

Abstract
Autophagy degrades the cytoplasmic contents engulfed by autophagosomes. Besides providing energy and building blocks during starvation via random degradation, autophagy selectively targets cytotoxic components to prevent a wide range of diseases. This preventive activity of autophagy is supported by many studies using animal models and reports identifying several mutations in autophagy-related genes that are associated with human genetic disorders, which have been published in the past decade. Here, we summarize the molecular mechanisms of autophagosome biogenesis involving the proteins responsible for these genetic disorders, demonstrating a role for autophagy in human health. These findings will help elucidate the underlying mechanisms of autophagy-related diseases and develop future medications.
Subject terms: Protein quality control, Macroautophagy
Introduction
Macroautophagy (hereafter autophagy) is a bulk intracellular degradation system that digests self-components, as its name is derived from the Greek words auto (self) and phagy (eating). Autophagy degrades the cytoplasmic contents engulfed by a characteristic double-membrane structure called an autophagosome, which is ~1 µm1. This nonselective degradation system is essential for survival during starved conditions as it provides cells with buck-up energy and building blocks required to retain cellular functions. Moreover, a low level of basal autophagy constitutively maintains the turnover of self-components even without any exogenous stresses. Furthermore, autophagy has been shown to target invading bacteria in a highly specific manner to support the viability of host cells, suggesting that autophagy can selectively degrade harmful substances that cannot be digested by other pathways such as proteasomal degradation pathway. The selective autophagy eliminates damaged organelles, such as mitochondria and lysosomes or protein aggregates, that play a causative role in a range of diseases, including neurodegeneration. Recent studies have shown that the number of the type of targets is larger than previously anticipated, calling attention to a role of autophagy as a biological defense mechanism2. Although autophagosome observation by electron microscope and biochemical analysis had been suggested for basic topology and workflow of autophagy since the 1960s, the underlying molecular mechanisms were not elucidated for many years due to the difficulty of analysis. Ohsumi et al. carried out a large-scale genetic screening of Saccharomyces cerevisiae mutants and identified a series of genes essential for autophagy, some of which are overlapped with genes independently identified by Thumm’s lab and Klionsky’s lab3. These results led to powerful genetic analyses and striking mechanistic findings and an establishment of basic mechanism of autophagy. These reports were followed by subsequent research on autophagy in mammals, which led to the identification of the role for autophagy in human health; this discovery was awarded the 2016 Nobel Prize in Physiology or Medicine.
Our current understanding of autophagy in physiology is strongly supported by studies using model experimental systems. Moreover, many human hereditary diseases caused by autophagy-related genes have been reported in the last decade, contributing to the elucidation of the role of autophagy in human physiology. In this paper, we summarize the molecular mechanism of mammalian autophagy and describe the current understanding of human hereditary disorders to elucidate the physiological significance of autophagy in humans for future studies. Although not mentioned in detail here, microautophagy, which directly delivers substrates into vacuoles or lysosomes, and chaperone-mediated autophagy (CMA), which transports substrates to lysosomes via activity of chaperones, are also involved in maintenance of cellular homeostasis4.
Molecular mechanism of autophagosome biogenesis
The most characteristic feature of autophagy is the engulfment of cytoplasmic components by a double-membrane structure called an autophagosome. Unlike other organelles such as mitochondria, autophagosomes are generated de novo upon induction by stresses such as starvation. First, an isolation membrane (phagophore) is generated at the autophagosome formation site, followed by elongation and closure of the edge of the membranes to form autophagosomes. By the subsequent fusion with lysosomes containing various hydrolases, autophagosomes mature to autolysosomes so that the contents with the inner membranes are degraded (Fig. 1). In an electron micrograph, autophagosomes appear as a double-membrane structure with a cytoplasmic fraction, and autolysosomes have a single-membrane structure with a high electron density. This dynamic process of degradation is carried out by several functional units consisting of autophagy-related proteins. Following the identification of the yeast autophagy-related gene group ATG, researchers have found that homologs of these genes exist in mammals and are functionally conserved (Table 1). These proteins are mainly divided into groups with four different functions: the ULK1 protein kinase complex, the transmembrane protein ATG9 in single-membrane vesicles called ATG9 vesicles, the phosphoinositide 3-kinase (PI3K) complex, and a series of proteins that carry out post-translational modification of Atg8 homologs. The ULK1 complex and ATG9 vesicles are required for the formation of isolation membranes, and the PI3K complex is needed for the recruitment of factors required for the modification of the downstream Atg8 homologs to the isolation membrane. Atg8 homologs play a role in coordinating the extension and closure of the isolation membrane and further fusion of autophagosomes with lysosomes. These proteins are essential for starvation-induced autophagy as well as selective autophagy. In the following section, we will summarize the molecular details of autophagy, which are required to understand the underlying mechanisms of each hereditary disorder associated with mutations in autophagy-related genes.
Fig. 1. A brief overview of autophagy.
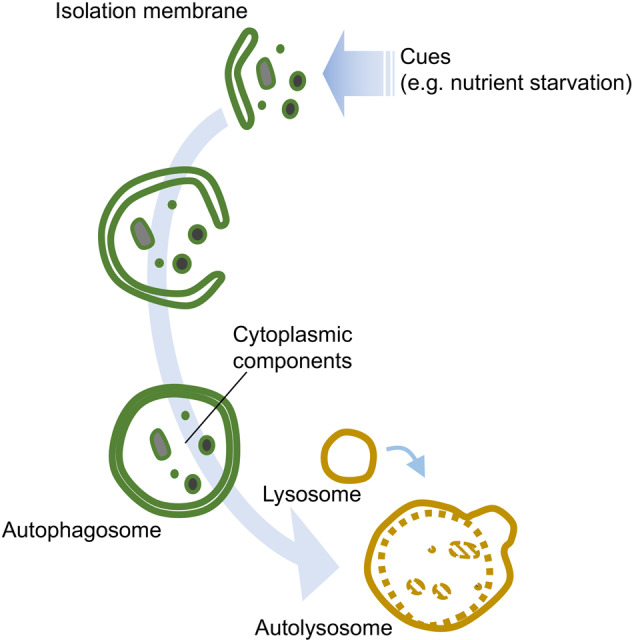
Upon cues such as a starvation signal, isolation membranes are generated de novo that extend to form autophagosomes sequestering cytoplasmic components. The contents are further digested by fusion with lysosomes containing a variety of hydrolases.
Table 1.
Homologs of autophagy-related genes.
| S. cerevisiae | Mammals | ||||||
|---|---|---|---|---|---|---|---|
| ULK1 complex | Atg1 | ULK1 | ULK2 | ULK3 | ULK4 | ||
| Atg13 | ATG13 | ||||||
| Atg17 | FIP200 | ||||||
| – | ATG101 | ||||||
| PI3K complex | VPS34 | VPS34 | |||||
| VPS15 | VPS15 | ||||||
| Atg6 | Beclin 1 | ||||||
| Atg14 | ATG14 | UVRAG | |||||
| – | Ambra1 | ||||||
| – | Rubicon | ||||||
| PI3P-binding proteins | Atg18 | WIPI1 | WIPI2 | WIPI3 (WDR45B) | WIPI4 (WDR45) | ||
| ATG9 vesicles | Atg9 | ATG9L1 | ATG9L2 | ||||
| LC3 conjugation system | Atg8 | LC3A | LC3B | LC3C | GABARAP | GABARAPL1 | GABARAPL2/GATE-16 |
| Atg4 | ATG4A | ATG4B | ATG4C | ATG4D | |||
| Atg3 | ATG3 | ||||||
| Atg7 | ATG7 | ||||||
| Atg10 | ATG10 | ||||||
| Atg5 | ATG5 | ||||||
| Atg12 | ATG12 | ||||||
| Others | Atg2 | ATG2A | ATG2B |
Induction of autophagy
Nutrient starvation is the best-characterized stressor inducing autophagy. The release from negative regulation by the mTOR complex triggers autophagic induction. Under normal nutrient conditions, ULK1 complex is inactivated due to its constitutive phosphorylation by the mTOR complex. The high amino acid concentration in the lysosome keeps the mTOR complex activated in a manner dependent on lysosomal proton pump v-ATPase complex, which promotes the binding of Rag GTPase with mTORC1. When cells are exposed to starvation conditions, decreased amino acid levels in the lysosomes makes mTORC1 to be released from the lysosomes, resulting in reduced phosphorylation of its targets5,6. Concomitantly, de-phosphorylation of ULK1 by phosphatase PP2A promotes activation of ULK1 complex7,8. In yeast, the phosphorylation changes the complex formation as phosphorylation of Atg13 by TOR inhibits the binding of Atg1 with Atg139. In mammalian cells, the phosphorylation does not halt the formation of the ULK1-ATG13-FIP200 complex but instead inhibits the kinase activity of ULK1 complex via regulating the binding of ULK1 complex with AMPK7,10. mTORC1 also regulates autophagy through transcription factors. Under normal nutrient conditions, the transcription factor TFEB is phosphorylated by mTORC1 that facilitates its localization in the cytoplasm through binding to the 14-3-3 protein. When this phosphorylation decreases during starvation, TFEB is transferred to the nucleus, which can induce the transcription of autophagy- and lysosome-related genes11,12. In addition to TFEB, another transcription factor, TFE3, also translocates to the nucleus in response to starvation to activate autophagy13. In summary, the mTOR pathway plays a central role in the control of autophagy. However, some pathways regulate autophagy independent of mTOR; and thus, the mechanism of autophagic activation is not always the same and varies in response to different kinds of cues14.
Origin of the isolation membrane
The source of autophagosomes has been a hot topic in the field of autophagy. The best-known source is the endoplasmic reticulum (ER). Autophagosomes have been proposed to be formed by extending the isolation membrane along with the ER, which is called the “cradle model”. This hypothesis is supported the three-dimensional structure generated by electron tomography15. It is generated from an extended ER membrane in a form of Ω-shaped structure called omegasome16. The shaping of omegasome and autophagosomes is supported by an assembly of actin inside these structures17. Consistent with the fact that mitochondria can also be a source, autophagosomes are formed at the contact sites between the ER and the mitochondria, as shown by simultaneous imaging of three organelle markers18,19. Other materials, such as cell membranes and lipid droplets, are also considered sources for autophagosomes20–22. ATG9 vesicles and the ER-Golgi intermediate compartment (ERGIC) are believed to be involved in supplying materials for isolation membranes23–26. To facilitate the source supply and the shaping of autophagosome, two upstream components play a major role: ULK1 complex and ATG9 recruit downstream factors and coordinate the events as described below.
Upstream factors of autophagy: membrane sources and formation of isolation membranes
In yeast, the pre-autophagosomal structure (PAS) is formed at the proximal area of the vacuole, where ATG proteins accumulate to generate isolation membranes27. The mechanism of their action was investigated by intracellular localization analysis of Atg proteins with fluorescent tags in the absence of each gene28. Based on this model, the hierarchy of the molecular actions of mammalian autophagy-related proteins was established. Mammalian cells do not form a clear PAS like yeast cells, but each autophagy-related protein accumulates at distinct sites where the isolation membrane is generated. These sites are observed by the formation of dots under fluorescence microscopy. As described above, phosphorylation of ULK1 complex is the most upstream target for autophagic induction and is located upstream of the cascade29. ATG9 is the only membrane protein in the core ATG proteins and is contained in a small membrane structure of ~30–60 nm called ATG9 vesicles. To initiate autophagosome nucleation, both ULK1 and ATG9 are localized at autophagosome formation sites that are supposed to be contact sites of ER and other organelles such as mitochondria19,30. Under the nutrient-rich conditions, ATG9 travels through plasma membranes, early endosomes, recycling endosomes and trans-Golgi network (TGN). Upon induction, a portion of it localizes to autophagosome formation site from recycling endosomes or TGN. It depends on TRAPPIII complex that mediates retrograde trafficking from endosomes to Golgi31–33. Although ATG9 vesicle itself does not become a membrane source, it facilitates the delivery of membrane sources for autophagosomes23. A recent study using a proteomics approach identified contents of ATG9 vesicles, showing that it is enriched in BAR-domain-containing proteins including Arfaptin 234. It supports the distribution of ATG9 vesicles and the delivery of PI4K and PI4KIIIβ to autophagosome formation sites.
Given that knockdown of ATG9 results in only a slight reduction in ULK1 dot formation23, and that ATG9 and ULK1 complex are independently targeted to depolarized mitochondria for Parkin-dependent mitophagy35, ULK1 and ATG9 are not essential for their initial recruitment to the site of autophagosome formation each other. However, ULK1 complex is needed for the trafficking of ATG9 vesicles as shown by the retention of ATG9 on TGN in the absence of ULK1 even in starved conditions36. It is mediated by phosphorylation of ATG9 on Ser14 by ULK1 upon starvation signal as is the case with yeast showing that Atg1 phosphorylates Atg9 to facilitate recruitment of downstream components to PAS37,38. Interestingly, knockdown of PI4KIIIβ leads to a reduction in dot formation of ATG13, a component of ULK1 complex, in starved condition, suggesting that ATG9 supports recruitment of ULK1 complex by promoting PI4P enrichment on the site of autophagosome formation34. Thus, it is most likely that those two major factors are required for their full activity to facilitate autophagy. It is not only at the step of initiation of isolation membrane formation but also elongation of the membrane. It has been known that ATG2A, which is required for supplying lipid to autophagosome membrane to facilitate the elongation37–40, works to establish ATG9 localization on mitochondria-associated ER membranes41,42. It is also the case of ULK1 complex because this complex phosphorylates a wide variety of substrates related to autophagosome biogenesis that are involved in the initiation step to the fusion step as well43.
Recently, it has been clear that PAS is a liquid-like condensate of Atg proteins. Upon starvation signal, the de-phosphorylation of Atg13 results in an establishment of Atg1 complex and formation of a liquid droplet of early PAS. This phase separation ensures Atg1 kinase activation and subsequent recruitment of other Atg proteins to make matured PAS44. As ATG9 is embedded in ATG9 vesicles that only transiently localize to the vicinity of autophagosome23,45, one can speculate that ATG9 vesicles exist as a distinct component from the liquid droplet, and ATG9 on the membrane might temporally contact with ATG2A, ULK1 or other autophagy-related proteins in the liquid droplet. Thus, it is important to understand a topological basis which ensures ATG9 vesicles to access to other ATG proteins in the liquid droplet and promote a range of events described above.
Taken together, coordinated actions of ATG9 and ULK1 complex are inevitable for autophagy through regulating membrane source supply and recruitment of downstream components, promoting proper autophagosome maturation.
Production of PI3P by the PI3K complex and extension of the isolation membrane
Phosphatidylinositol 3-phosphate (PI3P), generated by PI3K, is a phospholipid that plays an important role in the control of membrane trafficking. While yeast has only one PI3K (Vps34), mammalian cells have several PI3Ks, of which class III PI3K is involved in autophagy. This is composed of the complex containing ATG14 or UVRAG in addition to VPS34, VPS15, and Beclin 1 (VPS30 homolog)46–48. While the PI3K with UVRAG is involved in the autolysosome formation, that with ATG14 promotes both formation of isolation membrane and the later steps as well47–49. PI3P promotes the localization of proteins with PI3P-binding motifs such as FYVE domains to the membrane where PI3P is embedded. Isolation membranes require the recruitment of four ATG18 homologs, WIPI1-4 that interact with PI3P through FRRG motif for WIPI1 and WIPI2 or LRRG motif for WIPI3 and WIPI443. Since these paralogs may have not only a functionally redundant role as shown by localization of all of four paralogs on nascent autophagosomes upon starvation, but also specific roles as well, since phenotypes caused by the loss of each gene could not be fully compensated by the presence of the other paralogs. WIPI2 acts as a scaffold to recruit ATG16 to isolation membrane, WIPI3 binds to TSC complex and FIP200, and WIPI4 interacts with ATG2A to facilitate autophagosome formation37,39,40,50,51. Consistent with this idea, mutations in the WIPI4 (WDR45) gene cause static encephalopathy of childhood with neurodegeneration in adulthood (SENDA), a human genetic disease involving neurodegeneration52. WIPI2 gene is also known to be responsible for a hereditary disease showing neurological disorders (described below)53. FYVE domain-containing protein DFCP1 is often used as a marker of isolation membranes but is not functionally involved in autophagy itself16,54. The function of PI3K in the formation of the isolation membrane depends on ULK1 and ATG9 since the formation of ATG14, DFCP1 or WIPI2 dots decrease with deficiencies in the components of ULK1 complex or ATG923,29. By these actions, omegasome is formed from the ER, from which a bag-shaped isolation membrane is developed16.
Post-translational modification of LC3
The regulation of autophagosome formation and the subsequent events are governed by a characteristic reaction involving the post-translational modification of the Atg8 homolog LC3 with phosphatidylethanolamine (PE). It is mediated by a series of ubiquitin-like modification enzymes55–57(reviewed by N. Mizushima in detail58). This reaction is carried out via two pathways. The first pathway is a modification of the ubiquitin-like protein ATG12, which is covalently bound to ATG5 via the action of the E1-like enzyme ATG7 and the E2-like enzyme ATG10. Most ATG12 proteins exist in a form bound to ATG5. The ATG5-12 complex binds to ATG16. The ATG5-ATG12-ATG16 complex functions as an E3-like enzyme in the last step of another ubiquitin-like reaction to facilitate modification of LC3 with PE. The ubiquitin-like protein LC3, a homolog of the budding yeast Atg8, is first cleaved by the ATG4 protein at the C-terminus to expose the Gly residue. Through sequential reactions of ATG7 (E1), ATG3 (E2), and the ATG5-ATG12-ATG16 complex (E3), PE is added to the C-terminal glycine residue of LC3 to target it to the isolation membrane.
Membrane-bound LC3 with PE functions as a key molecule that controls critical steps in the molecular mechanism of autophagy: autophagosome closure59,60, movement of the autophagosomes61–64, fusion with the lysosomes and degradation of the inner membrane of the autophagosomes65–68. This molecule is also needed for the binding to adapter proteins in selective autophagy. LC3 homologs are mainly classified into the LC3 subfamily and GABARAP subfamily. Knockdown of the Atg8 homologs of each subfamily with siRNA reduced autophagic activity, which suggests some functional segregation69. Moreover, it is recently shown that LC3 subfamily and GABARAP subfamily have opposite roles for regulation of autophagosome formation via binding to ULK1 and ATG1370.
While other ATG proteins are located outside the isolation membrane and move away from the membrane after autophagosome formation, LC3 is located both outside and inside the isolation membrane and stays within the autophagosome. Therefore, LC3 itself is degraded by autophagy, making it a useful substrate to measure autophagic activity. As LC3-I (without PE) and LC3-II (LC3-PE) can be easily separated from each other by SDS-PAGE, an autophagic flux assay using LC3 is widely used as a standard method to measure autophagic activity71. LC3 is the best-known marker used to monitor the formation of autophagosomes by microscopic observation as well72. The use of fluorescent-tagged LC3 enables the observation of autophagosomes in living cells. Specifically, a tandem-tagged LC3 with both GFP and RFP has been developed to distinguish autophagosomes from autolysosomes, which is critical to determine the state of autophagic induction or inhibition. In the case of lysosomal inhibition, LC3 dots accumulate, which cannot be distinguished from elevated levels of LC3 due to autophagic induction. Because the GFP signal is quenched in lysosomes due to its pH-sensitive nature, RFP-GFP-LC3 shows green plus red for autophagosomes and red with no green signal for autolysosomes73 (the plasmid is available from Addgene, plasmid # 21074). Thus, a series of methods using LC3 as a marker for autophagosomes has substantially advanced research on mammalian autophagy71.
Fusion with lysosomes
Not only autophagosome formation but also the subsequent fusion with lysosomes is controlled in a highly orchestrated manner. The fusion event requires physical contact of both organelles, and autophagosomes dynamically travel throughout the cytosol because lysosomes are predominantly generated at a peripheral region of the nuclei, while autophagosome formation sites are not restricted to the area where lysosomes are present. Inhibition of LC3 by injection of an LC3 antibody prevented the movement of autophagosome, suggesting that LC3 is needed for this event61. It is mediated by a binding of LC3 with FYVE and coiled-coil domain containing protein (FYCO1), which tethers autophagosomes with kinesin and microtubules to facilitate the plus-end direction of movement (peripheral direction)64. The movement of autophagosomes in the perinuclear direction is mediated by the binding of autophagosomes with dynactin and dynein via Rab7, ORP1L, and RILP62,63. Interestingly, a lack of ORP1L caused decreased recruitment of the components required for the following fusion event described below to late endosomes or lysosomes63, suggesting that the movement and the fusion are not separated from each other but are instead regulated as a sequential event. To date, researchers have shown that SNARE complexes containing Stx17 and the HOPS complex are required for the tethering of autophagosomes with lysosomes to facilitate the formation of autolysosomes74. This process is also under the control of the abovementioned PI3P production by PI3K. Beclin 1/VPS34/VPS15 with UVRAG functions as a mammalian PI3K complex working at the fusion step. Interestingly, PI3K with UVRAG is controlled by binding to a protein called Rubicon48,75. Rubicon knock-down results in an upregulated autophagic flux and a promotion of the fusion process, suggesting that Rubicon is a negative regulator of the fusion step. Rubicon itself is regulated by other factors, including mTOR, indicating that it is a key regulatory factor of autophagy whose deregulation may cause deleterious effects in pathology76–78. Indeed, Rubicon protein showed excessive accumulation in fatty liver induced by a high-fat diet, and a genetic abrogation of Rubicon suppressed diet-induced liver diseases, suggesting that abnormal accumulation of Rubicon may be a cause of these diseases79. The tethering and fusion of autophagosomes and lysosomes are mediated by two regulatory factors, EPG5 and INPP5E, which are known to be responsible for two human hereditary disorders, Vici syndrome and Joubert syndrome, respectively. EPG5 is present in late endosomes and lysosomes, facilitating the fusion of both by promoting the assembly of the SNARE complex, which is required for autophagosome-lysosome fusion80. INPP5E is a phosphatase that converts PI(3, 5)P2 to PI3P. The downregulation of INPP5E resulted in a reduction in the autophagic flux associated with an accumulation of LC3 dots and reduced colocalization of LC3 with lysosomes, showing that INPP5E facilitates autophagy via regulation of autophagosome-lysosome fusion81,82. Consistent with the fact that actin polymerization facilitates autophagy17, PI(3,5)P2 regulates the endosomal actin dynamics by antagonizing cortactin binding to actin filaments83, and reduction in cortactin decreased autophagosome-lysosome fusion84, downregulation of INPP5E resulted in reduced activation of cortactin on lysosomes. This evidence suggests that the conversion of PI(3,5)P2 to PI3P by INPP5E on lysosomes may increase the stability of actin filaments in a cortactin-dependent manner, promoting the fusion of autophagosomes with lysosomes.
A physiological role for autophagy in humans; lessons from animal models and human hereditary disorders
The physiological significance of autophagy has been investigated using various model animals, including mice, zebrafish, fruit flies and nematodes. In the case of mice, these studies are mainly performed using models in which the genes essential for autophagy are specifically knocked out for each tissue because knockout mice with a systemic homozygous deletion in these genes show embryonically lethal or postnatal lethality85. To date, over 150 types of tissue-specific autophagy-related gene knockout mouse models have been generated, suggesting the important roles of these genes in maintaining homeostasis in each tissue. Moreover, studies on mice with an Atg5 homozygous deletion in a mosaic throughout the body86, tamoxifen-induced knockout mice after birth87, and mice that express Atg5 only in the nervous system88 suggest a comprehensive role for autophagy throughout the body. These findings emphasized that autophagy ensures the normal development of tissues and the maintenance of homeostasis of differentiated tissues, and the failure of this process leads to dysfunction of each organ in mammals.
What about its role in human beings? Direct evidence has been provided by human hereditary diseases. These diseases are caused by hypomorphic mutations in the genes essential for autophagy or loss-of-function mutations in genes related to but not essential for autophagy. No genetic disease linked to a lack of both alleles of genes essential for autophagy has been reported to date. This phenomenon is most likely because autophagic deficiency affects the survival of human individuals, as shown in the results of embryonic lethality or postnatal lethality in mice with systemic homozygous deletions in these genes. Here, we will summarize the features of each disease and discuss the physiological role of autophagy in human health.
SENDA
Neurodegeneration with brain iron accumulation (NBIA) is a rare degenerative disease (~1/1,000,000) characterized by iron deposition in the brain, and SENDA is one of its subtypes. Psychomotor development is delayed early in childhood. Motor function develops to some extent, but dystonia, parkinsonism, and dementia progress rapidly in adolescence or early adulthood89. Most SENDA is sporadic, and thus, de novo mutations are expected to be responsible for its symptoms. Saitsu et al. conducted an exome analysis of two SENDA families and identified mutations in WDR45 (WIPI4), a human homolog of Atg18, one of the core genes essential for yeast autophagy52 (Fig. 2). The WDR45 locus is located on the short arm of the X chromosome (Xp11.23). Although X chromosome inactivation should be heterogeneous in each cell, lymphoblastoid cell lines (LCLs) from 4 of the 5 patients showed expression of only the mutant form. Thus, this factor is believed to be related to the disease state, but the mechanism is still unclear. There are four Atg18 homologs, WIPI 1 to 4, in humans. The phenotype of the WDR45 neuron-specific knockout mice was slightly milder than that of the mice with knockout of the core genes such as ATG5 and ATG7, each of which has only one ortholog, suggesting some functional overlap among the paralogs90. However, while WIPI2 binds to ATG16, which is required for the PE reaction of LC3, WIPI4 binds ATG2A more strongly than the other WIPI proteins, showing some functional differences50,91. In a mutant of epg-6 (a nematode homolog of WIPI4), the accumulation of protein aggregates and the multi-layered membrane-like structures were observed, suggesting the role of WIPI4 in autophagosome formation in vivo92. A recent study showed that Wdr45 knockout mice have increased neuronal apoptosis due to an accumulation of ER proteins, which was associated with an increase in the unfolded protein response (UPR)93. An investigation of the link between ER stress and autophagy indicated that ER stress can be one of the cues that induces autophagy94.
Fig. 2. Multiple proteins involved in autophagosome biogenesis are responsible for the suppression of human hereditary disorders involving neurodegeneration.
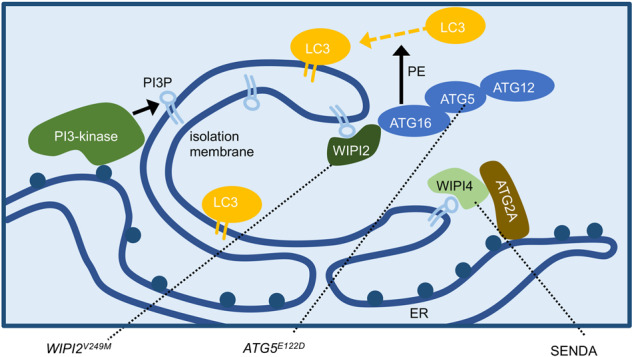
The formation of the isolation membrane is initiated at the contact sites of the mitochondria and the ER, followed by elongation of the membrane in a structure called the omegasome. PI3P production by PI3K facilitates the recruitment of WIPI2 that further promotes localization of ATG16 at isolation membranes for subsequent modification of LC3. WIPI4 also binds to PI3P to facilitate autophagosome biogenesis via binding to ATG2A, supporting the retention of the omegasome with the ER40 or a feeding of lipid to isolation membrane39.
Developmental abnormalities associated with the homozygous WIPI2V249M mutation
Since a loss of WIPI2 results in a defect in LC3 lipidation and the associated autophagosome formation, WIPI2 may have a distinct function in autophagy among human Atg18 paralogs54. Recently, individuals with a homozygous mutation in WIPI2 were reported to show developmental abnormalities and mental retardation53. Exome sequencing of two affected individuals identified the G745A mutation in exon 9 of the WIPI2 gene, which leads to a V249M amino acid change in the gene product. This mutation causes reduced binding activity of WIPI2 with ATG16L1, which is known to facilitate recruitment of ATG16L1 to the site of autophagosome formation and thus promote LC3 lipidation and autophagosome biogenesis50. The affected individuals analyzed for exome sequencing were aged 43 years and may have residual autophagosome biogenesis activity, as fibroblasts from the patient had mild but still induced LC3 lipidation activity in response to a starvation signal. The individuals with the mutation showed a complex developmental disorder including mental retardation, speech and language impairment, and cardiac, neurological, thyroid and skeletal abnormalities. Characteristic features of the disease are severe mental retardation associated with global brain volume loss and short stature. Cerebellar dysfunction is suggested, as fine/gross motor activities are delayed in patients. Cardiac abnormalities shown by electrocardiograms were observed53. The clinical features were similar at some degree in the patients with the WIPI2 mutation and those with SENDA although the patient with the WIPI2 mutation did not show iron deposition, a characteristic feature of SENDA.
Congenital ataxia with a homozygous ATG5E122D mutation
Mutations in the genes essential for autophagy among the core autophagy-related genes have not been shown to be related to human diseases until the identification of the ATG5E122D mutation. Exome analysis of two families with congenital ataxia (ataxia), intellectual disability, and growth retardation identified the chr6: 106,727,648 T > A mutation, a missense mutation that causes an E122D change in the ATG5 protein95. ATG5 is covalently bound to ATG12 to form a heterodimer of a ternary complex with ATG16, which promotes lipidation of LC355,58. Since the ATG5E122D mutation weakens the binding to ATG12 and decreases PE formation of LC3, basal autophagic activity was reduced in the LCLs derived from patients, and accumulation of p62 was observed95. These data showed that ATG5E122D is a hypomorphic allele whose homozygous mutation results in reduced but residual autophagic activity. The reduction in autophagy is most likely sufficient to support survival after birth but insufficient to maintain homeostasis in the CNS and suppress the symptoms of this condition. In contrast to SENDA, this disease did not show significant cerebral atrophy. Intriguingly, while cerebellar atrophy is a characteristic feature of the patients ATG5E122D, it is not a major feature of SENDA as nearly one third of the patients of SENDA show the symptom89. This controversy might be explained by the tissue-specific function of the WDR45 gene or genetic diversity (see below)96. No iron deposition in the cerebral area of the patient was reported.
Vici syndrome
Vici syndrome is an autosomal recessive disorder caused by mutations in the EPG5 gene, which has a regulatory role in autophagy at the fusion step between autophagosomes and lysosomes97. This condition affects not only the corpus callosum but also a wide range of organs and can include cardiomyopathy, cataracts, depigmentation, and combined immunodeficiency. Ectopic P-Granules Autophagy Protein 5 (EPG5) is a metazoan protein identified by screening Caenorhabditis elegans, and its homolog has not been found in yeast98. Systemic knockout mice that lack both alleles of Epg5 are born according to Mendelian inheritance and survive after birth99. This phenotype is clearly distinguished from those caused by the core autophagy genes, such as Atg5 and Atg7, which include neonatal lethality due to defects in neurological activity related to sacking defects and survival with starvation after birth88,100,101. This difference is probably due to the weak remaining autophagic activity that mitigates the phenotype in the Epg5 knockout mice99. The knockout mice began to show motor dysfunction after approximately 4 months and then showed symptoms similar to amyotrophic lateral sclerosis (ALS), such as muscle fiber atrophy and progressive paralysis99. Notably, the Epg5−/− mice showed only some features that are observed in patients with Vici syndrome. The mice showed agenesis of the corpus callosum and muscle atrophy but lacked several key symptoms of Vici syndrome, including facial dysmorphism, cataracts, and hypopigmentation. Additionally, Epg5 knockout mice can grow and reach sexual maturity in contrast to those with Vici syndrome, in whom death occurs at a median age of 42 months98,102. This observation that abrogation of EPG5 causes a less severe phenotype in mice than human support a speculative idea that human might depends on autophagy more than mice to maintain tissue homeostasis.
EPG5 promotes tethering of autophagosome and lysosome80 (Fig. 3). In EPG5 knockout cells, autophagosomes fuse with early endosomes, not lysosomes or late endosomes, and an enlarged autophagosome-like structure accumulates. Similar structural accumulation is observed in Epg5 knockout mice and tissues from patients with Vici syndrome. In addition, the accumulation of p62 and ubiquitin-positive dots was observed. Although further analysis is needed, the formation of these autophagosome-like structures and protein aggregates may contribute to the pathology of Vici syndrome.
Fig. 3. Two regulators of autohagosome–lysosome fusion, EPG5 and INPP5E, are responsible for human hereditary disorders.
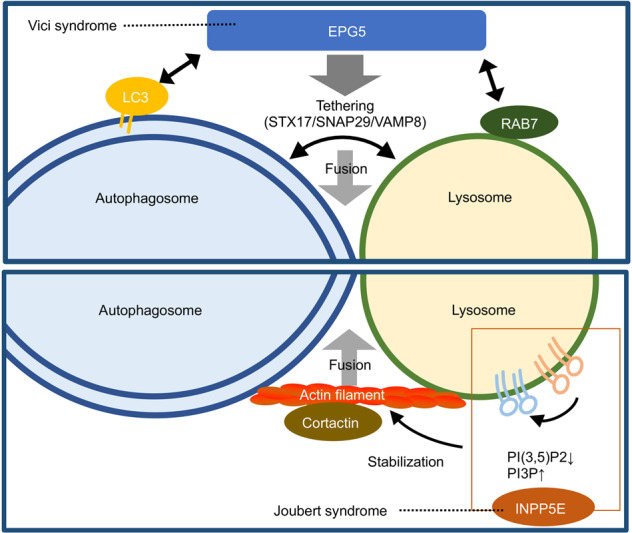
EPG5, which is responsible for Vici syndrome, is required for the tethering and fusion of autophagosomes and lysosomes. EPG5 facilitates the tethering by direct binding to and assembly of the SNARE complexes composed of STX17, SNAP29 and VAMP8. Jourbert syndrome patients have a mutation the in INPP5E gene. Its gene product INPP5E, a phosphatase, catalyzes the conversion of PI3P to PI(3, 5)P2 on lysosomes, resulting in of the binding of cortactin to actin filaments to stabilize this structure.
Joubert syndrome
Joubert syndrome is characterized by abnormal eye movements, decreased muscle tone, and delayed psychomotor development and brainstem dysplasia called the molar tooth sign. In addition to neuropathy, a wide range of symptoms, such as kidney damage and liver damage, are observed103. Bielas S et al. found that patients belonging to multiple families shown to have Joubert syndrome locus 1 (JBTS1) loci have mutations in the INPP5E gene104. INPP5E is an enzyme that generates PI3P by dephosphorylating the 5-position of PI (3,5)P2, which is a kind of inositol phospholipid (PIP). As described above, PI3P plays an important role in autophagosome formation and autophagosome maturation. Screening for factors related to autophagy among the factors involved in the metabolism of PIPs showed that INPP5E was required for autophagosome maturation81,82 (Fig. 3). Cells with mutations similar to the INPP5E mutation found in patients showed a significant decrease in autophagic activity, strongly suggesting that autophagic abnormalities may cause Joubert syndrome82. Cells from patients with Joubert syndrome showed abnormalities in the maintenance of cilia; thus, Joubert syndrome is believed to be associated with ciliopathy. Notably, autophagy was reported to be involved in the regulation of primary cilia formation and length, and conversely, signaling from cilia regulates autophagy105; thus, feedback of autophagy and cilia abnormalities may have affect the expression of the symptoms.
The physiological role of autophagy in homeostasis in humans
Although tissue-specific knockout of autophagy-related genes in mice has provided a clear-cut role for autophagy in tissue homeostasis, to develop clinical applications of autophagy induction or inhibition for related diseases, the physiological/pathological consequences of systemic changes in autophagy must be elucidated. In addition, recent studies have shown that autophagy declines with age. Knowledge of autophagy-associated human genetic diseases will help elucidate this issue. Consistent with the idea that systemic, amorphic mutations of genes essential for autophagy may result in lethality, patients with autophagy-related mutations retain residual autophagic activity, suggesting that the symptoms shown above are caused by partial decline in autophagic activity. Therefore not showing clear-cut mechanisms as shown in the tissue-specific knockout mice, those results provide physiologically relevant information. Based on these findings, we discuss the function of autophagy in human health, especially brain function, development, and tumorigenesis.
Brain function and development
The most striking shared feature in the disorders shown above is an aberration in the CNS. This finding is consistent with a role for autophagy in the prevention of neurodegeneration shown by mice with neuron-specific homozygous deletions in Atg5106. These mice showed accumulation of protein aggregates in the neurons, neuronal loss, motor dysfunction, and neurodegenerative disease symptoms. In the case of a systemic loss of autophagy in mice by inducible deletion of Atg7 after birth using the inducible Cre-lox system, abnormalities were observed in many tissues, such as the liver, brain, muscles, and pancreas87. However, except for some deaths caused by infection, most mice died ~3 months after the induction of deficiencies in autophagy due to neurodegeneration, supporting the idea that autophagy is critical for the CNS87. This phenomenon is not limited to mammals, as Drosophila Atg5 or Atg7 mutants also exhibited neurodegenerative disease-like symptoms, highlighting the general role of autophagy in maintaining neuronal homeostasis95,107.
The reason why autophagic deficiency primarily disrupts the CNS is unclear, but it may be due to two specific characteristics of the CNS. First, a defect in the clearance of cellular wastes may cause a strong deleterious effect in neurons, which rarely divide throughout life and thus are exposed to a cumulative toxic effect of the debris. In particular, the cells in the CNS may be strongly affected in long-lived humans compared to mice. Another reason may be the highly specified function of each cell in the CNS, which has less redundancy than other organs, such as the liver.
While the congenital diseases described above have some shared characteristics, there are significant differences in the severity and types of symptoms. Among them, Vici syndrome displays the most severe condition, followed by Joubert syndrome and SENDA. The ataxia associated with ATG5E122D and the developmental abnormalities associated with the homozygous WIPI2V249M mutation are relatively mild. Although still speculative due to a lack of a cross-cutting study involving the diseases shown here, the degree of severity may depend on the levels of residual autophagic activity in the CNS. Indeed, fibroblasts from patients with Vici syndrome showed almost no induction of autophagic flux by rapamycin and a strong accumulation of the autophagic substrate p62 and NBR1 in normal conditions97. LCLs from patients with the ATG5E122D mutation showed a decrease in LC3 lipidation and autophagic flux but still retained a detectable level of flux95. This finding was also observed for those with the WIPI2V249M mutation53. Among INPP5E knockout cell lines complemented by the transgenes with some mutations found in patients with Joubert syndrome, some of mild symptoms showed a slight but significant recovery in autophagic activity, but the severe symptoms did not show any recovery, showing a correlation between the enzymatic activity and the severity of symptoms82.
The characteristics of symptoms in the CNS also vary. SENDA is characterized by iron deposition in the CNS, which is not found in the other four diseases. While agenesis of the corpus callosum is typically found, and atrophy of cerebrum is not a major feature of the patients with Vici syndrome, patients with the ATG5E122D mutation show cerebellar hypoplasia without agenesis of the corpus. SENDA also shows cerebellar atrophy. This controversy may be explained by several reasons: (1) gene-specific roles other than autophagy, (2) genetic diversity and (3) tissue specificity of each gene.
Regarding (1) the gene-specific roles other than autophagy, the Vici syndrome-related protein EPG5 has a role in the regulation of endocytosis, which has a critical function in neural development and neurons. Similarly, the Joubert syndrome-associated protein INPP5E catalyzes the synthesis of PI3P from PI(3,5)P2, as PI(3,5)P2 and PI3P have physiological functions other than autophagy, including the regulation of endosomal function108. Deletion of FIG4, which facilitates the production of PI(3,5)P2, resulted in a depletion of PI(3,5)P2 and developmental defects including neurological abnormalities. Its homozygous mutation has been identified as a cause of Yunis-Varon syndrome, which is characterized by structural brain abnormalities and skeletal abnormalities109. Mutation of FIG4 was also found in mutant mice called “tremor mice” that show a severe tremor and abnormal gait, as well as in patients with the hereditary condition Charcot-Marie-Tooth disorder110. Therefore, it is likely that a defect in endocytosis may cause an additive effect on brain development, resulting in the complex symptoms found in Vici syndrome and Joubert syndrome. This phenomenon may also be the cause of symptoms in organs other than the brain, such as the immunological abnormalities in Vici syndrome and the liver disease in Joubert syndrome that are not found in SENDA or patients with ATG5E122D mutations. Notably, ATG5 also has roles in processes other than autophagy, such as cell death and pathogen resistance111,112, which could contribute to the symptoms of patients with the ATG5E122D mutation to some degree.
Regarding the #2 possibility, genetic diversity including quantitative trait loci (QTLs) is expected to contribute to variations in symptoms because autophagy is a multistep event requiring several different kinds of biological pathways. Membrane trafficking, including endocytosis, is a major contributor to autophagy because it regulates the maturation of autophagosomes. Genes involved in mitochondrial biogenesis, lysosomal biogenesis, and hormonal regulation are required for the proper regulation of autophagy. For instance, MFN2, a mitochondrial dynamin-related protein that is required for the tethering of the mitochondria with the ER113 and the formation of isolation membranes19, is known to be mutated in Charcot-Marie-Tooth neuropathy type 2A114. Multiple genetic variations in this wide range of factors may contribute to the differences in severity and tissue specificity. We expect that genetic variation in other genes may be related to the symptoms but independent of autophagy. For example, since genetic loci of BECN1 (encoding beclin 1) is close to a well-known tumor suppressor BRCA1, large deletions including both loci are found in the cancer genome115.
For the #3 possibility, the tissue specificity of each gene may also contribute to the symptoms. Mice with double systemic homozygous deletions in both Wdr45 and Wdr45b showed neonatal lethality, while each single knockout mouse was born normally, suggesting functional overlap between the genes96. Double knockout mice showed an increased accumulation of p62 aggregates in many brain regions and neurons, which was associated with suckling defects similar to those of Atg7 knockout mice100. Interestingly, these accumulations of the p62 aggregates were not detected in the liver and kidney in double knockout mice, suggesting that both genes may not be essential for autophagy in these tissues96. Therefore, the functional contribution of each autophagy-related gene may vary in different tissues.
In summary, the shared feature of dysfunction in the CNS in various diseases highlights the critical role for autophagy in the maintenance of homeostasis in neurons for CNS function. However, the possibilities shown above (which are not mutually exclusive) suggest that some abnormalities found in Vici syndrome, such as cataracts and severe immunological abnormalities, and Joubert syndrome, such as renal disease and liver disease, might be due to combined defects in autophagy and endocytosis, as well as other unidentified genetic factors(s). A comprehensive study using humanized mouse models for each disease with a defined genetic background may help elucidate the role of autophagy in the suppression of these diseases and human physiology as well.
Tumorigenesis
Several studies of tissue-specific knockout mice have identified a tumor-suppressive role for autophagy in many tissues, including the liver, lung, and pancreas86,116,117. Due to the prosurvival effect of autophagy on tumor cells, tumors arising from these mice usually do not develop into malignant tumors. However, these findings do not necessarily indicate that autophagy is dispensable for tumor suppression. Rather, an irreversible genetic abrogation of the genes essential for autophagy in these model systems clearly show both side of autophagy in tumorigenesis118. Nonetheless, the human genetic disorders with mutations in autophagy-related genes described here do not show a significant increase in any sign of neoplasm formation. Do these findings indicate that autophagy may not be important for tumor suppression in humans? Evidence from cell biology using human cells clearly shows that autophagy is required to suppress the genomic alterations that promote tumorigenesis. Autophagy promotes genomic integrity through the control of cellular senescence via both positive and negative regulation. Promoting cellular senescence in response to oncogenic stress119, autophagy also suppresses senescence by selective degradation of the transcription factor GATA4 in response to DNA damage120. Autophagy also maintains stemness of muscle stem cells by suppression of senescence caused by excess ROS production121. Alternatively, autophagy triggers cell death in response to telomeric DNA damage during replicative crisis122. These lines of evidence suggest that autophagy suppresses tumor-promoting genomic alterations in various processes. Therefore, we hypothesize that the controversy between human symptoms and the phenotypes of mice with tissue-specific, irreversible deletions in autophagy genes is most likely due to a major contribution of autophagy to the maintenance of the CNS in humans, which is vital for survival. Simply, a reduction in only a fraction of the CNS may immediately cause a serious defect, in contrast to carcinogenesis, which requires a long period to acquire multiple mutations for the development of a malignant tumor. Indeed, a mouse model with temporal systemic inhibition of autophagy followed by its restoration supports this idea. The mice with reduced expression of Atg5 by dox-inducible siRNA after birth expressed reduced viability (~6 months) and various signs of accelerated aging due to systemic degradation of tissue homeostasis. Interestingly, the restoration of autophagy in mice partially restored the shortened lifespan but accelerated the formation of spontaneous tumors123. We speculate that a systemic decrease in autophagy becomes a serious threat to survival, especially due to a defect in the CNS that precedes the formation of neoplasms, but a temporal but severe decrease in the autophagy of cells in a limited area, such as the damaged liver, may cause genomic alterations that facilitate tumorigenesis.
A complex tumor environment and the heterogeneity of cancer cells must be considered for the role of autophagy in tumorigenesis. Autophagy not only suppresses genomic instability or conversely supports cancer cell survival in a cell-autonomous manner but also controls tumor cell status in a non-cell autonomous manner. Autophagic activity in host mice is required for the maintenance of circulating arginine, which supports the growth of allografted cancer cells124, indicating that autophagic activity in the host supports cancer cell viability87,125. As circulating arginine is degraded by the arginase ARG1, which is released into blood flow due to liver damage in autophagy-deficient host mice, tumorigenesis in mice with autophagy knockout in each tissue may differ from mice with systemic loss of autophagy. Alternatively, autophagy in loser cells supports their elimination during cell competition126, which may cause selection pressure toward an expanded population of autophagy-deficient cells in a microenvironment involving heterogeneous autophagic activity, including model mice generated by an approach involving Cre recombinase. These non-cell autonomous factors need to be taken into account to determine the effect of systemic changes in autophagy in future studies.
In summary, autophagy suppresses tumorigenesis but, once cancer has developed, it promotes tumor malignancy. Thus, to interpret the effect of autophagy deficiency in animal models and pathology, it is essential to consider the effect of autophagy defect in non-cancerous and cancer cells independently. On top of that, it should be taken to account that there exists a non-cell autonomous effect of autophagy in multiple organs and tumor microenvironment.
Perspectives
Knowledge of the human genetics of autophagy is rapidly expanding as hereditary disorders with autophagy deficiency have been identified in the past decade, and we expect that many hereditary diseases for which other autophagy-related genes are responsible remain to be identified. We expect that the genes identified in the future will likely be biased. At present, genes encoding essential upstream regulatory factors for autophagy, such as FIP200 and ATG9, have not been associated with human hereditary diseases. This result could be partially due to functions specific to each gene, but data from cell biology have supported other possibilities. FIP200, ATG9, and the PI3K complex have essential roles in the formation of isolation membranes. However, cells deficient in the genes involved in LC3 lipidation, such as ATG5 and ATG12, show a small number of autophagosome-like structures67. These structures could be fused with a lysosome. Thus, it is suggested that the low residual autophagic activity observed in mice with knockout of the genes involved in LC3 lipidation could be sufficient to support embryonic development, although complete defects in autophagy result in embryonic lethality85. Therefore, hypomorphic alleles of the upstream factors could be identified as the cause of a hereditary disease with severe symptoms. Alternatively, similar to becn1+/− mice, which exhibit a high carcinogenic phenotype127,128, those with heterozygous mutations with haploinsufficiency may show some aspects of autophagy deficiency.
Since the molecular mechanism of autophagy has not been fully clarified, it is predicted that there are many unidentified factors related to autophagy. In future studies, the factors related to hereditary disorders could be recognized as autophagy-related disorders. One example is optineurin (OPTN), which is involved in selective autophagy. OPTN is known as one of the causative genes of ALS129. To date, the role of OPTN in autophagy is focused on the autophagy receptor, which helps sequester ubiquitinated substrates by isolation membranes via binding to both LC3 and ubiquitin130,131. However, recent reports have indicated that OPTN knockout cells have decreased autophagosome formation induced by starvation132. SQSTM1, a gene encoding p62, is one of the best-investigated substrates for autophagy. While it functions as a receptor protein to tether LC3 with targets for selective autophagy, several studies have suggested a possible involvement of p62 in autophagosome formation itself as well66,133. Interestingly, homozygous mutations in SQSTM1 were identified as a cause of human neurodegeneration with ataxia and other associated symptoms134. Of course, symptoms of the patients or phenotypes of model mice are expected to result from a combination of autophagy deficiency and aberrations other than autophagy, so it is requisite to carefully estimate the contribution of autophagy for suppression of each disease. To date, there are several powerful tools to estimate autophagy in vivo such as GFP-LC3 mice, GFP-LC3-RFP-LC3ΔG mice, and mt-keima mice71,135–137. However, it remains technically difficult to estimate autophagy activity in human. Moreover, a cavity of the model systems is that markers of autophagosome do not always necessarily indicate autophagy in vivo; LC3 is related to other events such as LC3-associated phagocytosis (LAP)138. Therefore, interpretation using both the animal models and the approach of cell biology is necessary to understand the role of autophagy in pathology.
The expansion of autophagy-related diseases may increase the demand for therapy. More strikingly, there are many lifestyle-related diseases with autophagic abnormalities. For example, autophagy is decreased in the fatty liver caused by excessive nutrition. This phenomenon is mainly due to the accumulation of Rubicon protein, which is a negative regulator of autophagy, and Rubicon knockout mice showed reduced hepatocyte cell death and fat accumulation associated with fatty liver79. In addition, autophagic activity gradually decreases as aging progresses and increased basal autophagy extends lifespan and healthspan in many organisms including mice139–141. Our group recently showed that the Rubicon protein level increases with aging and that Rubicon knockout reduces various diseases associated with aging142. In addition to the genetic diseases outlined here, determination of the pathology and molecular mechanisms of the changes in autophagic status in lifestyle-related diseases and the aging process is needed to develop novel strategies for human healthcare. In addition to basic research to elucidate the mechanisms of autophagy, further studies on these topics are needed in the future.
Acknowledgements
We thank all the lab members for helpful discussion. T.K. is supported by JSPS KAKENHI (19H03454) and Takeda Science Foundation. T.Y. is supported by AMED under grant number JP17gm5010001 and JP17gm0610005, MEXT/JSPS KAKENHI, JST CREST (JPMJCR17H6) and HFSP grant.
Author contributions
T.K. and T.Y. wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 2.Kawabata T, Yoshimori T. Beyond starvation: an update on the autophagic machinery and its functions. J. Mol. Cell. Cardiol. 2016;95:2–10. doi: 10.1016/j.yjmcc.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 3.Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24:9–23. doi: 10.1038/cr.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018;19:365–381. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zoncu R, et al. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science. 2011;334:678–683. doi: 10.1126/science.1207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1–Atg13–FIP200 complex required for autophagy. Mol. Biol. Cell. 2009;20:1981–1991. doi: 10.1091/mbc.e08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong P-M, Feng Y, Wang J, Shi R, Jiang X. Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 2015;6:8048. doi: 10.1038/ncomms9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kamada Y, et al. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol. Cell. Biol. 2010;30:1049–1058. doi: 10.1128/MCB.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Settembre C, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martina JA, et al. The nutrient-responsive transcription factor TFE3, promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. signal. 2014;7:ra9–ra9. doi: 10.1126/scisignal.2004754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 15.Hayashi-Nishino M, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat. Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 16.Axe EL, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mi N, et al. CapZ regulates autophagosomal membrane shaping by promoting actin assembly inside the isolation membrane. Nat. Cell Biol. 2015;17:1112–1123. doi: 10.1038/ncb3215. [DOI] [PubMed] [Google Scholar]
- 18.Hailey DW, et al. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamasaki M, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 20.Dupont N, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr. Biol. 2014;24:609–620. doi: 10.1016/j.cub.2014.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shpilka T, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. EMBO J. 2015;34:2117–2131. doi: 10.15252/embj.201490315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat. Cell Biol. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orsi A, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol. Biol. Cell. 2012;23:1860–1873. doi: 10.1091/mbc.e11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ge L, Melville D, Zhang M, Schekman R, Levine B. The ER–Golgi intermediate compartment is a key membrane source for the LC3 lipidation step of autophagosome biogenesis. ELife. 2013;2:e00947. doi: 10.7554/eLife.00947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ge L, Zhang M, Schekman R. Phosphatidylinositol 3-kinase and COPII generate LC3 lipidation vesicles from the ER-Golgi intermediate compartment. ELife. 2014;3:e04135. doi: 10.7554/eLife.04135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ge L, et al. Remodeling of ER-exit sites initiates a membrane supply pathway for autophagosome biogenesis. EMBO Rep. 2017;18:1586–1603. doi: 10.15252/embr.201744559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suzuki K, et al. The pre‐autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suzuki K, Kubota Y, Sekito T, Ohsumi Y. Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells. 2007;12:209–218. doi: 10.1111/j.1365-2443.2007.01050.x. [DOI] [PubMed] [Google Scholar]
- 29.Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. doi: 10.4161/auto.6.6.12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karanasios E, et al. Autophagy initiation by ULK complex assembly on ER tubulovesicular regions marked by ATG9 vesicles. Nat. Commun. 2016;7:12420–17. doi: 10.1038/ncomms12420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lynch-Day MA, et al. Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc. Natl Acad. Sci. USA. 2010;107:7811–7816. doi: 10.1073/pnas.1000063107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shirahama-Noda K, Kira S, Yoshimori T, Noda T. TRAPPIII is responsible for vesicular transport from early endosomes to Golgi, facilitating Atg9 cycling in autophagy. J. Cell. Sci. 2013;126:4963–4973. doi: 10.1242/jcs.131318. [DOI] [PubMed] [Google Scholar]
- 33.Imai K, et al. Atg9A trafficking through the recycling endosomes is required for autophagosome formation. J. Cell. Sci. 2016;129:3781–3791. doi: 10.1242/jcs.196196. [DOI] [PubMed] [Google Scholar]
- 34.Judith D, et al. ATG9A shapes the forming autophagosome through Arfaptin 2 and phosphatidylinositol 4-kinase IIIβ. J. Cell Biol. 2019;218:1634–1652. doi: 10.1083/jcb.201901115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J. Cell Sci. 2012;125:1488–1499. doi: 10.1242/jcs.094110. [DOI] [PubMed] [Google Scholar]
- 36.Young ARJ, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J. Cell Sci. 2006;119:3888–3900. doi: 10.1242/jcs.03172. [DOI] [PubMed] [Google Scholar]
- 37.Valverde DP, et al. ATG2 transports lipids to promote autophagosome biogenesis. J. Cell Biol. 2019;218:1787–1798. doi: 10.1083/jcb.201811139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Osawa T, et al. Atg2 mediates direct lipid transfer between membranes for autophagosome formation. Nat. Struct. Mol. Biol. 2019;26:281–288. doi: 10.1038/s41594-019-0203-4. [DOI] [PubMed] [Google Scholar]
- 39.Maeda, S., Otomo, C. & Otomo, T. The autophagic membrane tether ATG2A transfers lipids between membranes. ELife8, E3179 (2019). [DOI] [PMC free article] [PubMed]
- 40.Chowdhury S, et al. Insights into autophagosome biogenesis from structural and biochemical analyses of the ATG2A-WIPI4 complex. Proc. Natl Acad. Sci. USA. 2018;115:E9792–E9801. doi: 10.1073/pnas.1811874115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gómez-Sánchez R, et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J. Cell Biol. 2018;217:2743–2763. doi: 10.1083/jcb.201710116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tang Z, et al. TOM40 targets Atg2 to mitochondria-associated ER membranes for phagophore expansion. Cell Rep. 2019;28:1744–1757.e5. doi: 10.1016/j.celrep.2019.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J. Biol. Chem. 2018;293:5386–5395. doi: 10.1074/jbc.R117.810366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fujioka Y, et al. Phase separation organizes the site of autophagosome formation. Nature. 2020;578:301–305. doi: 10.1038/s41586-020-1977-6. [DOI] [PubMed] [Google Scholar]
- 45.Koyama-Honda I, Itakura E, Fujiwara TK, Mizushima N. Temporal analysis of recruitment of mammalian ATG proteins to the autophagosome formation site. Autophagy. 2013;9:1491–1499. doi: 10.4161/auto.25529. [DOI] [PubMed] [Google Scholar]
- 46.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 47.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 2008;19:5360–5372. doi: 10.1091/mbc.e08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsunaga K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat. Cell Biol. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 49.Diao J, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–566. doi: 10.1038/nature14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dooley HC, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol. Cell. 2014;55:238–252. doi: 10.1016/j.molcel.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bakula D, et al. WIPI3 and WIPI4 β-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat. Commun. 2017;8:15637–18. doi: 10.1038/ncomms15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saitsu H, et al. De novo mutations in the autophagy gene WDR45 cause static encephalopathy of childhood with neurodegeneration in adulthood. Nat. Genet. 2013;45:445–9–449e1. doi: 10.1038/ng.2562. [DOI] [PubMed] [Google Scholar]
- 53.Jelani M, et al. A mutation in the major autophagy gene, WIPI2, associated with global developmental abnormalities. Brain. 2019;142:1242–1254. doi: 10.1093/brain/awz075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Polson HEJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6:506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 55.Mizushima N, et al. A protein conjugation system essential for autophagy. Nature. 1998;395:395–398. doi: 10.1038/26506. [DOI] [PubMed] [Google Scholar]
- 56.Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 57.Mizushima N, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J. Cell Biol. 2001;152:657–668. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mizushima N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2019;63:1–10. doi: 10.1016/j.ceb.2019.12.001. [DOI] [PubMed] [Google Scholar]
- 59.Fujita N, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol. Biol. Cell. 2008;19:4651–4659. doi: 10.1091/mbc.e08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 61.Kimura S, Noda T, Yoshimori T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008;33:109–122. doi: 10.1247/csf.08005. [DOI] [PubMed] [Google Scholar]
- 62.Jordens I, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr. Biol. 2001;11:1680–1685. doi: 10.1016/S0960-9822(01)00531-0. [DOI] [PubMed] [Google Scholar]
- 63.Wijdeven RH, et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat. Commun. 2016;7:11808–14. doi: 10.1038/ncomms11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pankiv S, et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilkinson DS, et al. Phosphorylation of LC3 by the Hippo kinases STK3/STK4 is essential for autophagy. Mol. Cell. 2015;57:55–68. doi: 10.1016/j.molcel.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kishi-Itakura C, Koyama-Honda I, Itakura E, Mizushima N. Ultrastructural analysis of autophagosome organization using mammalian autophagy-deficient cells. J. Cell. Sci. 2014;127:4089–4102. doi: 10.1242/jcs.156034. [DOI] [PubMed] [Google Scholar]
- 67.Tsuboyama K, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science. 2016;354:1036–1041. doi: 10.1126/science.aaf6136. [DOI] [PubMed] [Google Scholar]
- 68.Koyama-Honda I, Tsuboyama K, Mizushima N. ATG conjugation-dependent degradation of the inner autophagosomal membrane is a key step for autophagosome maturation. Autophagy. 2017;13:1252–1253. doi: 10.1080/15548627.2017.1319041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Weidberg H, et al. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Grunwald DS, Otto NM, Park J-M, Song D, Kim D-H. GABARAPs and LC3s have opposite roles in regulating ULK1 for autophagy induction. Autophagy. 2020;16:600–614. doi: 10.1080/15548627.2019.1632620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12:1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kabeya Y, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2014;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 74.Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. doi: 10.1016/j.cell.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 75.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tan X, Thapa N, Sun Y, Anderson RA. A kinase-independent role for EGF receptor in autophagy initiation. Cell. 2015;160:145–160. doi: 10.1016/j.cell.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim Y-M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol. Cell. 2015;57:207–218. doi: 10.1016/j.molcel.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheng X, et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol. Cell. 2017;65:1029–1043.e5. doi: 10.1016/j.molcel.2017.02.010. [DOI] [PubMed] [Google Scholar]
- 79.Tanaka S, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994–2014. doi: 10.1002/hep.28820. [DOI] [PubMed] [Google Scholar]
- 80.Wang Z, et al. The vici syndrome protein EPG5 is a Rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell. 2016;63:781–795. doi: 10.1016/j.molcel.2016.08.021. [DOI] [PubMed] [Google Scholar]
- 81.Nakamura S, Hasegawa J, Yoshimori T. Regulation of lysosomal phosphoinositide balance by INPP5E is essential for autophagosome-lysosome fusion. Autophagy. 2016;12:2500–2501. doi: 10.1080/15548627.2016.1234568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hasegawa J, et al. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. EMBO J. 2016;35:1853–1867. doi: 10.15252/embj.201593148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hong NH, Qi A, Weaver AM. PI(3,5)P2 controls endosomal branched actin dynamics by regulating cortactin-actin interactions. J. Cell Biol. 2015;210:753–769. doi: 10.1083/jcb.201412127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lee J-Y, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010;29:969–980. doi: 10.1038/emboj.2009.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kuma A, Komatsu M, Mizushima N. Autophagy-monitoring and autophagy-deficient mice. Autophagy. 2017;13:1619–1628. doi: 10.1080/15548627.2017.1343770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Takamura A, et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Karsli-Uzunbas G, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Disco. 2014;4:914–927. doi: 10.1158/2159-8290.CD-14-0363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yoshii SR, et al. Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev. Cell. 2016;39:116–130. doi: 10.1016/j.devcel.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 89.Stige KE, Gjerde IO, Houge G, Knappskog PM, Tzoulis C. Beta-propeller protein-associated neurodegeneration: a case report and review of the literature. Clin. Case Rep. 2018;6:353–362. doi: 10.1002/ccr3.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao YG, et al. The autophagy gene Wdr45/Wipi4 regulates learning and memory function and axonal homeostasis. Autophagy. 2015;11:881–890. doi: 10.1080/15548627.2015.1047127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zheng J-X, et al. Architecture of the ATG2B-WDR45 complex and an aromatic Y/HF motif crucial for complex formation. Autophagy. 2017;13:1870–1883. doi: 10.1080/15548627.2017.1359381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu Q, et al. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev. Cell. 2011;21:343–357. doi: 10.1016/j.devcel.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 93.Wan, H. et al. WDR45 contributes to neurodegeneration through regulation of ER homeostasis and neuronal death. Autophagy.16, 531–547 (2019). [DOI] [PMC free article] [PubMed]
- 94.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J. Biol. Chem. 2006;281:30299–30304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim M, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. ELife. 2016;5:213. doi: 10.7554/eLife.12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ji C, et al. Role of Wdr45b in maintaining neural autophagy and cognitive function. Autophagy. 2019;7:1–11. doi: 10.1080/15548627.2019.1632621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cullup T, et al. Recessive mutations in EPG5 cause Vici syndrome, a multisystem disorder with defective autophagy. Nat. Genet. 2013;45:83–87. doi: 10.1038/ng.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhao YG, Zhao H, Sun H, Zhang H. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy. 2013;9:1258–1262. doi: 10.4161/auto.24856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao H, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J. Cell Biol. 2013;200:731–741. doi: 10.1083/jcb.201211014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kuma A, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 101.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Byrne S, Dionisi-Vici C, Smith L, Gautel M, Jungbluth H. Vici syndrome: a review. Orphanet J. Rare Dis. 2016;11:21–9. doi: 10.1186/s13023-016-0399-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J. Rare Dis. 2010;5:1–10. doi: 10.1186/1750-1172-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bielas SL, et al. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pampliega O, et al. Functional interaction between autophagy and ciliogenesis. Nature. 2013;502:194–200. doi: 10.1038/nature12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hara T, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- 107.Juhász G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev. 2007;21:3061–3066. doi: 10.1101/gad.1600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin N, Lang MJ, Weisman LS. Phosphatidylinositol 3,5-bisphosphate: regulation of cellular events in space and time. Biochem. Soc. Trans. 2016;44:177–184. doi: 10.1042/BST20150174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Campeau PM, et al. Yunis-Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. Am. J. Hum. Genet. 2013;92:781–791. doi: 10.1016/j.ajhg.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chow CY, et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448:68–72. doi: 10.1038/nature05876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Maskey D, et al. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 2013;4:2130. doi: 10.1038/ncomms3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kimmey JM, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015;528:565–569. doi: 10.1038/nature16451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.de Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- 114.Züchner S, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat. Genet. 2004;36:449–451. doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]
- 115.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 2014;12:485–490. doi: 10.1158/1541-7786.MCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rao S, et al. A dual role for autophagy in a murine model of lung cancer. Nat. Commun. 2014;5:3056. doi: 10.1038/ncomms4056. [DOI] [PubMed] [Google Scholar]
- 117.Rosenfeldt, M. T. et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature504, 296–300 (2017). [DOI] [PubMed]
- 118.Amaravadi R, Kimmelman AC, White E. Recent insights into the function of autophagy in cancer. Genes Dev. 2016;30:1913–1930. doi: 10.1101/gad.287524.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Young ARJ, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kang C, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349:aaa5612–aaa5612. doi: 10.1126/science.aaa5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.García-Prat L, et al. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 122.Nassour J, et al. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature. 2019;565:659–663. doi: 10.1038/s41586-019-0885-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cassidy LD, et al. Temporal inhibition of autophagy reveals segmental reversal of aging with increased cancer risk. Nat. Commun. 2020;11:307. doi: 10.1038/s41467-019-14187-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Poillet-Perez L, et al. Autophagy maintains tumour growth through circulating arginine. Nature. 2018;563:569–573. doi: 10.1038/s41586-018-0697-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang A, et al. Autophagy sustains pancreatic cancer growth through both cell-autonomous and nonautonomous mechanisms. Cancer Disco. 2018;8:276–287. doi: 10.1158/2159-8290.CD-17-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nagata R, Nakamura M, Sanaki Y, Igaki T. Cell competition is driven by autophagy. Dev. Cell. 2019;51:99–112.e4. doi: 10.1016/j.devcel.2019.08.018. [DOI] [PubMed] [Google Scholar]
- 127.Qu X, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl Acad. Sci. USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Maruyama H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465:223–226. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 130.Wild P, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011;333:228–233. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Korac J, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J. Cell. Sci. 2013;126:580–592. doi: 10.1242/jcs.114926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bansal M, et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. J. Biol. Chem. 2018;293:132–147. doi: 10.1074/jbc.M117.801944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Itakura, E. & Mizushima, N. p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 192, 17–27 (2011). [DOI] [PMC free article] [PubMed]
- 134.Haack TB, et al. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am. J. Hum. Genet. 2016;99:735–743. doi: 10.1016/j.ajhg.2016.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.e03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kaizuka T, et al. An autophagic flux probe that releases an internal control. Mol. Cell. 2016;64:835–849. doi: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 137.Sun N, et al. Measuring in vivo mitophagy. Mol. Cell. 2015;60:685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schaaf MBE, Keulers TG, Vooijs MA, Rouschop KMA. LC3/GABARAP family proteins: autophagy-(un)related functions. FASEB J. 2016;30:3961–3978. doi: 10.1096/fj.201600698R. [DOI] [PubMed] [Google Scholar]
- 139.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 140.Pyo J-O, et al. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013;4:2300–9. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fernández ÁF, et al. Disruption of the beclin 1-BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakamura S, et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun. 2019;10:847–11. doi: 10.1038/s41467-019-08729-6. [DOI] [PMC free article] [PubMed] [Google Scholar]