

Abstract
Our previous study delivered zinc finger nucleases to treat mice with mucopolysaccharidosis type I (MPS I), resulting in a phase I/II clinical trial (ClinicalTrials.gov: NCT02702115). However, in the clinical trial, the efficacy needs to be improved due to the low transgene expression level. To this end, we designed a proprietary system (PS) gene editing approach with CRISPR to insert a promoterless α-l-iduronidase (IDUA) cDNA sequence into the albumin locus of hepatocytes. In this study, adeno-associated virus 8 (AAV8) vectors delivering the PS gene editing system were injected into neonatal and adult MPS I mice. IDUA enzyme activity in the brain significantly increased, while storage levels were normalized. Neurobehavioral tests showed that treated mice had better memory and learning ability. Also, histological analysis showed efficacy reflected by the absence of foam cells in the liver and vacuolation in neuronal cells. No vector-associated toxicity or increased tumorigenesis risk was observed. Moreover, no off-target effects were detected through the unbiased genome-wide unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) analysis. In summary, these results showed the safety and efficacy of the PS in treating MPS I and paved the way for clinical studies. Additionally, as a therapeutic platform, the PS has the potential to treat other lysosomal diseases.
Graphical Abstract
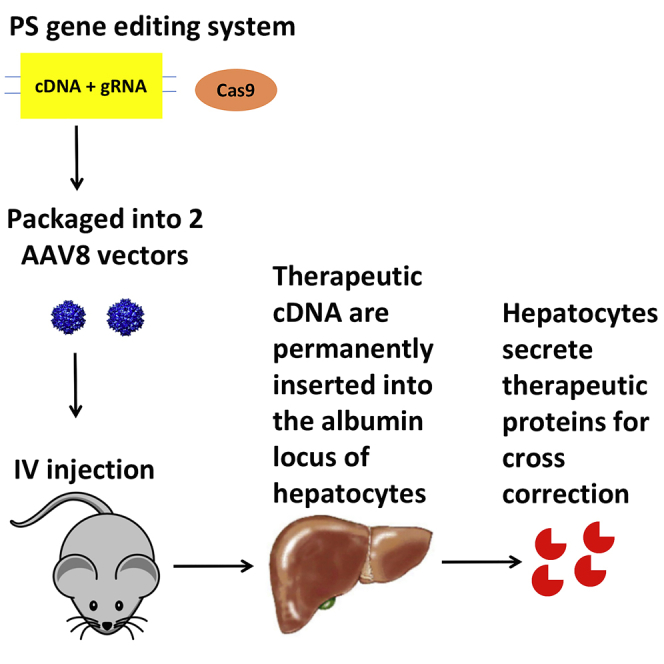
Based on our previous success using ZFN-mediated gene editing to treat MPS I, which resulted in a phase I/II clinical trial (NCT02702115), we designed a novel proprietary system gene editing system with CRISPR. It showed long-term safety and remarkably higher efficacy in correcting systemic and neurological symptoms.
Introduction
Mucopolysaccharidosis type I (MPS I) results from mutations in the gene encoding the lysosomal enzyme α-l-iduronidase (IDUA) and subsequent accumulation of glycosaminoglycan (GAG) heparan sulfate and dermatan sulfate.1 Without treatment, children with the most severe form uniformly die between 5 and 10 years of age. Both hematopoietic stem cell transplantation (HSCT) and enzyme replacement therapy (ERT) are available treatments for MPS I. However, these treatments have significant limitations. The procedure of HSCT is associated with morbidity or mortality.2, 3, 4 ERT is of limited use due to the need for frequent and life-long administration, high cost (>$200,000 annually), and negligible neurological benefits.5
Gene editing surmounts barriers to clinical relevance encountered by other methods, as it enables long-term transgene expression and minimizes insertional mutagenesis risk from random integration. Our previous study delivered zinc finger nucleases (ZFNs) to insert a therapeutic transgene into the albumin locus, which led to transgene expression without significantly affecting normal albumin expression. This strategy successfully treated neurological and systemic complications in mice with MPS I6 and II,7 resulting in two phase I/II clinical trials (ClinicalTrials.gov: NCT02702115 and NCT03041324). Progress reports from these clinical trials showed no drug-related adverse events; however, levels of transgene expression were low.8,9 As shown in clinical trials for treating MPS VI10 and hemophilia B,11 relatively low transgene expression is a major obstacle for gene therapy. The low transgene expression issue may be alleviated by increasing the dose. However, increasing the dose will bring about a higher risk of toxicity, more challenging vector production, and increased manufacturing costs. To this end, we designed a PS (proprietary system) gene editing approach with clustered regularly interspaced short palindromic repeats (CRISPR) technology. As opposed to three adeno-associated virus (AAV) vectors used in the study with ZFN, the PS only requires two vectors: one AAV vector encoding Cas9 and guide RNA (gRNA), and the other vector encoding a promoterless donor sequence. Assuming similar doses, AAV transduction, and nuclease targeting efficiency, the efficiency of successful genome editing by the PS is expected to be higher. In 2018, the US Food and Drug Administration (FDA) approved an investigational new drug (IND) application based on in vivo delivery of the CRISPR system for treating Leber congenital amaurosis type 10,12 which supports the use of CRISPR in patients. More recently, a study used a GOTI (genome-wide off-target analysis by two-cell embryo injection) method and identified very rare off-target events of Cas9 (similar to spontaneous mutations).13 These facts support the safety and potential clinical application of CRISPR/Cas9. In this study, we evaluated the safety and efficacy of the PS in MPS I mice and human hepatocytes.
Results
Study Design and Rationale
The design for the PS is similar to the ZFN system (Figure 1). Cas9 and gRNA mediate the insertion of a therapeutic transgene (promoterless cDNA) into the albumin intron 1 locus of hepatocytes. The splicing acceptor, IDUA cDNA, and poly(A) sequence can be inserted into the target locus through homology-directed repair (HDR). Through an alternative pathway of non-homologous end joining (NHEJ), the whole donor template, including inverted terminal repeats (ITRs), can be integrated at the double-strand break by end joining. Nevertheless, through the interaction between the splicing donor and splicing acceptor, the transcript will be the same: a hybrid of albumin exon 1 and the IDUA sequence. Since this insertion utilizes both HDR and NHEJ mechanisms, the PS can work in both dividing and non-dividing cells. The therapeutic transgene is expressed under the control of the very highly expressed endogenous albumin promoter. Systemic therapeutic benefits are achieved through cross-correction. Cross-correction is a phenomenon whereby lysosomal enzyme expressed from one cell can be secreted and taken up by another cell to degrade storage materials for metabolic correction.
Figure 1.

Molecular Mechanism of the PS Gene Editing Approach
First, Cas9 nuclease creates a double-strand break at the target locus. Through homology-directed repair, the splicing acceptor (SA) and human IDUA cDNA (IDUA), and the poly(A) (PA) sequence, are integrated. Through non-homologous end joining, the whole AAV sequence is integrated. Through the interaction between the splicing donor (SD) and SA, the fusion transcript is the same, which includes exon 1 of the albumin gene and the IDUA sequence. Exon 1 primarily encodes a signal peptide, which will be cleaved thereafter. The mature proteins are only the therapeutic IDUA proteins. ITR, inverted terminal repeat; HA, homology arm.
One potential challenge for the PS is to effectively treat neurological complications of MPS I through cross-correction. Nevertheless, the feasibility is supported by multiple preclinical studies with doses of ERT that are relatively high compared to the usual doses of ERT used to treat patients (Table 1). These studies showed that a high level of enzyme in circulation could facilitate entry of enzyme into the brain. This phenomenon was also observed in our previous ZFN studies in MPS I6 and II.7 In addition, hydrodynamic tail vein injection of a plasmid encoding the IDUA sequence into MPS I mice was performed. To eliminate any transgene expression in the CNS, the IDUA expression was restricted in the liver by using a liver-specific human α1-antitrypsin promoter. Two days after the injection, the mice were perfused and euthanized, and depletion of brain capillaries was performed. Interestingly, significant increases in IDUA activity and GAG reduction in the brain of injected mice were observed (Table 2). These results indicated that IDUA proteins were expressed in the liver, resulting in high blood IDUA levels and a small, but sufficient, amount of IDUA in the CNS. Of note, only 0.5% of wild-type activity is required to prevent neurological complications of MPS I.
Table 1.
Previous Preclinical Studies Show That High Doses of ERT Can Treat Neurological Complications
| Mouse Model | Dose (mg/kg) | Clinical Dose (mg/kg) | Brain Enzyme Activity (% Normal) | Brain Storage Teduced (% Affected) | References |
|---|---|---|---|---|---|
| α-Mannosidosis | 18.3 | 1 | N/A | 74 | 14 |
| α-Mannosidosis | 36.6 | 1 | 15 | 50 | 15 |
| Metachromatic leukodystrophy | 20 | N/A | N/A | 30 | 16 |
| Metachromatic leukodystrophy | 50 | N/A | N/A | 34 | 17 |
| Aspartylglycosaminuria | 10 | N/A | 10 | 20 | 18 |
| Krabbe disease | 6 | N/A | 7 | 18 | 19 |
| MPS II | 10 | 0.5 | 5 | N/A | 20 |
| MPS IIIA | 20 | N/A | 22 | 0 | 21 |
| MPS VII | 20 | 2 | 2.50 | N/A | 22 |
| MPS I | 20 | 0.58 | 97 | 63 | 23 |
N/A, not available.
Table 2.
IDUA Enzyme Activities and GAG Levels in the Brain of MPS I Mice after Hydrodynamic Injection of Plasmid Encoding IDUA cDNA under the Control of a Liver-Specific Promoter
| IDUA Enzyme Activity (nmol/h/mg protein) | GAG Levels (μg of GAG/mg of Protein) | |
|---|---|---|
| Treated MPS I (n = 3) | 0.33 ± 0.13 | 16.4 ± 0.9 |
| Control MPS I (n = 4) | 0 | 23.1 ± 2 |
The plasmids encoding IDUA cDNA under the control of a liver-specific hAAT promoter were intravenously injected into MPS I mice. Two days later, treated and control mice were perfused and tissues were collected. Enzyme activities and GAG levels in the brain were measured. Data are shown as mean ± standard errors.
Our previous study showed that one gRNA (5′-GTATCTTTGATGACAATAATGGGGGAT-3′) mediated cleavage at the target locus with the highest efficiency (11% indels).24 Then, the plasmid encoding SaCas9 and gRNA and the plasmid encoding promoterless IDUA cDNA donor were tested in MPS I mice through hydrodynamic injection. After 48 h, only the mice receiving both plasmids had significantly higher IDUA enzyme activities in the liver (2.7-fold those of wild-type levels) (Figure S1). Mice receiving the plasmid encoding promoterless cDNA donor had no increase in IDUA activities. These results showed the feasibility of the PS in editing hepatocytes and expressing IDUA enzyme.
Safety and Efficacy of the PS in Neonatal MPS I Mice
Neonatal MPS I mice received the test article (designated as PS822) intravenously (i.v.) at three different doses. Group assignment and doses are listed in Table 3. Blood samples were collected monthly for 10 months. Plasma enzyme activity increased significantly in all treated mice (the high-dose group achieved 500-fold that of wild-type levels) and was maintained for 10 months (Figure 2A). Neonatal MPS I mice were also injected with the donor vector alone (high dose, 3 × 1014 vector genomes [vg]/kg; middle dose, 3 × 1013 vg/kg; low dose, 3 × 1012 vg/kg). The enzyme activities in the donor-only groups were negligible compared to the corresponding Cas9+donor groups (Table S1). Therefore, the high transgene expression observed in Cas9+donor groups should be largely attributed to the integration at the target locus. In our previous ZFN studies, enzyme activity in the donor-only group were also observed.6,7 Notably, the enzyme activity observed in the donor-only groups do not necessarily come from the episomal expression. An alternative interpretation would be integration of the cDNA donor into the target locus without nuclease. Previous studies have shown the existence of this nuclease-free integration mechanism.25
Table 3.
Group Assignment for the Pharmacology and Toxicology Studies in Mice
| Group | Dose (AAV-IDUA and AAV-Cas9 in vg/kg) | Group Size |
|---|---|---|
| MPS I, high dose | 3 × 1014 and 5 × 1013 | 11 |
| MPS I, middle dose | 3 × 1013 and 5 × 1012 | 9 |
| MPS I, low dose | 3 × 1012 and 5 × 1011 | 13 |
| MPS I, untreated | N/A | 9 |
| Normal heterozygotes, untreated | N/A | 11 |
N/A, not available.
Figure 2.

The Efficacy of PS822 in Neonatal MPS I Mice
(A) Blood samples were collected from treated neonatal mice and controls monthly. Plasma IDUA enzyme activity increased significantly throughout 10 months. (B) Kaplan-Meier analysis showed the improved survival rate of treated neonatal mice. Three mice from the high-dose group were euthanized at 1 month post-dosing for early assessment and thus were not included in the Kaplan-Meier survival analysis. p < 0.05 when comparing each treatment group to untreated MPS I mice (log rank [Mantel-Cox] test for survival analysis. (C) Fear conditioning showed treated neonatal mice had better memory and learning ability. Mean ± SEM. ∗p < 0.05 when comparing treated to untreated MPS I mice; ∗∗p < 0.01. One-way ANOVA for multiple comparisons).
At 11 months post-dosing, tissues were collected from all mice. There were significant enzyme activities (Figure 3A) in tissues, including the brain of the high-dose and middle-dose mice in a dose-dependent manner. Notably, perfusion and brain capillary deletion were performed, largely obviating the possibility of contamination from enzyme in capillary endothelial cells and blood. Also, there was a significant positive correlation between plasma IDUA activity and brain IDUA activity (Figure S2A, p < 0.0001, R2 = 0.53), as well a significant negative correlation between plasma IDUA activity and brain GAG level (Figure S2B, p < 0.0001, R2 = 0.48). These results support our hypothesis that a constant high level of enzyme in the blood facilitates the entry of a small amount of enzyme into the brain. Furthermore, there was significant GAG storage reduction (Figure 3B) in the brain of all treated mice in a dose-dependent manner. The dose-dependent relationship indicates that the higher the transgene expression, the more the therapeutic benefits achieved. Notably, the GAG levels in the brains of mice from the middle-dose and high-dose groups were normalized. Fear conditioning showed that treated mice had better memory and learning ability (Figure 2C). Furthermore, mice from all three treated groups had significantly better survival rates (Figure 2B).
Figure 3.

Enzyme Activities and GAG Levels in Neonatal MPS I Mice after Treatment with PS822
(A and B) Enzyme activities (A) increased significantly and GAG storage levels (B) decreased significantly in tissues, including the brain of treated neonatal mice at 11 months post-dosing. Mean ± SEM. ∗p < 0.05 when comparing treated to untreated MPS I mice; ∗∗p < 0.01; ∗∗∗p < 0.0001. One-way ANOVA for multiple comparisons).
To further assess the efficacy, tissues were evaluated by light microscopy for evidence of the characteristic lesions associated with MPS I, that is, the presence of foam cells in the liver26 and of cytoplasmic vacuolation in neurons of the brain, with emphasis on Purkinje cell vacuolation.27,28 These lesions are a consequence of the engorgement of lysosomes with storage materials within the cytoplasm of macrophages and neurons, respectively. As shown in Table S2 and Figure 4 (upper panel), treatment with PS822 markedly reduced the incidence of foam cells in the liver of all treated groups. However, the efficacy of PS822 treatment with respect to Purkinje cell vacuolation was observed only in the high-dose group (Figure 4, middle panel). Purkinje cells, as the only output neurons of the cerebellar cortex, have been found to drive motor learning.29 More interestingly, previous studies have suggested the pivotal role of cerebellar Purkinje cells in fear conditioning,30,31 which assesses memory and learning. As mentioned earlier, the performance of untreated MPS I mice in fear conditioning was poor compared with normal mice, but it dramatically improved in treated MPS I mice. Therefore, the excessive storage accumulation in Purkinje cells may have led to the deficiency in memory, learning, and motor function, manifested as poor performances in behavior tests. To test whether the enzyme expressed from the liver can enter the CNS, immunolabeling for IDUA was performed with anti-IDUA polyclonal antibody. As shown in Figure 4 (lower panel), there appears to be a subtle but slightly increased intensity of signal in the choroid plexus of treated MPS I mice, which is suggestive of the presence of enzyme in the brain. More interestingly, occasional individual choroid plexus epithelial cells, the primary cerebrospinal fluid (CSF)-producing cells of the choroid plexus, were strongly immunopositive in all three examined animals, a finding that could be consistent with expression of IDUA proteins.
Figure 4.

Histology and IHC Analysis of Neonatal MPS I Mice after Treatment with PS822
(A–C) Columns show tissues from normal mice (A), untreated MPS I mice (B), and treated MPS I mice (C). The upper panel shows H&E staining of liver. Untreated MPS I mice have several foam cells (the hallmark of MPS I) present but these are absent in the treated MPS I and normal mice. The middle panel shows H&E staining of cerebellum. The Purkinje neurons in untreated MPS I mice have cytoplasmic cellular vacuolation (arrows), which is absent in high-dose MPS I treated and normal mice. The lower panel shows IHC for IDUA. There is a subtle but slightly increased intensity of signal in the choroid plexus of treated MPS I mice, which is suggestive of the presence of enzyme in the brain. Also, occasional individual choroid plexus epithelial cells (arrows) are strongly immunopositive for IDUA.
PS822 was well tolerated in mice, with no test article-related unscheduled deaths, and no clinical signs of toxicity. Moreover, the histopathological analysis identified benign liver tumor in only one mouse from the middle-dose group (Table 4). Previous studies showed that normal mice at this age developed tumors at the rate of 6%–13%.32,33 Therefore, there appears to be no significantly increased tumor risk in treated mice.
Table 4.
Tumor Rate in Each Group of Mice
| Group | Liver Tumor |
|---|---|
| MPS I, high dose | 0/11 |
| MPS I, middle dose | 1/11 |
| MPS I, low dose | 0/13 |
Quantitative polymerase chain reaction (qPCR) was performed to assess the biodistribution of AAV vectors (Table 5). AAV copy number was non-detectable in untreated MPS I and normal mice. In the low-dose group, AAV vector was detected only in the liver and heart, but not the brain and spleen. In the middle-dose group, AAV vector was detected in the liver, heart, and brain, but not in the spleen. In the high-dose group, AAV vector was detected in all tissues at a higher level than in the low-dose and middle-dose groups. More interestingly, AAV vector copy number in the liver of mice treated with a high dose but euthanized at 1 month post-dosing were higher than that in other mice from the high-dose group but euthanized at 11 months post-dosing. These results indicated the possibility of vector dilution as mice matured. Next-generation analysis showed that indel percentage at the target locus in the liver of mice from the high-dose group is approximately 8.04% ± 0.23% (Figure 5A). SaCas9 mRNA levels in tissues were also measured through qRT-PCR. The SaCas9 mRNA level in the liver of the high-dose group was reduced by 20-fold from 1 month to 11 months post-dosing (Figure 5B). This indicates the gradual loss of expression due to AAV vector dilution and/or promoter silencing. SaCas9 mRNA was not detected in the liver of the low-dose group. In addition, it was also non-detectable in other tissues (brain, spleen). This observation is expected because SaCas9 expression is limited to the liver through the liver-specific thyroxine-binding globulin (TBG) promoter and the liver tropism of the AAV8 vector. This further supports that IDUA enzyme entered the brain, instead of being expressed from edited brain cells. In addition, nested PCR was performed to confirm the integration of cDNA donor at the target locus (Figure S3). Liver samples from all three dose groups had the band at the expected size of 1,133 bp. Liver samples from untreated MPS I and normal mice, as well as brain samples from the high-dose group, had no bands indicating insertion.
Table 5.
AAV Copy Number in Tissues
| Group Assignment | AAV Copy Number in Tissues (100 vg/ng DNA) |
|||
|---|---|---|---|---|
| Brain | Liver | Heart | Spleen | |
| MPS I, untreated (n = 4) | ND | ND | ND | ND |
| Normal (n = 4) | ND | ND | ND | ND |
| MPS I, low dose (n = 6) | ND | 0.09 ± 0.01 | 0.29 ± 0.11 | ND |
| MPS I, middle dose (n = 5) | 0.07 ± 0.03 | 2.0 ± 0.78 | 1.0 ± 0.21 | ND |
| MPS I, high dose (n = 5) | 1.0 ± 0.27 | 51.2 ± 20.7 | 151.2 ± 53.1 | 0.07 ± 0.06 |
| MPS I, high dose, 1 month after dosing (n = 3) | N/A | 135.1 ± 64.6 | N/A | N/A |
Data are shown as mean ± SEM. ND, non-detectable; N/A, not available.
Figure 5.

Cutting Efficiency and Expression of Cas9, as well as Humoral Immune Response against IDUA Proteins
(A) Indel percentage in the liver of treated neonatal mice (high dose) was analyzed through next-generation sequencing. Untreated MPS I mice were included as controls. (B) SaCas9 mRNA level in the liver was quantified through qRT-PCR. The liver from treated neonatal MPS I mice (high, middle, or low dose) at 11 months post-dosing is depicted; RNA was also extracted from three mice from the high-dose group at 1 month post-dosing. (C) ELISA was performed with plasma samples from treated neonatal MPS I mice (high, middle, or low dose) at 30, 90, and 270 days post-dosing. In addition, ELISA was also performed with plasma samples from treated adult MPS I mice at 7 and 30 days post-dosing. The dashed line indicates the antibody level in untreated control mice (n = 6). (D) Plasma albumin levels were measured by ELISA with samples from untreated MPS I mice, neonatal MPS I mice treated with a high dose and low dose at 1 and 10 months post-dosing, as well as adult MPS I mice treated with a middle dose at 1 month post-dosing. Mean ± SEM. ∗p < 0.05 when comparing treated to untreated MPS I mice; ∗∗p < 0.01. One-way ANOVA for multiple comparisons).
Transgene Expression in Adult MPS I Mice
Previous studies have shown that the immune system of neonatal mice is naive, which makes it easier to treat neonatal MPS I mice.34 It also has been shown that Cas935 and IDUA proteins23 can elicit immune responses, which may affect the therapeutic efficacy. Therefore, it was crucial to assess PS822 in immunocompetent adult MPS I mice. Adult MPS I mice (n = 8, 4–6 weeks old) were i.v. injected with the middle dose of PS822 (total, 3.5 × 1013 vg/kg body weight). Plasma IDUA enzyme activity reached 4,950 ± 75 nmol/h/mL at 7 days post-dosing. This indicates that the transgene can be expressed in a very short time, highlighting the potential of PS822 in treating MPS I in a timely manner. Moreover, the plasma enzyme activities were even higher than what was observed in neonatal mice treated with the high dose (peaking at 3,990 ± 54 nmol/h/mL). This observation supports the high efficacy of PS822 in adult animals.
In addition, to assess the humoral immune response against IDUA proteins, ELISA with plasma samples from treated adult and neonatal mice was performed. As shown in Figure 5C, antibodies against IDUA proteins were only detected in treated neonatal mice from the low-dose and middle-dose groups at 270 days post-dosing. Notably, the level of antibodies was relatively low (absorbance, 0.11–0.17) compared with the level observed in our previous ERT studies in MPS I mice (0.7–3).23,36 Also, antibodies against IDUA proteins did not significantly affect the transgene expression even in adult mice. It also supports that while pulsatile ERT may trigger drug-neutralizing antibodies, it might be obviated by continuous delivery of enzyme by the PS gene editing approach.
In addition, ELISA showed that plasma albumin level in MPS I mice neonatally treated with the high dose decreased at 1 month post-dosing, but returned to the normal level at 10 months post-dosing (Figure 5D). No statistically significant reductions were observed in the low-dose group at 1 and 10 months post-dosing, or in MPS I mice treated as adults at 1 month post-dosing. In future studies in large animals and human patients, serum albumin level will be an important outcome and monitored closely.
Assessment of Human Albumin-Targeting Constructs
Since the target locus is not conserved between humans and mice, the test article PS822 that targets the human albumin intron 1 locus was designed. A total of 12 gRNAs were designed and transfected into human hepatocytes together with SaCas9. Sequencing results showed that none of these gRNAs mediated cleavage at the target locus. The protospacer adjacent motif (PAM) sequence of SaCas9 is 5′-NNGRRT-3′, while the PAM sequence of Streptococcus pyogenes Cas9 (SpCas9) is 5′-NGG-3. Statistically speaking, there will be more PAM sequence options in the target locus. Moreover, previous studies showed the feasibility of fitting SpCas9 into AAV vectors.37,38 Therefore, three gRNAs were designed and transfected together with SpCas9. Sequencing at the target locus showed that SpCas9 can be recruited to albumin intron 1 by gRNA3 (5′-TGTATTTGTGAAGTCTTACA-3′) and then cutting the DNA (Figure 6A). Then, gRNA3 was cloned into the donor plasmid encoding homology arms and IDUA cDNA. The donor plasmid and the plasmid encoding the TBG promoter and SpCas9 were cotransfected into HepG2 cells. After extracting genomic DNA from pooled cells, nested PCR was performed with two sets of primers (Figure 6B). The successful insertion into the target locus was confirmed by sequencing the amplicons. Additionally, the cell pellets and supernatants of cells cotransfected with Cas9 and donor plasmids had significantly higher enzyme activities (Figure 6C). These results showed that the test article PS822 efficiently inserted the therapeutic cDNA into the target locus.
Figure 6.

The Cutting Efficiency and Specificity of PS822 in Human Hepatocytes
(A) Plasmids encoding SpCas9 and three candidate gRNAs were transfected into human hepatocytes (Huh7 cell line), and the cleavage activity was measured through sequencing the target locus. Only gRNA3 showed significant cleavage, and it was selected as the candidate for further analyses. (B) The plasmid encoding SpCas9 and the donor plasmid encoding IDUA cDNA and gRNA3 were cotransfected into human hepatocytes (HepG2) cells. After 48 h, genomic DNA was extracted from collected cells, and nested PCR was performed. Cells without transfection and cells transfected with the donor plasmid only were used as controls. The gel image of the second round PCR is shown. Only cells transfected with both Cas9 and donor plasmids had the band at the expected size of 1,031 bp. (C) Cell pellets and supernatants were processed for IDUA enzyme assays. IDUA enzyme activities in cells transfected with Cas9 and donor plasmids had significantly higher IDUA enzyme activities in cell pellets and supernatants compared with untransfected cells and cells transfected with the donor plasmid only. (D) SpCas9 and gRNA3 ribonucleoprotein were cotransfected with double-strand oligonucleotide tag (dsTag) into Huh7 cells. Genomic DNA was extracted and used for library preparation. Deep sequencing was performed to search for the dsTag. (E) Only on-target cleavage was identified through GUIDE-seq.
One safety concern of any CRISPR-mediated gene editing is the possibility of off-target effects. Traditional off-target analysis involves two steps: (1) predicting potential off-target sites through in silico tools, and (2) sequencing the predicted sites. The in silico tools predict possible off-target sites across the genome based on the sequences of the genome and gRNA. Since genome-wide search criteria of these algorithms are not exhaustive, this method is intrinsically biased. In contrast, genome-wide unbiased identification of double-stranded breaks enabled by sequencing (GUIDE-seq) relies on the integration of a double-stranded oligodeoxynucleotide tag into a double-strand break created by Cas9, and then search the whole genome for these tags (Figure 6D). In this way, off-target sites can be identified in an unbiased manner.39 When tested in human hepatocytes (Huh7 cells), no off-target cleavage of PS822 was detected through GUIDE-seq (Figure 6E). These results laid a solid foundation for advancing the PS from preclinical mouse studies to the clinical studies.
Discussion
The Presence of Enzyme in the Brain
In this study, synthesized lysosomal enzymes were secreted and reached multiple tissues, including the brain, through cross-correction. The feasibility is supported by the ZFN study and many high-dose ERT studies from different groups (Table 1). These studies together showed that when there is a high and constant level of lysosomal enzyme in the blood, a small but sufficient amount can enter the brain. Possible mechanisms may include the following: (1) impaired integrity of the blood-brain barrier (BBB) due to disease; (2) fluid-phase pinocytosis; (3) extracellular pathway; and (4) residual mannose 6-phosphate receptor (M6PR) or other uncharacterized receptors. The correlation between plasma IDUA activity and brain IDUA activity (Figure S2) also strongly supports that the presence of enzyme in the brain is a result of a high and constant level of enzyme in the blood. Admittedly, this observation may be attributed to differences in BBB penetrability between mice and humans. Whether this strategy will work in human patients remains to be tested.
As shown in Figure 4, there was strong immunohistochemistry (IHC) immunoreactivity of occasional choroid plexus cells (not ependymal cells). This could be consistent with high levels of IDUA protein and a high degree of IDUA expression, but it does not prove the latter. If these cells indeed express IDUA proteins, there are several explanations. The first possibility is episomal expression of AAV vectors from transduced cells since AAV vector copy number was detected in the brain (Table 5). IDUA enzyme activity was detected in the donor-only groups (Table S1), albeit being very low. Episomal transgene expression could be driven by the cryptic promoter activity of ITRs.40,41 A previous study also indicated that choroid plexus epithelial cells might be susceptible to AAV8 vectors.42 The second possibility is transgene expression after integration into the target locus. In this scenario, Cas9 expression would be driven by ITRs, with insertion of the donor sequence into the albumin locus. However, this is unlikely because the transgene expression from transduced brain cells would still be under the control of the endogenous albumin promoter. Moreover, the Cas9 mRNA level in the brain is non-detectable, which makes the possibility of gene editing in the brain even lower. Notably, in the ZFN study, deep-sequencing analysis of the brain samples showed no gene editing events outside of the liver.6,7 The ZFN clinical trials showed no brain toxicity in all 12 patients.8,9 The third possibility is transgene expression after integration into an off-target locus. In this scenario, Cas9 expression would be driven by ITRs, with insertion of the donor sequence into an off-target site that is under the control of an endogenous promoter. This possibility is even more remote because it requires the occurrence of several unlikely events: AAV8 transduction in the brain, ITR-driven expression of Cas9, and off-target integration at a locus that happens to have an adjacent promoter. Further research is needed to elucidate the exact mechanism before advancing this approach to the clinical trial.
The Advantages of the PS over Other Therapies
The major advantage of the PS over the ZFN approach is its magnitude higher efficacy to achieve sufficient therapeutic benefits in human patients. Unlike what was observed in the mouse experiments, the ZFN system failed to provide sufficient therapeutic benefits due to its relatively low transgene expression in the MPS I and II clinical trials. As expected, it will be more challenging to achieve transgene expression above the therapeutic threshold in human patients. The PS is expected to overcome this issue by providing significantly higher enzyme levels. A comparison in plasma enzyme levels between this study and the ZFN study was performed. In neonatal mice, the middle dose of PS822 achieved 25-fold of the peak levels in the ZFN study at <50% of the ZFN dose (3.5 × 1013 versus 7.5 × 1013 vg/kg). In adult mice, the middle dose of the PS achieved 125-fold of the peak levels in the ZFN study at <50% of the ZFN dose. Notably, in the ZFN study, the mice were injected weekly with an immunosuppressant (cyclophosphamide), whereas no immunosuppressants were used in this study. IHC for IDUA expression was also conducted with the liver of mice treated with the high dose (Figure S4). There were many cells with much stronger signal than other cells. These IDUA-expressing cells constituted 24.1% of the cells observed, largely reflecting the integration rate at the target locus. In the ZFN study, based on the ratio between the albumin-transgene fusion transcript and the wild-type albumin transcript, the integration rate at the target locus was estimated to be less than 0.5%.43 Therefore, the higher efficiency of the PS than the ZFN system could be attributed to the higher possibility of hepatocytes being successfully edited (cDNA donor insertions at the target locus). Additionally, the PS will enable the usage of lower doses of AAV vector to treat lysosomal diseases, which minimizes toxicity, eases vector production, and reduces cost.
Compared with weekly ERT, the PS gene editing approach can also provide a significantly higher enzyme with a single administration. For a 75-kg patient with MPS I, the weekly ERT dose is approximately 43.5 mg, and the ERT half-life is 1.5–3.6 h. Both the ZFN system and the PS insert a therapeutic transgene in the albumin intron 1 locus, and then therapeutic proteins are expressed under the control of the endogenous albumin promoter. The albumin promoter is very highly expressed. Normal albumin levels in the blood are 40–50 mg/mL and synthesized from the liver at a rate of 105 g/week. Only 0.05% of albumin production needs to be co-opted to provide the same amount of IDUA enzyme provided by ERT. The ZFN study showed that at least 0.5% of hepatocytes had been edited for enzyme production.6 In this study using the PS, a magnitude higher transgene expression than that in the ZFN study was observed. Therefore, the PS is expected to provide a significantly higher enzyme than ERT with a single i.v. administration. Furthermore, pulsatile ERT may trigger drug-neutralizing antibodies,44 which could be obviated by continuous delivery of enzyme by PS gene editing. Such continuous enzyme delivery has been used to create immune tolerance from ERT and eliminate neutralizing antibodies against lysosomal enzymes. Thus, the PS is expected to achieve better safety and efficacy than ERT. Moreover, this study showed that the PS achieved significant neurological benefits in animal models of lysosomal diseases, which is another critical advantage over ERT.
The major advantage of the PS over traditional AAV gene therapy is its ability to provide potentially life-long therapeutic benefits, overcoming the issue of vector dilution, and will provide ongoing efficacy after the first few years following treatment. Most patients suffering from lysosomal diseases are children, and normal growth requires continuous cell divisions. AAV gene therapy faces this major problem of vector dilution, a significant issue that could become a major problem as children grow and mature. As shown in clinical trials of AAV gene therapy for hemophilia B, the transgene expression in humans declines over time. This could be due to the non-integrating nature of the AAV vector. It was shown that transgene expression from episomal AAV vectors was rapidly lost after one round of cell division,45 leading to a gradual decline of therapeutic effects. Unfortunately, secondary administration of AAV vectors often fails to rescue expression, due to the immune response to primary vector delivery.46 In addition, targeting the liver has additional advantages, including (1) substantial clinical experience with liver-targeting gene therapy, and (2) hepatic transgene expression known to induce immune tolerance.47,48
Safety of the PS
During the relatively long-term monitoring (11 months), no vector-associated adverse events or signs of severe immune responses were observed. Only one benign hepatic tumor was found in the middle-dose group, which was not statistically increased compared with controls or age-matched wild-type mice in previous studies.32,33 Moreover, if the tumor was a result of off-target effects, one would expect more cases of tumor in the high-dose group. Notably, even if the tumor was treatment-related, it could be due to AAV vectors instead of the CRISPR system. Previous studies have shown that AAV gene therapy could cause AAV vector integration and thus hepatocellular carcinoma.33,49,50 In this study, the mouse with the tumor was found dead in the cage during a weekend, and it was not certain how long it had been dead. In light of this, according to our standard protocol, the whole carcass was fixed in formalin for pathological analysis. Since it was the only benign liver tumor we observed, no sequencing analysis with the tumor tissue could be performed. GUIDE-seq was performed with human albumin-targeting constructs in human hepatocytes, and no off-target sites were identified, indicating at least the safety of the human constructs. More in-depth safety experiments focusing on the genotoxicity of the PS in large animal models (e.g., non-human primates) are warranted before advancing this technology to the clinical trial stage.
Another safety concern is the influence of the PS on normal albumin expression. Continuous cutting at the target locus could cause large deletions. A previous study used AAV delivery of the CRISPR to repair the endogenous mutation in mice with ornithine transcarbamylase (OTC) deficiency.51 Large deletions at the target locus caused a complete loss of residual OTC expression and thus death. In our study, the target locus is albumin intron 1, which does not have significant biological functions. Even if the deletion extends to the exon region of albumin, we would not expect losing expression of a subset of albumin expression to be lethal. We have measured albumin levels in plasma through ELISA at 1 and 11 months post-dosing. Albumin levels decreased only in the high-dose group at 1 month post-dosing, but they returned to normal at 10 months post-dosing. The return of albumin levels could be explained by transcriptional upregulation of albumin production. These results indicated that the influence on the normal albumin production was alleviated. No reduced albumin levels were observed in the low-dose group. In addition, all mice were monitored on a regular basis, and no signs similar to symptoms of hypoalbuminemia in humans were observed. The high dose was originally designed as the maximal tolerance dose, and it would not be used in the potential clinical trial. Moreover, to reduce the risk of continuous cutting at the target locus, when designing the construct for editing the human albumin locus, we introduced mutations in the region of the homology arms where are homologous to the target locus and the PAM site. Therefore, after one round of gene editing, the target locus has changed and would not be recognized by Cas9 anymore. Mutating the regions of homology arms has been shown to successfully avoid continuous cutting.52 Nevertheless, serum albumin levels will be an important outcome (“toxicity”) in future studies in large animals and, potentially, human patients, and proper mitigation methods (e.g., albumin infusion, dietary management) will be designed and used if necessary.
Contributions of HDR and NHEJ
The indel frequency in this study (8%) was much lower than that observed in the ZFN study (>40%). This could be explained by the fact that indel frequency is only a partial measure of gene editing rates. No indels are generated through HDR. Through an alternative pathway of NHEJ, the donor template can be integrated at the double-strand break in an indel-free manner by end joining.53 Previous studies have shown the integration of the AAV donor template at the target locus through this mechanism.43,52 Notably, the AAV integration at double-strand breaks could be a full or partial vector.54,55 Moreover, another study leveraged this mechanism and created a homology-independent targeted integration (HITI) method for in vivo gene editing.56 In this study, HITI could cause indels or be indel-free, but the indel-free mechanism was the predominant pathway. Therefore, while the indel frequency is lower in this study, the frequencies of the other two pathways might be high, both of which could lead to transgene expression (as shown in Figure 1).
Due to the large size of the insertion cassette (a total of 2.2–3.5 kb: homology arms, cDNA, poly(A), and potentially ITRs), it is difficult to measure the insertion frequency through next-generation sequencing (NGS). Two previous studies used ligation-mediated PCR coupled with unique molecular indices to determine the gene targeting efficiency.57,58 However, this method could not provide information about the relative frequency of HDR- and NHEJ-mediated insertion because all reads with the cDNA sequence at the target locus were defined as HDR events. Those authors also acknowledged that the real insertion frequency could be underestimated because some insertions might be too large to be captured by short PCR-amplicon NGS. Several studies have also shown that NHEJ-mediated insertions were more common than HDR-mediated insertions.52,59 In the first study, a comparison was performed between the donor template with or without homologous arms.52 There was no significant difference in the transgene expression level, indicating that NHEJ-mediated insertion would be the predominant mechanism. The second study found that the donor vector without homology arms produced 4- to 6-fold more edited cells than did the vector with homology arms.59
Materials and Methods
Animals and Injections
MPS I knockout mice (Idua−/−), a kind gift from Dr. Elizabeth Neufeld (University of California, Los Angeles [UCLA]), were generated by insertion of neomycin resistance gene into exon 6 of the 14-exon IDUA gene on the C57BL/6 background.60 MPS I mice (Idua−/−) and heterozygotes (Idua−/+) were genotyped by PCR. Neonatal MPS I mice were injected with AAV vectors (<30 μL) through the temporal facial vein on day 1 or 2. Adult MPS I mice were injected with AAV vectors (<100 μL) through the tail vein at the age of 4–6 weeks. Hydrodynamic injections of plasmids were performed in adult MPS I mice as previously described.24 All mouse care and handling procedures were in compliance with the rules of the Institutional Animal Care and Use Committee (IACUC) of the University of Minnesota.
Construct Design and Confirmation
The gRNAs targeting the mouse albumin locus were designed and confirmed in embryonic fibroblast cells as previously described.24 Briefly, in the Cas9 vector, SaCas9 was under the control of the human TBG promoter, and gRNA was under the control of the U6 promoter. In the donor vector, a splicing acceptor sequence (28 bp), the codon-optimized human IDUA cDNA (1,905 bp), and a poly(A) sequence (256 bp) were flanked by two homology arms (319 bp each). The homology arms were located exactly at the cleavage site (200 bp downstream of the last base pair of exon 1). The primers used to amplify the mouse albumin locus for sequencing were as follows and are described in Figure S3: forward prime, 5′-GGAACCAATGAAATGCGAGG-3′; reverse primer 1, 5′-GAGTAGTTTGCCTGTTTGCC-3′; reverse primer 2, 5′-CTACAACTTCACCCACCTGGA-3′.
To target the human albumin locus, gRNAs were designed and transfected into human hepatocytes (HepG2 or Huh7 cell line) together with the plasmid encoding SaCas9 or SpCas9. After 24–48 h, genomic DNA was extracted from pooled cells, and PCR was performed with the following primers: forward primer, 5′-GGTCAGAATTGTTTAGTGACTGT-3′; reverse primer, 5′-CCGATGAGCAACCTCACTCT-3′. The cleavage activities at the target locus were measured by sequencing the PCR amplicons. The gRNA that mediates efficient cleavage at the human albumin locus was packaged into the donor plasmid that encodes IDUA cDNA. Then, the donor plasmid was cotransfected with the plasmid encoding SpCas9 into HepG2 cells. After 48 h, genome DNA was extracted from collected cells. Nested PCR was performed with two sets of primers as follows: Nested-1 forward, 5′-TATACACAAGGGATTTAGTCAAAC-3′ and Nested-1 reverse, 5′-TGGGTAAGCCACCAAAGGAAAC-3′; Nested-2 forward, 5′-GGCAGCCAATGAAATACAAAGAT-3′ and Nested-2 reverse, 5′-ACCAGGTCCTTCCACTCGAACA-3′. The amplicons were confirmed by sequencing. Cell pellets and supernatants were also collected from IDUA enzyme assays.
GUIDE-Seq
An unbiased genome-wide off-target analysis, GUIDE-seq, was performed with the candidate gRNA targeting human albumin locus as previously described.39 Briefly, ribonucleoproteins were cotransfected with double-stranded oligodeoxynucleotides into Huh7 cells. After 48 h, genomic DNA was extracted from collected cells, and 5 μg of genomic DNA was used for library preparation and sequencing.
AAV Vector Production
AAV-IDUA-gRNA and AAV-SaCas9 were packaged into AAV8 vectors at the Children’s Hospital of Philadelphia Research Vector Core. The titer was verified by SDS-PAGE and silver staining. The core follows good laboratory practice (GLP) guidelines.
Depletion of Brain Capillaries
To rule out the possibility that enzyme activities in the brain come from capillary cells and blood, all mice were transcardially perfused with 35 mL of phosphate-buffered saline, and depletion of brain capillaries was performed as previously described.61
IDUA Enzyme Assay and GAG Assay
IDUA activity was determined by a fluorometric assay using 4-methylumbelliferyl α-l-iduronide (Glycosynth, #44076) as the substrate as previously described.62 Tissue GAG assays were conducted as described previously.63
Behavior Tests
Fear conditioning was performed according to an established protocol.64 Briefly, on the first day, the mice were put in an enclosed container and given stimuli, followed by electric shock. On the second day, the mice were only given the stimuli without electric shock, and the percentage of time the mice froze during the test was recorded. The percentage of time the mice froze indicated how robust the memory was. Each mouse underwent each test three times. Behavior tests were performed at the Mouse Behavior Core, University of Minnesota. The group assignment was blinded to the personnel performing behavior tests.
Histopathology and IHC
After fixation in 10% neutral buffered formalin, tissues from all mice were processed into paraffin using standard histology techniques, sectioned at a thickness of 4 μm, stained with hematoxylin and eosin (H&E), and evaluated by an American College of Veterinary Pathologists (ACVP)-board certified pathologist (M.G.O.) using light microscopy. For IDUA IHC preparations, 4-μm formalin-fixed, paraffin-embedded sections of tissue were deparaffinized, rehydrated, and subjected to heat-induced antigen retrieval (using 10 mM citrate buffer [pH 6.0]) in a steamer prior to performing the IHC procedure on a Dako Autostainer. IHC for IDUA was performed using a mouse anti-IDUA monoclonal antibody (R&D Systems, #AF4119) as primary antibody. Detection was achieved using a rabbit EnVision+ kit (Dako, #K4011), with diaminobenzidine (DAB) as the chromogen. All work was done at the University of Minnesota Masonic Cancer Center Comparative Pathology Laboratory.
ELISA
To detect antibodies against IDUA proteins, ELISA was conducted as described previously.23 In a 96-well microplate, 50 μL of diluted IDUA (4 μg/mL, R&D Systems, #4119GH) was incubated at 4°C overnight. After washing and blocking with Tris-buffered saline containing 1% bovine serum albumin at room temperature for 2 h, 100 μL of serum (1:50 dilutions) was incubated at room temperature for 1 h. After washing, the wells were incubated with 100 μL of anti-mouse immunoglobulin G (IgG) alkaline phosphatase conjugate (Abcam, #97027) at room temperature for 1 h. After washing, the wells were incubated with 200 μL of 1 mg/mL p-nitrophenyl phosphate chromogenic substrate (Sigma, #P7998) at room temperature for 1 h. Then, 50 μL of EDTA (0.1 M [pH 8.0]) was added to stop the reaction. Absorbance at 450 nm was quantified by using a BioTek plate reader.
To detect serum albumin levels, ELISA was performed with a mouse albumin ELISA kit (Abcam, #ab108791).
qPCR, qRT-PCR, and NGS
Total DNA from brain, heart, liver, and spleen of treated and control mice was extracted with the QIAamp DNA mini kit. qPCR was performed with PowerUp SYBR Green Master Mix (Thermo Fisher Scientific, #A25741) in MicroAmp 96-well plates (Applied Biosystems, #N8010560). Primers targeting the AAV ITR were as follows: forward primer, 5′-GGAACCCCTAGTGATGGAGTT-3′; reverse primer, 5′-CGGCCTCAGTGAGCGA-3′. Plasmids encoding the AAV ITR was used to make a standard curve. AAV copy number was expressed as 100 vg/ng DNA. To quantify SaCas9 mRNA levels, RNA was isolated using TRIzol (Thermo Fisher Scientific, #15596026) and reverse transcribed using a high-capacity cDNA reverse transcription kit (Thermo Fisher Scientific, #4368814). qPCR to measure SaCas9 were performed using the following primers: forward primer, 5′-CCGTCGTGAAGAGAAGCTTCATC-3′; reverse primer, 5′-CCACCTCATAGTTGAAGGGGTTG-3′. DNA was quantified in parallel samples using GAPDH primers as internal controls as follows: forward primer, 5′-CATCACTGCCACCCAGAAGACTG-3′; reverse primer, 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′.
To analyze the on-target cleavage activity, genomic DNA from the liver from treated and control mice was extracted. PCR was performed using primers around the target locus, and the amplicons were purified using DNA purification solid-phase reversible immobilization (SPRI) magnetic beads (ABM, #G950), processed for library preparation using a Nextera XT library preparation kit (Illumina, #FC-131-1024), and sequenced on the NextSeq platform. Indel percentage analysis was performed using CRISPResso.
Author Contributions
L.O., J.J., and C.B.W. designed the experiments; L.O., M.J.P., O.A., P.O., S.K., and M.G.O. conducted the experiments; L.O., M.G.O., and C.B.W. wrote the manuscript.
Conflicts of Interest
L.O. and C.B.W. are inventors of a pending patent (PCT/US2018/065747) based on the PS gene editing approach. The remaining authors declare no competing interests.
Acknowledgments
The authors would like to thank Dr. Michael Benneyworth (Mouse Behavior Core, University of Minnesota) for technical assistance in behavior tests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.03.018.
Supplemental Information
References
- 1.Neufeld E.F., Muenzer J. The mucopolysaccharidoses. In: Scriver C.R., Beaudet A.L., Sly W.S., Valle D., Childs B., Kinzler K.W., Vogelstein B., editors. Eighth Edition. Volume III. McGraw-Hill; 2001. pp. 3421–3452. (The Metabolic and Molecular Bases of Inherited Disease). [Google Scholar]
- 2.Hobbs J.R., Hugh-Jones K., Barrett A.J., Byrom N., Chambers D., Henry K., James D.C., Lucas C.F., Rogers T.R., Benson P.F. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet. 1981;2:709–712. doi: 10.1016/s0140-6736(81)91046-1. [DOI] [PubMed] [Google Scholar]
- 3.Whitley C.B., Ramsay N.K.C., Kersey J.H., Krivit W. Bone marrow transplantation for Hurler syndrome: assessment of metabolic correction. Birth Defects Orig. Artic. Ser. 1986;22:7–24. [PubMed] [Google Scholar]
- 4.Eisengart J.B., Rudser K.D., Xue Y., Orchard P., Miller W., Lund T., Van der Ploeg A., Mercer J., Jones S., Mengel K.E. Long-term outcomes of systemic therapies for Hurler syndrome: an international multicenter comparison. Genet. Med. 2018;20:1423–1429. doi: 10.1038/gim.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wraith J.E., Clarke L.A., Beck M., Kolodny E.H., Pastores G.M., Muenzer J., Rapoport D.M., Berger K.I., Swiedler S.J., Kakkis E.D. Enzyme replacement therapy for mucopolysaccharidosis I: a randomized, double-blinded, placebo-controlled, multinational study of recombinant human α-l-iduronidase (laronidase) J. Pediatr. 2004;144:581–588. doi: 10.1016/j.jpeds.2004.01.046. [DOI] [PubMed] [Google Scholar]
- 6.Ou L., DeKelver R.C., Rohde M., Tom S., Radeke R., St Martin S.J., Santiago Y., Sproul S., Przybilla M.J., Koniar B.L. ZFN-mediated in vivo genome editing corrects murine Hurler syndrome. Mol. Ther. 2019;27:178–187. doi: 10.1016/j.ymthe.2018.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laoharawee K., DeKelver R., Podetz-Petersen K.M., Rohde M., Sproul S., Nguyen H.O., Nguyen T., St Martin S., Ou L., Tom S. Dose-dependent prevention of metabolic and neurologic disease in murine MPS II by ZFN-mediated in vivo genome editing. Mol. Ther. 2018;26:1127–1136. doi: 10.1016/j.ymthe.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muenzer J., Prada C.E., Burton B., Lau H.A., Ficicioglu C., Foo C.W.P., Vaidya S.A., Whitley C.B., Harmatz P. CHAMPIONS: a phase 1/2 clinical trial with dose escalation of SB-913 ZFN-mediated in vivo human genome editing for treatment of MPS II (Hunter syndrome) Mol. Genet. Metab. 2019;126:S104. [Google Scholar]
- 9.Harmatz P., Lau H., Heldermon C., Leslie N., Foo C.W.P., Vaidya S.A., Whitley C.B. EMPOWERS: a phase 1/2 clinical trial of SB-318 ZFN-mediated in vivo human genome editing for treatment of MPS I (Hurler syndrome) Mol. Genet. Metab. 2019;126:S68. [Google Scholar]
- 10.Brunetti-Pierri N., Ferla R., Ginocchio V.M., Yildiz Y., Fecarotta S., Zancan S., Pecorella V., Graziano M., Dell’Anno M., Parenti G. Liver-directed gene therapy clinical trial for mucopolysaccharidosis type VI. Mol. Ther. 2019;27(Suppl 1):36–37. [Google Scholar]
- 11.Doshi B.S., Arruda V.R. Gene therapy for hemophilia: what does the future hold? Ther. Adv. Hematol. 2018;9:273–293. doi: 10.1177/2040620718791933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 13.Zuo E., Sun Y., Wei W., Yuan T., Ying W., Sun H., Yuan L., Steinmetz L.M., Li Y., Yang H. Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science. 2019;364:289–292. doi: 10.1126/science.aav9973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roces D.P., Lüllmann-Rauch R., Peng J., Balducci C., Andersson C., Tollersrud O., Fogh J., Orlacchio A., Beccari T., Saftig P., von Figura K. Efficacy of enzyme replacement therapy in α-mannosidosis mice: a preclinical animal study. Hum. Mol. Genet. 2004;13:1979–1988. doi: 10.1093/hmg/ddh220. [DOI] [PubMed] [Google Scholar]
- 15.Blanz J., Stroobants S., Lüllmann-Rauch R., Morelle W., Lüdemann M., D’Hooge R., Reuterwall H., Michalski J.C., Fogh J., Andersson C., Saftig P. Reversal of peripheral and central neural storage and ataxia after recombinant enzyme replacement therapy in α-mannosidosis mice. Hum. Mol. Genet. 2008;17:3437–3445. doi: 10.1093/hmg/ddn237. [DOI] [PubMed] [Google Scholar]
- 16.Matzner U., Herbst E., Hedayati K.K., Lüllmann-Rauch R., Wessig C., Schröder S., Eistrup C., Möller C., Fogh J., Gieselmann V. Enzyme replacement improves nervous system pathology and function in a mouse model for metachromatic leukodystrophy. Hum. Mol. Genet. 2005;14:1139–1152. doi: 10.1093/hmg/ddi126. [DOI] [PubMed] [Google Scholar]
- 17.Matzner U., Lüllmann-Rauch R., Stroobants S., Andersson C., Weigelt C., Eistrup C., Fogh J., D’Hooge R., Gieselmann V. Enzyme replacement improves ataxic gait and central nervous system histopathology in a mouse model of metachromatic leukodystrophy. Mol. Ther. 2009;17:600–606. doi: 10.1038/mt.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dunder U., Kaartinen V., Valtonen P., Väänänen E., Kosma V.M., Heisterkamp N., Groffen J., Mononen I. Enzyme replacement therapy in a mouse model of aspartylglycosaminuria. FASEB J. 2000;14:361–367. doi: 10.1096/fasebj.14.2.361. [DOI] [PubMed] [Google Scholar]
- 19.Lee W.C., Courtenay A., Troendle F.J., Stallings-Mann M.L., Dickey C.A., DeLucia M.W., Dickson D.W., Eckman C.B. Enzyme replacement therapy results in substantial improvements in early clinical phenotype in a mouse model of globoid cell leukodystrophy. FASEB J. 2005;19:1549–1551. doi: 10.1096/fj.05-3826fje. [DOI] [PubMed] [Google Scholar]
- 20.Polito V.A., Abbondante S., Polishchuk R.S., Nusco E., Salvia R., Cosma M.P. Correction of CNS defects in the MPSII mouse model via systemic enzyme replacement therapy. Hum. Mol. Genet. 2010;19:4871–4885. doi: 10.1093/hmg/ddq420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rozaklis T., Beard H., Hassiotis S., Garcia A.R., Tonini M., Luck A., Pan J., Lamsa J.C., Hopwood J.J., Hemsley K.M. Impact of high-dose, chemically modified sulfamidase on pathology in a murine model of MPS IIIA. Exp. Neurol. 2011;230:123–130. doi: 10.1016/j.expneurol.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 22.Vogler C., Levy B., Grubb J.H., Galvin N., Tan Y., Kakkis E., Pavloff N., Sly W.S. Overcoming the blood-brain barrier with high-dose enzyme replacement therapy in murine mucopolysaccharidosis VII. Proc. Natl. Acad. Sci. USA. 2005;102:14777–14782. doi: 10.1073/pnas.0506892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou L., Przybilla M.J., Koniar B., Whitley C.B. RTB lectin-mediated delivery of lysosomal α-l-iduronidase mitigates disease manifestations systemically including the central nervous system. Mol. Genet. Metab. 2018;123:105–111. doi: 10.1016/j.ymgme.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou L., Przybilla M.J., Tăbăran A.F., Overn P., O’Sullivan M.G., Jiang X., Sidhu R., Kell P.J., Ory D.S., Whitley C.B. A novel gene editing system to treat both Tay-Sachs and Sandhoff diseases. Gene Ther. 2020 doi: 10.1038/s41434-019-0120-5. Published online January 2, 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., Kay M.A. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendez D.C., Stover A.E., Rangel A.D., Brick D.J., Nethercott H.E., Torres M.A., Khalid O., Wong A.M., Cooper J.D., Jester J.V. A novel, long-lived, and highly engraftable immunodeficient mouse model of mucopolysaccharidosis type I. Mol. Ther. Methods Clin. Dev. 2015;2:14068. doi: 10.1038/mtm.2014.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferrer I., Cusí V., Pineda M., Galofré E., Vila J. Focal dendritic swellings in Purkinje cells in mucopolysaccharidoses types I, II and III. A Golgi and ultrastructural study. Neuropathol. Appl. Neurobiol. 1988;14:315–323. doi: 10.1111/j.1365-2990.1988.tb00891.x. [DOI] [PubMed] [Google Scholar]
- 28.Jones M.Z., Alroy J., Boyer P.J., Cavanagh K.T., Johnson K., Gage D., Vorro J., Render J.A., Common R.S., Leedle R.A. Caprine mucopolysaccharidosis-IIID: clinical, biochemical, morphological and immunohistochemical characteristics. J. Neuropathol. Exp. Neurol. 1998;57:148–157. doi: 10.1097/00005072-199802000-00006. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen-Vu T.D., Kimpo R.R., Rinaldi J.M., Kohli A., Zeng H., Deisseroth K., Raymond J.L. Cerebellar Purkinje cell activity drives motor learning. Nat. Neurosci. 2013;16:1734–1736. doi: 10.1038/nn.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu L., Scelfo B., Tempia F., Sacchetti B., Strata P. Membrane excitability and fear conditioning in cerebellar Purkinje cell. Neuroscience. 2006;140:801–810. doi: 10.1016/j.neuroscience.2006.02.040. [DOI] [PubMed] [Google Scholar]
- 31.Sacchetti B., Scelfo B., Tempia F., Strata P. Long-term synaptic changes induced in the cerebellar cortex by fear conditioning. Neuron. 2004;42:973–982. doi: 10.1016/j.neuron.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 32.Goldsworthy T.L., Fransson-Steen R. Quantitation of the cancer process in C57BL/6J, B6C3F1 and C3H/HeJ mice. Toxicol. Pathol. 2002;30:97–105. doi: 10.1080/01926230252824770. [DOI] [PubMed] [Google Scholar]
- 33.Donsante A., Miller D.G., Li Y., Vogler C., Brunt E.M., Russell D.W., Sands M.S. AAV vector integration sites in mouse hepatocellular carcinoma. Science. 2007;317:477. doi: 10.1126/science.1142658. [DOI] [PubMed] [Google Scholar]
- 34.Hinderer C., Bell P., Louboutin J.P., Katz N., Zhu Y., Lin G., Choa R., Bagel J., O’Donnell P., Fitzgerald C.A. Neonatal tolerance induction enables accurate evaluation of gene therapy for MPS I in a canine model. Mol. Genet. Metab. 2016;119:124–130. doi: 10.1016/j.ymgme.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou L., Herzog T., Koniar B.L., Gunther R., Whitley C.B. High-dose enzyme replacement therapy in murine Hurler syndrome. Mol. Genet. Metab. 2014;111:116–122. doi: 10.1016/j.ymgme.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishiyama J., Mikuni T., Yasuda R. Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron. 2017;96:755–768.e5. doi: 10.1016/j.neuron.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao H.K., Hatanaka F., Araoka T., Reddy P., Wu M.Z., Sui Y., Yamauchi T., Sakurai M., O’Keefe D.D., Núñez-Delicado E. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017;171:1495–1507.e15. doi: 10.1016/j.cell.2017.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aitken M.L., Greene K.E., Tonelli M.R., Burns J.L., Emerson J.C., Goss C.H., Gibson R.L. Analysis of sequential aliquots of hypertonic saline solution-induced sputum from clinically stable patients with cystic fibrosis. Chest. 2003;123:792–799. doi: 10.1378/chest.123.3.792. [DOI] [PubMed] [Google Scholar]
- 41.Flotte T.R., Afione S.A., Solow R., Drumm M.L., Markakis D., Guggino W.B., Zeitlin P.L., Carter B.J. Expression of the cystic fibrosis transmembrane conductance regulator from a novel adeno-associated virus promoter. J. Biol. Chem. 1993;268:3781–3790. [PubMed] [Google Scholar]
- 42.Churchill C.C. University of Minnesota; 2014. The impact of gene transfer of arginine decarboxylase to the central nervous system on opioid analgesic tolerance. PhD thesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharma R., Anguela X.M., Doyon Y., Wechsler T., DeKelver R.C., Sproul S., Paschon D.E., Miller J.C., Davidson R.J., Shivak D. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood. 2015;126:1777–1784. doi: 10.1182/blood-2014-12-615492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kishnani P.S., Dickson P.I., Muldowney L., Lee J.J., Rosenberg A., Abichandani R., Bluestone J.A., Burton B.K., Dewey M., Freitas A. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol. Genet. Metab. 2016;117:66–83. doi: 10.1016/j.ymgme.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Nakai H., Yant S.R., Storm T.A., Fuess S., Meuse L., Kay M.A. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J. Virol. 2001;75:6969–6976. doi: 10.1128/JVI.75.15.6969-6976.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calcedo R., Wilson J.M. Humoral immune response to AAV. Front. Immunol. 2013;4:341. doi: 10.3389/fimmu.2013.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finn J.D., Ozelo M.C., Sabatino D.E., Franck H.W., Merricks E.P., Crudele J.M., Zhou S., Kazazian H.H., Lillicrap D., Nichols T.C., Arruda V.R. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116:5842–5848. doi: 10.1182/blood-2010-06-288001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mingozzi F., Liu Y.L., Dobrzynski E., Kaufhold A., Liu J.H., Wang Y., Arruda V.R., High K.A., Herzog R.W. Induction of immune tolerance to coagulation factor IX antigen by in vivo hepatic gene transfer. J. Clin. Invest. 2003;111:1347–1356. doi: 10.1172/JCI16887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chandler R.J., LaFave M.C., Varshney G.K., Trivedi N.S., Carrillo-Carrasco N., Senac J.S., Wu W., Hoffmann V., Elkahloun A.G., Burgess S.M., Venditti C.P. Vector design influences hepatic genotoxicity after adeno-associated virus gene therapy. J. Clin. Invest. 2015;125:870–880. doi: 10.1172/JCI79213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walia J.S., Altaleb N., Bello A., Kruck C., LaFave M.C., Varshney G.K., Burgess S.M., Chowdhury B., Hurlbut D., Hemming R. Long-term correction of Sandhoff disease following intravenous delivery of rAAV9 to mouse neonates. Mol. Ther. 2015;23:414–422. doi: 10.1038/mt.2014.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ohmori T., Nagao Y., Mizukami H., Sakata A., Muramatsu S.I., Ozawa K., Tominaga S.I., Hanazono Y., Nishimura S., Nureki O., Sakata Y. CRISPR/Cas9-mediated genome editing via postnatal administration of AAV vector cures haemophilia B mice. Sci. Rep. 2017;7:4159. doi: 10.1038/s41598-017-04625-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhargava R., Sandhu M., Muk S., Lee G., Vaidehi N., Stark J.M. C-NHEJ without indels is robust and requires synergistic function of distinct XLF domains. Nat. Commun. 2018;9:2484. doi: 10.1038/s41467-018-04867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jarrett K.E., Lee C.M., Yeh Y.H., Hsu R.H., Gupta R., Zhang M., Rodriguez P.J., Lee C.S., Gillard B.K., Bissig K.D. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang L., Yang Y., Breton C.A., White J., Zhang J., Che Y., Saveliev A., McMenamin D., He Z., Latshaw C. CRISPR/Cas9-mediated in vivo gene targeting corrects hemostasis in newborn and adult factor IX-knockout mice. Blood. 2019;133:2745–2752. doi: 10.1182/blood.2019000790. [DOI] [PubMed] [Google Scholar]
- 58.Wang L., Yang Y., Breton C., Bell P., Li M., Zhang J., Che Y., Saveliev A., He Z., White J. A mutation-independent CRISPR-Cas9-mediated gene targeting approach to treat a murine model of ornithine transcarbamylase deficiency. Sci. Adv. 2020;6:eaax5701. doi: 10.1126/sciadv.aax5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen H., Shi M., Gilam A., Zheng Q., Zhang Y., Afrikanova I., Li J., Gluzman Z., Jiang R., Kong L.J., Chen-Tsai R.Y. Hemophilia A ameliorated in mice by CRISPR-based in vivo genome editing of human factor VIII. Sci. Rep. 2019;9:16838. doi: 10.1038/s41598-019-53198-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ohmi K., Greenberg D.S., Rajavel K.S., Ryazantsev S., Li H.H., Neufeld E.F. Activated microglia in cortex of mouse models of mucopolysaccharidoses I and IIIB. Proc. Natl. Acad. Sci. USA. 2003;100:1902–1907. doi: 10.1073/pnas.252784899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang D., El-Amouri S.S., Dai M., Kuan C.Y., Hui D.Y., Brady R.O., Pan D. Engineering a lysosomal enzyme with a derivative of receptor-binding domain of apoE enables delivery across the blood-brain barrier. Proc. Natl. Acad. Sci. USA. 2013;110:2999–3004. doi: 10.1073/pnas.1222742110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ou L., Herzog T.L., Wilmot C.M., Whitley C.B. Standardization of α-l-iduronidase enzyme assay with Michaelis-Menten kinetics. Mol. Genet. Metab. 2014;111:113–115. doi: 10.1016/j.ymgme.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ou L., Przybilla M.J., Koniar B.L., Whitley C.B. Elements of lentiviral vector design toward gene therapy for treating mucopolysaccharidosis I. Mol. Genet. Metab. Rep. 2016;8:87–93. doi: 10.1016/j.ymgmr.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin-Fernandez M., Jamison S., Robin L.M., Zhao Z., Martin E.D., Aguilar J., Benneyworth M.A., Marsicano G., Araque A. Synapse-specific astrocyte gating of amygdala-related behavior. Nat. Neurosci. 2017;20:1540–1548. doi: 10.1038/nn.4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.