

Abstract
We elucidated the anti-inflammatory mechanisms of IL-38 in allergic asthma. Human bronchial epithelial cells and eosinophils were cocultured upon stimulation with the viral RLR ligand poly (I:C)/LyoVec or infection-related cytokine TNF-α to induce expression of cytokines/chemokines/adhesion molecules. House dust mite (HDM)-induced allergic asthma and humanized allergic asthma NOD/SCID murine models were established to assess anti-inflammatory mechanisms in vivo. IL-38 significantly inhibited induced proinflammatory IL-6, IL-1β, CCL5, and CXCL10 production, and antiviral interferon-β and intercellular adhesion molecule-1 expression in the coculture system. Mass cytometry and RNA-sequencing analysis revealed that IL-38 could antagonize the activation of the intracellular STAT1, STAT3, p38 MAPK, ERK1/2, and NF-κB pathways, and upregulate the expression of the host defense-related gene POU2AF1 and anti-allergic response gene RGS13. Intraperitoneal injection of IL-38 into HDM-induced allergic asthma mice could ameliorate airway hyperreactivity by decreasing the accumulation of eosinophils in the lungs and inhibiting the expression of the Th2-related cytokines IL-4, IL-5, and IL-13 in the bronchoalveolar lavage fluid (BALF) and lung homogenates. Histological examination indicated lung inflammation was alleviated by reductions in cell infiltration and goblet cell hyperplasia, together with reduced Th2, Th17, and innate lymphoid type 2 cell numbers but increased proportions of regulatory T cells in the lungs, spleen, and lymph nodes. IL-38 administration suppressed airway hyperreactivity and asthma-related IL-4 and IL-5 expression in humanized mice, together with significantly decreased CCR3+ eosinophil numbers in the BALF and lungs, and a reduced percentage of human CD4+CRTH2+ Th2 cells in the lungs and mediastinal lymph nodes. Together, our results demonstrated the anti-inflammatory mechanisms of IL-38 and provided a basis for the development of a regulatory cytokine-based treatment for allergic asthma.
Keywords: allergic asthma, eosinophils, IL-38, ILC2s, Tregs
Subject terms: Mucosal immunology, Chemokines
Introduction
Asthma is a chronic bronchial inflammatory disease characterized by recurrent attacks of breathlessness and wheezing, with an increasing prevalence and global burdens of death and disability.1 Notably, allergic asthma, the most common form of asthma, presents with clinical hallmarks including chronic airway inflammation, airway remodeling, airway hyperresponsiveness (AHR), and an elevated serum allergen-specific IgE concentration. Allergic asthma patients are characterized by a T-helper (Th)2 predisposition with elevated levels of the asthma-related Th2 cytokines interleukin (IL)-4, IL-5, and IL-13.2 The typical Th2 cytokine IL-4 is important for driving isotype class switching in B cells for IgE production.3 IL-5 is an eosinophil-specific regulatory cytokine involved in the differentiation, activation, and survival of eosinophils,4 whereas IL-13 is critical for inducing allergic airway inflammation involving AHR, mucus production, and airway remodeling.5 Together with other proinflammatory cytokines/chemokines including tumor necrosis factor (TNF)-α, IL-6, CXCL8, and CXCL10, these cytokines contribute to the pathogenesis and exacerbation of allergic asthma.6 Notably, viral respiratory infections caused by influenza virus, respiratory syncytial virus, or rhinovirus are closely related to asthma exacerbations and wheezing illness, which are the major morbidity factors in allergic asthma patients.7 Although the essential molecular and cellular mechanisms linking viral infections and allergic airway inflammation are still poorly understood, previous studies have indicated that viral infection can impair epithelial barrier functions to enhance the entry and absorption of allergens that aggravate airway inflammation. Subsequently, the damaged epithelium will in turn promote viral replication to amplify the infection-induced allergic inflammatory cycle.7
Airway epithelial cells represent the frontline of innate immunity against inhaled viral, bacterial, and fungal pathogens, and play a pivotal role in orchestrating the pulmonary immune response. Epithelial cells recognize conserved molecular structures of microorganisms called pathogen-associated molecular patterns through pattern recognition receptors, including Toll-like receptors (TLRs), nucleotide-binding oligomerization domain-like receptors, and retinoic acid-inducible gene I (RIG-1)-like receptors (RLRs). The importance of airway epithelial cells in the release of cytokines, chemokines, and antimicrobial peptides that initiate and amplify airway inflammation has been identified.8 Eosinophils are the principal effector cells in allergic inflammation and they release a panel of cytokines, including granulocyte-macrophage colony-stimulating factor, transforming growth factor (TGF)-β and CXCL8, specific granular eosinophil cationic protein (ECP), and lipid mediators such as cysteinyl leukotrienes.9 The infiltration of eosinophils undergoing delayed apoptosis into the airway is mainly mediated by the eosinophil-specific chemokine CCL11 and the adhesion molecule intercellular adhesion molecule (ICAM)-1 expressed on epithelial cells, contributing to the development of bronchial asthma and allergic airway inflammation.10 We have previously demonstrated that the induction of allergic inflammation-related cytokines, chemokines, superoxide, and ECP, and the migration of eosinophils induced by TLR2, TLR5, and TLR7 ligands are differentially regulated by intracellular focal adhesion kinase, nuclear factor (NF)-κB, extracellular signal-regulated kinase (ERK), phosphoinositide 3-kinase-Akt, and p38 mitogen-activated protein kinase (MAPK).11 Bacterial infection-mediated activation of the intracytosolic bacterial sensors nucleotide oligomerization domain (NOD)1 and NOD2 was found to trigger allergic asthma via the activation of eosinophils interacting with bronchial epithelial cells in the inflamed airway.12 We have also elucidated the underlying molecular mechanisms involved in the interaction of eosinophils with epithelial cells that induces inflammatory cytokine and chemokine expression.13,14 However, the detailed viral infection-mediated intracellular mechanism underlying the activation of human eosinophils during allergic inflammation in allergic asthma has not been well elucidated.
IL-38 is a newly discovered cytokine in the IL-1 family and is expressed in the skin, tonsils, spleen, thymus, placenta, heart, and fetal liver.15 According to its biological function, IL-38 belongs to the IL-36 cytokine group.16 It is well known that IL-1Ra antagonizes IL-1α and IL-1β, and IL-36Ra inhibits IL-36α, IL-36β, and IL-36γ.17 Therefore, IL-38 is considered to act as a partial receptor antagonist of inflammatory IL-36.18 IL-38 suppresses the asthma-related production of CXCL8 induced by IL-36γ in cultures of peripheral blood mononuclear cells (PBMCs), implicating its potential inhibitory activity in the allergic inflammatory response.18 In addition, IL-38 can inhibit the production of the asthma-related Th17 cytokines IL-17 and IL-22, and preserve a low Th17 cell/regulatory T cell (Treg) balance.19 In clinical and animal studies, we and other groups have reported aberrant IL-38 expression in many inflammatory diseases including allergic asthma,20 rheumatoid arthritis, systemic lupus erythematosus (SLE), Crohn’s disease, psoriasis, and chronic obstructive pulmonary disease (COPD)21, and the protective and anti-inflammatory roles of IL-38 in a murine model of arthritis,22,23 SLE,24 psoriasis,25,26 obesity,27 and sepsis.28 Although IL-38 may be related to the IL-1R and IL-18R signaling pathways, the detailed intracellular signaling pathways involving IL-38 in eosinophil-mediated allergic inflammation have never been elucidated.
In view of the above information, the aim of our present study was to investigate the anti-inflammatory activities of IL-38 in a coculture of human primary bronchial epithelial cells and human eosinophils, and to identify the underlying intracellular signaling mechanisms. Moreover, the in vivo immunomodulatory role of IL-38 was further confirmed by using a house dust mite (HDM)-induced allergic asthma murine model and a humanized murine model of allergic asthma.
Results
Anti-inflammatory effect of IL-38 on a coculture of human primary bronchial epithelial cells and eosinophils upon stimulation by RIG-I/MDA5 ligand poly (I:C)/LyoVec or TNF-α
The exacerbation of allergic asthma is known to correlate with influenza viral infection. To investigate whether influenza virus can directly activate eosinophils in allergic inflammation, we stimulated eosinophils in vitro with the prevalent influenza virus type A subtypes H1 and H3, and the viral mimic dsRNA RLR ligand poly (I:C)/LyoVec. Significantly elevated production of granular ECP and proinflammatory TNF-α, IL-6, CCL2, IL-10, and IL-1β was observed in both the virus-infected and poly (I:C)/LyoVec-treated groups (all p < 0.05, Fig. 1a, b). These results demonstrated that eosinophils could be activated by both influenza viral particles and an RLR ligand/viral mimic, indicating that upper respiratory tract viral infection may exacerbate allergic inflammation through the activation of eosinophils. To further investigate the effect of IL-38 on allergic inflammation, viral poly (I:C)/LyoVec together with the infection-related proinflammatory cytokine TNF-α were applied in a coculture of human bronchial epithelial (HBE) cells and eosinophils to mimic the airway microenvironment in allergic asthma. To evaluate the potency and specificity of the effect of IL-38, heat-inactivated IL-38 (negative control) and dexamethasone (positive control) were included in our in vitro study. As shown in Fig. 2, significantly increased levels of the proinflammatory cytokines/chemokines IL-6, IL-1β, CCL5, and CXCL10, and the antiviral cytokine interferon (IFN)-β were observed upon stimulation by poly (I:C)/LyoVec or proinflammatory TNF-α in human epithelial cells and eosinophils alone, as well as in the coculture. Poly (I:C)/LyoVec-induced IL-6, CCL5, and IFN-β expressions were significantly downregulated by human recombinant IL-38 in the bronchial epithelial cells and coculture (all p < 0.05, Fig. 2a, b, e), whereas the release of proinflammatory Th1-related CXCL10 and IL-1β could be significantly suppressed by IL-38 in a dose-dependent manner in the bronchial epithelial cells, eosinophils, and coculture (all p < 0.05, Fig. 2c, d). Similarly, significantly decreased TNF-α-induced production of CXCL10 was observed in the bronchial epithelial cells, eosinophils, and coculture with IL-38 treatment (all p < 0.05, Fig. 2k). Upon stimulation with poly (I:C)/LyoVec and TNF-α, upregulated ICAM-1 cell surface expression was detected on the cocultured epithelial cells and eosinophils compared with the monocultured cells. The inhibitory role of IL-38 in ICAM-1 expression was more apparent in the bronchial epithelial cells and eosinophils in the coculture (Fig. 2h, m) than in those in the monocultures (Fig. 2g, l). Representative flow cytometry data for the effect of IL-38 on ICAM-1 expression in the coculture system and the gene expression of human IL-38 receptors in HBE cells and human eosinophils are shown in Supplementary Fig. S1 and S2, respectively. Altogether, the above results illustrated the in vitro anti-inflammatory activity of IL-38 in cocultures of human primary bronchial epithelial cells and eosinophils modeling airway inflammation.
Fig. 1.
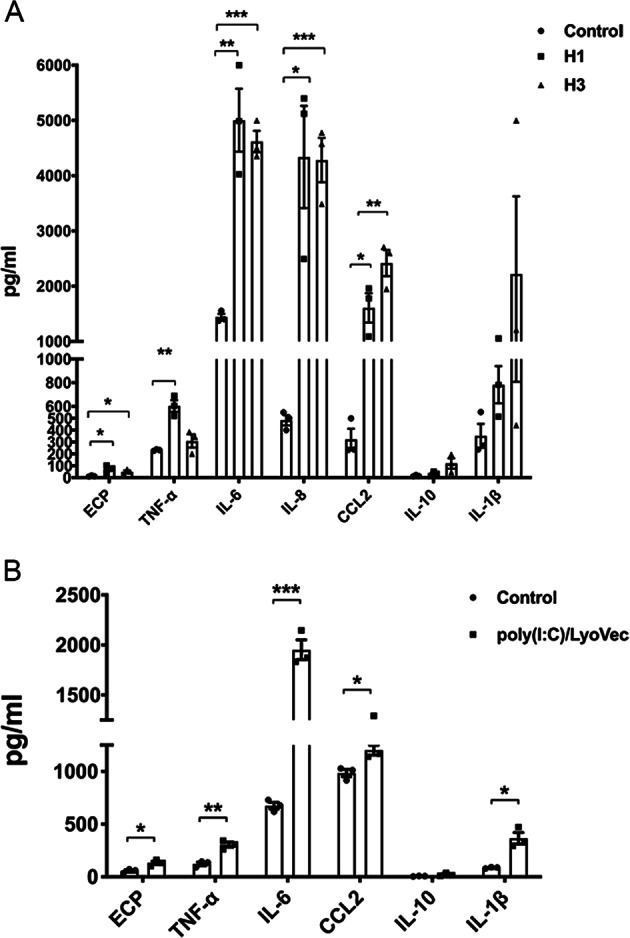
Effect of influenza virus H1, influenza virus H3, or poly (I:C)/LyoVec on eosinophils. Eosinophils (5 × 105) were added into tubes and then incubated with 0.5 MOI of H1 or H3 virus, or poly (I:C)/LyoVec (2 μg/ml) for 24 h. The release of cytokines and chemokines into the supernatant of a H1- or H3-treated eosinophils, or b poly (I:C)/LyoVec-treated eosinophils was determined. The results are shown as the mean ± SEM of triplicate independent experiments with a total of three donors. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups
Fig. 2.

Effect of IL-38 on cytokine/chemokine release in cocultures of human primary bronchial epithelial cells (HBE) and eosinophils (EOS) upon stimulation by poly (I:C)/LyoVec or TNF-α. Primary bronchial epithelial cells (1 × 105) and purified eosinophils (3 × 105) were cocultured with or without human IL-38 pretreatment for 10 min, followed by poly (I:C)/LyoVec (2 μg/ml) or TNF-α (20 ng/ml) treatment for an additional 20 h. Release of a IL-6, b CCL5, c CXCL10, d IL-1β, and e IFN-β into the supernatant of the poly (I:C)/LyoVec-treated coculture was determined. f Gating strategies for bronchial epithelial cells and eosinophils are shown. Bronchial epithelial cells and eosinophils in the cocultures were gated based on the FSC and SSC parameters. The cell surface expression of ICAM-1 on g HBE/EOS single-cultured cells and h cocultured cells was analyzed by flow cytometry. The levels of i IL-6, j CCL5, and k CXCL10 in the supernatant of the TNF-α-treated coculture were measured. The expression of ICAM-1 on l HBE/EOS single-cultured cells and m cocultured cells after stimulation with TNF-α was determined by flow cytometry. A negative control of 56 °C heat-inactivated human IL-38 (100 ng/ml) and a positive control of dexamethasone (1 μM) were included. Abbreviations: HBE, human primary bronchial epithelial cells; PLV, poly (I:C)/LyoVec; EOS, eosinophils; HBE-EOS, coculture of human bronchial epithelial cells and eosinophils; Co-HBE, human primary bronchial epithelial cells in coculture; and Co-EOS, eosinophils in coculture. The results are shown as the mean ± SEM of triplicate independent experiments with a total of three donors. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups
IL-38 suppressed poly (I:C)/LyoVec- and TNF-α-induced inflammatory mediators via downregulation of the intracellular p38, signal transducers and activators of transcription (STAT)1, STAT3, ERK, and NF-κB signaling pathways in cocultures
To address the specific intracellular signaling pathways responsible for the inhibitory activity of IL-38 in cocultured HBE cells and eosinophils activated by poly (I:C)/LyoVec or TNF-α, high-throughput mass cytometry, which combines time-of-flight mass spectrometry with metal-tagged antibodies, was used to investigate intracellular pathways. As shown in Fig. 3, heatmap data analysis generated in Cytobank (Fig. 3a) indicated the increase or reduction in the level of each phosphoepitope (calculated as the arcsinh difference of the 95th percentile) (Fig. 3b). Poly (I:C)/LyoVec-upregulated phosphorylation of signal transducers and activators of transcription (STAT)-1, p38, STAT3, and ERK1/2 was significantly inhibited by IL-38 in cocultures and TNF-α-induced STAT1, p38, and ERK1/2 activation was also suppressed by IL-38 (all p < 0.05, Fig. 3c). To further confirm the signal transduction pathways mediating the anti-inflammatory effect of IL-38 in the coculture system, human primary bronchial epithelial cells and eosinophils were analyzed by flow cytometry, and showed results consistent with those of the mass cytometry analysis (Fig. 3e–i). Moreover, poly (I:C)/LyoVec-induced pIκBα was significantly suppressed by IL-38 in bronchial epithelial cells and eosinophils (Fig. 3i). Representative flow cytometry data for the signaling pathways are shown in Supplementary Fig. S2. Furthermore, RNA-sequencing (RNA-seq) was applied to identify IL-38 target genes in cocultures at the transcriptional level. Cluster analysis of differentially expressed genes (DEGs) was used to construct a heatmap according to the criteria of P < 0.05 (Fig. 4a). Excluding pseudogenes and uncategorized genes, eight immune-related genes including POU2AF1, RGS13, PHLPP2, LYZ, BMF, MT1G, MUC15, and S1PR3 were identified, and gene descriptions are included in Table 1. The expression level of each DEG was generated in a log2 scale heatmap (Fig. 4b). Real-time quantitative PCR (qPCR) was applied to further validate these target genes in three biological replicates and the result was consistent with the RNA-seq finding (Fig. 4c). IL-38 significantly upregulated POU2AF1, RGS13, LYZ, and MT1G expression in the poly (I:C)/LyoVec-treated coculture. Moreover, IL-38 induced elevated expression of POU2AF1 and RGS13 in the TNF-α-treated coculture (Fig. 4b, c).
Fig. 3.

Mass cytometric profiling of intracellular signaling pathways. Cocultured cells were pretreated with human IL-38 (100 ng/ml) for 10 min and then stimulated with poly (I:C)/LyoVec (2 μg/ml) or human TNF-α (20 ng/ml) for an additional 20 min. The phosphorylated signaling molecules in the stimulated cells were stained by using the Maxpar® Phospho Panel Kit. a A heatmap was generated by Cytobank. b, c The change in each phosphoepitope (calculated as the arcsinh difference of the 95th percentile) was quantified for analysis. d Gating strategies for bronchial epithelial cells and eosinophils from cocultures are shown. The intracellular levels of e phosphorylated p38, f phosphorylated STAT1, g phosphorylated STAT3, h phosphorylated ERK1/2, and i phosphorylated IκBα in cells were measured by intracellular staining with specific antibodies and analyzed by flow cytometry. The absolute MFI values for the phosphorylated signals were used for analysis. Heat-inactivated human IL-38 (100 ng/ml) served as a negative control. The results are shown as the mean ± SEM of triplicate independent experiments with a total of three donors. Abbreviations: Co-HBE, human primary bronchial epithelial cells in coculture; Co-EOS, eosinophils in coculture *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups
Fig. 4.
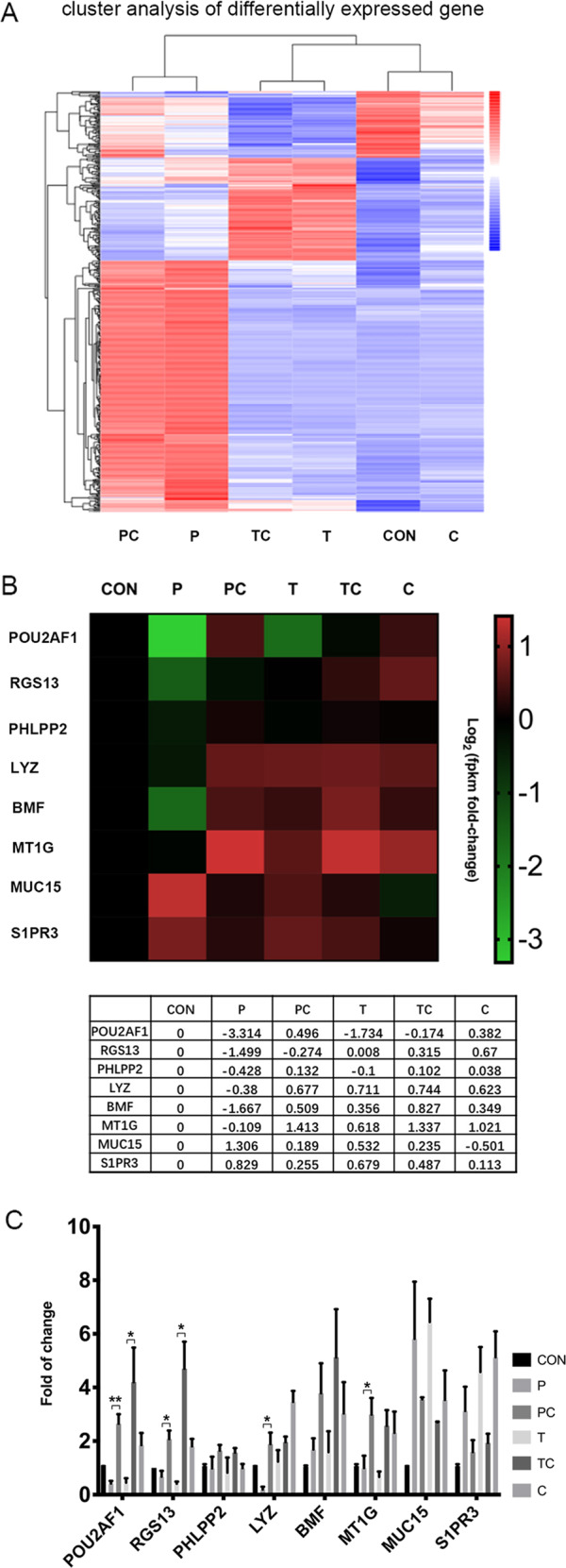
Identification of IL-38 target genes through transcriptional profiling. Cocultured cells were pretreated with human IL-38 (100 ng/ml) for 10 min and then stimulated with poly (I:C)/LyoVec (2 μg/ml) or human TNF-α (20 ng/ml) for 20 h. Total RNA was extracted from each coculture and transcriptome analysis was performed to identify IL-38 target genes. a A heatmap of the DEGs in different groups is shown. b The fold change for differentially expressed genes is displayed in a log2-scale heatmap and the corresponding data are shown in Table. c RNA-seq results were confirmed using real-time qPCR. The GAPDH housekeeping gene was used as the reference gene. The results are shown as the mean ± SEM of triplicate independent experiments with a total of three donors. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups. Abbreviations: DEG, differentially expressed gene; PC, poly (I:C)/LyoVec + IL-38-treated group; P, poly (I:C)/LyoVec-treated group; TC, TNF-α + IL-38-treated group; T, TNF-α-treated group; CON, unstimulated group; and C, IL-38-treated group
Table 1.
Gene descriptions of eight identified immune-related genes
| Gene symbol | Gene description | Gene type |
|---|---|---|
| POU2AF1 | POU class 2-associating factor 1 | Protein coding |
| RGS13 | Regulator of G-protein signaling 13 | Protein coding |
| PHLPP2 | PH domain and leucine-rich repeat protein phosphatase 2 | Protein coding |
| LYZ | Lysozyme | Protein coding |
| BMF | Bcl 2-modifying factor | Protein coding |
| MT1G | Metallothionein 1G | Protein coding |
| MUC15 | Mucin 15 cell surface associated | Protein coding |
| S1PR3 | Sphingosine-1-phosphate receptor 3 | Protein coding |
Intraperitoneal injection of murine recombinant IL-38 ameliorated HDM-induced allergic airway inflammation in mice
To investigate whether exogenous IL-38 application can exert any anti-inflammatory effect on allergic asthma, an in vivo protocol for an HDM (the common inhaled allergen that causes allergic asthma)-induced allergic asthma murine model with IL-38 treatment was established (Fig. 5a). The mice that received recombinant murine IL-38 (800 ng) showed a significant decrease in AHR as indicated by Penh values compared with untreated HDM-induced allergic asthma mice (Fig. 5b). In parallel, the treated mice displayed reduced total inflammatory infiltrating cell numbers, especially significantly decreased eosinophil numbers in the BALF, after intraperitoneal (i.p.) injection of IL-38 (Fig. 5c–g). Significantly higher levels of the eosinophil chemotactic protein CCL11 and allergy-related inflammatory cytokines including IL-4, IL-5, and IL-13, and TGF-β in the serum, the BALF, and lung homogenates were observed in the untreated HDM group compared with the IL-38-treated group (Fig. 5h–j). Moreover, histological examinations of lung tissue samples performed by using hematoxylin and eosin (H&E) or periodic acid-Schiff (PAS) staining showed that IL-38 alleviated inflammatory cell infiltration, mucus production, and airway wall thickening during an asthma attack (Fig. 5k, l). To further investigate the effect of IL-38 on inflammatory cell infiltration, immunostaining was used to analyze the spatial distributions of eosinophils as evidenced by the localization of eosinophil granule major basic protein (MBP) (Fig. 5m). The results showed that IL-38 decreased eosinophil infiltration into the lungs compared with HDM alone, which was also in concordance with the results in Fig. 5g. In view of the above findings, it is suggested that exogenous IL-38 application is capable of relieving allergic airway inflammation and exerting anti-allergic inflammatory activities in HDM-induced allergic asthma mice.
Fig. 5.

In vivo effects of IL-38 on an HDM-induced allergic asthma murine model. a The timeline protocol of the HDM-induced allergic asthma model is shown. b Airway obstruction was measured as Penh values; Penh (% baseline) was calculated by dividing the methacholine-induced Penh value by the PBS baseline value (n = 5). c The total cell number and d–g differential cell counts in the bronchoalveolar lavage fluid (BALF) were determined. The release of cytokines and chemokines was measured in h the serum, i the BALF, and j lung homogenates. k, l Representative lung sections stained with H&E or PAS were examined at ×100 or ×400 magnification (n = 5). m Immunostaining analysis of eosinophils in mouse lung tissue samples is shown. Eosinophils were identified using anti-MBP (red) antibodies and nuclei were visualized by DAPI (blue) staining. The lung sections were examined at ×200 magnification. The results are shown as the mean ± SEM of triplicate independent experiments with a total of four to six mice. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups. Abbreviations: PAS, periodic acid-Schiff; H&E, hematoxylin and eosin
Intraperitoneal injection of murine IL-38 diminished the proportion of Th2 cells and enhanced the frequency of Tregs in HDM-induced allergic asthma mice
To explore the underlying T-cell-mediated anti-allergic mechanisms of IL-38, the changes in CD4+ Th subsets in mice after injection of IL-38 were investigated. The percentage changes in Th1, Th2, and Th17 cells, and Tregs in the lung tissue (Fig. 6b–f), splenocyte (Fig. 6g–k), and mediastinal lymph node (MLN) samples (Fig. 6l–p), as well as the change in group 2 innate lymphoid cells (ILC2s; Fig. 6q) in mice were analyzed by flow cytometry. In the IL-38-treated group, a significantly reduced proportion of Th2 cells and an increased frequency of Treg cells were observed in lung cells, splenocytes, and lymphocytes when compared with the HDM group. The populations of Th17 cells in the lungs and spleen showed a moderated decrease in the mice that received IL-38 injection. In addition, a remarkably reduced ILC2 number was observed in the spleen of IL-38-treated mice compared with that of HDM mice (Fig. 6q). As macrophages are the main source of inflammatory mediators, we next investigated the phenotypic change in the macrophage population in mouse lung tissue. Our results showed that a dramatically increased percentage of M2 macrophages (F4/80+CD11c−CD206+) was observed in the HDM group compared with the IL-38-treated group, whereas there was no significant difference in M1 macrophages (F4/80+CD11c+CD206−) among the three groups (Fig. 6r–t). Furthermore, HDM-induced allergic asthma mice exhibited significantly elevated ex vivo production of the asthma-related Th2 cytokines IL-4 and IL-5, and proinflammatory cytokines/chemokines IL-6, IL-17, and CCL5 in HDM-restimulated splenocytes (Fig. 6u–z). Reduced IL-4 and IL-5 production was observed in HDM-stimulated splenocytes from IL-38-treated mice. Notably, the ex vivo concentration of the anti-inflammatory Treg-related cytokine IL-10 was markedly increased in the IL-38-treated group upon HDM restimulation. Moreover, we compared the expression levels of IL-38 receptors including IL-1R1, IL-36R, and IL-1RAPL1 in mouse lung tissue samples among the three groups using real-time qPCR. The expression of the receptors IL-1R1 and IL-36R was significantly increased in the IL-38-treated group, whereas IL-1RAPL1 expression was slightly increased in the HDM group compared with the IL-38 group (Supplementary Fig. S4). These results therefore indicated that IL-38 displayed anti-inflammatory activities in allergic asthma by downregulating Th2 cells, ILC2s, and probably Th17 cells but upregulating Treg functions.
Fig. 6.


Effects of IL-38 on CD4+ Th subsets in mouse lung tissue, splenocyte, and mediastinal lymph node samples. a Th1, Th2, Th17, and Treg cells were gated as indicated in the protocol. The frequencies of Th1, Th2, Th17, and Treg cells in b–f lung tissue, g–k spleen, and l–p mediastinal lymph node samples, as well as q the frequency of ILC2s (Lin−CD45+CD127+T1/ST2+CD25+ cells) in the mouse spleen were analyzed using flow cytometry (n = 4). q Gating strategies for ILC2s are shown (CD45 single-stained cells were used as a control for staining). r Gating strategies for M1 and M2 macrophages are shown. s, t Representative flow cytometry data for M1 and M2 macrophages in mouse lung tissue samples and the percentages of M1 and M2 macrophages in the lungs are shown. Ex vivo production of u IL-4, v IL-5, w IL-6, x IL-10, y IL-17, and z CCL5 was measured in the supernatant of mouse splenocytes after restimulation with HDM (30 μg/ml) for 72 h. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups. Control_re, HDM_re, and HDM + IL-38_re indicate splenocytes from the control, HDM, and HDM + IL-38 groups restimulated with HDM, respectively
Anti-inflammatory activities of human IL-38 in humanized HDM-induced allergic asthma mice
In our translational study, humanized non-obese diabetic/severe combined immunodeficiency (NOD/SCID) mice with allergic asthma were employed as a preclinical murine model to further evaluate the therapeutic effect of IL-38 on human allergic asthma. The NOD/SCID mice were reconstituted by i.p. injection of human PBMCs from allergic asthma patients and exposed to HDM at the indicated time (Fig. 7a). The accumulation of CD45+ human leukocytes in the lungs was analyzed on Day 20 to ensure stable reconstitution with human PBMCs in the mice (Fig. 7c). Mice treated with human recombinant IL-38 showed reduced AHR compared with untreated asthmatic mice as reflected by reduced increases in Penh values (Fig. 7b). The production of the asthma-related cytokine IL-4 in the BALF and lung homogenates was significantly decreased in the IL-38-treated group (Fig. 7d, e). Histopathological examination of lung tissue samples indicated that IL-38 ameliorated inflammatory cell infiltration and mucus-secreting goblet cell accumulation (Fig. 7f, g), and immunostaining results showed decreased eosinophil infiltration in the IL-38-treated group (Fig. 7h). Furthermore, reduced populations of CCR3+ eosinophils in the BALF and lungs were observed in the IL-38-treated mice (Fig. 7j–m). It is well known that Th2 lymphocytes participate in the pathogenesis of allergic asthma and CD4+CRTH2+ cells are reported to be a reliable marker of human central memory Th2 cells.29 Therefore, the percentage of CD4+CRTH2+ cells was examined to show that the frequency of CD4+CRTH2+ cells was significantly decreased in the lungs and MLNs in the IL-38-treated group (Fig. 7n–r).
Fig. 7.
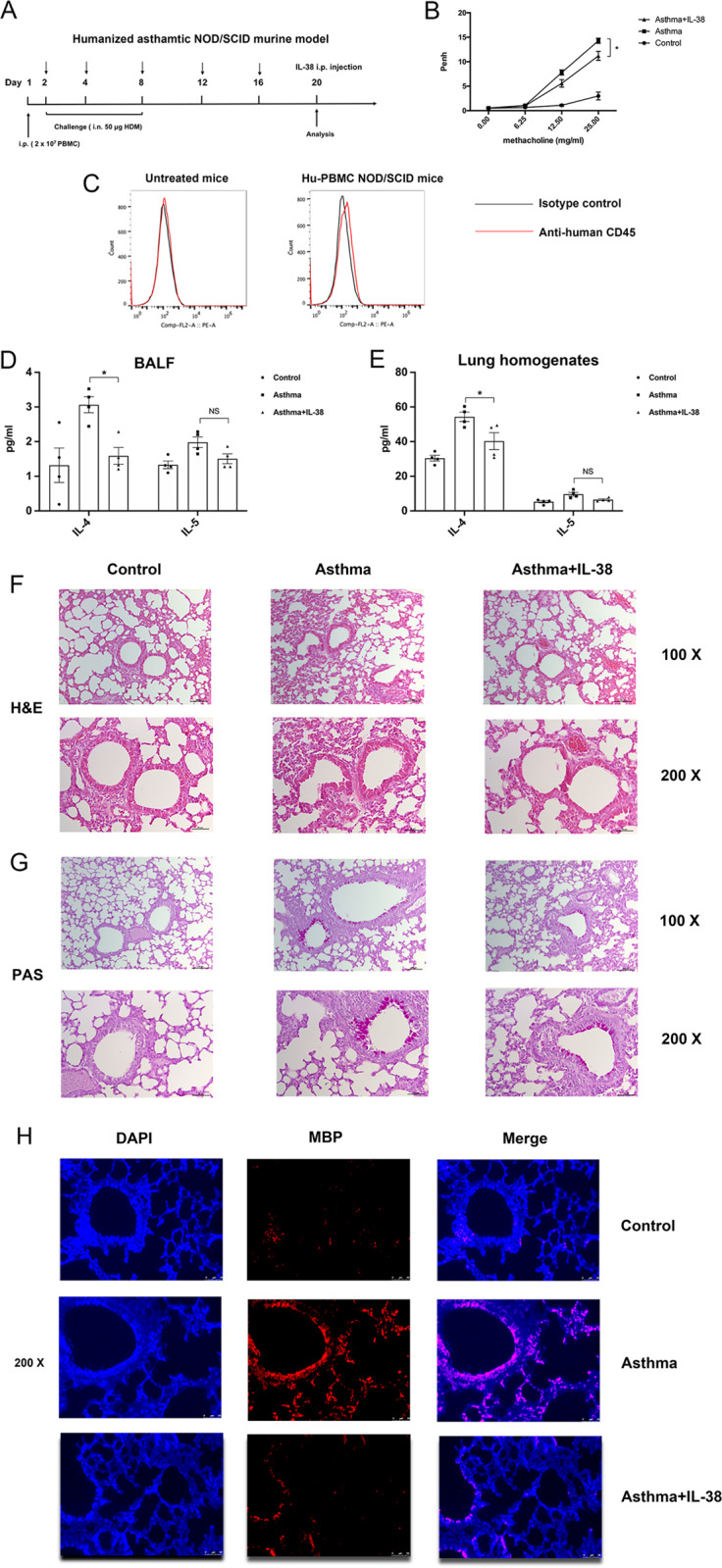

In vivo effects of IL-38 on humanized HDM-induced allergic asthma mice. a The timeline protocol of the humanized allergic asthma model is shown. b Airway obstruction was measured as Penh values; Penh (% baseline) was calculated by dividing the methacholine-induced Penh value by the PBS baseline value (n = 3). c Human CD45+ cells were assessed on day 20 in the lungs of human PBMC-reconstituted NOD/SCID mice. The secretion of human IL-4 and IL-5 into d the BALF and e lung homogenates was determined. f, g Representative lung sections stained with H&E or PAS were examined at ×100 or ×200 magnification (n = 3). h Immunostaining analysis of eosinophils in mouse lung tissue samples and lung sections was examined at ×200 magnification. i Gating strategies for CCR3+ eosinophils and CD4+CRTH2+ cells are shown. Flow cytometric identification of CCR3+ eosinophils in single-cell suspensions generated from the j, k BALF and l, m lungs is shown. Flow cytometric analysis of CD4+CRTH2+ cells in n the lungs, o MLN, p spleen, and q thymus is shown. r Results are expressed as percentages for CD4+ Th cells. The results are shown as the mean ± SEM of triplicate independent experiments with a total of four mice. *P < 0.05, **P < 0.01, and ***P < 0.001 when compared between the denoted groups. Abbreviation: MLN, mediastinal lymph nodes
Discussion
IL-38 has been considered a potential inhibitor of the IL-1 family (IL-1F), as it has sequence homology with the antagonists IL-1RA and IL-36RA.30 The anti-inflammatory properties of IL-38 have been investigated in autoimmune diseases, particularly RA22 and SLE.31 In addition, IL-38 can exert anti-inflammatory effects on human macrophages by suppressing proinflammatory mediator release, resulting in reduced Th17 cell activation.19 Our group previously reported significantly higher expression of circulating IL-38 in allergic asthma children than in controls, as well as the associated clinical implications, thereby indicating that IL-38 might be involved in the immunopathogenesis of allergic asthma.20
Allergic asthma is characterized by infiltration of inflammatory cells, including eosinophils, and secretion of inflammatory mediators, which lead to AHR. Notably, eosinophils can be activated by Th2 cytokines or virus infection-related proteins contributing to asthma exacerbation, and cytotoxic molecules released by eosinophil degranulation, including ECP and eosinophil peroxidase, are detrimental to airway epithelial cells, leading to tissue damage and airway remodeling.10 As the bronchial epithelial cell–eosinophil interaction is the major contributor to tissue remodeling and asthma exacerbation, our results showed that IL-38 exhibited anti-inflammatory activity in a coculture of human primary bronchial epithelial cells and eosinophils modeling allergic asthma inflammation by downregulating the inflammatory cytokine, chemokine, and ICAM-1 expression induced by the RLR ligand poly (I:C)/LyoVec or proinflammatory cytokine TNF-α. Notably, the viral infection-related Th1 chemokine CXCL10 could be significantly inhibited by IL-38 in the coculture system, even at a minimal concentration (10 ng/ml), in a dose-dependent manner. ICAM-1 has been considered a marker of airway inflammation, which is closely associated with the disease severity of asthma.32 Adhesion of eosinophils to epithelial cells via ICAM-1 can activate eosinophils to contribute to asthma exacerbation.33 In our study, ICAM-1 expression was markedly inhibited by IL-38 in cocultured epithelial cells and eosinophils, thereby indicating that IL-38 may display a regulatory effect on airway inflammation.
It is worth noting that IL-1 family cytokines generally need to be N-terminally processed to become bioactive.30 However, there is no caspase-1 consensus cleavage site in IL-38. Thus, the maturation process of IL-38 is still poorly understood. Previous studies indicate that both full-length and truncated IL-38 exert anti-inflammatory effects. For example, both forms can limit IL-6 and IL-8 production in human inflammatory macrophages19 and reduce IL-17A expression induced in Candida albicans-stimulated human PBMCs.25 Of note, mature IL-38 seems to exert a stronger effect and limits proinflammatory mediators over a much broader concentration range compared with full-length IL-38.25 Here, in our in vitro study, full-length human recombinant IL-38 was used and our results are comparable with those of previous studies19,25,26.
To explore signature intracellular signal transduction pathways involved in the suppressive activity mediated by IL-38, high-dimensional mass cytometry was utilized to screen for the regulation of seven essential signaling pathways related to the allergic inflammatory immune response in the coculture system. Together with flow cytometric analysis, mass cytometry indicated that the central signal transduction cascades were suppressed by IL-38 in both bronchial epithelial cells and eosinophils. It has been reported that the activation of STAT1 promotes allergen-induced airway inflammation and is linked to the expression level of ICAM-1 on epithelial cells, thereby contributing to the development of diseases.34 In allergic asthma, STAT3 functions as an epithelial mediator and directly regulates inflammatory responses by inducing mediators and adhesion molecules.35 In addition, MAPK pathways, including the ERK, JNK, and p38 pathways, participate in the regulation of inflammatory cytokine expression and appear to be involved in allergic asthma.36 Overall, IL-38 can diminish the production of inflammatory cytokines and chemokines, and adhesion molecules induced by poly (I:C)/LyoVec or TNF-α, which is regulated by the intracellular STAT1, STAT3, p38, ERK1/2, and NF-κB signaling pathways, in cocultured human primary epithelial cells and eosinophils. It was notable that IL-38 induced a significant elevation in POU2AF1 gene expression in poly (I:C)/LyoVec- and TNF-α-stimulated cocultures, and the expression of POU2AF1 in airway epithelium was closely related to airway host defense.37 Moreover, IL-38 upregulated the expression of RGS13, which can inhibit allergic responses in mice by inhibiting IgE receptor-mediated mast cell degranulation.38 Consistently, IL-38 has been shown to inhibit the in vitro release of the allergic inflammatory cytokines IL-17 and IL-22 from fungus-activated human PBMCs.18 Therefore, IL-38 can exhibit a suppressive effect on allergic inflammation.
Using an HDM-induced asthma murine model, IL-38 was found to ameliorate the infiltration of inflammatory cells, especially eosinophils, into the airway, which was mediated by a striking reduction in the secretion of the Th2 cytokines IL-4 and IL-5, and the principal eosinophil chemoattractant CCL11. It is widely believed that the Th2 response initiates, perpetuates, and exacerbates asthma.39 Our study and other studies have shown that Th17 cells provoke the development of asthma and play vital roles in airway remodeling and collagen deposition.40,41 ILC2s have been reported to participate in the central pathogenesis of allergic asthma, contributing to type 2 inflammatory responses.42 Hyperactive immune responses mediated by Th1, Th2, and Th17 cells and ILC2s can lead to tissue inflammation; therefore, Treg cells are crucial for maintaining immune homeostasis and preventing the development of inflammatory diseases. Treg cells such as Tr1, Tr2, and Tr35 cells can suppress allergic asthma responses by downregulating airway inflammation and preventing tissue remodeling.43 Our previous study also demonstrated that Tr35 cell-related IL-35 could inhibit the development of allergic airway inflammation by suppressing the differentiation of inflammatory dendritic cells in the inflamed site and their accumulation in the draining lymph nodes.44 Our present study showed significantly decreased Th2 cell and ILC2 numbers, slightly reduced Th17 cell numbers, and markedly elevated Treg cell numbers in the lungs, spleen, and draining lymph nodes of IL-38-treated allergic asthma mice. These results were further confirmed by reduced ex vivo release of IL-4 and IL-5 from HDM-stimulated splenocytes obtained from IL-38-treated mice, which exhibited fewer Th2 cells than control group mice (Fig. 6u–z). Moreover, the Treg-related suppressive cytokine IL-10 can downregulate allergen-specific Th1 and Th2 cell proliferation.45 Therefore, the elevated IL-10 levels in supernatants of splenocytes from IL-38-treated mice upon restimulation with HDM suggested that IL-38 could upregulate the splenic Treg cell population via IL-10. An emerging study has shown that TGF-β plays dual roles in allergic disease43. TGF-β promotes the conversion of naive T cells into Treg cells to perform regulatory functions through the suppression of Th1 and Th2 cell development.46 On the other hand, TGF-β participates in the pathogenesis of airway remodeling in asthma patients47 and facilitates inflammatory immune responses by promoting Th17 cell development in the presence of IL-6.48 In our study, elevated TGF-β expression in asthmatic mice was closely associated with the proinflammatory effect of TGF-β characterized by increased numbers of Th17 cells rather than Treg cells. Taken together, our results indicate that exogenous IL-38 plays an anti-inflammatory role in decreasing TGF-β release, thereby reducing the percentage of Th17 cells. Macrophage polarization plays an important role in the development of asthma. Upon exposure to different microenvironments, macrophages can be polarized into classically activated macrophages (M1) or alternatively activated macrophages (M2).49 In our study, higher levels of M2 cells were found in HDM-group mice, thereby indicating a predominantly M2 response in allergic asthma, which is in concordance with a previous study.50 By mimicking human allergic asthma, the humanized murine model of asthma further confirmed the in vivo anti-inflammatory activity of IL-38, as evidenced by IL-38 exhibiting a regulatory effect on allergic inflammation by downregulating the infiltration of eosinophils into the airway and decreasing the frequencies of Th2 cells in the lungs and MLNs.
A recent study suggested that IL-38-knockout (KO) C57BL/6 mice might attenuate eosinophilic airway inflammation.51 The difference in effect on allergic asthma between knocking out IL-38 and the present exogenous application of IL-38 may be due to differences in mouse strains, allergic asthma models, and IL-38 application. BALB/c and C57BL/6 mice actually display different distributions of proinflammatory cytokines and inflammatory cells, as well as varying degrees of airway responsiveness.52 Therefore, Th2-prone BALB/c mice, which have greater levels of asthma-related cytokines, were used in our study.53 Notably, OVA is hardly relevant to human asthma and OVA-challenged mice can only develop limited and moderated lung inflammation.54 In our present study, using an HDM-induced allergic asthma model established in BALB/c mice could better mimic the key features of asthma. In addition, previously generated IL-38-KO mice actually do not share the same genetic background as wild-type mice, as no IL-38-KO mice have been generated on the C57BL/6NCrl background.51 In this regard, further studies may be required to study IL-38 in airway allergic inflammation, e.g., the application of CRISPR/Cas9 to generate IL-38-knockin mice.
In conclusion, our study elucidated the in vitro anti-inflammatory role of IL-38 in viral/TNF-α-activated cocultures of HBE cells and eosinophils via the intracellular p38, STAT1, STAT3, ERK, and NF-κB pathways by modulating a panel of genes, including POU2AF1 and RGS13, expressed in allergic airway inflammation. Furthermore, in vivo and ex vivo anti-inflammatory activities of IL-38 in HDM-induced and humanized allergic asthma murine models were demonstrated, and IL-38 was shown to ameliorate airway obstruction, decrease eosinophil accumulation and Th2-related cytokine release, and attenuate Th2, ILC2, and Th17 cell proliferation, while also promoting Treg cell differentiation (Fig. 8). Therefore, our results demonstrated the regulatory role of IL-38 in allergic inflammation in allergic asthma and provided a biochemical basis for the development of this regulatory cytokine as a novel therapeutic modality for the treatment of allergic disease.
Fig. 8.

Graphical summary of the anti-inflammatory activities of IL-38 in allergic airway inflammation. In murine allergic asthma, IL-38 can inhibit the accumulation of eosinophils, ILC2s, and Th2 cells, and the release of CCL11, ECP, and the Th2-related cytokines IL-4, IL-5, and IL-13. Moreover, IL-38 can promote Tregs, which are regulated by IL-10 to maintain immune homeostasis. In our in vitro study, we focused on the interaction between eosinophils and bronchial epithelial cells. The activation of cocultured human primary epithelial cells and eosinophils by the viral mimic dsRNA RLR ligand poly (I:C)/LyoVec or proinflammatory TNF-α could be suppressed by IL-38, and this suppression was mediated by downregulation of the p38, STAT1, STAT3, ERK1/2, and NF-κB pathways, as well as upregulation of the expression of the airway host defense gene POU2AF1 and anti-allergic response gene RGS13 in bronchial epithelial cells, leading to significantly reduced expression of ICAM-1 and proinflammatory cytokines and chemokines. In addition, IL-38 was capable of decreasing the phosphorylation of p38, STAT1, STAT3, ERK1/2, and IκBα in eosinophils in the coculture system, thereby ameliorating allergic airway inflammation
Materials and methods
Mice
Inbred male BALB/c mice (6–8 weeks old) and NOD/SCID mice were bred under specific pathogen-free conditions and were maintained at the Laboratory Animal Services Center, The Chinese University of Hong Kong. Experiments were conducted following the principles outlined in the Animal Experimentation Ethics Committee Guide for the Care and Use of Laboratory Animals, as approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong.
Patients
Adult patients (aged 30–40 years) diagnosed with allergic asthma caused by HDM sensitization (n = 3) and sex- and age-matched non-atopic healthy control subjects (n = 3) were recruited from the Prince of Wales Hospital, Hong Kong. EDTA-treated blood (50 ml) obtained from each subject was diluted with phosphate-buffered saline (PBS) and PBMCs were then collected using Ficoll gradient centrifugation (GE Healthcare Bio-Sciences, Piscataway, NJ, USA). The experimental protocol used in our study was approved by the Clinical Research Ethics Committee, The Chinese University of Hong Kong-New Territories East Cluster Hospitals, according to the 1964 Declaration of Helsinki and its later amendments. Informed written consent was obtained from all subjects.
Purification of eosinophils from human buffy coats
Fresh human buffy coats were obtained from healthy volunteers at the Hong Kong Red Cross Blood Transfusion Service for the purification of primary human eosinophils. PBS-diluted fresh human blood buffy coats were centrifuged in a 1.082 g/ml isotonic Percoll solution (GE Healthcare Life Sciences) for 25 min at 1800 r.p.m. After red blood cell lysis, the obtained granulocyte fraction was collected to isolate eosinophils with anti-CD16 antibody-coated magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany), followed by depletion of CD16-positive cells by loading the cells onto a LS+ column (Miltenyi Biotec) within a magnetic field. The flow-through fraction containing eosinophils with a purity of at least 99% as assessed by a Hemacolor rapid blood smear stain (Merck Diagnostica, Darmstadt, Germany) was collected.13
Activation of eosinophils by influenza virus
Purified eosinophils (5 × 105) were prepared in tubes and then stimulated with 0.5 multiplicity of infection of influenza virus subtype H1 or H3 at 37 °C and 5% CO2 for 24 h. The cell culture supernatants were obtained for quantification of ECP (Thermo Fisher Scientific, Inc., Waltham, MA USA), TNF-α, IL-6, IL-8, CCL2, IL-10, and IL-1β levels (Millipore Corp., Billerica, MA, USA).
Identification and transcriptional profile of IL-38 target genes using RNA-sequencing
After optimization, cocultured primary HBE cells and eosinophils were pretreated with full-length recombinant human IL-38 (aa 1–152) (Adipogen Life Sciences, San Diego, CA, USA) (100 ng/ml) for 10 min and stimulated with low-molecular-weight poly (I:C)/LyoVec (2 μg/ml) or human TNF-α (20 ng/ml) for an additional 20 h. Total RNA was extracted from the cocultured cells using QIAzol reagent (Qiagen, Inc., Valencia, CA, USA) and the RNA samples were sent to Novogene Ltd., Beijing, China, for library preparation and transcriptomic sequencing with next-generation sequencing. mRNA was purified from total RNA using poly-T oligo-attached magnetic beads and fragmented randomly by the addition of a fragmentation buffer for library construction. Sequencing libraries were created using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (Illumina Corp., San Diego, CA, USA). The library preparations were sequenced on an Illumina HiSeq 4000 platform to generate 150 bp paired-end reads. The sequenced reads/raw reads containing low-quality reads or reads containing an adaptor were filtered to obtain clean reads and these clean reads were subsequently mapped to a reference genome by HISAT (version 2.0.4). To analyze gene expression levels, fragments per kilobase of transcript per million mapped reads were calculated by HTSeq (version 0.6.1). Differential gene expression analysis of six experiments was performed using DESeq2 (version 1.10.1, Bioconductor) and genes with P < 0.05 were regarded as differentially expressed. Cluster analysis of the DEGs was applied to describe the expression patterns of the DEGs under different treatments.
Validation of IL-38 receptors and RNA-seq results by real-time quantitative PCR
Total RNA was extracted from HBE cells, eosinophils, cocultured cells, and mouse lung tissue samples using QIAzol reagent (Qiagen, Valencia, CA, USA) and reverse transcribed into complementary DNA using PrimeScriptTM RT Master Mix (Takara Bio, Inc., Shiga, Japan). The expression of human IL-38 receptors, mouse IL-38 receptors, and target genes identified in RNA-seq data were quantified using a SYBR® Premix Ex TaqTM kit (Takara Bio, Inc., Shiga, Japan). The housekeeping gene GAPDH was considered a reference gene and the 2−ΔCt method (Ct, target gene-Ct, GAPDH) was used to calculate relative gene expression. The murine IL-1RAPL1 gene was detected by quantitative primer assays (Qiagen). All primer sequences are shown in Table 2.
Table 2.
Sequences of primers used for real-time qPCR
| Gene | Primer sequence (5′−3′) |
|---|---|
| Human- POU2AF1 | Forward CGTGTGAAGGAGCCAGTGAA |
| Reverse AGGCAGGAAGGACCCACT | |
| Human- RGS13 | Forward ATGAGCAGGCGGAATTGTTGGA |
| Reverse GAAACTGTTGTTGGACTGCATA | |
| Human- PHLPP2 | Forward CTAAGTTGCAACGACTTGAC |
| Reverse TCCATCAAACATGCCATACA | |
| Human-LYZ | Forward CTCTCATTGTTCTGGGGC |
| Reverse ACGGACAACCCTCTTTGC | |
| Human-BMF | Forward ATGGAGCCATCTCAGTGTGTG |
| Reverse CCCCGTTCCTGTTCTCTTCT | |
| Human-MT1G | Forward CTTCTCGCTTGGGAACTCTA |
| Reverse AGGGGTCAAGATTGTAGCAAA | |
| Human-MUC15 | Forward CTACACCTGCTCTGTCTTCA |
| Reverse GCAATGAGACACCCAGAATA | |
| Huamn-S1PR3 | Forward ACAACCGCATGTACTTTTTCA |
| Reverse TACTGCCCTCCCTGAGGAACC | |
| Human-IL-1R1 | Forward CTGAGAAGCTGGACCCCTTG |
| Reverse GCATTTATCAGCCTCCAGAGA | |
| Human-IL-36R | Forward GCTGGAGTGTCCACAGCATA |
| Reverse GCGATAAGCCCTCCTATCAA | |
| Human-IL-1RAPL1 | Forward AACTTGGCATTTGTGAATGGGAT |
| Reverse CCATCGGCGGAGCCTCTTTT | |
| Human-GAPDH | Forward CTCTGCTCCTCCTGTTCGAC |
| Reverse ACGACCAAATCCGTTGACTC | |
| Mouse-IL-1R1 | Forward GTGCTACTGGGGCTCATTTGT |
| Reverse GGAGTAAGAGGACACTTGCGAAT | |
| Mouse-IL-36R | Forward AAACACCTAGCAAAAGCCCAG |
| Reverse AGACTGCCCGATTTTCCTATG | |
| Mouse-GAPDH | Forward GCAAAGTGGAGATTGTTGCCA |
| Reverse CCTTGACTGTGCCGTTGAATTT |
HDM-induced allergic asthma murine model
To induce allergic airway inflammation, mice were sensitized by intranasal (i.n.) instillation of 1 μg HDM protein (Greer laboratories, Lenoir, NC, USA) or PBS on day 1, followed by i.n. challenge with 10 μg HDM protein or PBS daily between days 8 and 12. An optimized dose of recombinant murine IL-38 (aa 3–152) (800 ng) (Adipogen Life Sciences, San Diego, CA, USA) was given by i.p. injection just before the sensitization and challenge. The mice were examined 1 day after the last challenge. For monitoring AHR, Penh values were measured by exposing conscious mice to aerosolized PBS to establish a baseline value, followed by exposure to increasing concentrations of aerosolized methacholine using a whole-body plethysmograph (Buxco-Force Pulmonary Maneuvers, Buxco Research Systems, Wilmington, NC, USA).44 Lung tissue samples were obtained from killed mice, fixed with 4% paraformaldehyde, and embedded in paraffin. Sections (5 μm) were prepared for hematoxylin and PAS staining using a H&E staining kit (Beyotime, Inc., Jiangsu, China) or PAS staining kit (Sigma-Aldrich Corp., Saint Louis, MO, USA), respectively. For immunostaining analysis, lung sections were first subjected to antigen retrieval with a citric acid buffer (10 mM, pH 6.0), blocked with 5% fetal bovine serum (FBS) (Gibco Invitrogen Corp, Carlsbad, CA, USA), incubated with a primary rat anti-mouse MBP antibody (specific for eosinophils, provided by James J. Lee, PhD, Mayo Clinic, Scottsdale, AZ, USA) and then incubated with a secondary Cy3-conjugated goat anti-rat immunoglobulin G antibody (Beyotime Co., Shanghai, China). After staining, the lung sections were examined by light microscopy (Leica DM6000B, Leica Microsystems, Inc., Buffalo Grove, IL, USA). Bronchoalveolar lavage was performed by injecting 1 ml PBS into the lungs through a secured tracheal cannula. Then, the BALF was centrifuged to obtain the supernatant for the quantification of cytokine/chemokine levels and the cell pellets were resuspended in PBS for flow cytometric analysis.
Humanized allergic asthma NOD/SCID mouse model
SCID mice were reconstituted by i.p. injection of purified human PBMCs (2 × 107) obtained from allergic asthma patients or control subjects on day 1. On days 2, 4, and 8, the mice were i.n. treated with 50 μg HDM. Recombinant human IL-38 (aa 1–152) (800 ng) (Adipogen Life Sciences, San Diego, CA, USA) or PBS was i.p injected on days 2, 4, 8, 12, and 16. All the mice were killed for analysis on day 20.
Preparation of single-cell suspensions and flow cytometric analysis
Single-cell suspensions were prepared from the lungs, spleen, thymus, and MLNs of mice. The spleen, thymus, and MLNs were mechanically disrupted using a 70 μm cell strainer (Corning, Inc., New York, NY, USA). Dissected lung tissue samples were digested with RPMI 1640 medium (Gibco Invitrogen Corp., Carlsbad, CA, USA) containing Collagenase D (Sigma-Aldrich) and DNase I (Sigma-Aldrich) for 30 min at 37 °C. Single-cell suspensions from the lungs were obtained after filtration with a 70 μm cell strainer. The cells were then fixed with a Fixation/Permeabilization working solution, followed by treatment with a permeabilization buffer (Invitrogen, Inc., Carlsbad, CA, USA). Subsequently, a mouse Treg cell staining kit (Invitrogen, CA, USA) was used for flow cytometric analysis of Treg cells. Single cells were stained with anti-mouse IFN-γ, IL-4, and IL-17 antibodies (BioLegend, Inc., San Diego, CA, USA) to identify Th1, Th2, and Th17 cells, respectively. After the last wash, the different Th-cell subsets were analyzed by a FACSVia flow cytometer (BD Biosciences, San Jose, CA, USA). For ILC2 staining, single-cell suspensions from mouse spleens were stained with anti-mouse Lineage, CD45, CD127, T1/ST2, and CD25 antibodies at room temperature for 30 min,55 and the cells were washed twice with PBS before being analyzed by using a Navios EX Flow Cytometer (Beckman Coulter, Inc., Miami, FL, USA). To detect the phenotype of macrophages, anti-mouse F4/80, anti-mouse CD11c, and anti-mouse CD206 antibodies (BioLegend, Inc., San Diego, CA, USA) were used and the stained cells were analyzed by a FACSCalibur flow cytometer (BD Biosciences). The specific markers for M1 and M2 macrophages were defined in previous studies.56 For the humanized allergic asthma NOD/SCID mouse model, an anti-mouse CCR3 antibody was used to detect murine eosinophils in mouse BALF and lung tissue samples, and an anti-human CD45 antibody was utilized to detect human PBMCs in mouse lung tissue samples. Anti-human CD3 and anti-human CRTH2 antibodies were used to detect human T cells in the humanized mice. After immunofluorescence staining, cells were analyzed by using a BD FACSVia flow cytometer.
Coculture of human primary bronchial epithelial cells and eosinophils
Primary HBE cells were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and were cultured in bronchial epithelial cell medium (ScienCell). For coculture studies, the medium was replaced with RPMI 1640 medium containing 10% FBS (Thermo Fisher, Waltham, MA USA). A negative control of 56 °C heat-inactivated human IL-38 (100 ng/ml) and positive control of dexamethasone (1 μM) (Sigma-Aldrich) were included in our in vitro study. Primary bronchial epithelial cells (1 × 105) and purified eosinophils (3 × 105) were cocultured with or without human IL-38 (100 ng/ml) pretreatment for 10 min, followed by poly (I:C)/LyoVec or human TNF-α (R&D Systems, Minneapolis, MN, USA) stimulation for an additional 20 h. To analyze the expression of ICAM-1 using a mouse anti-human ICAM-1 antibody (BioLegend), mixed bronchial epithelial cells and eosinophils were differentially analyzed by using a FACSCalibur flow cytometer (BD Biosciences) and distinguished based on the forward-scatter and side-scatter parameters.
Mass cytometric analysis of intracellular signaling pathways
Cocultured cells were pretreated with human IL-38 (100 ng/ml) or heat-inactivated human IL-38 (100 ng/ml) for 10 min and stimulated with poly (I:C)/LyoVec (2 μg/ml) or human TNF-α (20 ng/ml) for 20 min. The cocultured cells were first blocked with heparin sodium salt (Sigma-Aldrich) to reduce nonspecific eosinophil staining57 and then the cells were labeled with metal-tagged antibodies using a Maxpar® signaling I panel kit (Fluidigm, Inc., San Francisco, CA, USA). After staining, the cells were acquired by a Helios CyTOF mass cytometer (Fluidigm) and the data were analyzed by the cloud-based analytical platform Cytobank (Cytobank, Inc., Santa Clara, CA, USA).
Flow cytometric analysis of intracellular signaling pathways
Cocultured cells were pretreated with human recombinant IL-38 (100 ng/ml) for 10 min and then stimulated with poly (I:C)/LyoVec (2 μg/ml) or human TNF-α (20 ng/ml) for 20 min. The primary bronchial epithelial cells and eosinophils were collected and fixed with a fixation buffer (Biolegend, Inc., San Diego, CA, USA) for 30 min at room temperature, followed by incubation in an intracellular staining permeabilization wash buffer (Thermo Fisher) at room temperature for 30 min. The cells were then stained with mouse anti-human fluorescent dye-conjugated antibodies specific for phosphorylated p38, phosphorylated ERK1/2, phosphorylated STAT1, phosphorylated STAT3, and phosphorylated IκBα or the corresponding isotypic control antibodies (BD Pharmingen, San Diego, CA, USA). After washing, the cells were analyzed with a BD FACSCalibur flow cytometer.
Quantification of cytokine and chemokine concentrations
The concentrations of human IL-4 and IL-5 in culture supernatants were analyzed using an ELISA kit (Biolegend, Inc., San Diego, CA, USA) and the human IFN-β level in cell culture supernatants was detected by using an ELISA kit (Invitrogen, CA, USA). Human IL-6, IL-1β, CCL5, and CXCL10 levels in culture supernatants and murine IL-4, IL-5, IL-6, IL-13, IL-17, CCL5, CCL11, and TGF-β levels in the serum, the BALF, lung homogenates, and splenic culture supernatants were quantified with a Human or Mouse Cytokine Milliplex MAP assay kit (Millipore Corporation, Billerica, MA, USA) by using a Bio-Plex 200 system (Bio-Rad Laboratories, Hercules, CA, USA).
Statistics
All numerical data are expressed as the mean ± SEM. Intergroup differences were assessed by one-way analysis of variance or Student’s t-test using GraphPad PRISM software version 6.01. A significance level of p < 0.05 was considered significantly different.
Supplementary information
Acknowledgements
This work was supported by Direct Grant for Research 2016/2017 and 2018/2019 (Medicine Panel), project codes 4054327 and 4054391, respectively, and The Chinese University of Hong Kong, Hong Kong, and Grant from Hong Kong Institute of Allergy 2018/2019 (project code: 6904815). This work was supported in part by grants from the University of Macau (MYRG 2018-00033-FHS) and the Macau Science and Technology Development Fund (FDCT102/2015/A3) to E.C. We would also like to acknowledge Kaeling Tan for her help of the daily operation of the Helios Mass Cytometer at the Genomics, Bioinformatics and Single Cell Analysis Core at Faculty of Health Sciences, University of Macau.
Author contributions
X.S., C.K.W. and P.K.-S.C. designed experiments. X.S., T.H., E.C.-W.C., T.N.-T.I., V.W.-H.T., I.M.T.C. and M.S.-M.T. performed the experiments and analyzed the data. X.S., C.-K.W. and C.W.-K.L. drafted and revised the manuscript.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version of this article (10.1038/s41423-019-0300-7) contains supplementary material.
References
- 1.Wenzel SE. Asthma: defining of the persistent adult phenotypes. Lancet. 2006;368:804–813. doi: 10.1016/S0140-6736(06)69290-8. [DOI] [PubMed] [Google Scholar]
- 2.Schatz M, Rosenwasser L. The allergic asthma phenotype. J. Allergy Clin. Immunol. Pract. 2019;2:645–648. doi: 10.1016/j.jaip.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Foster PS, et al. Modeling TH2 responses and airway inflammation to understand fundamental mechanisms regulating the pathogenesis of asthma. Immunol. Rev. 2017;278:20–40. doi: 10.1111/imr.12549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matucci A, Vultaggio A, Maggi E, Kasujee I. Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question. Respir. Res. 2018;19:1–10. doi: 10.1186/s12931-018-0813-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mckenzie GJ, et al. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–432. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 6.Yang T, et al. Characteristics of proinflammatory cytokines and chemokines in airways of asthmatics: relationships with disease severity and infiltration of inflammatory cells. Chin. Med. J. (Engl.) 2017;130:2033–2040. doi: 10.4103/0366-6999.213428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J. Allergy Clin. Immunol. 2018;140:895–906. doi: 10.1016/j.jaci.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hallstrand TS, et al. Airway epithelial regulation of pulmonary immune homeostasis and inflammation. Clin. Immunol. 2014;151:1–15. doi: 10.1016/j.clim.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Possa SS, Leick EA, Prado CM, Martins MA. Tibério IFLC. Eosinophilic inflammation in allergic asthma. Front. Pharm. 2013;4:1–9. doi: 10.3389/fphar.2013.00046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fulkerson PC, Rothenberg ME. Targeting eosinophils in allergy, inflammation and beyond. Nat. Rev. Drug Discov. 2013;12:117–129. doi: 10.1038/nrd3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shamri R, Young KM, Weller PF. PI3K, ERK, p38 MAPK and integrins regulate CCR3-mediated secretion of mouse and human eosinophil-associated RNases. Allergy. 2013;68:880–889. doi: 10.1111/all.12163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wong CK, et al. NOD-like receptors mediated activation of eosinophils interacting with bronchial epithelial cells: a link between innate immunity and allergic asthma. Cell Mol. Immunol. 2013;10:317–329. doi: 10.1038/cmi.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu J, et al. Anti-allergic inflammatory activity of interleukin-37 is mediated by novel signaling cascades in human eosinophils. Front Immunol. 2018;9:1445. doi: 10.3389/fimmu.2018.01445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiao D, et al. Activation of eosinophils interacting with bronchial epithelial cells by antimicrobial peptide LL-37: implications in allergic asthma. Sci. Rep. 2017;4:1–13. doi: 10.1038/s41598-017-02085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin H, et al. Cloning and characterization of IL-1HY2, a novel interleukin-1 family member. J. Biol. Chem. 2001;276:20597–20602. doi: 10.1074/jbc.M010095200. [DOI] [PubMed] [Google Scholar]
- 16.van de Veerdonk FL, de Graaf DM, Joosten LA, Dinarello CA. Biology of IL-38 and its role in disease. Immunol. Rev. 2018;281:191–196. doi: 10.1111/imr.12612. [DOI] [PubMed] [Google Scholar]
- 17.Towne JE, et al. Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) J. Biol. Chem. 2011;286:42594–42602. doi: 10.1074/jbc.M111.267922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Veerdonk FL, et al. IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl Acad. Sci. USA. 2012;109:3001–3005. doi: 10.1073/pnas.1121534109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mora J, et al. Interleukin-38 is released from apoptotic cells to limit inflammatory macrophage responses. J. Mol. Cell Biol. 2016;8:426–438. doi: 10.1093/jmcb/mjw006. [DOI] [PubMed] [Google Scholar]
- 20.Chu M, et al. Aberrant expression of novel cytokine IL-38 and regulatory T lymphocytes in childhood asthma. Molecules. 2016;21:933. doi: 10.3390/molecules21070933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu WD, Huang AF. Role of interleukin-38 in chronic inflammatory diseases: a comprehensive review. Front. Immunol. 2018;9:1462. doi: 10.3389/fimmu.2018.01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takenaka S, et al. IL-38: a new factor in rheumatoid arthritis. Biochem. Biophys. Rep. 2015;4:386–391. doi: 10.1016/j.bbrep.2015.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boutet MA, et al. IL-38 overexpression induces anti-inflammatory effects in mice arthritis models and in human macrophages in vitro. Ann. Rheum. Dis. 2017;76:1300–1308. doi: 10.1136/annrheumdis-2016-210630. [DOI] [PubMed] [Google Scholar]
- 24.Chu M, et al. Immunobiology in vivo anti-inflammatory activities of novel cytokine IL-38 in Murphy Roths Large (MRL)/lpr mice. Immunobiology. 2017;222:483–493. doi: 10.1016/j.imbio.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 25.Han Y, et al. IL-38 ameliorates skin inflammation and limits IL-17 production from γδ T cells. Cell Rep. 2019;27:835–846. doi: 10.1016/j.celrep.2019.03.082. [DOI] [PubMed] [Google Scholar]
- 26.Mercurio L, et al. IL-38 has an anti-inflammatory action in psoriasis and its expression correlates with disease severity and therapeutic response to anti-IL-17A treatment. Cell Death Dis. 2018;9:1104. doi: 10.1038/s41419-018-1143-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu K, et al. Hydrodynamic delivery of IL-38 gene alleviates obesity-induced inflammation and insulin resistance. Biochem. Biophys. Res. Commun. 2019;508:198–202. doi: 10.1016/j.bbrc.2018.11.114. [DOI] [PubMed] [Google Scholar]
- 28.Xu F, et al. Interleukin 38 protects against lethal sepsis. J. Infect. Dis. 2018;218:1175–1184. doi: 10.1093/infdis/jiy289. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, et al. A novel subset of CD4+TH2 memory / effector cells that produce inflammatory IL-17 cytokine and promote the exacerbation of chronic allergic asthma. J. Exp. Med. 2010;207:2479–2491. doi: 10.1084/jem.20101376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garraud T, Harel M, Boutet MA, Le Goff B, Blanchard F. The enigmatic role of IL-38 in inflammatory diseases. Cytokine Growth Factor Rev. 2018;39:26–35. doi: 10.1016/j.cytogfr.2018.01.001. [DOI] [PubMed] [Google Scholar]
- 31.Chu M, et al. In vivo anti-inflammatory activities of novel cytokine IL-38 in Murphy Roths Large (MRL)/lpr mice. Immunobiology. 2017;222:483–493. doi: 10.1016/j.imbio.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Gorska-Ciebiada M, Ciebiada M, Gorska MM, Gorski P. Grzelewska-Rzymowska I. ICAM-1 and TNF-α in patients with persistent asthma - relationship with the disease severity. J. Allergy Clin. Immunol. 2006;117:s258. doi: 10.1016/S1081-1206(10)61372-5. [DOI] [PubMed] [Google Scholar]
- 33.Nagata M, Sedgwick JB, Bates ME, Kits H, Busse WW. Eosinophil adhesion to vascular cell adhesion molecule-1 activates superoxide anion generation. J. Immunol. 1995;155:2194–2102. [PubMed] [Google Scholar]
- 34.Sampath D, et al. Constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asthma Find the latest version: constitutive activation of an epithelial signal transducer and activator of transcription (STAT) pathway in asth. J. Clin. Invest. 1999;103:1353–1361. doi: 10.1172/JCI6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simeone-penney MC, et al. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J. Immunol. 2007;178:6191–6199. doi: 10.4049/jimmunol.178.10.6191. [DOI] [PubMed] [Google Scholar]
- 36.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 37.Zhou H, et al. POU2AF1 functions in the human airway epithelium to regulate expression of host defense genes. J. Immunol. 2016;196:3159–3167. doi: 10.4049/jimmunol.1502400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bansal G, et al. RGS13 controls G-protein-coupled receptor-evoked responses of human mast cells. J. Immunol. 2008;181:7882–7890. doi: 10.4049/jimmunol.181.11.7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cho SH, Stanciu LA, Holgate ST, Johnston SL. Increased interleukin-4, interleukin-5, and interferon-gamma in airway CD4+ and CD8+ T cells in atopic asthma. Am. J. Respir. Crit. Care Med. 2005;171:224–230. doi: 10.1164/rccm.200310-1416OC. [DOI] [PubMed] [Google Scholar]
- 40.Molet S, et al. IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J. Allergy Clin. Immunol. 2001;108:430–438. doi: 10.1067/mai.2001.117929. [DOI] [PubMed] [Google Scholar]
- 41.Wong CK, et al. Activation of peripheral Th17 lymphocytes in patients with asthma. Immunol. Invest. 2009;38:652–664. doi: 10.1080/08820130903062756. [DOI] [PubMed] [Google Scholar]
- 42.Aron JL, Akbari O. Regulatory T cells and type 2 innate lymphoid cell-dependent asthma. Allergy. 2017;72:1148–1155. doi: 10.1111/all.13139. [DOI] [PubMed] [Google Scholar]
- 43.Kearley J, Robinson DS, Lloyd CM. CD4+CD25+ regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. J. Allergy Clin. Immunol. 2008;122:617–624. doi: 10.1016/j.jaci.2008.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong J, et al. Amelioration of allergic airway inflammation in mice by regulatory IL-35 through dampening inflammatory dendritic cells. Allergy. 2015;70:921–932. doi: 10.1111/all.12631. [DOI] [PubMed] [Google Scholar]
- 45.Timmann C, et al. Promoter haplotypes of the interleukin-10 gene influence proliferation of peripheral blood cells in response to helminth antigen. Genes Immun. 2004;5:256–260. doi: 10.1038/sj.gene.6364094. [DOI] [PubMed] [Google Scholar]
- 46.Li MO, Wan YY, Sanjabi S, Robertson AL, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu. Rev. Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 47.Flood-page P, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Invest. 2003;112:1029–1036. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mangan PR, et al. Transforming growth factor-beta induces development of the TH 17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 49.Saradna A, Do DC, Kumar S, Fu QL, Gao P. Macrophage polarization and allergic asthma. Transl. Res. 2018;191:1–14. doi: 10.1016/j.trsl.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robbe P, et al. Distinct macrophage phenotypes in allergic and nonallergic lung inflammation. Am. J. Physiol. Lung Cell Mol. Physiol. 2015;308:L358–L367. doi: 10.1152/ajplung.00341.2014. [DOI] [PubMed] [Google Scholar]
- 51.Matsuoka M, et al. Attenuated airway eosinophilic inflammations in IL-38 knockout mouse model. Kurum. Med. J. 2018;65:37–46. doi: 10.2739/kurumemedj.MS652009. [DOI] [PubMed] [Google Scholar]
- 52.De Sanctis GT, Daheshia M, Daser A. Genetics of airway hyperresponsiveness. J. Allergy Clin. Immunol. 2001;108:11–20. doi: 10.1067/mai.2001.116429. [DOI] [PubMed] [Google Scholar]
- 53.Gueders MM, et al. Mouse models of asthma: a comparison between C57BL/6 and BALB/c strains regarding bronchial responsiveness, inflammation, and cytokine production. Inflamm. Res. 2009;58:845–854. doi: 10.1007/s00011-009-0054-2. [DOI] [PubMed] [Google Scholar]
- 54.Nials AT, Uddin S. Mouse models of allergic asthma: acute and chronic allergen challenge. Dis. Model Mech. 2008;1:213–220. doi: 10.1242/dmm.000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klein Wolterink RG, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol. 2012;42:1106–1116. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 56.Fujisaka S, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58:2574–2582. doi: 10.2337/db08-1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rahman AH, Tordesillas L, Berin MC. Heparin reduces nonspecific eosinophil staining artifacts in mass cytometry experiments. Cytom. A. 2016;89:601–607. doi: 10.1002/cyto.a.22826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.