

Summary
Extrahepatic portosystemic shunt belongs to a family of rare vascular abnormalities. The clinical importance and manifestations of this vascular abnormality range from asymptomatic cases to liver or metabolic dysfunctions of various degrees. Congenital extrahepatic portosystemic shunt, also termed as Abernethy malformation, is a very rare congenital vascular malformation in which splenomesenteric blood drains into a systemic vein, bypassing the liver through a complete or partial extrahepatic shunt. So far, limited cases of congenital extrahepatic portosystemic shunt have been reported. In this review, incidence, mechanisms, complications, diagnoses and treatments of congenital extrahepatic portosystemic shunt are described.
Keywords: Congenital extrahepatic portosystemic shunt, incidence, mechanism, complications, diagnosis, treatment, review
1. Introduction
Extrahepatic portosystemic shunt belongs to a family of rare vascular abnormalities. Clinical importance of this vascular abnormality lies in its broad spectrum of symptoms and complications, ranging from incidentally discovered asymptomatic cases to liver or metabolic dysfunctions of various degrees and even severe clinical scenarios, which are caused by variations in sites or types of shunt (1). Congenital extrahepatic portosystemic shunt (CEPS), also termed as Abernethy malformation, is a very rare congenital vascular malformation in which splenomesenteric blood drains directly into a systemic vein, bypassing the liver through a complete or partial extrahepatic shunt. Since it was first documented in 1793 by John Abernethy (2), the number of reported cases has progressively increased (3). As is proposed by Morgan and Superina, CEPS is classified into two variants based on absence (type 1) or presence (type 2) of intrahepatic portal supply (4,5). Type 1 is described as complete portosystemic shunts (Figure A, end-to-side, a complete extrahepatic shunt) and the liver is not perfused with portal blood; whereas type 2 is defined as a certain proportion of portal blood perfused to the liver and the remaining portal flow bypasses the liver and is diverted into a systemic vein through a partial shunt (Figure B, side-to-side, a partial extrahepatic shunt) (4,6). This classification has been widely recognized and referred by a lot of studies because of its practical understanding of pathophysiologic implications and managements of included cases. In this review, we mainly present incidence, mechanisms, complications, diagnoses and treatments of CEPS.
Figure 1.
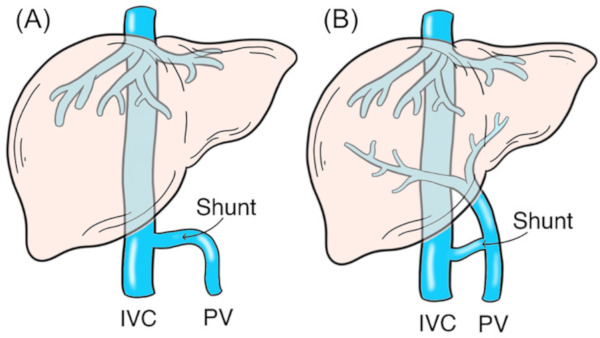
Classification of congenital extrahepatic portosystemic shunt. (A), Type 1 is described as complete portosystemic shunts (end-to-side) and the liver is not perfused with portal blood; (B), Type 2 is defined as a certain proportion of portal blood perfused to the liver and the remaining portal flow bypassed the liver and diverted into a systemic vein through a partial shunt (side-to-side). IVC, inferior vena cava; PV, portal vein.
2. Incidence
Knowledge about CEPS is scarce given that the low incidence of this malformation has prevented realization of large studies. Several issues stay unanswered, such as the actual incidence of CEPS. With limited sources, incidence of CEPS was once described close to 1/30,000 births and the prevalence proportion of permanent CEPS was 1/50,000, which came from results of a nationwide galactosemia survey. Because CEPS gave rise to increased levels of bile acids and galactoses, the incidence was extrapolated. Of note, only 7 CEPS cases were diagnosed and biases might remain in that survey (7,8). To a certain degree, the assumption that CEPS could be diagnosed through increases in blood galactoses after birth yet requires further validations, and this incidence ought to be accepted with suspicions of a certain degree (7). No remarkable gender predominance was found in CEPS (9); however, it has been described in certain researches that type 1 shunts feature a significant female predilection of 61-78%, while no clear explanation is given (7,10). Thus, future studies to systematically evaluate presence of CEPS are warranted.
3. Mechanism
Rather limited researches have explored etiology mechanisms of CEPS. Two possible explanations have dominated literatures on development of CEPS: i) A genetic origin and complex congenital malformation processes: such malformations are considered to originate from an insult occurring in week 4-10 of embryological development, which is a crucial stage for hepatic and systemic vessel formations. And this to certain degree give evidence to the fair association with cardiac and other vascular anomalies (7); ii) The absence of ductus venosus during fetal stage: abnormal vessels may develop in association with occlusions or agenesis of ductus venosus. Through abnormal vessels, oxygenated blood from the umbilical vein flows into the fetal heart. Afterwards, certain vessels may persist and develop into anabnormal shunt, resulting in hypoplasia of the portal venous system (11,12). As is shown in literatures, absence of ductus venosus has been documented among some CEPS children (13,14).
Etiology mechanisms of CEPS differ significantly from that of acquired extrahepatic portosystemic shunts. As to acquired extrahepatic portosystemic shunts, due to liver cirrhosis, collateral vessels would generate or reopen to compensate for blocked or narrowed portal veins, so as to lower pressures on the portal tree. Gradually, this will result in an acquired extrahepatic portosystemic shunt (15,16). Hence, variations of therapeutic strategies should be noted for CEPS and acquired extrahepatic portosystemic shunts.
4. Complications
Spectrums of clinical variants of CEPS ranges from completely asymptomatic forms to severe forms of hepatic encephalopathy, hepatopulmonary syndromes (HPS) and pulmonary arterial hypertension (PaHT).
4.1. Hepatic encephalopathy
Hepatic encephalopathy is a common clinical manifestation in symptomatic patients, with a reported incidence of as high as 17-30% (7,14,17,18). Blood ammonia produced by gastrointestinal tracts directly bypasses the liver and flows into inferior vena cava, then it is metabolized in astrocytes to glutamine, which in turn has deleterious effects on the brain. This phenomenon is more frequently seen among older patients. Most children with CEPS are asymptomatic, which proves that children are more resistant to hepatic encephalopathies than adults. Partially, this might be caused by ageing of brain, decreased blood-brain barriers and losses of brain reserves, all of which make patients more vulnerable to harmful metabolic products such as ammonia and facilitate the revelations of hepatic encephalopathies (19-21). In a previous international observational study, Baiges, A. described that 29% of patients with CEPS presented hepatic encephalopathies at a certain time during the study. Median age at hepatic encephalopathy onset or diagnosis was 12 years (range, 5-65). Different patterns of hepatic encephalopathy were documented with a fair predominance of chronic hepatic encephalopathy: 73.7% featured persistent hepatic encephalopathy with permanent cognitive impairment, 11.0% featured recurrent hepatic encephalopathy, and 15.3% featured episodic hepatic encephalopathy. In most cases, in accordance with West-Haven standard, episodes of hepatic encephalopathies were of moderate intensities (20).
4.2. Pulmonary complications
Patients with CEPS will be frequently associated with severe pulmonary complications, such as HPS (7,22) and PaHT (14,23). Developments of HPS and PaHT can be linked with vasoactive mediators from intestines (22,24,25). Such mediators skip hepatic circulations and directly approach the pulmonary vascular bed, causing pulmonary circulation imbalances and inducing a long-term pulmonary vasoconstriction in PaHT (9,24) or, reversely, a pulmonary vasodilation in HPS (26).
HPS is a clinical relationship between hepatic diseases and existence of pulmonary vascular dilatations, which is characterized by presence of dyspnea and hypoxia and can result in arterial oxygenation abnormalities of a certain range. Prevalence of HPS among patients with CEPS is still not clear, because mild HPS are often asymptomatic and a comprehensive study on lungs cannot always be carried out on all patients. Hence, an actual prevalence cannot be accurately predicted. In addition, it is recognized that CEPS should be considered as one of the etiologies of HPS (27). Since the first case report about CEPS with HPS by Papagiannis (28), there have been more than 20 similar cases described in literatures (22,29,30). A baseline comprehensive assessment on pulmonary complications is suggested to be undergone. In follow-up durations, transcutaneous oxygen saturation measurements can be of help in early and primary detections of HPS.
It has been demonstrated that PaHT is a considerable issue among CEPS patients, inducing symptomatic dyspnea in about 80% of cases; it has been shown by data that PaHT occurred in 13-66% of children with CEPS (17,23). When carefully screened, PaHT is also manifested on nearly 11% of asymptomatic patients (20). Rarity of such disease and the small amount of patient series have led to a rather broad percentage range. Despite virous hypotheses and reports, PaHT mechanisms stay controversial. From histological perspective, the lungs present microthrombotic lesions, and it is considered that PaHT resulted from recurrent microemboli originating in the mesenteric circulation and travelling through a portosystemic shunt directly to the lungs (23). Symptoms of PaHT vary from disturbed consciousness to syncope. Right ventricular hypertrophies, decompressions of left ventricle or an increased estimated right ventricular systolic pressure are vital signs of an echocardiography. Notably, PaHT is not seemingly related to severity or degree of a shunt, while it features a decreased outcome, with a reported morality of nearly 50% (7). As PaHT can occur in a broad range of age, periodic and regular surveillances for PaHT may be of a great significance during follow-ups.
4.3. Liver nodules
An abnormal shunt of blood leads to non-specific liver disturbances because of uneven portal flow perfusions and arterialisations (compensatory increases in hepatic arterial flows). In ischemic liver parenchyma, absent or reduced portal flows result in lacks of nutrition and fatty degenerations in hepatocytes; thusly, liver dysfunctions occur, certain normal hepatocytes diminish, and hepatic atrophy follows; in well perfused areas, regenerative nodule generated. Altogether, these elements contribute to abnormal hepatic developments and functions, which will bring an incentive to nodule generations (21,31,32).
Nodular liver tumors are commonly seen in as many as 40-65% of patients with CEPS. Although most of such tumors are benign, among other neoplastic lesions, malignancies such as hepatocellular carcinomas and adenomas have also been reported (20,21,33,34). In a case series involving 26 CEPS patients with liver nodules, 70% of them had focal nodular hyperplasia or regenerative nodular hyperplasia, 20% had hepatocellular carcinoma, and 10% had adenomas; also, it has been shown in researches that 21% of patients had single nodules and 79% had multiple nodules (9,14,20,21,31). Hence, it should be noted that performances of a rigorous and periodic screening of liver nodules for patients with CEPS is of significant needs and values. Furthermore, researchers have also advocated that it is sensible to term cirrhosis strategies of hepatocellular carcinoma surveillance every half year (20).
5. Diagnosis
CEPS can be diagnosed at any age, which is often in childhood during the setting of neonatal cholestasis, hypergalactosemia, failures to thrive, psychomotor delays or other congenital defects. Prenatal diagnoses will be considered through ultrasound detections of abnormal communication vessels between portal and peripheral venous systems or an enlarged umbilical vein (9,14,35,36). In adulthood, CEPS could be incidentally diagnosed through abdominal ultrasound, computed tomography (CT) or magnetic resonance imaging (MRI) in the screening of unexplained abdominal pains, abnormal liver functions and hepatic nodules, or etc. (37-39). Of note, for accurate diagnoses of CEPS, one should first excluded potential acquired portosystemic shunts, such as those patients with hepatic cirrhosis, with or without concomitant presentations of portal hypertension (40), and surgically created portosystemic shunts (41). Generally, non-congenital or acquired shunts are small, tortuous peripheral vessels, and CEPS patients will not present hepatic cirrhosis or portal hypertension imaging features (ascites, varices, or splenomegaly) (42,43).
Ultrasound is widely utilized in abdominal diseases for its safe and fast imaging properties. Primarily, it can be a useful diagnostic tool to identify presence of portosystemic shunts. Doppler ultrasound is of special diagnostic values for its abilities to assess vessel flow directions. As is reported previously (35), clinically asymptomatic shunts have been occasionally diagnosed in children through ultrasound during galactosaemia test. Ponziani FR, et al. have summarized concomitant presence of five ultrasound signs to strengthen the suspicion of CEPS (39), including: i) Solid focal lesions in liver parenchyma; ii) Portal trunk absence/hypoplasia; iii) Deficiencies of intrahepatic portal vessels and flow signals; iv) Existence of porto/systemic shunts; v) Hepatic artery hypertrophies. However, ultrasound might fail to accurately detect inapparent or small shunts. Hence, anomalies identified by ultrasound should be further confirmed through other imaging methods (44).
Radiological methods like CT or MRI are preferred investigations to confirm a portosystemic shunt. Asymptomatic CEPS are often diagnosed through incidental presentations on CT or MRI scans in abdominal imaging. CT and MRI are also of great values in further classifications of shunts and measurements of accompanying anomalies (10). Postprocessing technologies, such as multiplanar reformations, have supplied additional information about diagnoses and managements.
A Contrast-enhanced CT can document anatomies, locations and sizes of abnormal shunts. With techniques like maximum intensity projection and multiplanar reconstruction, much information will be provided by CT, including shunt courses, sizes and orientations, which help both radiologists and surgeons to make or choose suitable treatment regimens.
Furthermore, MR can be used to visualize shunts and avoid radiation exposures for both radiologists and patients. Particularly, in assessments of associated liver nodules, MRI includes diffusion sequences and shows unique advantages over CT (10). Compared to CT, MRI would be preferred as the first-choice test. A research team from Canada analyzed 61 reported cases of CEPS and recommended MRI as a superior tool in diagnoses and classifications of CEPS as well as examinations of associated cardiovascular and hepatic abnormalities (42). Furthermore, MRI angiography serves as a reliable and noninvasive examination for hepatic vascular anatomies. Despite this, CT can be reserved for patients who are noncooperative or have specific contraindications. Traditionally, conventional angiography is regarded as a golden standard for detections of portomesenteric vasculatures. However, improvements in CT and MRI techniques have changed the situation. Currently, conventional angiography is not necessarily a must in CEPS diagnoses, for CT and MRI can provide accurate diagnoses in most cases.
According to existing findings on nuclear medicine researches, iodine-123 iodoamphetamine could also be used to measure shunt dynamics (10,19).
Furthermore, serum ammonia level is useful, although it is a non-specific and investigative adjunct. In 66-100% of CEPS cases, a heightened level of serum ammonia is found (7). Hence, concentrations of ammonia without known hepatic cirrhosis or portal hypertensions ought to imply subsequent examinations for CEPS (45). In certain cases, liver biopsies may illustrate small portal venules within portal triads, which is indicative of type 2 shunt (42).
6. Treatment
Currently, there is no standard therapeutic approach to treat extrahepatic portosystemic shunts due to rarity of such diseases (46). Several approaches including shunt closures through surgical or radiological interventions and liver transplantations have been proposed, but clear comparisons among different treatment strategies are still unavailable. Treatment strategies are decided according to shunt types, locations, symptoms severities and related complications. There still remains debate regarding therapeutic strategies for asymptomatic cases (1,46). Conservative managements including lifestyle changes, such as protein restrictions as well as lactulose and non-absorbable antibiotics administrations, may be recommended in asymptomatic shunts. Yet, presence of shunts could potentially develop clinical implications, such as supplying a route for intestinal toxic materials to bypass hepatic circulations, immune surveillances as well as offering a possible route for lung tumor metastases of gastrointestinal tumors (1,19). A Japanese research team has previously observed natural courses of 51 patients with CEPS and found that spontaneous shunt closures were never evident. Hence, early detections and suitable therapies are vital for a good prognosis (32).
6.1. Shunt closure for type 2 CEPS
Discrepancies regarding management of patients with asymptomatic type 2 did exist. Researchers from University of Catania chose active surveillances (46). However, more experts urge early and active interventions to be mandatory (9,47). When a clear diagnosis of extrahepatic shunt is made, it is important to remark that shunt closures facilitating progressive redirections of portal blood flowing to livers are possible and essential for such patients. As is proved in treatments of Abernethy malformations, shunt closures are especially useful in improving hepatic encephalopathies. Baiges A, et al. suggested that shunt closures had a huge efficacy in managing most shunt complications and, most interestingly, preventing their occurrences (3); this is in agreement with findings of Papamichail M. et al., who proved that early occlusions would reverse associated complications (48). Sanada Y, et al. and Pathak A, et al. had also demonstrated that patients with CEPS and hepatic encephalopathies can benefit by early shunt occlusion surgeries (49,50).
Therefore, shunt closures must always be considered for symptomatic patients and should also be regarded as a prophylactic treatment early in evolutions of the disease to prevent developments of severe complications. Earlier studies have shown that for shunt occlusions, either interventional embolization or surgical ligations (open or laparoscopic surgical techniques) can be choices of treatment that lead to rapid ameliorations of symptoms and normalizations of ammonia levels (25,51). Likewise, multiple results have proved that the associated HPS could also be resolved by shunt closures (25,26,52,53). The choice from an interventional embolization or a surgical ligation depends upon medical expertise, shunt vessel anatomies and sizes as well as induvial general conditions (48,54,55). For patients with wide and short shunt vessels or those who fail to receive embolization, a surgical ligation will be preferred (54,56).
6.2. Liver transplantation for type 1 CEPS
Shunt closures are not a feasible option for patients with type 1 CEPS, as the shunt stands for the only drainage path of mesenteric and splenic venous blood. Thusly, liver transplantation serves as effective therapeutic approaches for both liver and pulmonary complications. Literatures have described the successful application of liver transplantation for CEPS patients with severe complications (including refractory encephalopathy, CEPS associated with biliary atresia, or patients with severe HPS) (37,57,58).
Timing of liver transplantations for type 1 CEPS is still a matter of debate. Results from Japan show that prophylactic liver transplantations should be justified prior to occurrences of severe pulmonary complications (HPS or PaHT), because such complications would complicate or even preclude liver transplantations (57). However, Guerin F, et al. found that asymptomatic patients with type 1 CEPS ought not to receive liver transplantations as a prophylactic option, which would make them experience prolonged periods of immunosuppression (7). Sakamoto S, et al. reviewed a collection of 34 transplantation cases of CEPS and concluded that presence of pulmonary complications was an early indication of liver transplantations; in the review, 30 out of 34 CEPS cases with liver transplantations stayed alive after a median follow-up period of one and a half years, indicating an encouraging outcome (6). Sanada Y, et al. also revealed that liver transplantations could be potentially curative for patients with symptomatic type 1 CEPS (49). In most reported cases, liver transplantations provide a complete resolution for associated complications. Technical difficulties of portal system anatomies and reconstructions are main challenges (39).
6.3. Management of liver nodules
When liver nodules are identified in patients with CEPS, differential diagnoses of malignant and benign tumors will be crucial for determination of following treatments. For benign tumors, conservative treatments and regular follow-ups should be suggested. In cases of malignant tumors, subsequent surgical interventions like biopsies will be adequate.
At last, a close surveillance is indicated for patients with such vascular malformations. Long-term follow-ups will create a good clinical compliance and provide a comprehensive understanding and management of disease processes.
7. Conclusion
Differential diagnoses between CEPS and acquired portosystemic shunts are of much importance. When a clear diagnosis of CEPS is made, it is important to remark that active interventions are possible and essential for such patients. As to type 1 CEPS, liver transplantation serves as an effective therapeutic approach for both liver and pulmonary complications; as with type 2 CEPS, shunt closures must always be considered for symptomatic patients and should also be regarded as a prophylactic treatment early in evolutions of the disease, so as to prevent developments of severe complications. Yet, knowledge about CEPS is scarce due to its low incidence. Future studies on systematical explorations on CEPS are warranted.
Acknowledgements
This study was supported by Beijing Natural Science Foundation (NO.7204309) and Beijing New Star of Science and Technology Foundation (NO.2017B503).
References
- 1. Matthews TJ, Trochsler MI, Bridgewater FH, Maddern GJ. Systematic review of congenital and acquired portal-systemic shunts in otherwise normal livers. Br J Surg. 2014; 101:1509-1517. [DOI] [PubMed] [Google Scholar]
- 2. Abernethy J. Account of two instances of uncommon formation in the viscera of the human body: from the philosophical transactions of the royal society of London. Med Facts Obs. 1797; 7:100-108. [PMC free article] [PubMed] [Google Scholar]
- 3. Baiges A, Turon F, Simon-Talero M, et al. Congenital Extrahepatic Portosystemic Shunts (Abernethy Malformation): An International Observational Study. Hepatology. 2019. [DOI] [PubMed] [Google Scholar]
- 4. Mathai SV, Kondray V, Salloum E, Kukreja K, Tavri S. Role of interventional radiology in the diagnosis and management of congenital extrahepatic portosystemic shunts: Two case reports. Indian J Radiol Imaging. 2019; 29:219-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morgan G, Superina R. Congenital absence of the portal vein: two cases and a proposed classification system for portasystemic vascular anomalies. J Pediatr Surg. 1994; 29:1239-1241. [DOI] [PubMed] [Google Scholar]
- 6. Sakamoto S, Shigeta T, Fukuda A, Tanaka H, Nakazawa A, Nosaka S, Uemoto S, Kasahara M. The role of liver transplantation for congenital extrahepatic portosystemic shunt. Transplantation. 2012; 93:1282-1287. [DOI] [PubMed] [Google Scholar]
- 7. Guerin F, Blanc T, Gauthier F, Abella SF, Branchereau S. Congenital portosystemic vascular malformations. Semin Pediatr Surg. 2012; 21:233-244. [DOI] [PubMed] [Google Scholar]
- 8. Sakura N, Mizoguchi N, Eguchi T, Ono H, Mawatari H, Naitou K, Ito K. Elevated plasma bile acids in hypergalactosaemic neonates: a diagnostic clue to portosystemic shunts. Eur J Pediatr. 1997; 156:716-718. [DOI] [PubMed] [Google Scholar]
- 9. Sokollik C, Bandsma RH, Gana JC, van den Heuvel M, Ling SC. Congenital portosystemic shunt: characterization of a multisystem disease. J Pediatr Gastroenterol Nutr. 2013; 56:675-681. [DOI] [PubMed] [Google Scholar]
- 10. Alonso-Gamarra E, Parron M, Perez A, Prieto C, Hierro L, Lopez-Santamaria M. Clinical and radiologic manifestations of congenital extrahepatic portosystemic shunts: a comprehensive review. Radiographics. 2011; 31:707-722. [DOI] [PubMed] [Google Scholar]
- 11. Acherman RJ, Evans WN, Galindo A, Collazos JC, Rothman A, Mayman GA, Luna CF, Rollins R, Kip KT, Berthody DP, Restrepo H. Diagnosis of absent ductus venosus in a population referred for fetal echocardiography: association with a persistent portosystemic shunt requiring postnatal device occlusion. J Ultrasound Med. 2007; 26:1077-1082. [DOI] [PubMed] [Google Scholar]
- 12. Shen O, Valsky DV, Messing B, Cohen SM, Lipschuetz M, Yagel S. Shunt diameter in agenesis of the ductus venosus with extrahepatic portosystemic shunt impacts on prognosis. Ultrasound Obstet Gynecol. 2011; 37:184-190. [DOI] [PubMed] [Google Scholar]
- 13. Manning N, Impey L, Lindsell D, Lakhoo K. Prenatally diagnosed portocaval shunt and postnatal outcome: a case report. Prenat Diagn. 2004; 24:537-540. [DOI] [PubMed] [Google Scholar]
- 14. Franchi-Abella S, Branchereau S, Lambert V, Fabre M, Steimberg C, Losay J, Riou J-Y, Pariente D, Gauthier F, Jacquemin E, Bernard O. Complications of Congenital Portosystemic Shunts in Children: Therapeutic Options and Outcomes. Journal of Pediatric Gastroenterology and Nutrition. 2010. [DOI] [PubMed] [Google Scholar]
- 15. Wu Q, Shen L, Chu J, Ma X, Jin B, Meng F, Chen J, Wang Y, Wu L, Han J, Zhang W, Ma W, Wang H, Li H. Characterization of uncommon portosystemic collateral circulations in patients with hepatic cirrhosis. Oncol Lett. 2015; 9:347-350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zardi EM, Uwechie V, Caccavo D, Pellegrino NM, Cacciapaglia F, Di Matteo F, Dobrina A, Laghi V, Afeltra A. Portosystemic shunts in a large cohort of patients with liver cirrhosis: detection rate and clinical relevance. J Gastroenterol. 2009; 44:76-83. [DOI] [PubMed] [Google Scholar]
- 17. Kim MJ, Ko JS, Seo JK, Yang HR, Chang JY, Kim GB, Cheon JE, Kim WS. Clinical features of congenital portosystemic shunt in children. Eur J Pediatr. 2012; 171:395-400. [DOI] [PubMed] [Google Scholar]
- 18. Lautz TB, Tantemsapya N, Rowell E, Superina RA. Management and classification of type II congenital portosystemic shunts. J Pediatr Surg. 2011; 46:308-314. [DOI] [PubMed] [Google Scholar]
- 19. Diyar R, Wells T, Haboubi H. Congenital extrahepatic portosystemic shunt. Br J Hosp Med (Lond). 2015; 76:424-425. [DOI] [PubMed] [Google Scholar]
- 20. Baiges A, Turon F, Simon-Talero M, et al. Congenital Extrahepatic Portosystemic Shunts (Abernethy Malformation): An International Observational Study. Hepatology. 2020; 71:658-669. [DOI] [PubMed] [Google Scholar]
- 21. Sorkin T, Strautnieks S, Foskett P, Peddu P, Thompson RJ, Heaton N, Quaglia A. Multiple beta-catenin mutations in hepatocellular lesions arising in Abernethy malformation. Hum Pathol. 2016; 53:153-158. [DOI] [PubMed] [Google Scholar]
- 22. Fu L, Wang Q, Wu J, Guo Y, Huang M, Liu T, Chen Q, Li F. Congenital extrahepatic portosystemic shunt: an underdiagnosed but treatable cause of hepatopulmonary syndrome. Eur J Pediatr. 2016; 175:195-201. [DOI] [PubMed] [Google Scholar]
- 23. Ohno T, Muneuchi J, Ihara K, Yuge T, Kanaya Y, Yamaki S, Hara T. Pulmonary hypertension in patients with congenital portosystemic venous shunt: a previously unrecognized association. Pediatrics. 2008; 121:e892-899. [DOI] [PubMed] [Google Scholar]
- 24. Yi JE, Jung HO, Youn HJ, Choi JY, Chun HJ, Lee JY. A case of pulmonary arterial hypertension associated with congenital extrahepatic portocaval shunt. J Korean Med Sci. 2014; 29:604-608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Morikawa N, Honna T, Kuroda T, Kitano Y, Fuchimoto Y, Kawashima N, Kawasaki K. Resolution of hepatopulmonary syndrome after ligation of a portosystemic shunt in a pediatric patient with an Abernethy malformation. J Pediatr Surg. 2008; 43:e35-38. [DOI] [PubMed] [Google Scholar]
- 26. Alvarez AE, Ribeiro AF, Hessel G, Baracat J, Ribeiro JD. Abernethy malformation: one of the etiologies of hepatopulmonary syndrome. Pediatr Pulmonol. 2002; 34:391-394. [DOI] [PubMed] [Google Scholar]
- 27. Alvarez AE, Ribeiro AnF, Hessel G, Baracat J, Ribeiro JD. Abernethy malformation: One of the etiologies of hepatopulmonary syndrome. Pediatric Pulmonology. 2002; 34:391-394. [DOI] [PubMed] [Google Scholar]
- 28. Papagiannis J, Kanter RJ, Effman EL, Pratt PC, Marcille R, Browning IB, 3rd, Armstrong BE. Polysplenia with pulmonary arteriovenous malformations. Pediatr Cardiol. 1993; 14:127-129. [DOI] [PubMed] [Google Scholar]
- 29. Ohnishi Y, Ueda M, Doi H, Kasahara M, Haga H, Kamei H, Ogawa K, Ogura Y, Yoshitoshi EY, Tanaka K. Successful liver transplantation for congenital absence of the portal vein complicated by intrapulmonary shunt and brain abscess. Journal of Pediatric Surgery. 2005; 40:e1-e3. [DOI] [PubMed] [Google Scholar]
- 30. Bernard O, Franchi-Abella S, Branchereau S, Pariente D, Gauthier F, Jacquemin E. Congenital portosystemic shunts in children: recognition, evaluation, and management. Semin Liver Dis. 2012; 32:273-287. [DOI] [PubMed] [Google Scholar]
- 31. Sharma R, Suddle A, Quaglia A, Peddu P, Karani J, Satyadas T, Heaton N. Congenital extrahepatic portosystemic shunt complicated by the development of hepatocellular carcinoma. Hepatobiliary Pancreat Dis Int. 2015; 14:552-557. [DOI] [PubMed] [Google Scholar]
- 32. Uchino T, Matsuda I, Endo F. The long-term prognosis of congenital portosystemic venous shunt. J Pediatr. 1999; 135:254-256. [DOI] [PubMed] [Google Scholar]
- 33. Benedict M, Rodriguez-Davalos M, Emre S, Walther Z, Morotti R. Congenital Extrahepatic Portosystemic Shunt (Abernethy Malformation Type Ib) With Associated Hepatocellular Carcinoma: Case Report and Literature Review. Pediatr Dev Pathol. 2017; 20:354-362. [DOI] [PubMed] [Google Scholar]
- 34. Song P, Cai Y, Tang H, Li C, Huang J. The clinical management of hepatocellular carcinoma worldwide: A concise review and comparison of current guidelines from 2001 to 2017. Biosci Trends. 2017; 11:389-398. [DOI] [PubMed] [Google Scholar]
- 35. Venkat-Raman N, Murphy KW, Ghaus K, Teoh TG, Higham JM, Carvalho JS. Congenital absence of portal vein in the fetus: a case report. Ultrasound Obstet Gynecol. 2001; 17:71-75. [DOI] [PubMed] [Google Scholar]
- 36. Athanasiadis A, Karavida A, Chondromatidou S, Tsitouridis J, Tarlatzis B. Prenatal diagnosis of Abernethy malformation by three-dimensional ultrasonography. Ultrasound Obstet Gynecol. 2015; 46:638-639. [DOI] [PubMed] [Google Scholar]
- 37. Kobayashi N, Niwa T, Kirikoshi H, Fujita K, Yoneda M, Saito S, Nakajima A. Clinical classification of congenital extrahepatic portosystemic shunts. Hepatol Res. 2010; 40:585-593. [DOI] [PubMed] [Google Scholar]
- 38. Barchetti F, Pellegrino L, Al-Ansari N, De Marco V, Scarpato P, Ialongo P. Congenital absence of the portal vein in a middle-aged man. Surg Radiol Anat. 2011; 33:369-372. [DOI] [PubMed] [Google Scholar]
- 39. Ponziani FR, Faccia M, Zocco MA, Giannelli V, Pellicelli A, Ettorre GM, De Matthaeis N, Pizzolante F, De Gaetano AM, Riccardi L, Pompili M, Rapaccini GL. Congenital extrahepatic portosystemic shunt: description of four cases and review of the literature. J Ultrasound. 2019; 22:349-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Konstas AA, Digumarthy SR, Avery LL, Wallace KL, Lisovsky M, Misdraji J, Hahn PF. Congenital portosystemic shunts: imaging findings and clinical presentations in 11 patients. Eur J Radiol. 2011; 80:175-181. [DOI] [PubMed] [Google Scholar]
- 41. Hu GH, Shen LG, Yang J, Mei JH, Zhu YF. Insight into congenital absence of the portal vein: is it rare? World J Gastroenterol. 2008; 14:5969-5979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murray CP, Yoo SJ, Babyn PS. Congenital extrahepatic portosystemic shunts. Pediatr Radiol. 2003; 33:614-620. [DOI] [PubMed] [Google Scholar]
- 43. Lisovsky M, Konstas AA, Misdraji J. Congenital extrahepatic portosystemic shunts (Abernethy malformation): a histopathologic evaluation. Am J Surg Pathol. 2011; 35:1381-1390. [DOI] [PubMed] [Google Scholar]
- 44. Altavilla G, Cusatelli P. Ultrastructural analysis of the liver with portal vein agenesis: a case report. Ultrastruct Pathol. 1998; 22:477-483. [DOI] [PubMed] [Google Scholar]
- 45. Lux D, Naito A, Harikrishnan S. Congenital extrahepatic portosystemic shunt with progressive myelopathy and encephalopathy. Pract Neurol. 2019; 19:368-371. [DOI] [PubMed] [Google Scholar]
- 46. Timpanaro T, Passanisi S, Sauna A, Trombatore C, Pennisi M, Petrillo G, Smilari P, Greco F. Congenital portosystemic shunt: our experience. Case Rep Pediatr. 2015; 2015:691618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shah A, Aziz A, Awwad A, Ramjas G, Higashi Y. Incidental radiological diagnosis of asymptomatic Abernethy malformations-two case reports. BJR Case Rep. 2017; 3:20150496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Papamichail M, Ali A, Quaglia A, Karani J, Heaton N. Liver resection for the treatment of a congenital intrahepatic portosystemic venous shunt. Hepatobiliary Pancreat Dis Int. 2016; 15:329-333. [DOI] [PubMed] [Google Scholar]
- 49. Sanada Y, Urahashi T, Ihara Y, et al. The role of operative intervention in management of congenital extrahepatic portosystemic shunt. Surgery. 2012; 151:404-411. [DOI] [PubMed] [Google Scholar]
- 50. Pathak A, Agarwal N, Mandliya J, Gehlot P, Dhaneria M. Abernethy malformation: a case report. BMC Pediatr. 2012; 12:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kimura T, Soh H, Hasegawa T, Sasaki T, Kuroda S, Yuri E, Tomoda K, Fukuzawa M. Laparoscopic correction of congenital portosystemic shunt in children. Surg Laparosc Endosc Percutan Tech. 2004; 14:285-288. [DOI] [PubMed] [Google Scholar]
- 52. Emre S, Arnon R, Cohen E, Morotti RA, Vaysman D, Shneider BL. Resolution of hepatopulmonary syndrome after auxiliary partial orthotopic liver transplantation in Abernethy malformation. A case report. Liver Transpl. 2007; 13:1662-1668. [DOI] [PubMed] [Google Scholar]
- 53. Kuo MD, Miller FJ, Lavine JE, Peterson M, Finch M. Exploiting phenotypic plasticity for the treatment of hepatopulmonary shunting in Abernethy malformation. J Vasc Interv Radiol. 2010; 21:917-922. [DOI] [PubMed] [Google Scholar]
- 54. Papamichail M, Pizanias M, Heaton N. Congenital portosystemic venous shunt. Eur J Pediatr. 2018; 177:285-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kanazawa H, Nosaka S, Miyazaki O, Sakamoto S, Fukuda A, Shigeta T, Nakazawa A, Kasahara M. The classification based on intrahepatic portal system for congenital portosystemic shunts. J Pediatr Surg. 2015; 50:688-695. [DOI] [PubMed] [Google Scholar]
- 56. Kamimatsuse A, Onitake Y, Kamei N, Tajima G, Sakura N, Sueda T, Hiyama E. Surgical intervention for patent ductus venosus. Pediatr Surg Int. 2010; 26:1025-1030. [DOI] [PubMed] [Google Scholar]
- 57. Shinkai M, Ohhama Y, Nishi T, Yamamoto H, Fujita S, Take H, Adachi M, Tachibana K, Aida N, Kato K, Tanaka Y, Takemiya S. Congenital absence of the portal vein and role of liver transplantation in children. J Pediatr Surg. 2001; 36:1026-1031. [DOI] [PubMed] [Google Scholar]
- 58. Ohnishi Y, Ueda M, Doi H, Kasahara M, Haga H, Kamei H, Ogawa K, Ogura Y, Yoshitoshi EY, Tanaka K. Successful liver transplantation for congenital absence of the portal vein complicated by intrapulmonary shunt and brain abscess. J Pediatr Surg. 2005; 40:e1-3. [DOI] [PubMed] [Google Scholar]