

Abstract
Adeno-associated viral (AAV) vectors are a leading candidate for the delivery of CRISPR-Cas9 for therapeutic genome editing in vivo. However, AAV-based delivery involves persistent expression of the Cas9 nuclease, a bacterial protein. Recent studies indicate a high prevalence of neutralizing antibodies and T cells specific to the commonly used Cas9 orthologs from Streptococcus pyogenes (SpCas9) and Staphylococcus aureus (SaCas9) in humans. We tested in a mouse model whether pre-existing immunity to SaCas9 would pose a barrier to liver genome editing with AAV packaging CRISPR-Cas9. Although efficient genome editing occurred in mouse liver with pre-existing SaCas9 immunity, this was accompanied by an increased proportion of CD8+ T cells in the liver. This cytotoxic T cell response was characterized by hepatocyte apoptosis, loss of recombinant AAV genomes, and complete elimination of genome-edited cells, and was followed by compensatory liver regeneration. Our results raise important efficacy and safety concerns for CRISPR-Cas9-based in vivo genome editing in the liver.
Keywords: gene therapy, SaCas9, AAV-CRISPR, CD8+ T cell, immune response, somatic genome editing, hepatocytes, adeno-associated virus, pre-existing immunity, liver
Graphical Abstract
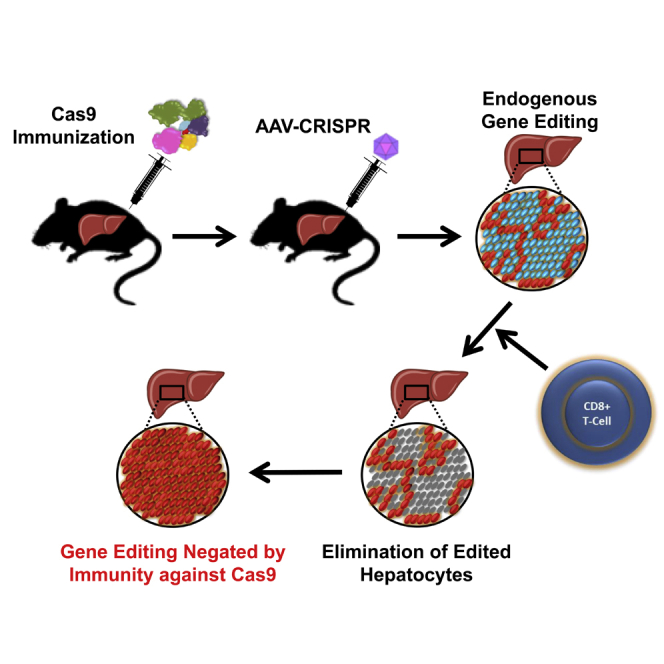
Li and colleagues demonstrated that pre-existing immunity to Cas9 poses a significant barrier to liver-directed genome editing with AAV-CRISPR. They found that AAV expression of Cas9 elicited a robust CD8+ T cell response, resulting in elimination of edited hepatocytes. This raises important safety and efficacy concerns for in vivo editing with CRISPR-Cas9.
Introduction
Adeno-associated viral (AAV) vectors are a leading candidate for the delivery of CRISPR-Cas9 for therapeutic genome editing in humans. These vectors have a strong track record of safety in clinical trials and an ability to transduce multiple tissues with high efficiency. AAV-based delivery of CRISPR-Cas9 (AAV-CRISPR) has been used to correct disease-relevant genes in preclinical models of human diseases, including Duchenne muscular dystrophy, hypercholesterolemia, and urea cycle disorders.1, 2, 3, 4, 5, 6 The gene-editing-based approaches are generally well tolerated in mice, producing long-term correction of pathology without obvious adverse effects from sustained Cas9 expression.7 Nonetheless, there are indications that AAV-CRISPR genome editing may not be completely benign to the host. For example, it has been reported that delivery of AAV-CRISPR to mouse skeletal muscle leads to infiltration by both CD4+ helper and CD8+ cytotoxic T cells, and the production of circulating immunoglobulins.8 Further, a recent paper by Moreno et al.9 indicates that immunological memory to AAV, as well as Cas9, is a major barrier to repeated dosing, which can in some cases be overcome by using divergent AAV serotypes or Cas9 orthologs.
Pre-existing exposure to Cas9, a bacterial protein, could be a serious obstacle to therapeutic gene editing in humans. Recently, two groups reported a remarkably high prevalence of pre-existing immunity toward Streptococcus pyogenes (Sp) and Staphylococcus aureus (Sa) Cas9, the two most commonly used Cas9 orthologs.10,11 These studies reported that 78% of humans exhibit an immune response toward SaCas910 and 58%–96% toward SpCas9.10,11 In addition to neutralizing antibodies, many individuals also have T cell memory against Cas9. Pre-existing immunity to Cas9 could have multiple adverse effects in CRISPR-Cas9-based therapeutic genome editing, including blocking delivery, triggering acute inflammation, or initiating destruction of the edited cells by the immune system. Thus, pre-existing immunity to Cas9 could be a major safety concern, depending on the severity of the response, which could vary widely among individuals. Despite the progress in somatic genome editing, major questions remain concerning its feasibility and safety in solid organs, such as the liver. In this work, we show that pre-existing immunity to Cas9 poses a significant barrier to liver-directed genome editing with AAV-CRISPR. Our results indicate that mice can mount a strong memory T cell response to small amounts of SaCas9 protein delivered, and pre-immunization against SaCas9 did not directly block AAV-CRISPR transduction or genome editing in the liver. However, AAV expression of SaCas9 elicited a robust CD8+ T cell response, resulting in elimination of gene-edited hepatocytes, followed by compensatory liver regeneration over a period of just 12 weeks. These results raise important efficacy and safety concerns for therapeutic liver-directed genome editing, particularly in the setting of pre-existing immunity to Cas9.
Results
Establishing Immune Memory in Mice against SaCas9 Protein
Unlike humans, mice raised in specific pathogen-free facilities do not have pre-existing immunity to SaCas9. To establish immune memory against SaCas9 protein, we performed a classical delayed-type hypersensitivity (DTH) assay.12 We immunized C57BL/6 mice against either 25 μg SaCas9 or 100 μg ovalbumin (as control) and challenged them a week later in the pinna of one ear with 16 μg of either ovalbumin or SaCas9 and in the pinna of the other ear with saline (Figure 1A). Ear thickness was recorded 24 h later, and swelling was used as a measure of memory T lymphocyte-mediated inflammation. As expected,13 immunization and subsequent challenge with ovalbumin led to a positive DTH reaction, whereas ovalbumin-immunized mice challenged with SaCas9 displayed no such reaction (Figures 1B and 1C). Mice immunized and challenged with SaCas9 developed a substantial DTH reaction, resulting in a doubling of the challenged ear thickness relative to that of mice immunized and challenged with ovalbumin. Histology of the ears of these animals revealed edema and immune infiltrates, providing clear evidence of immune memory to the antigen (Figure 1C). These data show that C57BL/6 mice can rapidly mount a strong memory T lymphocyte response to small amounts of SaCas9.
Figure 1.

Induction of Immune Memory against SaCas9 in C57BL/6 Mice
(A) Mice were initially immunized with either purified ovalbumin (Ova) or SaCas9 protein. One week after immunization, mice were challenged with either ovalbumin or SaCas9, and ear thickness was measured after 24 h. (B) Histological analysis of the left ear of mice in (A) stained with hematoxylin and eosin. ∗p < 0.05, ∗∗p < 0.01; ns, not significant. Scale bar, 50 μm. (C) Representative H&E staining of mouse ears.
Induction of a Cytotoxic CD8+ T Cell Response to SaCas9-Expressing Hepatocytes
We next tested whether pre-existing immunity to SaCas9 would provoke a cytotoxic T cell response against hepatocytes transduced with AAV8 vectors expressing a SaCas9 transgene. Two groups of mice were immunized against 100 μg ovalbumin and 25 μg SaCas9 protein, respectively. One week later, all the animals received both an AAV-CRISPR targeting the low-density lipoprotein receptor (Ldlr) gene14 and a second AAV vector expressing GFP to track the transduced hepatocytes. Ldlr was targeted because it is a non-essential gene in the liver, and we previously reported a highly efficient guide RNA (gRNA),15 allowing us to track the fate of the gene-edited hepatocytes by deep sequencing. Cohorts of randomly selected mice from each group were then euthanized for liver analysis at 1, 2, 4, 6, and 12 weeks after AAV administration (Figure 2A). Livers were digested and subjected to flow cytometry to identify T cells. We found that the proportion of CD4+ helper T cells relative to the total T cells steadily decreased over time, with no significant difference between the two groups (Figures S1A and S1B). However, there was a significant increase in the proportion of CD8+ cytotoxic T cells in the liver of mice immunized against SaCas9 beginning at 1 week after AAV injection, which persisted for 4 weeks (Figure 2B). An increased abundance of CD8+ T cells in the liver was also confirmed by qPCR analysis (Figure 2C). The percentage of T cells and the amount of CD8+ and CD4+ T cells relative to the total amount of T cells in the spleen remained unchanged throughout the 12-week period between groups (Figures S2A–S2C). To determine the functional consequences of a higher proportion of CD8+ T cells in the liver, we performed TUNEL staining to identify apoptotic cells. We observed a marked increase in TUNEL-positive cells in livers of mice that were pre-immunized against SaCas9 relative to the ovalbumin controls, which coincided with the increased ratio of CD8+ T cells (Figure 2D; Figure S3A). Alanine transaminase (ALT) activity, a general marker of liver damage, showed similar kinetics with a peak at 2 weeks after AAV delivery, which returned to normal levels by 12 weeks (Figure 2E). Liver sections were also stained for Ki-67, an established marker of cell proliferation. As expected, many infiltrating lymphocytes stained positive for Ki-67, and the amount of Ki-67-positive cells was markedly increased in the group of animals pre-immunized against SaCas9 compared with the control group (Figure 2F; Figure S3B). In addition, hepatocytes throughout the liver were also positive for Ki-67, indicating extensive regeneration in response to injury. Transcript levels of Ki-67 showed a biphasic response, possibly reflecting an initial wave of CD8+ T cell expansion followed by hepatocyte proliferation (Figure 2G).
Figure 2.

AAV-CRISPR Gene Editing of Mice Immunized against SaCas9 Elicits a Robust CD8+ T Cell Response
(A) Mice were immunized with either ovalbumin or SaCas9 protein 1 week prior to co-delivery of AAV vectors encoding a gRNA targeting Ldlr with SaCas9 and GFP. Tissues were harvested at 1, 2, 4, 6, and 12 weeks post-injection. (B) CD8+ T cells as a percentage of total lymphocytes in the liver were measured by flow cytometry. (C) CD8 mRNA expression between mice immunized against ovalbumin and SaCas9 measured by qPCR. (D) Representative TUNEL staining in mouse liver sections. (E) Serum alanine transaminase (ALT) activity levels indicating liver damage. (F) Representative Ki-67 staining of mouse liver sections. (G) Ki-67 mRNA expression in mouse liver. Group sizes in (C), (E), and (G) are n = 7, except for ovalbumin 6 weeks (n = 6) and SaCas9 1 week (n = 6).
Pre-existing Immunity to SaCas9 Eliminates AAV-CRISPR-Transduced Hepatocytes In Vivo
To better understand the impact of a shift toward a more CD8+ T cell-rich environment on liver genome editing, we first measured the copy number of the episomal AAV transgenes delivered. Both GFP and SaCas9 transgenes persisted in the livers of both pre-immunized and control mice groups through 6 weeks, indicating efficient co-transduction with both AAV-CRISPR and AAV-GFP vectors regardless of pre-immunization. Interestingly, the mice that were pre-immunized against SaCas9 showed a significant decrease in the copy number of both AAV transgenes between 6 and 12 weeks post-delivery. AAV-GFP was reduced by 37-fold (Figures 3A and 3B) and AAV-CRISPR genomes were reduced by 36-fold (Figures 3C and 3D) relative to the ovalbumin controls at 12 weeks. The loss of episomal AAV genomes suggests replacement of gene-edited hepatocytes through liver regeneration, most likely from neighboring hepatocytes that escaped AAV-CRISPR transduction. To test this, we examined the endogenous target of our AAV-CRISPR vector (Ldlr), which should have been knocked out through Cas9-induced inactivating indel mutations in the coding sequence. Western blot analysis showed higher LDLR protein levels at the end of the 12-week experiment in the mice that were pre-immunized against SaCas9, indicating the expansion of hepatocytes that escaped genome editing (Figure 3E). Likewise, SaCas9 protein was undetectable at 12 weeks by western blot, and the protein levels of the co-delivered GFP transgene were dramatically reduced (Figure 3E). Immunohistochemistry analysis corroborated the loss of GFP protein at 12 weeks (Figure S4). Next generation sequencing of the Ldlr gene showed a higher rate of indel formation over 6 weeks in mice pre-immunized with ovalbumin versus SaCas9 (Figure 3F). By week 12, robust editing was still observed in mice immunized against ovalbumin (20.5%), whereas those immunized against SaCas9 had only 3.4% detectable indels. These data demonstrate the loss of genome-edited hepatocytes by CD8+ T cells following transduction with AAV-CRISPR in animals with pre-existing immunity to SaCas9.
Figure 3.

Pre-existing Immunity to SaCas9 Results in Elimination of Gene-Edited Hepatocytes following AAV Transduction
(A) AAV genome copies of GFP transgene measured via qPCR. (B) Fold change of GFP genome copies relative to ovalbumin-treated mice. (C) AAV genome copies of SaCas9 transgene measured via qPCR. (D) Fold change of SaCas9 genome copies relative to ovalbumin-treated mice. (E) Representative western blots of LDLR, SaCas9 (hemagglutinin, HA-Tag), and GFP in mouse liver. Asterisk indicates non-specific band by the HA-Tag antibody. (F) Ldlr indels in mouse liver samples were measured via deep sequencing.
Administration of AAV-CRISPR to SaCas9-Immunized Mice Stimulates a Memory T Cell Response
Although our studies demonstrated a shift toward a higher proportion of CD8+ T cells per total lymphocytes in the liver, the phenotype of these T cells is unclear. To better understand the specificity of the immune response, we pre-immunized mice with 25 μg SaCas9 and injected them 1 week later with either saline, AAV-GFP, or AAV-CRISPR. Livers and spleens were then harvested 2 weeks later, when the greatest proportion of CD8+ T cells was previously observed (Figure 4A). Although the percentage of total T cells did not increase per total lymphocyte count in the liver (Figure 4B), the proportion of CD8+ T cells was significantly increased only in those mice treated with AAV-CRISPR (Figure 4C). Of these CD8+ T cells, 85% demonstrate a memory phenotype compared with GFP (42%) and saline (63%) (Figure 4D). The percentage of activated CD8+ T cells was also significantly higher in the AAV-CRISPR group versus AAV-GFP and saline-treated mice (Figure 4E). Upon closer inspection of the memory CD8+ T cell population, we observed a significant increase in the AAV-CRISPR-treated group, with 94% of memory CD8+ T cells being activated compared with the AAV-GFP (85%) and saline (84%) groups (Figure 4F). An increase in regulatory T cells (Tregs) in the liver was seen only in the group injected with AAV-CRISPR (Figure 4G). The percentage of CD4+ T cells decreased in mice injected with AAV-CRISPR and increased in the mice injected with AAV-GFP compared with saline (Figure S5A). Like the memory CD8+ T cell response, a similar trend was observed in the memory CD4+ T cells (Figure S5B). The percentage of activated CD4+ T cells was lower in the AAV-CRISPR mice compared with the AAV-GFP group, although the percentage of activated memory CD4+ T cells was significantly increased in the AAV-CRISPR-treated mice compared with AAV-GFP and saline (Figures S5C and S5D). The CD8+ and CD4+ T cell populations within the spleen showed no significant differences between treatment conditions (Figures S6A–S6F and S7A–S7D).
Figure 4.

CD8+ T Cell and Treg Responses in SaCas9-Immunized Mice after Treatment with Either AAV-GFP or AAV-CRISPR
(A) Mice were immunized with SaCas9 protein 1 week prior to delivery of an AAV vector encoding a gRNA targeting Ldlr with SaCas9 or GFP. Tissues were harvested at 2 weeks post-injection. (B) T cells as a percentage of total lymphocytes in the liver were measured via flow cytometry. (C) CD8+ T cells as a percentage of T cells. (D) Percentage of memory CD8+ to total CD8+ T cells. (E) Percentage of activated CD8+ T cells to total CD8+ T cell population. (F) Percentage of activated memory CD8+ T cells to total CD8+ T cell population. (G) Tregs as a percentage of total lymphocytes in the liver measured via flow cytometry. n = 6 per condition.
Discussion
Reports of pre-existing immunity toward the most common Cas9 variants have raised concern about CRISPR-Cas9-based therapeutic genome editing; however, the consequences of such pre-existing immunity remain unknown. Here we show that in vivo transduction with AAV-CRISPR is accompanied by a strong CD8+ T cell response in the liver in the setting of pre-existing immunity. Our findings demonstrate that SaCas9 is an immunogenic protein that can trigger a robust memory T cell response in mice. Although AAV transduction, SaCas9 expression, and gene editing were not initially impeded by pre-immunization, CD8+ T cell activation in the liver resulted in a gradual but near-complete elimination of gene-edited hepatocytes. Livers were able to regenerate through proliferation of unedited hepatocytes over time, but the effects of genome editing largely disappeared.
Recent studies by Charlesworth et al.10 and Wagner et al.11 have reported a high prevalence of pre-existing immunity to Cas9 in humans, ranging from 78% for SaCas910 and 58%–96% for SpCas9.10,11 There is some debate about the frequency of anti-Cas9 immunity, with other groups finding values of 2.5%–10%.16,17 These differences may be related to the sensitivity of the assays, as well as the characteristics of the populations studied. It is also possible that some fraction of anti-Cas9 signal could arise from cross-reactivity to other epitopes. Ferdosi et al.17 identified two HLA-A∗02:01 epitopes specific to SpCas9: SpCas9_240-248 and SpCas9_615-623. Interestingly, neither peptide resembled known epitopes in the Immune Epitope Database and Analysis Program (IEDB) database but have similarity to other Cas9 orthologs (33/38 hits) and other bacterial proteins (5/38 hits). This suggests that there is not complete immune orthogonality between different species of Cas9. It also indicates that exposure to other bacterial proteins with related sequences to Cas9 may also confer anti-Cas9 immunity. Although our study focused on SaCas9, which is easily deliverable with AAV, it is likely that AAV expression of SpCas9 would have similar effects. Regardless of the source of anti-Cas9 reactivity and its ultimate frequency in the population, it is critical that we understand its implications in the setting of genome editing.
The severity of the immune response to SaCas9 was particularly noteworthy in these experiments. For example, the DTH challenge experiment used ovalbumin as a positive control. Ovalbumin is a smaller protein than SaCas9 (43 versus 130 kDa), so on a molar basis there was approximately a 12-fold excess of ovalbumin (100 μg) used for immunization relative to SaCas9 (25 μg). The strong response suggests that SaCas9 is at least as immunogenic as ovalbumin (Figure 1B). It should be noted that ovalbumin, although one of the most common antigens used for inducing memory T cell responses in DTH, is considered mildly immunogenic. Other antigens, such as keyhole limpet hemocyanin, are used to elicit much stronger immune responses. It is possible that there is some evolutionary pressure in mammals to more readily detect Cas9 and other bacterial proteins, which might be mediated by HLA diversity. It will be interesting to determine whether different HLA haplotypes exhibit a greater frequency of pre-existing immunity to Cas9 in humans, as well as the consequences of these antigen recognition preferences for genome-editing therapies. This is an important topic that merits further exploration.
Immune responses to AAV capsids are an important factor influencing the effectiveness of AAV gene therapy in humans. Neutralizing antibodies to AAV can prevent the initial transduction event, and immunosuppression can be used to mitigate subsequent anti-capsid T cell responses should they occur.18 Although we recognize that anti-AAV immunity remains an important area of research, the goal of this study is to interrogate potential immune response to Cas9 when expressed as an AAV transgene, specifically in the setting of pre-existing Cas9 immunity. Thakore et al.19 previously reported evidence of CD8+ T cell infiltration of the liver and ALT elevations with AAV-CRISPR. Interestingly, this response did not prevent long-term transcriptional repression by dCas9KRAB. A key difference is that the previous study used mice housed under specific pathogen-free conditions and therefore unlikely to have ever been in contact with S. aureus or SaCas9. In contrast, our study involved AAV overexpression of Cas9 following exposure to the antigen. The elimination of the gene-edited hepatocytes we observed was most likely due to CD8+ T cells in the livers of these animals. Although we did find an increase in activated CD8+ T cells in the livers of mice injected with AAV-GFP compared with saline, similar to what was reported by Thakore et al.,19 the mice injected with AAV-CRISPR had significantly higher amounts of activated CD8+ T cells compared with AAV-GFP (Figure 4). The memory CD8+ T cells were also significantly lower in the AAV-GFP group, indicating that the increase in activated T cells in the AAV-GFP group is a non-specific response to AAV. The increase in activated memory CD8+ T cells and Tregs in the AAV-CRISPR-treated group further indicates that the immune memory responses we observe are specific to SaCas9.
In terms of kinetics, it is interesting that the peak of CD8+ T cells in the liver occurs at 2 weeks, yet the loss of gene-edited hepatocytes is not complete until the 12-week time point. There are several factors that might explain these discordant data. The first is that there is still a significant increase in CD8+ T cells at 4 weeks, and the loss of indels begins to achieve statistical significance at 6 weeks. As such, inflammation, liver injury, and hepatocyte proliferation are linked but dynamic processes that happen over a period of time for different cells. Based on our experimental time points, we cannot pinpoint the exact window between 6 and 12 weeks when the hepatocytes are fully replaced by new cells. It is expected that T cell killing should precede the drop in indels, with some delay required for clearance of apoptotic hepatocytes, which might still be detected by deep sequencing. Aside from this, it also is possible that there could be a brief period of suppression by Tregs and/or that time may be needed for the CD8+ T cells to become functionally competent.20 This is an intriguing possibility given other recent data and the known lag in CD8+ T cell response to AAV capsid in humans receiving liver-directed gene therapies.21
Wagner et al.11 reported a high prevalence of Cas9-reactive Tregs, which could potentially dampen the immune response and spare Cas9-expressing cells in the context of pre-existing immunity in humans. However, although these Tregs reduced proliferation and thus effector cytokine release, they did not prevent killing of Cas9-expressing target cells, showing only a modest effect compared with polyclonal natural regulatory T cells (nTregs). Interestingly, we also observed a significant increase in Tregs in the mice expressing SaCas9, but this response did not prevent the elimination of gene-edited hepatocytes. Therefore, it seems unlikely that the normal activation of Cas9-specific Tregs would provide a buffer to protect Cas9-expressing cells, at least in the context of the liver. However, this is an important area of research, and targeted strategies to promote immune tolerance through Tregs may still be beneficial.
Our studies used AAV8, a serotype with a high tropism for the liver in mice. Expression of SaCas9 was further restricted to this tissue with a liver-specific promoter.22 Although the overall health of the mice was not adversely affected by removal of gene-edited hepatocytes, the effects of pre-existing immunity on other organs that are non-regenerative, such as heart, skeletal muscle, or the central nervous system, would require further study. This is especially true in the context of other AAV serotypes or more ubiquitously expressed promoters. Another concern with pre-existing immunity against Cas9 is the risk for subsequent autoimmunity. An immune reaction to a foreign antigen, such as a pathogen, can lead to tissue damage and the exposure of previously hidden autoantigens. In a susceptible genetic background, this can lead to epitope spreading and the development of a destructive autoimmune response.23,24 It is important to note that our studies involved deliberate immunization of the animals to SaCas9 protein a week before AAV injection. This experimental design allowed us to interrogate the consequences of a memory T cell response in a controlled setting. However, actual immune responses in humans will certainly be more variable considering the timing, localization, and nature of previous Cas9 exposure, as well as the genetic and environmental variation between individuals. Therefore, our findings do not definitively preclude Cas9 editing in humans with pre-existing immunity, but certainly raise points of caution to consider.
Potential solutions for evading the immune response toward CRISPR-Cas9 are being explored. One option is to use more exotic bacterial CRISPR-Cas systems, such as those found in hot springs,25 to which humans would have no previous environmental exposure. Other approaches have focused on engineering Cas9 variants to evade epitopes recognized by the immune system.17 However, studies have shown that antibody responses toward these epitopes are highly variable, making it difficult to engineer a broadly applicable Cas9 that still retains activity.8 Complementary to the investigation of novel Cas proteins, new delivery vectors are also being explored. Self-deleting AAV-CRISPR systems are a promising avenue that would capitalize on the high efficiency of AAV delivery.26, 27, 28 It remains to be seen whether SaCas9 self-removal would prevent presentation of SaCas9 peptides on MHC class I and subsequent loss of edited cells. Other promising approaches include transient delivery of Cas9 mRNA or Cas9 protein with targeting moieties or nanoparticle systems29, 30, 31 that render a short period of Cas9 exposure and could be re-administered. However, challenges exist in manufacturing, efficiency, and safety to translate non-viral delivery approaches to somatic genome editing in humans.
In this study, we show that pre-existing immunity to SaCas9 poses a significant barrier to liver-directed genome editing with AAV-CRISPR. Cytotoxic CD8+ T cells become the dominant population of T lymphocytes in the liver, which eliminate the gene-edited hepatocytes over a short period of just 12 weeks, thus negating the intended effects. It remains to be determined whether a similar or more severe response occurs in humans. Our new discoveries raise important efficacy and safety concerns for CRISPR-Cas9-based in vivo genome editing, illustrating the importance of considering anti-Cas9 immunity in clinical trial designs and the need to develop new approaches for in vivo delivery of genome-editing machinery.
Materials and Methods
AAV Production
Plasmids with a gRNA targeting the Ldlr locus and SaCas9 or emerald green fluorescent protein (EmGFP) were packaged into AAV8 as previously described.15,26 The plasmid encoding EmGFP is identical to 1162_pAAV-HLP-EmGFP-SpA that was previously reported, albeit driven by a chicken beta actin (CB) promoter instead of the hybrid liver-specific promoter (HLP). The HLP promoter was chosen for SaCas9 to restrict expression to hepatocytes. The CB promoter was used to express GFP, because this also gives robust expression in liver and would minimize the risk for competition with HLP for transcription factor binding. All clones were sequence verified, and inverted terminal repeat (ITR) sequence fidelity was verified by independent digests using XmaI, SnaBI, and PvuII. The adenoviral helper plasmid pAdDeltaF6 (PL_F_PVDAF6) and AAV packaging plasmid pAAV2/8 (PL-T-PV0007) were obtained from the University of Pennsylvania Vector Core. The triple-transfection method32 was used to generate AAVs in HEK293T cells using polyethylenimine (PEI). Cell pellets were purified using a single cesium chloride (CsCl) density gradient centrifugation.33 Fractions positive for vector genomes were pooled and dialyzed in 100,000 molecular weight cutoff (MWCO) cassettes against three washes of PBS at 4°C overnight to remove CsCl. Purified AAVs were then concentrated using an Amicon 100-kDa MWCO centrifugal filtration (UFC510024) device. AAV titers were calculated after DNase digestion using qPCR against a standard curve and primers specific to SaCas9 and GFP before storage at −80°C until use.
Animals
Male C57BL/6J mice, 6–8 weeks of age, were obtained from Jackson Laboratories and kept with a light cycle from 7 a.m. to 9 p.m. Animals were allowed free access to food and water and maintained on a standard chow diet. Blood was collected via retro-orbital bleeding using heparinized Natelson collection tubes, and plasma was isolated by centrifugation at 10,000 × g for 20 min at 4°C. All experiments were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee (IACUC) and performed in accordance with institutional guidelines under protocol numbers AN-6243, AN-7243, and AN-5198.
DTH Assay
Emulsions were prepared with either SaCas9 or ovalbumin in complete Freund’s adjuvant immediately before immunization.34 Mice were immunized subcutaneously in two sites in the flanks with either 25 μg SaCas9 (ABM, K144) or 100 μg ovalbumin per mouse. One week later, the mice were challenged with saline in the pinna of one ear and with 16 μg of either SaCas9 or ovalbumin dissolved in saline in the pinna of the other ear. Ear thickness was measured 24 h after challenge using a spring-loaded micrometer (Mitutoyo), and the difference in left and right ear thickness was calculated for each animal.34
Immunization and AAV Administration
Purified recombinant SaCas9 protein was a kind gift of New England Biolabs (EnGen Sau Cas9, M0654T).35 Mice were immunized subcutaneously in two sites in the flanks with either 25 μg SaCas9 or 100 μg ovalbumin per mouse as in the DTH assays. One week later, AAV8 vectors were co-injected intraperitoneally (i.p.) at 5 × 1011 genome copies (GCs) of the Ldlr gRNA and SaCas9 and 2 × 1011 GCs of GFP. AAVs were diluted in 300 μL sterile PBS and delivered via i.p. injection. Mice were euthanized at the indicated times after AAV injection for blood and tissue collection. One mouse from each of the following conditions was excluded from the study in Figures 2 and 3 because of failed i.p. injection based on the absence of detectable AAV-GFP genomes in the liver: 2-week ovalbumin, 4-week SaCas9, 6-week ovalbumin, and 12-week ovalbumin. A total of 74 mice were immunized in this study, with an additional 12 mice injected with saline as controls for flow cytometry.
Flow Cytometry
Single-cell suspensions were prepared from the spleen and a piece of the liver of each mouse using a 100-μm cell strainer. Cells were washed with fluorescence-activated cell sorting (FACS)-PBS (PBS + 2% bovine serum albumin [BSA] + 2% goat serum) and distributed into 1.1-mL staining tubes. Cells were incubated for 20 min on ice, in the dark, with fluorophore-conjugated antibodies against surface markers (Table S1). For Foxp3-stained cells, following surface marker staining, cells were fixed, permeabilized, and stained using a Foxp3 staining buffer kit (catalog no. 00-5523-00; Thermo Fisher) according to the manufacturer’s instructions with a fluorophore-conjugated antibody against Foxp3. Cells were then washed with FACS-PBS and fixed with 1% paraformaldehyde in PBS. Data were acquired on a BD FACSCanto II equipped with 407-, 488-, and 635-nm lasers and the FACSDiva software. Data were analyzed with FlowJo software.
Western Blots
Liver lysates were prepared by homogenizing liver pieces in 10 vol of radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1.0% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate) supplemented with Complete Protease Inhibitor cocktail (reference #11836153001; Roche) at a frequency of 2.5 Hz, four times, in a benchtop bead mill homogenizer. Samples were cleared by centrifugation at 15,000 × g, and the supernatant was collected. Protein concentrations were determined using the bicinchoninic acid (BCA) protein assay (catalog no. 23225; Pierce) according to the manufacturer’s instructions. Liver lysates (70 μg protein) were diluted in 4× lithium dodecyl sulfate (LDS) buffer (Ref. NP0007; Life Technologies) supplemented with 5% beta-mercaptoethanol to a 20 μL final volume. Samples were denatured by heating to 95°C for 5 min and cooled on ice until gels were loaded. Proteins were resolved by SDS-PAGE on 4%–12% gradient gels (Ref. NP0322BOX, WGF1402BX10; Invitrogen) using 2-(N-morpholino)ethanesulfonic acid, 4-morpholineethanesulfonic acid (MES) running buffer (Ref. NP0002; Life Technologies) and transferred to polyvinylidene fluoride (PVDF) membranes. Blocking was carried out for 1-h rocking at 50 rpm on a shaking platform with a 2:1 ratio of Odyssey Blocking Solution (P/N 927-40000; Li-Cor) to PBS with 0.05% Tween 20 (PBS-T) or 5% dehydrated non-fat milk in PBS-T. Primary antibodies were either diluted in a solution of PBS-T supplemented with 0.1% BSA or 5% milk in PBS-T. Primary antibodies were then detected using goat anti-rabbit 680 nm (RL6111440020.5; Rockland Immunochemical) and goat anti-mouse 800 nm secondary antibodies (RL6111450020.5; Rockland Immunochemical) diluted in PBS-T + 0.1% BSA for 2 h. Fluorescent imaging was performed on an Odyssey Classic Imager (Li-Cor). All western blots were performed in a similar manner, and antibody catalog numbers and dilutions are provided in Table S2.
Indel Quantification by Deep Sequencing
Genomic DNA extracted from mouse livers was amplified using locus-specific primers containing common adaptor sequences, and a second round of PCR amplification was used to add sample barcodes as previously described.26 Amplicons were purified using SPRI magnetic beads, pooled in equimolar amounts, and sequenced using the Illumina MiSeq platform with paired-end 250 kits. Alignment of sequence reads to reference sequences was performed using the Borrows-Wheeler Aligner (BWA) as previously described,36 and indels within a 2-bp window were quantified.
ALT Assay
ALT units were measured using the Teco ALT (SGPT) Kinetic Liquid Kit (A524-150) with slight modifications. Plasma was diluted 1:10, and 10 μL was added to a pre-warmed 96-well plate at 37°C containing the working solution. Signal was read at 340 nm using the following procedure: waiting 1 min, reading 5 flashes at a 1-min read, waiting 1 min, reading five flashes at a 2-min read, and repeating the following seven more times for a total of 10 reads. Units per liter activity was measured using the following formula: (Absorbance)/0.00622 × (total volume)/(sample volume).
qPCR Analysis
DNA was isolated using the QIAGEN Blood and Tissue Kit (69504), and RNA was isolated using the QIAGEN RNeasy Mini Kit (74106). cDNA was generated using the iScript cDNA synthesis kit (170-8891) and 1 μg RNA. Fifty nanograms of DNA and 4 μL of a 1:10 dilution of cDNA were used in each qPCR. All primers are listed in Table S3. Fold change and relative expression of all genes were calculated using the ΔΔCt method.37
Histology
Mouse livers were fixed overnight in 10% formalin, washed in 70% ethanol for 24 h, and dehydrated in 100% ethanol. Paraffin embedding, sectioning, and antibody staining were performed by the Texas Digestive Diseases Morphology Core. Slides were imaged at ×200 magnification on a Nikon Ci-L bright-field microscope.
Statistics
All data are shown as the mean ± standard deviation. Comparisons involving two groups were evaluated by a two-tailed Student’s t test. For comparisons involving three or more groups, a one-way ANOVA was applied, with Tukey’s post-test used to test for significant differences among groups. In all cases, significance was assigned at p < 0.05.
Author Contributions
A.L., C.M.L., G.B., C.B., and W.R.L. conceived the project and designed the studies; A.E.H. produced the viral vectors; M.R.T., A.L., A.E.H., M.D.G., K.E.J., A.M.D., C.B., and W.R.L. performed the in vivo experiments; T.H.D., A.L., and C.M.L. performed analysis of genome editing; A.L., C.B., W.R.L., and G.B. wrote the manuscript, which was revised and approved by all authors.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) (grants HL132840 and U42OD026645 to W.R.L. and grant UG3HL151545 to G.B. and W.R.L.), the Cancer Prevention and Research Institute of Texas (grant RR140081 to G.B.), the American Heart Association (grant 19PRE34380467 to A.M.D. and grant 19POST34430092 to M.D.G.), NIH T32 (grant HL007676 to M.R.T. and grants GM08231 and HL007676 to K.E.J.). This work was also supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NIH (grants CA125123 and RR024574) and the Cancer Prevention and Research Institute of Texas (grant RP180672).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.017.
Contributor Information
Gang Bao, Email: gang.bao@rice.edu.
Christine Beeton, Email: beeton@bcm.edu.
William R. Lagor, Email: william.lagor@bcm.edu.
Supplemental Information
References
- 1.Tabebordbar M., Zhu K., Cheng J.K.W., Chew W.L., Widrick J.J., Yan W.X., Maesner C., Wu E.Y., Xiao R., Ran F.A. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351:407–411. doi: 10.1126/science.aad5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Long C., Amoasii L., Mireault A.A., McAnally J.R., Li H., Sanchez-Ortiz E., Bhattacharyya S., Shelton J.M., Bassel-Duby R., Olson E.N. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351:400–403. doi: 10.1126/science.aad5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson C.E., Hakim C.H., Ousterout D.G., Thakore P.I., Moreb E.A., Castellanos Rivera R.M., Madhavan S., Pan X., Ran F.A., Yan W.X. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351:403–407. doi: 10.1126/science.aad5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ran F.A., Cong L., Yan W.X., Scott D.A., Gootenberg J.S., Kriz A.J., Zetsche B., Shalem O., Wu X., Makarova K.S. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520:186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Pérez J.A., Sontheimer E.J. A Compact, High-Accuracy Cas9 with a Dinucleotide PAM for In Vivo Genome Editing. Mol. Cell. 2019;73:714–726.e4. doi: 10.1016/j.molcel.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang Y., Wang L., Bell P., McMenamin D., He Z., White J., Yu H., Xu C., Morizono H., Musunuru K. A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice. Nat. Biotechnol. 2016;34:334–338. doi: 10.1038/nbt.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chew W.L., Tabebordbar M., Cheng J.K.W., Mali P., Wu E.Y., Ng A.H.M., Zhu K., Wagers A.J., Church G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods. 2016;13:868–874. doi: 10.1038/nmeth.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moreno A.M., Palmer N., Alemán F., Chen G., Pla A., Jiang N., Leong Chew W., Law M., Mali P. Immune-orthogonal orthologues of AAV capsids and of Cas9 circumvent the immune response to the administration of gene therapy. Nat. Biomed. Eng. 2019;3:806–816. doi: 10.1038/s41551-019-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2019;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 12.Black C.A. Delayed type hypersensitivity: current theories with an historic perspective. Dermatol. Online J. 1999;5:7. [PubMed] [Google Scholar]
- 13.Matheu M.P., Beeton C., Garcia A., Chi V., Rangaraju S., Safrina O., Monaghan K., Uemura M.I., Li D., Pal S. Imaging of effector memory T cells during a delayed-type hypersensitivity reaction and suppression by Kv1.3 channel block. Immunity. 2008;29:602–614. doi: 10.1016/j.immuni.2008.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarrett K.E., Lee C.M., Yeh Y.H., Hsu R.H., Gupta R., Zhang M., Rodriguez P.J., Lee C.S., Gillard B.K., Bissig K.D. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jarrett K.E., Lee C., De Giorgi M., Hurley A., Gillard B.K., Doerfler A.M., Li A., Pownall H.J., Bao G., Lagor W.R. Somatic editing of Ldlr with adeno-associated viral-CRISPR is an efficient tool for atherosclerosis research. Arterioscler. Thromb. Vasc. Biol. 2018;38:1997–2006. doi: 10.1161/ATVBAHA.118.311221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simhadri V.L., McGill J., McMahon S., Wang J., Jiang H., Sauna Z.E. Prevalence of Pre-existing Antibodies to CRISPR-Associated Nuclease Cas9 in the USA Population. Mol. Ther. Methods Clin. Dev. 2018;10:105–112. doi: 10.1016/j.omtm.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferdosi S.R., Ewaisha R., Moghadam F., Krishna S., Park J.G., Ebrahimkhani M.R., Kiani S., Anderson K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019;10:1842. doi: 10.1038/s41467-019-09693-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Louis Jeune V., Joergensen J.A., Hajjar R.J., Weber T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods. 2013;24:59–67. doi: 10.1089/hgtb.2012.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thakore P.I., Kwon J.B., Nelson C.E., Rouse D.C., Gemberling M.P., Oliver M.L., Gersbach C.A. RNA-guided transcriptional silencing in vivo with S. aureus CRISPR-Cas9 repressors. Nat. Commun. 2018;9:1674. doi: 10.1038/s41467-018-04048-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar S.R.P., Hoffman B.E., Terhorst C., de Jong Y.P., Herzog R.W. The Balance between CD8+ T Cell-Mediated Clearance of AAV-Encoded Antigen in the Liver and Tolerance Is Dependent on the Vector Dose. Mol. Ther. 2017;25:880–891. doi: 10.1016/j.ymthe.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E.J., Ragni M.V., Manno C.S., Sommer J., Jiang H. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 22.McIntosh J., Lenting P.J., Rosales C., Lee D., Rabbanian S., Raj D., Patel N., Tuddenham E.G., Christophe O.D., McVey J.H. Therapeutic levels of FVIII following a single peripheral vein administration of rAAV vector encoding a novel human factor VIII variant. Blood. 2013;121:3335–3344. doi: 10.1182/blood-2012-10-462200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderlugt C.L., Miller S.D. Epitope spreading in immune-mediated diseases: implications for immunotherapy. Nat. Rev. Immunol. 2002;2:85–95. doi: 10.1038/nri724. [DOI] [PubMed] [Google Scholar]
- 24.Getts D.R., Chastain E.M.L., Terry R.L., Miller S.D. Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 2013;255:197–209. doi: 10.1111/imr.12091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schmidt S.T., Yu F.B., Blainey P.C., May A.P., Quake S.R. Nucleic acid cleavage with a hyperthermophilic Cas9 from an uncultured Ignavibacterium. Proc. Natl. Acad. Sci. USA. 2019;116:23100–23105. doi: 10.1073/pnas.1904273116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li A., Lee C.M., Hurley A.E., Jarrett K.E., De Giorgi M., Lu W., Balderrama K.S., Doerfler A.M., Deshmukh H., Ray A. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2018;12:111–122. doi: 10.1016/j.omtm.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Domenger C., Grimm D. Next-generation AAV vectors-do not judge a virus (only) by its cover. Hum. Mol. Genet. 2019;28(R1):R3–R14. doi: 10.1093/hmg/ddz148. [DOI] [PubMed] [Google Scholar]
- 28.Li F., Hung S.S.C., Mohd Khalid M.K.N., Wang J.-H., Chrysostomou V., Wong V.H.Y., Singh V., Wing K., Tu L., Bender J.A. Utility of self-destructing CRISPR/Cas constructs for targeted gene editing in the retina. Hum. Gene Ther. 2019;30:1349–1360. doi: 10.1089/hum.2019.021. [DOI] [PubMed] [Google Scholar]
- 29.Finn J.D., Smith A.R., Patel M.C., Shaw L., Youniss M.R., van Heteren J., Dirstine T., Ciullo C., Lescarbeau R., Seitzer J. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018;22:2227–2235. doi: 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- 30.Yin H., Song C.Q., Dorkin J.R., Zhu L.J., Li Y., Wu Q., Park A., Yang J., Suresh S., Bizhanova A. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol. 2016;34:328–333. doi: 10.1038/nbt.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin H., Song C.Q., Suresh S., Wu Q., Walsh S., Rhym L.H., Mintzer E., Bolukbasi M.F., Zhu L.J., Kauffman K. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao X., Li J., Samulski R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lagor W.R., Johnston J.C., Lock M., Vandenberghe L.H., Rader D.J. Adeno-associated viruses as liver-directed gene delivery vehicles: focus on lipoprotein metabolism. Methods Mol. Biol. 2013;1027:273–307. doi: 10.1007/978-1-60327-369-5_13. [DOI] [PubMed] [Google Scholar]
- 34.Beeton C., Chandy K.G. Induction and monitoring of adoptive delayed-type hypersensitivity in rats. J. Vis. Exp. 2007;8:e325. doi: 10.3791/325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yourik P., Fuchs R.T., Mabuchi M., Curcuru J.L., Robb G.B. Staphylococcus aureus Cas9 is a multiple-turnover enzyme. RNA. 2019;25:35–44. doi: 10.1261/rna.067355.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin Y., Cradick T.J., Brown M.T., Deshmukh H., Ranjan P., Sarode N., Wile B.M., Vertino P.M., Stewart F.J., Bao G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014;42:7473–7485. doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.