light-treated prostate cancer PC3 cells; effects of iNOS/NO on cell resistance and on proliferation and invasion of surviving cells. Cells at ~60% confluency were sensitized with PIX by incubating with 1 mM ALA for 30 min in the dark. (A, upper panels): mitochondrial localization of PpIX. After being switched to ALA-free medium, cells were treated with MitoTracker Green (MTG, 0.1 μg/ml, 10 min), then examined by confocal fluorescence microscopy: PpIX (λex 488 nm, λem 620–650 nm); MTG (λex 490 nm, λem 516 nm). (A, lower panels): iNOS upregulation. AKA-treated cells were either dark-incubated for 20 h (ALA) or irradiated (ALA/hν, ~1 J/cm2). After 0–20 h of post-hν incubation, cells were lysed and analyzed for iNOS by Western blotting. Numbers in lower row indicate iNOS band intensity relative to β-actin and normalized to controls. (B): NO upregulaton. After ALA treatment, cells wither served as dark controls (DC) or were irradiated in absence (ALA/hν) or presence of 20 μM 1400W (ALA/W/hν). After 22 h in dark, all cells were treated with NO probe DAF-2DA (20 μM, 30 min), then examined by fluorescence microscopy (λex 488 nm, λem 650 nm). Numbers indicate integrated fluorescence intensity, means ± SD (n=3). (C): Resistance to photokilling and enhanced proliferation of surviving cells. After irradiation, cells were switched to 10% serum-medium; where indicated, 1400W or cPTIO was present throughout; DC, dark controls (ALA-only). Plotted values are means ± SEM (n=3). (D): Increased invasiveness of viable cells after exposure to ALA alone, ALA/light, or ALA/1400W/light, as assessed with a CytoSelect® system. Plotted values are means ± SEM (n=3); *P<0.01 vs. ALA; **P<0.05 vs. ALA/ hν. (Reproduced from Refs. 27 and 28, with permission.)
Other studies in the authors’ laboratory have shown that iNOS/NO also plays a key role in enhancing resistance of glioblastoma cells to ALA/light-induced killing as well as stimulating migratory and invasive aggressiveness of surviving cells. PDT, particularly ALA-based PDT, has demonstrated considerable promise over conventional chemotherapy and radiotherapy in treating glioblastoma, the most difficult and lethal of all brain malignancies [30]. Despite PDT’s advantages for glioblastoma, limitations exist, a key one being that significant basal iNOS can be expressed in this malignancy, often serving as a marker of poor prognosis [31,32]. Using human glioblastoma U87 and U251 cells, Fahey et al. [33] showed that these cells, when sensitized with ALA-induced PpIX in mitochondria, could increase their iNOS/NO levels after irradiation. These increases were not as great as those in PC3 cells (Fig. 2A), but nevertheless substantial, e.g. up to 4-fold over pre-existing levels over a 20 h post-hν incubation period. Unlike PC3 cells, U87 and U251 cells also expressed significant nNOS, but its level did not change after a photodynamic challenge [33]. Photodynamic apoptosis of U87 and U251 cells was strongly enhanced by 1400W or cPTIO, implying that NO from iNOS (but not nNOS) was signaling for hyper-resistance. Like PC3 cells, U87 cells that survived a photochallenge, proliferated, migrated, and invaded more rapidly than controls, and in 1400W- or cPTIO-inhibitable fashion, again signifying NO involvement. One can predict from these findings that iNOS/NO would be antagonistic to clinical PDT against glioblastoma, and that suppressing this (see Sect. 7) could be crucial for improving PDT efficacy.
Recently, Rapozzi et al. [34] have also asked how NO might influence the efficacy of antitumor PDT. They reported that endogenous iNOS/NO can modulate pheophorbide a-sensitized photokilling of melanoma cells in two distinct ways, depending on PDT intensity. For example, low level NO induced by modest photodynamic pressure signaled for cytoprotection via upregulation of anti-apoptotic NF-κB and Snail, but down-regulation of pro-apoptotic RKIP. However, higher level NO produced by greater photodynamic pressure signaled for less protection through down-regulation of NF-κB and Snail, but upregulation of RKIP [34]. Thus, an intriguing pro- versus anti-PDT signaling role for NO was revealed based on the level of applied PDT pressure. Of related interest is a study by Della Pietra et al. [35] showing that PC3 cells subjected to relatively modest photodynamic action exploit the NF-κB/Snail/RKIP axis and iNOS/NO to proliferate and migrate more aggressively. In these studies [34,35], switching from greater cytoprotection to greater cytotoxicity was attributed to relatively low vs. high NO steady state levels, but the underlying signaling mechanisms remain to be defined.
4. Signaling events leading to upregulation of pro-survival iNOS/NO under PDT stress
How NO might promote PDT resistance by quenching lipid-derived radicals was discussed in Sect. 2. However, this occurs by a chemical reaction that is effectively irreversible rather than by a signaling mechanism, e.g. S-nitrosation of key effector proteins [36], which is typically reversible. Relatively little is known about such signaling in the context of PDT (see above) or any other cancer therapy for that matter. In contrast, there is considerable evidence on the signaling mechanisms that underlie iNOS upregulation in a cancer cell experiencing photodynamic stress. For iNOS induction in at least two cancer lines, breast COH-BR1 and glioblastoma U87, it is now clear that activation of transcription factor NF-κB is required. As supporting evidence for ALA/light-challenged COH-BR1 cells, Bay11–7082, an inhibitor of phosphorylation and proteasomal removal on NF-κB regulatory subunit IκB: (i) prevented translocation of NF-κB subunit p65 to the nucleus for transcriptional activity, (ii) suppressed iNOS upregulation after the photochallenge, and (iii) increased the extent of apoptotic photokilling [37]. These findings established a functional link between photostress activation of NF-κB, greater iNOS transcriptional activity, NO elevation, hyper-resistance to photokilling, and hyper-aggressiveness of resistant cells. Evidence about upstream events has also been obtained. For example, the pro-tumor kinases PI3K and Akt underwent rapid phosphorylation-activation in ALA/light-treated COH-BR1 and U87 cells [37,38]. PI3K inhibitors (Wortmannin, LY294002) strongly suppressed Akt activation, NF-κB activation, and iNOS upregulation while enhancing apoptosis [37,38]. For COH-BR1 cells, soluble guanylate cyclase (sGC) inhibitor ODQ failed to increase ALA/light-provoked apoptosis, thereby arguing against NO/sGC/cGMP-mediated activation of protein kinase G, a pro-survival/pro-growth effector. However, pro-apoptotic Bax was upregulated and anti-apoptotic Bcl-xL downregulated after a photochallenge, and 1400W or cPTIO strongly stimulated these responses, consistent with NO-mediated resistance to apoptosis [39]. More recent evidence on upstream signaling revealed that Akt activation, iNOS upregulation, and viability loss in ALA/light-treated U87 cells were all strongly inhibited by added L-histidine, consistent with primary 1O2 generation by photoactivated mitochondrial PpIX [38]. Transacetylase p300 not only underwent greater Akt-dependent activation after a photochallenge, but greater interaction with NF-κB-p65, which exhibited greater acetylation at K310. Moreover, photostressed U87 cells exhibited greater inactivating disulfide formation in tumor-suppressor PTEN [38]. This would have favored activation of Akt and p300, leading to greater iNOS transcription via interaction of p65-acK310 with bromodomain protein Brd4 [40]. In addition, deacetylase Sirt1 was down-regulated by photostress, consistent with an observed increase in p65-acK310 level [38], which would promote iNOS transcription. These findings on two different cancer lines, COH-BR1 and U87, provided new mechanistic insights into how these (and by inference the other lines discussed) can exploit iNOS/NO to not only resist a PDT challenge, but become more aggressive if surviving it.
5. iNOS/NO-imposed resistance to PDT in a human tumor xenograft model
Much of the above evidence for iNOS/NO’s anti-PDT effects in vitro has been confirmed at the in vivo level in the authors’ laboratory [41]. Severe combined immunodeficient (SCID) female mice bearing human breast carcinoma MDA-MB-231 tumor xenografts were subjected to ALA-PDT using a 633 nm LED source for optimal light penetration into tumor tissue. An iNOS-specific inhibitor (1400W or GW274150) was administered immediately after ALA and then again at 1-day intervals after irradiation. PDT-treated mice exhibited a significant slowdown in tumor growth relative to light-only controls over a 12-day post-hν period (Fig. 3B). More importantly, however, each iNOS inhibitor slowed tumor growth even more after PDT, but had no significant effect on the light-only controls (Fig. 3A). Western blot analyses on post-PDT tumor samples indicated that iNOS was progressively upregulated after irradiation, reaching ~5-times the starting level after six days (Fig. 3C). In contrast, light control samples showed no change in iNOS from a low starting level. In agreement with these results, a large 1400W-inhibitable increase in NO-derived nitrite was observed in post-PDT tumors compared with light controls (Fig. 3D). The fact that 1400W had no effect on control tumor progression (Fig. 3A), but further suppressed post-PDT tumor progression (Fig. 3B) suggests that basal iNOS/NO was too low for any significant pro-tumor activity whereas upregulated iNOS/NO was highly effective in this regard. Other MDA-MB-231 xenograft data revealed that pro-apoptotic Bax was downregulated after PDT, whereas anti-apoptotic Bcl-xL Survivin, and S100A4 were upregulated, each response being GW274150-inhibitable, demonstrating an iNOS/NO regulatory role [41]. This was the first known evidence that PDT efficacy in an in vivo human tumor model can be compromised by endogenous iNOS/NO. It remains to be determined whether these in vivo findings will hold if animals with intact immune systems and bearing syngeneic tumors are used as PDT models. An important consideration in this case would be that myeloid-derived suppressor cells (MDSCs) might supply relatively high level NO that acts protectively by inactivating any PDT-acquired anti-tumor immunity [42]. It is important to point out that in vitro experiments with MDA-MB-231 cells revealed that upregulated iNOS/NO after ALA/light treatment not only signaled for greater resistance as observed in vivo, but also greater growth, migration and invasion of surviving cells [41], similar to the effects observed with COH-BR1, PC3, and U87 cells. Thus, the pro-tumor effects of iNOS/NO in response to PDT stress appear to be widely applicable.
Figure 3.
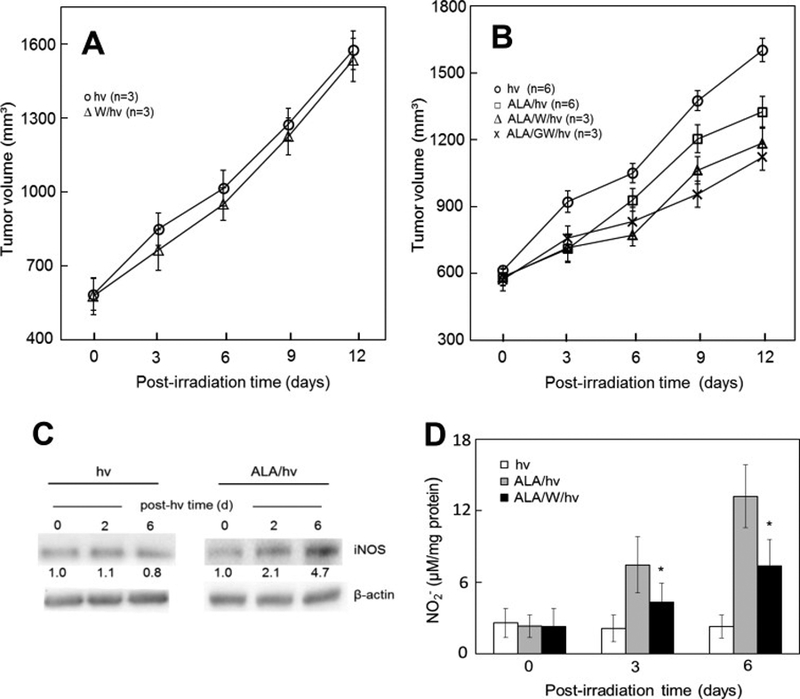
Antagonistic effects of iNOS/NO on ALA-PDT in a tumor xenograft model. (A) Light-only controls: female SCID mice bearing human breast MDA-MB-231 tumors were injected i.p. with PBS or 1400W (10 mg/kg) in PBS; after 4 h, the mice were anesthetized, placed in opaque restraints with cutouts for tumor exposure, and irradiated using a 633-nm light source and fluence ~95 J/cm2; after irradiation, mice were reinjected with 1400W once per day until termination; plotted tumor volumes are means ± SEM (n=3). (B) ALA-PDT: Tumor-bearing mice were injected i.p. with ALA (100 mg/kg) or PBS (light-only controls), followed by 1400W (10mg/kg) or GW274150 (25 mg/kg); after 4 h in the dark, the mice were anesthetized, placed in restraints with cutouts, and irradiated as in (A); after PDT, mice were kept in subdued light and reinjected with each iNOS inhibitor once daily; measured tumor volumes are means ± SEM (n=3–6). (C) Western blots showing iNOS levels in tumor samples from light control and ALA-PDT mice; number below each band indicates iNOS intensity relative to β-actin and normalized to day-0. (D) Nitrite levels in tumor samples after ALA-PDT in the absence vs. presence of 1400W; values determined by Griess assay are means ± SEM (n=3). (Reproduced from Ref. 41, with permission.)
6. NO-mediated bystander effects in PDT
Bazak et al. [43] have recently described another manifestation of how iNOS/NO can antagonize PDT, viz. bystander effects. In general, such effects can occur when cells targeted by stress-inducing physical or chemical agents send pro-death or pro-survival signals to non-or minimally-targeted counterparts (bystander cells). In many cases, bystanders make no physical contact with targeted cells, so signaling molecules simply diffuse through the medium from the latter to the former. The bulk of the research in this area has involved X-ray or gamma-ray ionizing radiation (IR), which produces a variety of cancer-promoting versus cancer-suppressing effects ranging from DNA damage/mutations and apoptotic cell death to accelerated growth and migration/invasion [44,45]. Cells targeted by IR have been shown to generate a variety of mobile signaling molecules, including cytokines, H2O2, fatty acid peroxides, and NO, the latter receiving the greatest attention for non-contacting cancer cells [45–49]. An interesting dichotomy in responses has been observed for NO; for some cell types, increased bystander proliferation has been described while for others, carcinogenic transformation via defective homologous recombination repair has been reported [46–49]. The possibility of PDT-induced bystander effects has been recognized for at least 20 years [50], but questions such as identity of signaling mediator(s) and types of responses set forth have remained largely unsettled. Bazak et al. [43] postulated that in any given tumor, non-uniformity in sensitizer delivery due to vascular irregularities as well as non-uniformity in sensitizer photoexcitation would produce a situation in which cells heavily targeted by PDT might send NO-based signals to non- or only weakly-targeted counterparts (bystanders). A novel experimental approach was devised, based on impermeable silicone-edged rings to separate ALA/light-targeted cells on a culture dish (outside rings) from non-targeted bystanders of the same cell type (inside rings). Initial experiments were carried out with prostate cancer PC3 cells. Several hours after irradiation, the bystander rings were removed, leaving a small gap between cell populations such that their only contact was via mobile species traversing the medium. Bazak et al. [43] found that after irradiation, iNOS and NO were significantly upregulated in the bystander as well as targeted cell populations. cPTIO suppressed both responses, consistent with NO diffusing from targeted to bystander cells and driving iNOS induction in the latter. Like surviving targeted cells, bystanders exhibited a striking 1400W- and cPTIO-inhibitable increase in proliferation and migration rate, indicating that targeted cell NO was also increasing bystander aggressiveness. Conditioned medium from targeted cells had no significant effect on bystander iNOS level or growth/migration rate [43], ruling out any signaling effects of relatively long-lived species like NO-derived nitrite/nitrate, H2O2, and cytokines such as TNFα. Consistent with the enhanced bystander aggressiveness was the phosphorylation-activation of two protein kinases (Akt and ERK1/2) and upregulation of cyclooxygenase-2 (COX-2) - each of these associated with tumor cell persistence/progression [43]. The described NO-mediated bystander effects, which are the first to be recognized in the context of PDT, exemplify a feed-forward field effect of NO similar to that described for ionizing radiation [49]. Induction of such effects in PDT-surviving cells could promote tumor growth/expansion unless mitigated pharmacologically, e.g. by inhibitors of iNOS activity or transcription/translation.
7. Inhibitors of iNOS activity vs. transcription as potential PDT adjuvants
Several cancer researchers have advocated pharmacologic inhibitors of iNOS enzymatic activity for retarding growth and metastatic expansion of malignant tumors known to exploit iNOS/NO for these purposes [51–54]. Such inhibitors could be used alone or in combination with chemo- or radiotherapeutic regimens in order to improve treatment efficacy. Based on the work discussed in this review, iNOS activity inhibitors might also prove beneficial for anti-tumor PDT at the clinical level. Although inhibitors such as L-NAME and L-NNA have already been successfully tested in PDT animal models [22–24], these compounds are not specific for iNOS and their effects could be far from optimal, given that iNOS appears to be the main source of pro-tumor NO after a PDT challenge. However, even if iNOS-specific inhibitors were considered, a major clinical concern would be whether normal physiologic functions of iNOS/NO might be compromised, e.g. combatting microbial infections. Nevertheless, at least two activity inhibitors, L-NIL [53] and GW274150 [54] have tested safely in clinical trials for asthma alleviation, but no cancer-related trials have been conducted yet.
Another possibility for reducing the negative effects of upregulated iNOS/NO after PDT would be to suppress its expression at the transcriptional level. Recent studies by the authors [40] showed that accelerated growth and migration of PDT-surviving glioblastoma cells in vitro could be nearly abolished by JQ1, a bromo/extra-terminal domain (BET) inhibitor of the epigenetic “reader” Brd4. Brd4, along with p65-ack310, was found to play a crucial role in iNOS transcription [39]. This was the first evidence for targeting iNOS transcription in cancer cells with a BET inhibitor. JQ1 was found to suppress iNOS upregulation much more powerfully (i.e. at far lower concentrations) than 1400W or GW274150. It may have also targeted Survivin, Bcl-xL, and MMP-9 transcription, but an indirect effect via iNOS was also likely, since NO is known to signal for induction of these proteins [28,39,55]. BET inhibitors like JQ1 are undergoing clinical evaluation for a variety of malignancies, and their testing in conjunction with PDT is anticipated soon.
8. Summary and conclusions
PDT has many advantages over other anti-cancer therapies, but like them, it is often confronted with resistance mechanisms (either pre-existing or acquired) which reduce treatment efficacy. As is clear from this review, iNOS/NO plays a major role not only in PDT resistance, but also the hyper-aggressiveness of cells that survive the challenge or in non-challenged bystanders (Fig. 4). Although both pre-existing and stress-induced iNOS could be implicated in these responses, the induced enzyme plays an almost exclusive role in the several different cancer types discussed. Recognizing this is important because up to now, most therapy-based studies have considered only pre-existing (basal) iNOS and not the possibility of upregulation during treatment or that the latter might be more important in affecting treatment efficacy. Whether iNOS upregulation is unique to PDT or if other oxidant-based therapies might share this antagonistic response is not yet clear. Concerns about more aggressive and possibly more metastatic phenotypes of PDT-surviving cells would be alleviated through pharmacologic interventions with inhibitors of iNOS activity or transcription, as discussed in Sect. 7, and it is hoped that promising interventions in either of these categories will soon be forthcoming.
Figure 4.
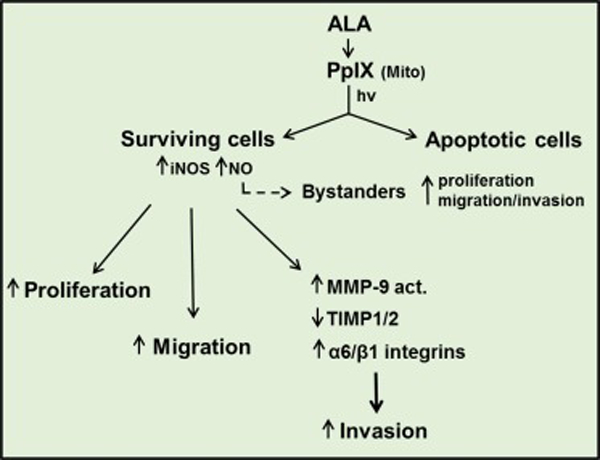
Scheme showing enhanced NO-mediated proliferation, migratory, and invasive aggressiveness of cancer cells that withstand an ALA-PDT challenge. These responses pertain not only to cells that are direct targets of photodynamic action, but also to non-targeted bystander cells, which are accessible to NO from targeted cells.
Acknowledgements
Studies in the authors’ laboratory were supported by NIH/NCI grant CA70823 and Rock River Pilot Grant Award FP14869 from the MCW Cancer Center. Magda Niziolek-Kierecka, Mariusz Zareba, and Reshma Bhowmick are thanked for their valuable contributions to much of the work described. Witold Korytowski, Neil Hogg, and Mladin Korbelik are also thanked for their helpful input along the way.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- [1].Dougherty TJ, Grindey GB, Fiel R, Weishaupt KR, Boyle DG, Photoradiation therapy II: Cure of animal tumors with hematoporphyrin and light. J. Natl. Cancer Inst 55 (1975) 115–121. [DOI] [PubMed] [Google Scholar]
- [2].Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J, Peng Q, Photodynamic therapy. J. Natl. Cancer Inst 90 (1998) 889–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO, Hahn SM, Hamblin MR, Juzeniene A, Kessel D, Korbelik M, Moan J, Mroz P, Nowis D, Piette J, Wilson BC, Golab J, Photodynamic therapy of cancer: an update. CA Cancer J Clin. 61 (2011) 250–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Spikes JD, Photosensitization, in: Smith KC (Ed.) The Science of Photobiology, Plenum Press, New York: (1989) pp. 79–110. [Google Scholar]
- [5].Kennedy JC, Pottier RH, Endogenous protoporphyrin IX, a clinically useful photosensitizer for photodynamic therapy, J. Photochem. Photobiol. B 14 (1992) 275–292. [DOI] [PubMed] [Google Scholar]
- [6].Peng Q, Berg K, Moan J, Kongshaug M, Nesland JM, 5-aminolevulinic acid-based photodynamic therapy: principles and experimental research, Photochem. Photobiol 65 (1997) 235–251. [DOI] [PubMed] [Google Scholar]
- [7].Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II, Therapy resistance mediated by cancer stem cells, Semin Cancer Biol. 53 (2018) 156–167. [DOI] [PubMed] [Google Scholar]
- [8].Casas A, Perotti C, Fukuda H and del C Batlle AM, Photodynamic therapy of activated and resting lymphocytes and its antioxidant adaptive response, Lasers Med. Sci 17 (2002) 42–50. [DOI] [PubMed] [Google Scholar]
- [9].Dolgachev V, Oberley LW, Huang TT, Kraniak JM, Tainsky MA, Hanada K and Separovic D, A role for manganese superoxide dismutase in apoptosis after photosensitization, Biochem. Biophys. Res. Commun 332 (2005) 411–417. [DOI] [PubMed] [Google Scholar]
- [10].Kriska T, Korytowski W, Girotti AW, Hyperresistance to photosensitized lipid peroxidation and apoptotic killing in 5-aminolevulinate-treated tumor cells overexpressing mitochondrial GPX4, Free Radic. Biol. Med 33 (2002) 1389–1402. [DOI] [PubMed] [Google Scholar]
- [11].Palasuberniam P, Yang X, Kraus D, Jones P, Myers KA, Chen B, ABCG2 transporter inhibitor restores the sensitivity of triple negative breast cancer cells to aminolevulinic acid-mediated photodynamic therapy. Sci. Rep 5 (2015) 13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Girotti AW, Role of endogenous nitric oxide in hyper-aggressiveness of tumor cells that survive a photodynamic therapy challenge. Crit. Rev. Oncog 21 (2016) 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wink DA, Mitchell JB, chemical biology of nitric oxide: insights into the regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide, Free Radic. Biol. Med 25 (1998) 434–456. [DOI] [PubMed] [Google Scholar]
- [14].Ridnour LA, Thomas DD, Donzelli S, Espey MG, Roberts DD, Wink DA et al. , The biphasic nature of nitric oxide responses in tumor biology, Antiox. Redox Signal. 8 (2006) 1329–1337. [DOI] [PubMed] [Google Scholar]
- [15].Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S et al. , The chemical biology of nitric oxide: implications in cellular signaling, Free Radic Biol Med. 45 (2008:18–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rubbo H, Radi R, Trujillo M, Telleri R, Kalyanaraman B, Barnes S, Kirk M, Freeman BA, Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation: formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem 269 (1994) 26066–26075. [PubMed] [Google Scholar]
- [17].Rubbo H, Parthasarathy S, Barnes S, Kirk M, Kalyanaraman B, Freeman BA, Nitric oxide inhibition of lipoxygenase-dependent liposome and low-density lipoprotein oxidation: termination of radical chain propagation reactions and formation of nitrogen-containing oxidized lipid derivatives, Arch. Biochem. Biophys, 324 (1995) 15–25. [DOI] [PubMed] [Google Scholar]
- [18].Niziolek M, Korytowski W, Girotti AW, Nitric oxide inhibition of free radical-mediated lipid peroxidation in photodynamically treated membranes and cells, Free Radic. Biol. Med 34 (2003) 997–1005. [DOI] [PubMed] [Google Scholar]
- [19].Niziolek M, Korytowski W, Girotti AW, Chain-breaking antioxidant and cytoprotective action of nitric oxide on photodynamically stressed tumor cells, Photochem. Photobiol. 78 (2003) 262–270. [DOI] [PubMed] [Google Scholar]
- [20].Niziolek M, Korytowski W, Girotti AW, Self-sensitized photodegradation of membrane-bound protoporphyrin mediated by chain lipid peroxidation: inhibition by nitric oxide with sustained singlet oxygen damage, Photochem. Photobiol. 81 (2005) 299–305. [DOI] [PubMed] [Google Scholar]
- [21].Niziolek M, Korytowski W, Girotti AW, Nitric oxide-induced resistance to lethal photooxidative damage in a breast tumor cell line, Free Radic. Biol. Med 40 (2006) 1323–1331. [DOI] [PubMed] [Google Scholar]
- [22].Henderson BW, Sitnik TM, Vaughan LA, Potentiation of photodynamic therapy antitumor activity in mice by nitric oxide synthase inhibition is fluence rate-dependent, Photochem. Photobiol. 70 (1999) 64–71. [PubMed] [Google Scholar]
- [23].Korbelik M, Parkins CS, Shibuya H, Cecic I, Stratford RML, Chaplin DJ, Nitric oxide production by tumor tissue: impact on the response to photodynamic therapy, Br. J. Cancer 82 (2000) 1835–1843, [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Reeves KL, Reeve MWR, Brown NJ, The role of nitric oxide in the treatment of tumors with aminolaevulinic acid-induced photodynamic therapy, J. Photochem. Photobiol B: Bilogy 101 (2010) 224–232. [DOI] [PubMed] [Google Scholar]
- [25].Bhowmick R, Girotti AW, Cytoprotective induction of nitric oxide synthase in a cellular model of 5-aminolevulinic acid-based photodynamic therapy, Free Radic. Biol. Med 48 (2010) 1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bhowmick R, Girotti AW, Rapid upregulation of cytoprotective nitric oxide in breast tumor cells subjected to a photodynamic therapy-like oxidative challenge, Photochem. Photobiol. 87 (2011) 378–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bhowmick R, Girotti AW, Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model, Cancer Lett. 343 (2014) 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Fahey JM, Girotti AW, Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: role of nitric oxide, Nitric Oxide 49 (2015) 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Keefer LK, Nims RW, Davies KM, Wink DA, “NONOates” (1-substituted diazen-1-ium-1,2 diolates) as nitric oxide donors: convenient nitric oxide dosage forms, Methods Enzymol. 268 (1996) 281–293. [DOI] [PubMed] [Google Scholar]
- [30].Wen PY, Kesari S, Malignant gliomas in adults. N. Engl. J. Med 359 (2008) 492–507. [DOI] [PubMed] [Google Scholar]
- [31].Kostourou JV, Cartwright JE, Johnstone AP, Boult JKR, Cullis ER, Whitley GSJ et al. The role of tumor-derived iNOS in tumor progression and angiogenesis, Br. J. Cancer 104 (2011) 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Wyler CE, Wu Q, Yan Y, McSwords MM, Chandler-Militello D, Misuraca L et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthase-2. Cell 146 (2011) 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fahey JM, Emmer JV, Korytowski W, Girotti AW, Antagonistic effects of endogenous nitric oxide in a glioblastoma photodynamic therapy model, Photochem. Photobiol 92 (2016) 842–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rapozzi V, Umezawa K, Xodo LE, Role of NF-kB/Snail/RKIP loop in the response of tumor cells to photodynamic therapy, Lasers Surg. Med. 43 (2011) 575–585. [DOI] [PubMed] [Google Scholar]
- [35].Della Pietra E, Simonella F, Bonavida B, Xodo LE, Rapozzi V, Repeated sub-optimal photodynamic treatments with pheophorbide-a induce an epithelial-mesenchymal transition in prostate cancer cells via nitric oxide. Nitric Oxide 45 (2015) 43–53. [DOI] [PubMed] [Google Scholar]
- [36].Thomas DD, Jord’heuil D, S-nitrosation: current concepts and new developments, Antiox., Red. Signal 17 (2012) 924–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Bhowmick R, Girotti AW, Cytoprotective signaling associated with nitric oxide upregulation in tumor cells subjected to photodynamic therapy-like oxidative stress, Free Radic. Biol. Med 57 (2013) 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Fahey JM, Korytowski W, Girotti AW, Upstream signaling events leading ot elevated production of pro-survival nitric oxide in photodynamically-challenged glioblastoma cells, Free Radic. Biol. Med 137 (2019) 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bhowmick R, Girotti AW, Signaling events in apoptotic photokilling of 5-aminolevulinic acid-treated tumor cells: inhibitory effects of nitric oxide, Free Radic. Biol. Med 47 (2009) 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Fahey JM, Stancill JS, Smith BC, Girotti AW, Nitric oxide antagonism to glioblastoma photodynamic therapy and mitigation thereof by BET bromodomain inhibitor JQ1, J. Biol. Chem 393 (2018) 5345–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Fahey JM, Girotti AW, Nitric oxide-mediated resistance to photodynamic therapy in a human breast tumor xenograft model: improved outcome with NOS2 inhibitors, Nitric Oxide 62 (2017) 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fionda C, Abruzzese MP, Santoni A, Cippitelli M, Immunoregulation and effector activities of nitric oxide and reactive nitrogen species in cancer, Curr. Med. Chem 23 (2016) 2618–2636. [DOI] [PubMed] [Google Scholar]
- [43].Bazak J, Fahey JM, Wawak K, Korytowski W, Girotti AW, Enhanced aggressiveness of bystander cells in an anti-tumor photodynamic therapy model: role of nitric oxide produced by targeted cells, Free Radic. Biol. Med 102 (2017) 111–121. [DOI] [PubMed] [Google Scholar]
- [44].Azzam EI, de Toledo SM, Little JB, Stress signaling from irradiated to non-irradiated cells, Curr. Cancer Drug Targets 4 (2006) 53–64. [DOI] [PubMed] [Google Scholar]
- [45].Hei TK, Zhou H, Chai Y, Ponnaiya B, Ivanov VN, Radiation-induced non-targeted response: mechanism and potential clinical implications, Curr. Mol. Pharmacol 4 (2011) 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Matsumoto H, Hayashi S, Hatashita M, Ohnishi K, Shioura H, Ohtsubo T et al. Induction of radioresistance by a nitric oxide-mediated bystander effect, Radiat. Res 155 (2001) 387–396. [DOI] [PubMed] [Google Scholar]
- [47].Matsumoto H, Takahashi A, Ohnishi T, Nitric oxide radicals choreograph a radioadaptive response, Cancer Res. 67 (2007) 8574–8579. [DOI] [PubMed] [Google Scholar]
- [48].Han W, Chen S, Yu KN, Wu L, Nitric oxide-mediated DNA double strand breaks induced by proliferating bystander cells after alpha particle irradiation, Mutat. Res 684 (2010) 81–89. [DOI] [PubMed] [Google Scholar]
- [49].Yakovlev VA, Role of nitric oxide in the radiation-induced bystander effect, Redox Biol. 6 (2015) 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dahle J, Bagdonas S, Kaalhus O, Olsen G, Seen HB, Moan J, The bystander effect in photodynamic inactivation of cells, Biochim. Biophys. Acta 1475 (2000) 273–280. [DOI] [PubMed] [Google Scholar]
- [51].Crowell JA, Steele VE, Sigman CC, Fay JR, Is inducible nitric oxide synthase a target for chemoprevention? Mol. Cancer Ther 2 (2003) 815–823. [PubMed] [Google Scholar]
- [52].Eyler CE, Wu Q, Yan K, MacSwords JM, Chandler-Militello D, Misuraca et KL et al. Glioma stem cell proliferation and tumor growth are promoted by nitric oxide synthans-2, Cell 146 (2011) 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hansel TT, Kharitonov SA, Donnelly LE, Erin EM, Currie MG, Moore WM et al. A selective inhibitor of inducible nitric oxide synthase inhibits exhaled breath nitric oxide in healthy volunteers and asthmatics, FASEB J. 17 (2003) 1298–1317. [DOI] [PubMed] [Google Scholar]
- [54].Singh D, Richards D, Knowles RC, Schwartz S, Woodcock A, Langley S et al. Selective inducible nitric oxide synthase inhibition has no effect on allergen challenge in asthma. Am. J. Respir. Crit. Care Med 176 (2007) 988–993. [DOI] [PubMed] [Google Scholar]
- [55].Engels K, Knauer SK, Loibl S, Fetz V, Harter P, Schweitzer A et al. NO signaling confers cytorotectivity through the surviving networks in ovarian carcinomas, Cancer Res. 68 (2008) 5159–5166. [DOI] [PubMed] [Google Scholar]