

Abstract
TFEB (transcription factor EB) and TFE3 (transcription factor E3) are “master regulators” of autophagy and lysosomal biogenesis. The stress response p38 mitogen-activated protein (MAP) kinases affect multiple intracellular responses including inflammation, cell growth, differentiation, cell death, senescence, tumorigenesis, and autophagy. Small molecule p38 MAP kinase inhibitors such as SB202190 are widely used in dissection of related signal transduction mechanisms including redox biology and autophagy. Here, we initially aimed to investigate the links between p38 MAP kinase and TFEB/TFE3-mediated autophagy and lysosomal biogenesis. Unexpectedly, we found that only SB202190, rather than several other p38 inhibitors, promotes TFEB and TFE3 to translocate from the cytosol into the nucleus and subsequently enhances autophagy and lysosomal biogenesis. In addition, siRNA-mediated Tfeb and Tfe3 knockdown effectively attenuated SB202190-induced gene expression and lysosomal biogenesis. Mechanistical studies showed that TFEB and TFE3 activation in response to SB202190 is dependent on PPP3/calcineurin rather than on the inhibition of p38 or MTOR signaling, the main pathway for regulating TFEB and TFE3 activation. Importantly, SB202190 increased intracellular calcium levels, and calcium chelator BAPTAP-AM blocked SB202190-induced TFEB and TFE3 activation as well as autophagy and lysosomal biogenesis. Moreover, endoplasmic reticulum (ER) calcium is required for TFEB and TFE3 activation in response to SB202190. In summary, we identified a previously uncharacterized role of SB202190 in activating TFEB- and TFE3-dependent autophagy and lysosomal biogenesis via ER calcium release and subsequent calcium-dependent PPP3/calcineurin activation, leading to dephosphorylation of TFEB and TFE3. Given the importance of p38 MAP kinase invarious conditions including oxidative stress, the findings collectively indicate that SB202190 should not be used as a specific inhibitor for elucidating the p38 MAP kinase biological functions due to its potential effect on activating autophagy-lysosomal axis.
Keywords: p38 inhibitor, SB202190, SB203580, TFEB, TFE3, Autophagy and lysosomal biogenesis
Graphical abstract
Highlights
-
•
A stress response p38 MAP kinase inhibitor SB202190 promoted TFEB/TFE3-dependent autophagy and lysosomal biogenesis.
-
•
SB202190-induced TFEB/TFE3 activation is independent of p38 MAP kinase inhibition.
-
•
SB202190-induced TFEB/TFE3 activation is independent of mTOR inhibition.
-
•
ER calcium-induced PPP3/calcineurin is required for TFEB/TFE3 activation in response to SB202190.
Abbreviations
- MAPK14/p38 α
Mitogen-Activated Protein Kinase 14
- MAPK11/p38 β
Mitogen-Activated Protein Kinase 11
- Baf A1
bafilomycin A1
- CLEAR
coordinated lysosomal expression and regulation
- CQ
chloroquine
- CTSD
cathepsin D
- CTSB
cathepsin B
- ER
endoplasmic reticulum
- EIF4EBP1/4EBP1
Eukaryotic translation initiation factor 4E-binding protein 1
- GPN
glycyl-l-phenylalanine-β- naphthylamide
- LAMP1
lysosomal-associated membrane protein 1
- MAP1LC3B/LC3B
microtubule-associated protein 1 light chain 3B
- MTORC1
mechanistic target of rapamycin (serine/threonine kinase) complex 1
- RPS6KB1/p70S6K
ribosomal protein S6 kinase
- SQSTM1/p62
sequestosome 1
- tfLC3
tandem fluorescent LC3
- TFE3
transcription factor E3
- TFEB
transcription factor EB
- TG
thapsigargin
1. Introduction
Macroautophagy (henceforth autophagy) facilities the degradation of long-lived proteins, protein aggregates, cellular organelles (e.g., mitochondria), and infectious agents via lysosomes [[1], [2], [3], [4], [5]]. Autophagy lysosomal axis dysregulation has been linked to a variety of diseases such as cancers, neurodegenerative diseases, and inflammatory disorders [6,7]. TFEB (transcription factor EB), TFE3 (transcription factor E3) and MITF (melanogenesis-associated transcription factor) are members of the microphthalmia-transcription factor E (MiT/TFE) subfamily [[8], [9], [10]]. Among them, TFEB and TFE3 play crucial roles in controlling autophagy and lysosomal biogenesis [[8], [9], [10], [11], [12]]. In normal states, TFEB and TFE3 are phosphorylated and predominately retained in the cytoplasm bound to 14-3-3 proteins [10,11,13]. Under metabolic stress conditions, TFEB and TFE3 translocate from the cytoplasm into the nucleus [10,11,13,14]. In the nucleus, TFEB recognizes and binds to promoter regions containing the CLEAR (coordinated lysosomal expression and regulation) sequence to activate the expression of multiple autophagy- and lysosomal-related genes [[9], [10], [11],13,15]. A similar mode of action has been ascribed to TFE3. In the nucleus, TFE3 binds to the E-box sequence motif CANNTG, which partially overlaps with the CLEAR sequence to regulate the expression of multiple autophagy- and lysosomal-related genes [10,12]. As a result, activated TFEB and TFE3 not only promote autophagy but also enhance lysosomal biogenesis.
The phosphorylation status of TFEB and TFE3 is critical for their cytosol and nucleus localization. MTORC1 (mechanistic target of rapamycin (serine/threonine kinase) complex 1) is the main kinase to phosphorylate TFEB at Ser142 and Ser211 [11,14,16,17], to phosphorylate TFE3 at Ser321 [10], and to promote the interaction of TFEB and TFE3 with 14-3-3 proteins and their cytosol retention. Inactivation of MTORC1 by starvation or Torin 1 leads to dephosphorylation of TFEB and TFE3, and dissociation of the TFEB (TFE3)/14-3-3 complex; free TFEB and TFE3 then translocate into the nucleus [12,16,17]. Additionally, PPP3/calcineurin was recently identified as a phosphatase to dephosphorylate TFEB and TFE3, which promotes TFEB and TFE3-mediated autophagy and lysosomal biogenesis [18,19]. For instance, lysosomal calcium channel TRPML1 (also known as ML1 or mucolipin-1)-mediated PPP3/calcineurin activation contributes to starvation-induced TFEB nucleus translocation [18,20]. PPP3/calcineurin activation rather than MTOR inhibition is involved in ER stress-induced TFE3 nucleus translocation [19].
The p38 mitogen-activated protein kinases (MAPK) are a class of evolutionarily conserved serine/threonine protein kinases that link extracellular signals to the intracellular machinery to regulate a variety of cellular processes including redox homeostasis [[21], [22], [23]]. p38α (also known as MAPK14 or SAPK2a), p38β (MAPK11, SAPK2b), p38γ (MAPK12, SAPK3, ERK6), and p38δ (MAPK13, SAPK4) are four p38 kinase family members. Among them, p38α and p38β are ubiquitously expressed, whereas p38γ and p38δ are expressed in a tissue-specific manner [21,22]. The p38 is regarded as a stress-activated protein kinase because it is frequently activated in response to a wide range of environmental stresses (e.g. redox stress, ultraviolet irradiation, cytokines, heat shock and osmotic shock) to induce inflammation response [21,22,24], which is a key process in the host defense system. Excessive inflammation contributes to the pathogenesis of multiple human diseases, making the p38 pathway inhibitors potential drugs for inflammation-related diseases [21,22]. Additionally, p38 also plays crucial roles in the regulation of the cell cycle, promotion of cell apoptosis, and induction of cell death, differentiation, senescence, and autophagy [21,22,25,26]. Thus, targeting p38 for the development of novel therapeutics against multiple chronic and acute pathologies is being tested.
Given the importance of the p38 MAP kinase pathway, small molecule p38 protein kinase inhibitors are commonly used as tools for dissecting p38-related signal transduction mechanisms, in both physiological and pathological conditions, and are being developed for the treatment of cancers and inflammatory diseases [22,27,28]. Among these inhibitors, SB202190 [4-(4-fluorophenyl)-2-(4-hydroxyphenyl)-5-(4-pyridyl)1H-imidazole] is one of the most widely used inhibitors of the p38 pathway [29,30]. SB202190 acts as an inhibitor of p38α and p38β via competition with ATP for the same binding site on p38 [31,32]. A single residue difference between p38 and other MAPKs, such as Jun N-terminal kinase and ERK, determines its specificity as evidenced by the crystal structure [29]. Due to its specificity, SB202190 is widely used to elucidate p38-related signal transduction mechanisms including oxidative stress conditions. However, the off-target effects of SB202190 also have been reported including inhibition of CK1d, GAK, GSK3, RIP2 and inhibition of TGFβ receptors and Raf [33,34]. Importantly, recent studies indicate that p38 MAPK pathway may be linked to autophagy [26,[35], [36], [37], [38], [39]]. For instance, a study reported that SB202190 increased both LC3B-II and SQSTM1/p62 levels, and concluded that SB202190 induced a defective autophagy [36,40]. Additionally, SB202190 was reported to activate the AMPK-FoxO3A-dependent autophagy pathway [37].
Because recent studies suggest that the p38 MAP kinase pathway plays important roles in regulating autophagy [25,26,35], and because TFEB and TFE3 are master regulators of autophagy and lysosomal biogenesis, we initially aimed to investigate whether p38 MAPK modulates TFEB and TFE3 pathways by using several p38 MAPK inhibitors. However, we found that only SB202190, among several p38 inhibitors, activated TFEB- and TFE3-dependent autophagy and lysosomal biogenesis. Given the wide use of SB202190 (especially in redox biology study) as well as the importance and multiple functions of the pathway, in this study, we characterized the role and underlying mechanisms of SB202190 in activating TFEB/TFE3-mediated autophagy and lysosomal biogenesis. Our results revealed a novel and unexpected role of SB202190, the stress response p38 MAP kinase inhibitor, in activating lysosomal biogenesis and autophagy induction. The findings from this study call for caution in using and interpreting results when utilizing SB202190 because it can promote autophagy and lysosomal biogenesis apart from its well-characterized ability to inhibit p38.
2. Materials and methods
2.1. Reagents and antibodies
The p38 MAP kinase inhibitors SB202190 (S1077), SB203580 (S1076), BIRB-796 (S1574), SB239063 (S7741) were ordered from Selleckchem. Chloroquine (C6628) was purchased from Sigma-Aldrich. Torin 1 (2273–5) was purchased from BioVision Inc. FK-506 (sc-24649A), Cyclosporin A (sc-3503), Bafilomycin A1 (sc-201550), Anti-β-actin/ACTB (sc-47778) were purchased from Santa Cruz Biotechnology. Tfeb siRNA (L-009798-00-0005), and non-target siRNA were purchased from Dharmacon. LysoTracker® Red DND-99 (L-7528), DMEM (11965–126), FBS (10270–106), Opti-MEM I (31985–070) were purchased from Life Technologies. Flag MAPK14/Flag p38 alpha (agf) was a gift from Roger Davis (Addgene plasmid #20352). Anti-Flag M2 (F1804) and anti-SQSTM1/p62 (P0067) antibodies, TFE3 siRNA (EHU157921), MAPK14 (p38 MAPK alpha) siRNA (EHU081441) were purchased from Sigma-Aldrich. Anti-phospho-P70S6K/RPS6KB1 (Thr389) (9234), anti-P70S6K/RPS6KB1 (9202), anti-phospho- EIF4EBP1/4EBP1 (Thr37/46) (2855), anti-4E-BP1 (53H11) (9644), anti-phospho-p38 MAPK (Thr180/Tyr182) (9211), anti-p38 MAPK (D13E1) (8690) and anti-H3F3A/histone H3 (D1H2) (4499) antibodies were purchased from Cell Signaling Technology. Anti-LAMP1 (ab24170) antibody was purchased from Abcam. HRP-conjugated goat anti-mouse (115-035-003) and goat anti-rabbit (111-035-003) secondary antibodies were purchased from Jackson ImmunoResearch. Anti-LC3B (NB100-2220) antibodies were purchased from Novus Biologicals. Anti-TFE3 (HPA023881) was purchased from Sigma-Aldrich. Anti-TFEB (A303-673A) was purchased from Bethyl Laboratories, Inc. Anti-GAPDH antibody (GTX100118) was obtained from GeneTex. Alexa Fluor®488 goat anti-mouse IgG (A-11001), Alexa Fluor®488 goat anti-Rabbit IgG (A-11034), Alexa Fluor 488 (A-11008) and Alexa Fluor®594 goat anti-rabbit IgG (A-11012) were purchased from Life Technologies.
2.2. Cell culture
HeLa cells, HEK293 cells, HCT116 cells, DLD-1 cells and PC12 cells were cultured in DMEM supplemented with 10% FBS. HeLa cells stably expressing 3x-Flag-TFEB [8,16,41] (CF-7) were maintained in DMEM supplemented with 10% FBS and 200 μg/ml G418. All cells were maintained with cell culture medium containing 50 U of penicillin-streptomycin mixture (Invitrogen) at 37 °C, gassed with 5% CO2.
2.3. Cell transfection
For knockdown experiments, HeLa cells were transfected with indicated siRNA using Lipofectamine RNAiMAX (13778030, Invitrogen); for overexpression, cells were transfected by using Lipofectamine 3000 (L3000015) according to the manufacturer's instructions. After transfection of cells with siRNAs or plasmids for 24–72 h, cells were treated with indicated drugs.
2.4. Western blotting analysis and immunoprecipitation
Cells were washed twice with ice-cold phosphate-buffered saline (PBS) [137 mM NaCl, 10 mM Na2HPO4, 2 mM KH2PO4, 2.7 mM KCl (pH 7.4)] and then lysed on ice in 1XRIPA Lysis Buffer (9803, Life Technologies) with complete protease inhibitor mixture (04693124001, Roche Applied Science) and phosphatase inhibitor (B15001, Biotool). Anti-Flag antibody was added to the whole cell lysates and Dynabeads® Protein G (Life Technologies, 10003D) was used for immunoprecipitation. Protein concentrations were determined by Bio-Rad Bradford assays using bovine serum albumin (BSA) as standard. Equal volumes of Laemmli 2 × buffer [4% SDS, 10% β-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue, 0.125 M tris-HCl (pH 6.8)] and protein lysate were mixed and denatured in boiling water for 10 min. Proteins were separated by 10–15% SDS-PAGE, transferred, and blotted with the antibodies described. The blots were then incubated with primary and secondary antibodies. The protein signals were detected by the ECL kit (32106, Pierce) and captured by X-ray film (Fujifilm). Western blot results were quantified by using ImageJ software.
2.5. Isolation of the cytosol and the nucleus fractions
Cytosol and nucleus extracts were prepared according to a previous protocol [42]. In brief, at the end of drug treatment, the cells were washed with ice-cold PBS, centrifuged, and re-suspended in cold lysis buffer containing 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), pH 8.0, 1 mM ethylenediaminetetraacetic acid (EDTA), 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM PMSF, 0.5 mg/ml benzamidine, 0.1 mg/ml leupeptin, and 1.2 mg/ml aprotinin. The cells were allowed to swell on ice for 15 min. NP-40 (10% (v/v)) was subsequently added to the cell suspensions. The samples were vortexed vigorously for 10 s. The homogenates were centrifuged for 50 s at 16,000×g, and the supernatant was used as cytosolic extract. The nuclear pellet was re-suspended in cold extraction buffer containing 20 mM HEPES, pH 8.0, 1 mM EDTA, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM PMSF, 0.5 mg/ml benzamidine, 0.1 mg/ml leupeptin, 1.2 mg/ml aprotinin, and 20% glycerol. All the protein fractions were stored at −30 °C until use.
2.6. Single-cell Ca2+ imaging
Single-cell Ca2+ imaging was used to measure cytoplasmic Ca2+ concentration according to our previous protocol [43]. Briefly, CF-7 cells were plated and cultured on coverslips to 80% confluency. Cells were then loaded with 2 μM Fura-2 AM in Hepes-buffered Hanks’ balanced salt solution (Hepes-HBSS) with 1% BSA for 30 min at room temperature. Fura-2 AM-loaded cells were mounted in an open bath imaging chamber (RC-21B; Warner Instruments) and visualized with a Nikon Eclipse Ti2 microscope (Nikon) using a 20X objective (Nikon CFI S Fluor Objective). Cells were alternately excited at 340 and 380 nm, and fluorescence emitted at 510 nm was captured by ORCA-Flash 4.0 LT PLUS Digital CMOS camera (Hamamatsu Photonics) every 5s controlled by Micro-manager [44]. Ratios of emitted fluorescence were determined pixel-to-pixel using ImageJ (NIH). During the Ca2+ imaging, ER or lysosomal Ca2+ stores were depleted by perfusing 1 μM TG (Thapsigargin) or GPN (glycyl-l-phenylalanine-β-naphthylamide, 400 μM) in Ca2+-free Hepes-HBSS, respectively and followed by re-addition of indicated concentration of SB202190. All the experiments were repeated 3 times, with at least 50 cells analyzed in each experiment as described by our previous report [43].
2.7. Determination of lysosomal contents using LysoTracker
The lysosomal contents were estimated using LysoTracker Red DND99 (L7528, Thermo Fisher Scientific) following manufacturer's instructions. Briefly, during last 1 h of drug treatment, cells were loaded with 75 nM LysoTracker Red DND99. After removing excess dye, cells were harvested and the fluorescence intensity was observed under a flow cytometry assay (BD Accuri C6 Plus Flow Cytometer). FlowJo was used to analyse the data.
2.8. Immunocytochemistry
Cells were seeded on coverslips placed in 24-well plates. At the end of drug treatment, slides were fixed with 3.7% paraformaldehyde, permeabilized in 0.2% Triton X-100, and blocked with 3% BSA. After blocking, the slides were stained with anti-Flag (1:600) or anti-TFE3 (1:600) antibodies overnight at 4 °C. Alexa Fluor®594 (red) or Alexa Fluor®488 (green) secondary antibodies (1:1000) were added for 1 h at room temperature. After nuclear staining with DAPI, the slides were mounted with FluorSave reagent (345789, Calbiochem). Cells were visualized using the API DeltaVision Personal Imaging System.
The TFEB and TFE3 nuclear translocation analysis was done according to a previous protocol with some modification [41,45]. Briefly, images were acquired randomly from at least ten different fields per sample. Quantitation was done from each image for each sample, and around 200 cells per treatment from three independent experiments were performed to generate the graphed values. NIH ImageJ software was used to perform the analysis of TFEB localization on the different images. The ratio value resulting from the average intensity of nuclear TFEB/TFE3 fluorescence was divided by the average of the cytosolic intensity of TFEB/TFE3 fluorescence by using imageJ. The results were normalized using negative (DMEM medium with 10% FBS) control samples in the same plate. The data are represented by the percentage of nuclear translocation at the different concentrations of each compound using Prism software (GraphPad software).
2.9. Quantitative real-time PCR
Total RNA was extracted from cells using the Trizol reagent (15596026, Thermo Fisher Scientific). Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368814). Autophagy- and lysosomal-related geneprimers were used according to our previous study [41] and synthesized by Life Technologies. The oligonucleotide sequences are listed in Table S1. Real-time PCR was carried out with the Fast SYBR Green Master Mix (Life Technologies, 4385612) using the ViiA™ 7 Real-Time PCR System (Life Technologies, Carlsbad, CA, USA). Fold changes were calculated using the ΔΔCT method, and the results were normalized against an internal control (GAPDH).
2.10. Statistical analysis
All data were analyzed by GraphPad Prism (GraphPad Software). Unless specified, data are presented as means ± SEM. Unpaired Student's t-test was used when comparing drug treatment with vehicle group within the same cell type. Multiple comparisons between groups were performed by one-way ANOVA. A probability value of P < 0.05 was considered to be statistically significant.
3. Results
3.1. TFE3 and TFEB translocate from the cytosol to the nucleus in response to SB202190
In the normal state, TFEB and TFE3 are predominantly localized in the cytoplasm. After activation, TFEB and TFE3 translocate into the nucleus and transcriptionally induce the expression of multiple genes responsible for autophagy and lysosomal functions [[8], [9], [10], [11], [12],46]. Initially, we aimed to investigate the links between the p38 MAP kinase and TFEB and TFE3 by using p38 small molecule inhibitors. Surprisingly, immunofluorescence results showed that there was a striking and dose-dependent nuclear accumulation of TFEB in CF-7 cells (HeLa cells stabling expressing 3XFlag-TFEB) [8] after 3-h exposure to p38 MAP kinase inhibitor SB202190 (Fig. 1A–B). To confirm this result, we isolated cytosolic and nuclear fractions and detected TFEB levels by Western blotting (Fig. 1C). Consistent with immunofluorescence results, SB202190 reduced the levels of TFEB in the cytosol and increased TFEB contents in the nucleus. As expected, a well-known TFEB activator Torin 1 [16] robustly promoted TFEB translocation from the cytoplasm into the nucleus (Fig. 1A–C) as reflected by immunostaining and Western blotting assays. Interestingly, endogenous TFE3 also translocated from the cytosol into the nucleus in response to SB202190 in a dose-dependent manner in HeLa cells as evidenced by immunostaining (Fig. 1D–E) and Western blotting assay after subcellular fractionation analysis (Fig. 1F). Overall, these results suggest that SB202190 promotes both TFEB and TFE3 nuclear accumulation. To test whether SB202190-induced TFEB nucleus translocation is specific to HeLa cells, we examined the effects of SB202190 on subcellular distribution of TFEB and TFE3 in HEK 293 cells, a cell line commonly used to determine cellular signaling pathways. Immunostaining results demonstrated that SB202190 effectively promoted the translocation of Flag-TFEB and endogenous TFE3 from the cytosol into the nucleus (Fig. 1G–H). Additionally, SB202190 enhanced the nuclear accumulation of TFEB in multiple cell lines such as human colorectal carcinoma cell line HCT116, DLD-1 cells (Fig. S1), and a well-used neuronal cell line, PC12 (adrenal phaeochromocytoma cells) (Fig. S1). Together, these results indicate that SB202190 activated TFEB in multiple cells rather than in a cell type-specific manner.
Fig. 1.

The p38 MAP kinase inhibitor SB202190 promoted the translocation of TFEB and TFE3 from the cytoplasm into the nucleus. (A) After being treated with SB202190 at indicated concentrations or a positive control Torin 1 (250 nm) for 3 h, HeLa cells stably expressing 3XFlag-TFEB (CF-7) were fixed and stained with the anti-Flag antibody (green) and DAPI (blue). Representative images are shown. (B) Quantification of the number of cells with nuclear TFEB localization. Data are presented as mean ± SEM of 3 replicates in a representative experiment. At least 200 cells were analyzed in each treatment group. (C) The expression of Flag-TFEB in the cytosol (Cyt.) and the nucleus (Nuc.) were detected by Western blotting after being treated with indicated compounds for 3 h. (D) After being treated with SB202190 at indicated concentrations or a positive control Torin 1 (250 nm) for 3 h, cells were fixed and stained with anti-TFE3 antibody (green) and DAPI (blue). Representative images are shown. (E) Quantification of the number of cells with nuclear TFE3 localization. (F) The expression of endogenous TFE3 in the cytosol (Cyt.) and nucleus (Nuc.) were detected by Western blotting after being treated with indicated compounds for 3 h. (G) SB202190 promoted TFEB translocation from the cytoplasm into the nucleus in HEK293 cells. After transiently transfection of cells with Flag-TFEB plasmids for 48 h, followed by treating cells with SB202190 (20 μM) or a positive control Torin 1 (250 nm) for 3 h, cells were fixed and stained with the anti-Flag antibody (green) and DAPI (blue). Representative images are shown. (H) After being treated with SB202190 (10 μM) for 3 h, cells were fixed and stained with the TFE3 antibody (green) and DAPI (blue). Representative images are shown. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.2. SB202190 enhances autophagy and lysosomal biogenesis
TFEB and TFE3 are critical regulators of the autophagy-lysosome system [12]. To investigate whether SB202190 treatment, under conditions that result in TFEB and TFE3 activation, could induce autophagy, we first treated HeLa cells with different concentrations (0–20 μM) of SB202190 and found that there was a significant and dose-dependent increase in the expression of LC3B-II, a widely used marker of autophagy [47] (Fig. 2A). Additionally, to measure whether SB202190 induces autophagy flux, we inhibited autophagosomal degradation by adding the lysosomal inhibitor chloroquine (CQ), or autophagosome-lysosome fusion inhibitor bafilomycin A1 (Baf A1). We observed a significant increase in LC3B-II protein levels after SB202190 treatment (Fig. 2B–C), suggesting that SB202190 enhances autophagy rather than blocks lysosomal degradation [47]. Moreover, both yellow- and red-only puncta (Fig. 2D) increased after treatment of SB202190 in HeLa cells stably expressing mRFP-GFP-LC3 (tfLC3) [48], indicating that SB202190 increases the formation of both autophagosomes and autolysosomes. These results demonstrated that SB202190 promotes autophagy flux.
Fig. 2.

SB202190 enhanced autophagy and lysosomal biogenesis. (A) SB202190 increased LC3B-II levels in a dose-dependent manner. After HeLa cells were treated with indicated concentrations of SB202190 or a positive control Torin 1 (250 nM) for 16 h, the expression of LC3B was detected by Western blotting. (B) HeLa cells were treated with SB202190 (10 μM) or a positive control Torin 1 (250 nM) for 16 h in the presence or absence of the lysosomal inhibitor chloroquine (CQ, 50 μM) (CQ was added into cells at last 3 h for drug treatment), then the expression of LC3B-II was detected and quantified. (C) HeLa cells were treated with SB202190 (10 μM) or a positive control Torin 1 (250 nM) for 16 h in the presence or absence of the v-ATPase inhibitor bafilomycin A1 to inhibit autophagy-lysosome fusion (Baf A1, 50 nM) (Baf A1 was added into cells at last 3 h for drug treatment). The expression of LC3B-II was detected and quantified. (D) After the treatment of HeLa cells stably expressing tf-LC3 plasmids with SB202190 (10 μM) for 16 h, the signal was captured by a fluorescence microscope and representative images are shown. (E) SB202190 increased LAMP1 levels in a dose-dependent manner. After HeLa cells were treated with indicated concentrations of SB202190 or a positive control, Torin 1 (250 nM) for 16 h, the expression of LAMP1 was detected by western blotting and quantified. (F) After HeLa cells were treated with SB202190 (10 μM) or Torin 1 (250 nM) for 16 h, cells were loaded with LysoTracker Red DND-99 (75 nm) for 1 h and the intensity was recorded by flow cytometer. Representative data are shown from three independent experiments. Quantitative data are presented as the mean ± SEM from at least 3 independent experiments. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Because TFEB and TFE3 have been established as critical regulators of lysosomal biogenesis by promoting the expression of multiple lysosomal-related genes, we attempted to test whether SB202190 treatment, under conditions that result in TFEB and TFE3 activation, promotes lysosomal biogenesis. We first measured the effect of SB202190 on the expression of a lysosomal marker, LAMP1 (lysosomal-associated membrane protein 1). As showed in Fig. 2E, SB202190 significantly increased LAMP1 levels in a dose-dependent manner. A well-characterized TFEB and TFE3 activator Torin 1 was used as a positive control (Fig. 2E). Similar, SB202190 also increased the expression of LC3B-II and LAMP1 in a time-dependent manner (Fig. S2). Moreover, we found that SB202190 increased fluorescent intensity compared with vehicle control cells as reflected by flow cytometry assay after staining of cells with LysoTracker Red DND99 (Fig. 2F), suggesting that SB202190 enhances lysosome contents.
3.3. TFEB and TFE3 are necessary for SB202190-induced autophagy and lysosomal biogenesis
Because TFEB and TFE3 transcriptionally promote the expression of multiple autophagy- and lysosomal-related genes, we wondered whether SB202190-induced TFEB and TFE3 nuclear accumulation is accompanied by the enhanced expression of multiple autophagy-lysosomal axis related genes. As showed in Fig. 3A, SB202190 significantly increased the mRNA levels of several autophagy-lysosomal genes such as Map1lc3, Uvrag, Ctsd, Ctsb, Atp6ve01, Atg16l, Mcoln1, Gls, Vps18, and Atp6v1h as reflected by q-PCR assay. Together, these results demonstrate that SB202190 transcriptionally regulates the expression of multiple autophagy- and lysosomal-related genes and enhances autophagy and lysosomal biogenesis.
Fig. 3.

TFEB and TFE3 are necessary for SB202190-induced autophagy and lysosomal biogenesis. (A) SB202190 increased the expression of multiple autophagy-lysosomal pathway related genes. After HeLa cells were treated with SB202190 (10 μM) for 12 h, the expression of several autophagy- and lysosome-related genes were detected by real-time PCR. (B) Quantification data of TFEB and TFE3 levels after transfection of HeLa cells with TFEB and TFE3 siRNAs for 48 h. (C) Knockdown of the expression of Tfeb and Tfe3 attenuated SB202190-induced expression of several autophagy and lysosome-related genes as reflected by real-time PCR assay. (D) Tfeb or/and Tfe3 knockdown attenuated SB202190-induced expression of LAMP1, SQSTM1/p62 and LC3B-II levels in HeLa cells. HeLa cells were treated with SB202190 (10 μM) for 16 h after transfection of cells with indicated SiRNAs for 48 h. The expressions of LAMP1, SQSTM1/p62, MAP1LC3, TFEB, and TFE3 were detected. (E) Quantification data showed that depletion of TFEB/TFE3 partially but significantly attenuated SB202190-induced expression of LC3B-II levels. Quantitative data are presented as the mean ± SEM from at least 3 independent experiments.
To determine whether TFEB and TFE3 are specifically required for SB202190-induced autophagy and lysosomal biogenesis, we knocked down Tfeb and Tfe3 in HeLa cells by transfecting cells with their specific siRNAs (Fig. 3B). We found that the transcription increases of several autophagy- and lysosome-related genes (Map1lc3, Sqstm1, Ctsd, Atp6v1h and Uvrag) in response to SB202190 was significantly impaired in cells depleted of TFE3 and TFEB (Fig. 3C). Interestingly, the increased lysosomal marker LAMP1 and LC3B-II levels after SB202190 treatment were also attenuated in HeLa cells depleted of TFEB or/and TFE3 (Fig. 3D and quantification of LC3B-II levels in Fig. 3E), indicating that TFEB and TFE3 are required for SB202190-induced autophagy and lysosomal biogenesis. Notably, TFEB and TFE3 transcriptionally upregulated the expression of autophagy adaptor SQSTM1/p62 levels [13]; our results demonstrated that both SQSTM1/p62 mRNA and protein levels were significantly increased upon SB202190 treatment, and these upregulations were remarkably impaired in cells depleted of TFE3 and/or TFE3 (Fig. 3D). These results demonstrate that TFEB and TFE3 are required for upregulation of autophagy adaptor protein SQSTM1/p62 levels in response to SB202190. These results exclude the possibility that SB202190 inhibited SQSTM1/p62 degradation. Overall, these results indicate that TFE3/TFEB are necessary for SB202190-induced autophagy and lysosomal biogenesis.
3.4. SB202190-induced TFEB and TFE3 activation and lysosomal biogenesis is independent of p38 MAP kinase inhibition
Because SB202190 is a p38 MAP kinase inhibitor, we next asked whether the p38 MAPK inhibition is sufficient for TFEB and TFE3 activation and subsequently induces autophagy and lysosomal biogenesis. To answer this question, we analyzed TFEB subcellular localization after treating cells with several p38 inhibitors such as SB202190, SB203580, BIRB-796, and SB239063 at the concentration of 10 μM for 3 h. As shown in Fig. 4A–B, only SB202190 promoted TFEB and TFE3 to translocate from the cytosol into the nucleus. Though, BIRB-796 showed much greater potency for inhibition of anisomycin-induced p38 activation (Fig. S3A). These results suggest that SB202190 activated TFEB and TFE3 may be independent of p38 inhibition. Moreover, only SB202190 raised the expression of LC3B-II levels (Fig. 4C). Importantly, depletion of MAPK14/p38 by siRNA did not promote TFEB and TFE3 (Fig. 4E–F) to translocate from the cytosol into the nucleus though p38 siRNA effectively inhibited the phosphorylation of p38 MAP kinase and it's downstream targets such as p-HSP27, p-ATF4 in basal conditions (Figs. S3B–C) or activation by anisomycin (Fig. S3C), further confirming that p38 inhibition is not sufficient for activating TFEB and TFE3. In agreement with these results, SB202190 increased LAMP1 and LC3B-II levels in cells transiently expressing a dominant-negative form of p38 MAPK (Figs. S3D–E), in which the Thr 180 and Tyr 182 sites were replaced with Ala and Phe, respectively [49]. Overall, our results not only indicated that p38 MAP kinase inhibition is insufficient for promoting TFEB and TFEB to translocate from the cytosol into the nucleus, but also demonstrated that SB202190-induced TFEB and TFE3 activation and subsequently increased autophagy and lysosome biogenesis are independent of p38 MAP kinase inhibition.
Fig. 4.

SB202190-induced TFEB and TFE3 nuclear accumulation is independent of p38 MAP kinase inhibition. (A) After CF-7 cells were treated with p38 MAP kinase inhibitors SB202190, BIRB-796, SB203580, and SB239063 at the concentration of 10 μM for 3 h, cellular distribution of TFEB was detected by immunostaining. (B) After HeLa cells were treated with p38 MAP kinase inhibitors SB202190, BIRB796, SB203580, and SB239063 at the concentration of 10 μM for 3 h, cellular distribution of endogenous TFE3 was detected by immunostaining. (C) Western blotting results showed that SB202190, rather than other p38 MAP kinase inhibitors (10 μM for 16 h) robustly increases LC3B-II and SQSTM1/p62 levels in HeLa cells. (D) SB202190 still increased the expression of LAMP1 and LC3B-II levels after knockdown of Mapk14/p38α. (E-F) After HeLa were transfected with non-target of MAPK14/p38α specific siRNA for 48 h, followed by treatment with SB202190 for 3 h, the cellular distribution of TFEB (E) and TFE3 (F) were detected by immunostaining assay.
3.5. PPP3/calcineurin activation rather than MTOR signaling is required for SB202190-induced TFEB/TFE3 activation as well as autophagy and lysosomal biogenesis
Because phosphorylation status is critical in regulating the subcellular localization of TFEB and TFE3 [14], we tested whether SB202190 promoted the nuclear accumulation of TFEB and TFE3 is accompanied by their dephosphorylation. As shown in Fig. 5A, Fig. 5B, and Fig. S4, there were pronounced gel-shift in response to different concentrations of SB202190 (0–20 μM) or 20 μM of SB202190 for different durations (0–3 h), indicating that TFEB and TFE3 are dephosphorylated [8]. Moreover, SB202190 inhibited the phosphorylation of TFEB at Ser211 in a time-and dose-dependent manner (Figs. S5A–B), and SB202190 attenuated the interaction of TFEB with 14-3-3 [11,17] (Fig. S6), further supporting the role of SB202190 in promoting TFEB translocation from the cytosol into the nucleus.
Fig. 5.

PPP3/calcineurin rather than MTOR signaling pathway is critical for SB202190-induced TFEB and TFE3 activation. (A-B) There were pronounced gel-shift of Flag-TFEB in CF-7 cells (A), or endogenous TFE3 in HeLa cells (B) in response to different concentrations of SB202190 (0–20 μM) or 20 μM of SB202190 for different durations (0–3 h), indicating that SB202190 caused TFEB and TFE3 dephosphorylation. (C-D) SB202190 did not inhibit MTOR signaling pathway. After cells were treated with indicated concentrations of SB202190 for 3 h, (at the time point that SB202190 robustly promotes TFEB/TFE3 to translocate into the nucleus) or 20 μM SB202190 for indicated durations, the expression of p-MTOR, MTOR, p-RPS6KB1/P70S6K, RPS6KB1/P70S6K, p-EIF4EBP1/p-4EBP1 and EIF4EBP1/4EBP1 were detected by Western blotting; Torin 1 (250 nm) was used as a positive control. (E-F) PPP3/calcineurin inhibitors FK506 plus CsA attenuated the translocation of TFEB from the cytoplasm into the nucleus in response to SB202190. After pretreatment of CF-7 cells with FK506 (10 μM) plus CsA (20 μM) for 30 min followed by adding SB202190 into cells for another 3 h, the cytoplasm and the nucleus distribution of TFEB were detected by immunostaining or Western blotting analysis. (G) FK506 plus CsA partially attenuated the gel-shift of Flag-TFEB in response to SB202190. After pretreatment of CF-7 cells with FK506 (10 μM) and CsA (20 μM) for 30 min followed by adding SB202190 (10 μM) into cells for another 3 h, the phosphorylation status of Flag-TFEB was determined. (H–I) FK506 and CsA attenuated the translocation of endogenous TFE3 from the cytoplasm into the nucleus in response to SB202190. After pretreatment of HeLa cells with FK506 (10 μM) plus CsA (20 μM) for 30 min followed by adding SB202190 into cells for another 3 h, the cytoplasm and the nucleus distribution of endogenous TFE3 were detected by immunostaining or Western blotting analysis. (J) FK506 plus CsA partially attenuated the gel-shift of endogenous TFE3 in response to SB202190. After pretreatment of HeLa cells with FK506 (10 μM) and CsA (20 μM) for 30 min followed by adding SB202190 (10 μM) into cells for another 3 h, the phosphorylation status of TFE3 was detected. Quantification of the number of cells with nuclear TFEB/TFE3 localization is presented as mean ± SEM of 3 replicates in a representative experiment. At least 200 cells were analyzed in each treatment group.
It is well-established that MTOR inhibition is important for regulating the translocation of TFEB and TFE3 from the cytosol into the nucleus by phosphorylation of TFEB and TFE3 [11,16,17]. To test whether SB202190-mediated TFEB and TFE3 nuclear accumulation are through inhibition of MTOR activities, we examined the effects of SB202190 on the phosphorylation of RPS6KB1/P70S6K and EIF4EBP1/4EBP1, two well-known downstream targets of the MTOR pathway. We found that SB202190 did not inhibit the phosphorylation of RPS6KB1/P70S6K and EIF4EBP1/4EBP1 exposure to different concentrations (0–20 μM) of SB202190 for 3 h (Fig. 5C), at the time point that SB202190 robustly promotes TFEB and TFE3 nuclear accumulation and dephosphorylation. Moreover, we incubated cells with SB202190 for various times (0–3 h) and observed that SB202190 did not obviously inhibit the phosphorylation of either RPS6KB1/P70S6K or EIF4EBP1/4EBP1 (Fig. 5D), indicating that SB202190-promoted TFEB nuclear translocation is MTOR-independent. In contrast, as expected, a well-characterized MTOR inhibitor and TFEB and TFE3 activator remarkably inhibited the phosphorylation of RPS6KB1/P70S6K and EIF4EBP1/4EBP1.
Because PPP3/calcineurin, an endogenous serine/threonine phosphatase, has been reported to dephosphorylate TFEB and TFE3 [16,17,50], and thereby promote their translocation from cytosol to nucleus, we next asked whether PPP3/calcineurin is required for SB202190-induced TFEB and TFE3 activation. As shown in Fig. 5E, pretreatment of cells with PPP3/calcineurin inhibitor FK506 and CsA (Cyclosporin A) [51] significantly inhibited the nuclear accumulation of TFEB in response to SB202190. This result was further verified by Western blotting assay after isolation of cytosolic and nuclear fractions Fig. 5F. Interestingly, PPP3/calcineurin inhibitors FK506 and CsA also partially inhibited SB202190-induced TFEB gel-shift (Fig. 5G), indicating that PPP3/calcineurin inhibitors inhibit the dephosphorylation of TFEB. Similarly, PPP3/calcineurin inhibitor FK506 plus CsA also attenuated SB202190-induced accumulation of TFE3 in the nucleus as reflected by immunostaining and Western blotting assay Fig. 5H–I. Importantly, PPP3/calcineurin inhibitors also partially attenuated the dephosphorylation of TFE3 exposured to SB202190 (Fig. 5J). Together, these results demonstrate that PPP3/calcineurin rather than MTOR signaling is required for SB202190-induced dephosphorylation and subsequently nuclear accumulation of TFEB and TFE3.
3.6. SB202190 induces lysosomal biogenesis by modulating intracellular Ca2+ levels
Recent studies have demonstrated that intracellular Ca2+ levels modulate autophagy and lysosomal biogenesis via calcineurin-mediated TFEB and TFE3 activation [52] and PPP3/calcineurin is a calcium-dependent enzyme [53]. Therefore, we asked whether SB202190 could increase intracellular Ca2+ levels and subsequently activate TFEB/TFE3-mediated autophagy and lysosomal biogenesis. To answer this question, by using single-cell Ca2+ imaging technology, we found that, in a Ca2+-free buffer, SB202190 elicited a transient and significant increase in intracellular Ca2+ concentration in CF-7 cells in a dose-dependent manner (Fig. 6A). We next assessed whether the SB202190-mediated increase in cytoplasmic Ca2+ levels played a critical role in modulating the accumulation of TFEB and TFE3 in the nucleus. As evidenced by immunostaining results in CF-7 cells and HeLa cells and as shown in Fig. 6B–C and Fig. S7, Ca2+ chelator BAPTA-AM almost completely blocked SB202190-induced translocation of TFEB from the cytoplasm into the nucleus. This phenomenon was further validated by the detection of nuclear TFEB contents by using Western blotting after subcellular fractionation Fig. 6E. Moreover, Ca2+ chelator BAPTA-AM [54] also dramatically attenuated the nuclear accumulation of endogenous TFE3 upon SB202190 treatment as reflected by immunostaining and Western blotting assay Fig. 6D and F. These results indicate that intracellular Ca2+ is required for TFEB and TFE3 activation in response to SB202190.
Fig. 6.

Calcium is required for SB202190-induced TFEB and TFE3 activation as well as autophagy and lysosomal biogenesis. (A) SB202190 increased cytoplasmic Ca2+ levels. Cytoplasmic Ca2+ concentration in CF-7 cells was detected by using Fura-2 AM probe in Ca2+-free HBSS, followed by addition of indicated concentrations of SB202190. The peak value of 340/380 nm excitation ratio for fura-2 was quantified. At least 50 cells were recorded in each experiment and the experiments were repeated for 3 times. Data are presented as mean ± SEM of 3 replicates. (B) Calcium chelator BAPTA-AM reduced the translocation of TFEB from the cytoplasm into the nucleus in response to SB202190. After retreatment of CF-7 cells with BAPTA-AM (20 μM) for 30 min followed by being treated with SB202190 for another 3 h, the nucleus distribution of TFEB was detected by immunostaining. (C) Quantification of the number of cells with nuclear TFEB localization. Data are presented as mean ± SEM of 3 replicates in a representative experiment. At least 200 cells were analyzed in each treatment group. (D) Calcium chelator BAPTA-AM attenuated SB202190-induced accumulation of endogenous TFE3 in the nucleus. (E) Quantification of the number of cells with nuclear TFE3 localization. Data are presented as mean ± SEM of 3 replicates in a representative experiment. (F-G) Western blotting assay showed that calcium chelator BAPTA-AM reduces SB202190-induced translocation of TFEB and TFE3 from the cytoplasm into the nucleus. (H) Calcium chelator BAPTA-AM blocks SB202190-induced increase in lysosome contents as evidenced by Lysotracker Red staining. After pretreatment of HeLa cells with calcium chelator BAPTA-AM (20 μM) for 30 min followed by treatment with SB202190 for another 16 h, the lysosome contents were determined by flow cytometry after cells were loaded with LysoTracker Red DND-99 (75 nm, for 1 h). (I) Pretreatment of HeLa cell with BAPTA-AM reduced SB202190-inducedexpression of LAMP1, LC3B-II, and SQSTM1/p62 levels. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Next, we asked whether autophagy and lysosomal biogenesis are dependent on the SB202190-mediated increase in intracellular Ca2+ levels. To achieve this goal, HeLa cells were treated with SB202190 in the presence or absence of Ca2+ chelator BAPTA-AM. As shown in Fig. 6I, pretreatment of cells with the Ca2+ chelator inhibited the increase of LC3B-II levels in response to SB202190. This result was further confirmed by Western blotting analysis of LAMP1 and SQSTM1/p62 levels upon SB202190 treatment in the presence of BAPTA-AM (Fig. 6I). Consistently, lysotracker red staining results showed that BAPTA-AM almost completely blocked the increase of lysosomal contents in response to SB202190 (Fig. 6H). Together, these results clearly demonstrate that intracellular Ca2+ is required for SB202190-induced TFEB and TFE3 activation as well as for subsequent autophagy and lysosomal biogenesis.
3.7. ER calcium is required for SB202190-induced TFEB/TFE3 activation, autophagy and lysosomal biogenesis
The release of Ca2+ from the endoplasmic reticulum (ER) or lysosomes contributes to the increase of intracellular Ca2+ levels and subsequent activation of TFEB and TFE3 in response to SB202190 [55]. To identify the internal store that contributes to TFEB and TFE3 activation in response to SB202190, we treated CF-7 cells with TG (thapsigargin), a non-competitive inhibitor of the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), to effectively deplete the ER calcium stores. As shown in Fig. 7A, pretreatment of cells with TG effectively blocked SB202190-induced increase of intracellular Ca2+ levels as evidenced by single cell calcium imaging, suggesting that the ER may be a main internal calcium store that contributes to the increase of intracellular calcium in response to SB202190. Interestingly, SB202190 still significantly increased intracellular calcium levels after depletion of lysosomal calcium by GPN (glycyl-l-phenylalanine-β-naphthylamide) (Fig. S8), further confirming that the ER is the main source contributing to the increase of intracellular Ca2+ levels in response to SB202190.
Fig. 7.

ER calcium contributes to SB202190-induced TFEB and TFE3 activation as well as autophagy and lysosomal biogenesis. (A) Depleting ER Ca2+ stores with thapsigargin (TG) reduced SB202190-induced increase of cytoplasmic calcium levels. In contrast, SB202190 still significantly increases cytoplasmic calcium levels after depleting lysosomal Ca2+ stores with GPN (glycyl-l-phenylalanine-β-naphthylamide). The peak value of 340/380 nm excitation ratio for Fura-2 was quantified. At least 50 cells were recorded in each experiment and the experiments were repeated for 3 times. Data are presented as mean ± SEM of 3 replicates. (B) Depleting ER calcium stores with TG prevented the translocation of TFEB from the cytoplasm into the nucleus in response to SB202190. After pretreatment of CF-7 cells with TG (1 μM) for 30 min followed by being treated with SB202190 for another 3 h, the nucleus distribution of TFEB was detected by immunostaining. (C) Quantification of the number of cells with nuclear TFEB localization. Data are presented as mean ± SEM of 3 replicates in a representative experiment. At least 200 cells were analyzed in each treatment group. (D) TG attenuated SB202190-induced accumulation of endogenous TFE3 in the nucleus. (E) Quantification of the number of cells with nuclear TFE3 localization. Data are presented as mean ± SEM of 3 replicates in a representative experiment. (F-G) Effects of SB202190 on the expression of TFEB and endogenous TFE3 in the cytosolic (Cyt.) and nuclear (Nuc.) fractions were determined by Western blotting in the presence or the absence of TG, indicating that TG reduced SB202190-induced translocation of TFEB and TFE3 from the cytoplasm into the nucleus. (H) TG reduces SB202190-induced increase of lysosome contents as evidenced by flow cytometry assay after cells were loaded with LysoTracker Red DND-99 (75 nm, for 1 h). (I) Pretreatment of HeLa cell with TG (1 μM) reduced SB202190-induced expression of LAMP1, LC3B-II, and SQSTM1/p62 levels. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We next analyzed whether depletion of ER calcium by TG could comprise SB202190-mediated TFEB and TFE3 activation as well as autophagy and lysosomal biogenesis. As shown in Fig. 7B–C, pretreatment of cells with TG for 30 min significantly attenuated the nuclear accumulation of TFEB after exposure to SB202190. This result was further confirmed by subcellular fractionation of the cytosol and nucleus, followed by detection of TFEB levels in response to SB202190 in the presence or absence of TG (Fig. 7F). Interestingly, depletion of ER calcium by TG compromised SB202190-induced translocation of TFE3 from the cytosol into the nucleus (Fig. 7D–E, G). These results suggest that ER calcium contributes to the activation of TFEB and TFE3 in response to SB20190. In contrast, under the same conditions, depletion of ER calcium by TG did not block Torin 1-induced nuclear accumulation of TFEB (Fig. S9A) and TFE3 (Fig. S9B). These results further support the notion that SB202190 has different mechanisms for activating TFEB/TFE3, even though PPP3/calcineurin is reported to play a role in regulating Torin 1-mediated TFEB activation [18]. The results further indicate that ER calcium is required for activating TFEB and TFE3 in response to SB202190. We next asked whether depletion of ER calcium by TG could attenuate SB202190-induced autophagy and lysosomal functions. As shown in Fig. 7I, consistently, pretreatment of cells with TG attenuated the increase of LC3B-II levels in response to SB202190. This result was further verified by Western blotting analysis of LAMP1 and SQSTM1/p62 levels, which are upregulated by TFEB/TFE3 activation upon SB202190 treatment (Fig. 7I). Finally, lysotracker red staining results showed that TG almost completely eliminated the increase in lysosomal contents upon SB202190 treatment (Fig. 7G). Interestingly, from the perspective of chemical structures, it can be found that among several p38 inhibitors such as SB202190, SB203580 and BIRB 796 (Fig. S10A), SB202190 will have more chances to destabilize the ER membranes, which may induce the release of calcium and promote ER stress (Fig. S10B). Together, these results clearly demonstrate that ER Ca2+ is required for SB202190-induced TFEB and TFE3 activation and subsequent autophagy and lysosomal biogenesis.
4. Discussion
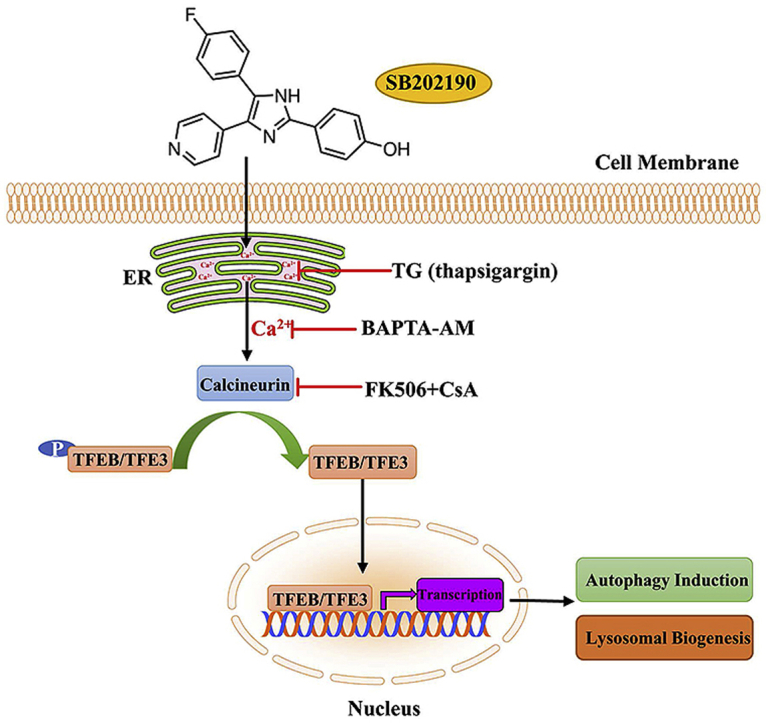
In this study, for the first time, we demonstrated that the p38 MAP kinase specific inhibitor SB202190 has a novel role in activating TFEB and TFE3, thereby promoting autophagy and lysosomal biogenesis. Analysis of the mechanism of this chain of events has revealed that p38 MAP kinase inhibition and the MTOR signaling are not required for the activation of TFEB and TFE3 in response to SB202190. Instead, calcium-dependent serine/threonine protein phosphatase PPP3/calcineurin contributes to SB202190-induced TFEB and TFE3 activation. We also showed that SB202190 causes an increase in intracellular calcium levels and that the calcium chelator BAPTA-AM blocks the activation of TFEB and TFE3 upon SB202190 treatment. Furthermore, we demonstrated that ER calcium rather than lysosomal calcium is required for SB202190-induced activation of TFEB and TFE3 (see Fig. 8). Overall, we found that the release of calcium from the ER and subsequent calcium-dependent PPP3/calcineurin activation, instead of p38 MAP kinase inhibition or MTOR signaling, are required for SB202190-induced TFEB and TFE3 activation, autophagy and lysosomal biogenesis. These results provided novel insights into the role and mechanism of SB202190 in activating autophagy and the lysosomal pathway, apart from its known function as a p38 MAP kinase inhibitor. Given the importance of p38 MAPK in a variety of conditions including in response to oxidative stress, our data suggested the cautionary use of SB202190 to elucidate p38 kinase biological functions due to its potential effect on activating autophagy-lysosomal axis.
Fig. 8.

Schematic illustration of the mechanism of action of SB202190 on activating TFEB/TFE3-mediated autophagy and lysosomal biogenesis. SB202190 causes the release of calcium from ER. Intracellular calcium subsequently activates the phosphatase PPP3/calcineurin, thereby de-phosphorylating and promoting TFEB and TFE3 nuclear accumulation. In the nucleus, TFEB and TFE3 transcriptionally regulate the expression of multiple autophagy- and lysosomal-related genes to enhance autophagy and lysosomal biogenesis. PPP3/calcineurin inhibitors FK506 and CsA, calcium chelator BAPTA-AM, or depleting ER calcium by TG (thapsigargin) all effectively attenuate TFEB and TFE3 activation in response to SB202190. Taken together, SB202190 activates TFEB- and TFE3-dependent autophagy and lysosomal biogenesis via ER calcium release and subsequent calcium-dependent PPP3/calcineurin activation, leading to dephosphorylation and activation of TFEB and TFE3.
The p38 pathways are noted for its activation by a wide range of stresses including cytokines, radiation, osmotic shock, mechanical injury, heat stress, and oxidative damage, and p38 dysregulation is implicated in a variety of diseases, and p38 inhibitors are widely used as a research tool and drugs targeting of p38 for multiple diseases are being tested [22,27,28]. Though SB202190 is regarded as a p38 MAP kinase “specific” inhibitor, it has several other p38 MAP kinase inhibition-independent functions such as activating the JNK pathway, MEK/MAPK pathway [56], AMPK-FoxO3A pathway [37], inhibiting CK1d, GAK, GSK3, RIP2 and inhibiting TGFβ receptors and Raf [33,34], indicating that SB202190 has other targets in addition to p38 MAP kinase. Our finding showed that SB202190 enhances autophagy, which is consistent with previous reports that SB202190 promotes autophagy flux [38,57]. However, here we also demonstrated that SB202190 enhances lysosomal functions. Though p38 MAP kinase is reported to play an important role in regulating autophagy in several different conditions [25,35], p38 MAP kinase inhibition does not affect TFEB- and TFE3-mediated lysosomal biogenesis. In other words, the activation of TFEB and TFE3, and subsequent autophagy and lysosomal biogenesis are SB202190-specific rather than via p38 MAP kinase inhibition. Interestingly, another pyridinyl imidazole inhibitor SB203580 did not effectively activate TFEB/TFE3 at its normal concentrations (e.g. 10 and 20 μM) for effectively inhibition of p38 MAP kinase. However, whether high concentration of SB203580 could also activate TFEB are unknown. It is well established that TFEB and TFE3 transcriptionally regulate multiple autophagy- and lysosomal-related genes including autophagy substrate SQSTM1/p62 [13]. Our results indicated that SB202190 significantly upregulates the expression of several autophagy- and lysosomal-related genes including SQSTM1/p62 at both mRNA and protein levels via TFEB and TFE3 since Tfeb or/and Tfe3 knockdown attenuated SB202190-induced expression of several autophagy and lysosomal pathway related genes including SQSTM1/p62.
TFEB protein post-translational modification plays a key role in regulating TFEB's activities and functions [14,58,59]. MTORC1 is a main kinase for controlling TFEB and TFE3 activation by regulating their phosphorylation [14,16,17,46]. Interestingly, SB202190-induced TFEB and TFE3 activation is independent of inhibiting MTORC1 activity, judged by the phosphorylation status of its well-characterized downstream targets RPS6KB1/P70S6K and EIF4EBP1/4EBP1. As such, TFEB and TFE3 activation by SB202190 appears to be mechanistically distinct from the response to starvation or MTORC1 inhibitors. We further showed that Ser 211 of TFEB is important for its nuclear accumulation. Although SB202190 causes TFE3 dephosphorylation as reflected by gel-shift, the dephosphorylation site (s) are unknown for lack of commercially available p-TFE3 antibodies in the current stage. Nevertheless, future studies are required for determination of specific dephosphorylation site for SB202190-induced TFE3 activation.
PPP3/calcineurin is a calcium-dependent serine/threonine phosphatase that dephosphorylates and activates TFEB/TFE3 [18,20]. Interestingly, PPP3/calcineurin activities are required for SB202190-induced TFEB/TFE3 activation because PPP3/calcineurin specific inhibitors attenuated the nuclear accumulation and dephosphorylation of TFEB/TFEB in response to SB202190. However, PPP3/calcineurin only partially inhibited SB202190-induced TFEB and TFE3 activation, indicating that there might be uncharacterized phosphatase/kinase(s) responsible for TFEB and TFE3 dephosphorylation and activation in response to SB202190. Future studies are required to identify the potential phosphatase/kinase(s). We further showed that SB202190 increases intracellular calcium levels, and chelator calcium with BAPTA-AM compromised SB202190-induced TFEB/TFE3 dephosphorylation, activation, autophagy induction, and lysosomal biogenesis, suggesting that SB202190 activates PPP3/calcineurin in a calcium-dependent manner. Interestingly, previous study indicated that lysosomal calcium channel TRPML1-mediated lysosomal calcium release and subsequent PPP3/calcineurin activation are crucial for TFEB activation in starvation conditions [18,20]. Here, we found that SB202190 seems to promote ER calcium release rather than lysosomal calcium release, which accounted for PPP3/calcineurin-mediated TFEB and TFE3 activation because depletion of ER calcium, rather than lysosomal calcium attenuated SB202190-induced increase of intracellular calcium levels and attenuated TFEB and TFE3 activation. In contrast, depletion of ER calcium did not block Torin 1-induced TFEB and TFE3 activation, indicating that SB202190 is different from Torin 1 in activating TFEB and TFE3. These results highlight the importance of ER calcium in TFEB and TFE3 activation in response to SB202190. Indeed, a recent study indicated that several calcium sources such as lysosomes, the ER, and the mitochondria can contribute to TFEB activation [55]. Nevertheless, it remains unclear how SB202190 promotes the release of calcium from ER; future studies are required to determine which channel such as inositol 1,4,5-trisphosphate receptor (InsP3R), ryanodine receptor (RyR), or other calcium channels [60] in the ER is activated in response to SB202190. Additionally, the specificity of SB202190, compared with other p38 MAP kinase inhibitors, in modulating intracellular Ca2+ levels is still unclear. Nevertheless, if one compares the chemical structures of p38 MAPK inhibitors SB202190, SB203580, and BIRB 796, it can be found that SB202190 may easily destabilize the ER membranes and subsequently promotes the release of ER calcium (Fig. S10A). Moreover, among several p38 inhibitors, only SB202190 increased ER stress (Fig. S10B), which probably also via destabilization of the ER membranes. However, though calcium chelator BAPTA-AM effectively inhibited SB202190-induced TFEB/TFE3 activation and subsequent lysosomal biogenesis and autophagy, BAPTA-AM did not block SB202190-induced ER stress (data not shown). Additionally, calcium image results showed that SB202190 almost immediately induced calcium release, and SB202190-induced ER stress at around 3–6 h (data not shown), suggesting that SB202190-induced calcium release may be independent of its effects on activating ER stress. These results excluded the possibility that SB202190-induced ER stress contributes to calcium release and subsequent TFEB/TEF3 activation. Moreover, time point experiments showed that SB202190-induced TFEB/TFE3 activation was earlier than its effect on the induction of ER stress. Overall, these data indicated that SB202190-induced ER stress may not contribute to its effects on activation of TFEB/TFE3-mediated lysosomal biogenesis and autophagy. Nevertheless, future studies are required to further characterize the interplay between the effects of SB202190-induced ER stress, calcium release, and autophagy induction.
Importantly, Ca2+ is a second messenger that is involved in multiple cellular activities, including redox biology, protein synthesis, cell metabolism, cell cycle progression, cell apoptosis, and gene expression [52,53,60]. Interestingly, calcium also can lead to the production of high amounts of reactive oxygen species (ROS), and ROS can further target ER-based calcium channels leading to increased release of calcium and further increased ROS levels [61]. Future studies are required to determine the interplay between SB202190-induced calcium and ROS production. Because SB20190 increased intracellular Ca2+ levels, it is reasonable to postulate that SB202190 may have other biological functions in the downstream of calcium pathways including ROS biology. Indeed, we found that SB202190 also induced ER stress as reflected by the upregulated expression of a well-known ER stress marker ATF4. Future studies are greatly needed to answer these questions.
Notably, Ca2+ also can induce autophagy via Ca2+/calmodulin-dependent kinase kinase-β (CaMKKβ)-dependent activation of AMPK that ultimately leads to the inhibition of MTORC1 [52,62]. However, we found that exposure of cells with SB202190, under the conditions that TFEB and TFEB accumulated in the nucleus, did not dramatically cause MTOR inhibition, excluding the possibility that SB202190-induced TFEB and TFE3 activation and subsequent autophagy induction are dependent on the calcium-MTOR pathway. However, Tfeb and Tfe3 knockdown only partially but significantly (about 30% reduction) inhibited prolonged (e.g. 16 h) SB202190 treatment-induced increases of LC3B-II levels, but almost completely inhibited the increase of LAMP1 and SQSTM1/p62, suggesting that there might be a TFEB- and/or TFE3-independent way to promote LC3B-II lipidation in response to SB202190. We postulate that prolonged treatment of cells with SB202190 may cause a decrease of MTORC1 activity and subsequently enhance LC3B-II lipidation via the calcium/CaMKKβ pathway. Another possibility is that SB202190-induced ER stress may also partially contribute to LC3 lipidation because ER stress has been linked to autophagy induction [63].
Overall, our results indicated that SB202190 induces autophagy and lysosomal biogenesis via the activation of TFEB and TFE3, and that this activation is dependent on ER calcium-induced PPP3/calcineurin rather than p38 MAP kinase inhibition or mTOR signaling. The precise mechanism that SB202190-induced TFEB/TFE3 activation may be due to its effects on direct destabilization of the ER membranes. Given that the autophagy-lysosomal pathway participates in both physiological, pathological conditions and is involved in multiple diseases such as redox biology, cancer [64], immune-related diseases [[65], [66], [67]], and neurodegenerative diseases [[68], [69], [70]], this study calls for avoiding use or using caution when utilizing p38 MAP kinase inhibitor SB202190, especially for cell culture studies.
Funding
This study was supported by Shenzhen Science and Technology Innovation Commission (SZSTI, 201803023000787), the National Natural Science Foundation of China (81703487, and 81773926), the Hong Kong General Research Fund (GRF/HKBU12101417, GRF/HKBU12100618), the Hong Kong Health and Medical Research Fund (HMRF14150811, HMRF15163481), and research funds from Hong Kong Baptist University (HKBU/RC-IRCs/17–18/03, HKBU/RC-IRMS/15–16/04, FRGI/17–18/041, FRGII/17–18/021).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Prof. Han-Ming Shen from the National University of Singapore, Singapore, for his suggestions and critical reading of the manuscript. We also thank Dr. Martha Dahlen for the English editing of this manuscript.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101445.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Bento C.F., Renna M., Ghislat G., Puri C., Ashkenazi A., Vicinanza M., Menzies F.M., Rubinsztein D.C. Mammalian autophagy: how does it work? Annu. Rev. Biochem. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 2.Hurley J.H., Young L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017;86:225–244. doi: 10.1146/annurev-biochem-061516-044820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ktistakis N.T., Tooze S.A. Digesting the expanding mechanisms of autophagy. Trends Cell Biol. 2016;26(8):624–635. doi: 10.1016/j.tcb.2016.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura S., Yoshimori T. New insights into autophagosome–lysosome fusion. J. Cell Sci. 2017;130(7):1209–1216. doi: 10.1242/jcs.196352. [DOI] [PubMed] [Google Scholar]
- 5.Lamb C.A., Yoshimori T., Tooze S.A. The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 2013;14(12):759. doi: 10.1038/nrm3696. [DOI] [PubMed] [Google Scholar]
- 6.Choi A.M., Ryter S.W., Levine B. Autophagy in human health and disease. N. Engl. J. Med. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 7.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Settembre C., Di Malta C., Polito V.A., Arencibia M.G., Vetrini F., Erdin S., Erdin S.U., Huynh T., Medina D., Colella P. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332(6036):1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sardiello M., Palmieri M., di Ronza A., Medina D.L., Valenza M., Gennarino V.A., Di Malta C., Donaudy F., Embrione V., Polishchuk R.S. A gene network regulating lysosomal biogenesis and function. Science. 2009;325(5939):473–477. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 10.Martina J.A., Diab H.I., Lishu L., Jeong-A L., Patange S., Raben N., Puertollano R. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 2014;7(309) doi: 10.1126/scisignal.2004754. ra9-ra9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martina J.A., Chen Y., Gucek M., Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8(6):903–914. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Raben N., Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 2016;32:255–278. doi: 10.1146/annurev-cellbio-111315-125407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Palmieri M., Impey S., Kang H., di Ronza A., Pelz C., Sardiello M., Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011;20(19):3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 14.Puertollano R., Ferguson S.M., Brugarolas J., Ballabio A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018;37(11) doi: 10.15252/embj.201798804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settembre C., Medina D.L. TFEB and the CLEAR network. Methods Cell Biol. 2015;126:45–62. doi: 10.1016/bs.mcb.2014.11.011. [DOI] [PubMed] [Google Scholar]
- 16.Settembre C., Zoncu R., Medina D.L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M.C. A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31(5):1095–1108. doi: 10.1038/emboj.2012.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roczniak-Ferguson A., Petit C.S., Froehlich F., Qian S., Ky J., Angarola B., Walther T.C., Ferguson S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012;5(228) doi: 10.1126/scisignal.2002790. ra42-ra42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17(3):288. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martina J.A., Diab H.I., Brady O.A., Puertollano R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016;35(5):479–495. doi: 10.15252/embj.201593428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang W., Gao Q., Yang M., Zhang X., Yu L., Lawas M., Li X., Bryant-Genevier M., Southall N.T., Marugan J. Up-regulation of lysosomal TRPML1 channels is essential for lysosomal adaptation to nutrient starvation. Proc. Natl. Acad. Sci. Unit. States Am. 2015;112(11):E1373–E1381. doi: 10.1073/pnas.1419669112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ono K., Han J. The p38 signal transduction pathway activation and function. Cell. Signal. 2000;12(1):1–13. doi: 10.1016/s0898-6568(99)00071-6. [DOI] [PubMed] [Google Scholar]
- 22.Goldstein D.M., Gabriel T. Pathway to the clinic: inhibition of P38 MAP kinase. A review of ten chemotypes selected for development. Curr. Top. Med. Chem. 2005;5(10):1017–1029. doi: 10.2174/1568026054985939. [DOI] [PubMed] [Google Scholar]
- 23.Kevil C.G., Oshima T., Alexander J.S. The role of p38 MAP kinase in hydrogen peroxide mediated endothelial solute permeability. Endothelium. 2001;8(2):107–116. doi: 10.3109/10623320109165320. [DOI] [PubMed] [Google Scholar]
- 24.Klotz L.O., Pellieux C., Briviba K., Pierlot C., Aubry J.M., Sies H. Mitogen‐activated protein kinase (p38‐, JNK‐, ERK‐) activation pattern induced by extracellular and intracellular singlet oxygen and UVA. Eur. J. Biochem. 1999;260(3):917–922. doi: 10.1046/j.1432-1327.1999.00255.x. [DOI] [PubMed] [Google Scholar]
- 25.He Y., She H., Zhang T., Xu H., Cheng L., Yepes M., Zhao Y., Mao Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018;217(1):315–328. doi: 10.1083/jcb.201701049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Webber J.L. Regulation of autophagy by p38α MAPK. Autophagy. 2010;6(2):292–293. doi: 10.4161/auto.6.2.11128. [DOI] [PubMed] [Google Scholar]
- 27.Yang C.-b., Pei W.-j., Zhao J., Cheng Y.-y., Zheng X.-h., Rong J.-h. Bornyl caffeate induces apoptosis in human breast cancer MCF-7 cells via the ROS-and JNK-mediated pathways. Acta Pharmacol. Sin. 2014;35(1):113. doi: 10.1038/aps.2013.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang C., Zhao J., Pei W., Zheng X., Rong J. Biochemical mechanisms of bornyl caffeate induced cytotoxicity in rat pheochromocytoma PC12 cells. Chem. Biol. Interact. 2014;219:133–142. doi: 10.1016/j.cbi.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 29.Fabian M.A., Biggs W.H., III, Treiber D.K., Atteridge C.E., Azimioara M.D., Benedetti M.G., Carter T.A., Ciceri P., Edeen P.T., Floyd M. A small molecule–kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005;23(3):329. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- 30.Nemoto S., Xiang J., Huang S., Lin A. Induction of apoptosis by SB202190 through inhibition of p38β mitogen-activated protein kinase. J. Biol. Chem. 1998;273(26):16415–16420. doi: 10.1074/jbc.273.26.16415. [DOI] [PubMed] [Google Scholar]
- 31.Davidson W., Frego L., Peet G.W., Kroe R.R., Labadia M.E., Lukas S.M., Snow R.J., Jakes S., Grygon C.A., Pargellis C. Discovery and characterization of a substrate selective p38α inhibitor. Biochemistry. 2004;43(37):11658–11671. doi: 10.1021/bi0495073. [DOI] [PubMed] [Google Scholar]
- 32.English J.M., Cobb M.H. Pharmacological inhibitors of MAPK pathways. Trends Pharmacol. Sci. 2002;23(1):40–45. doi: 10.1016/s0165-6147(00)01865-4. [DOI] [PubMed] [Google Scholar]
- 33.Lali F.V., Hunt A.E., Turner S.J., Foxwell B.M. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 2000;275(10):7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- 34.Bain J., Plater L., Elliott M., Shpiro N., Hastie C.J., Mclauchlan H., Klevernic I., Arthur J.S.C., Alessi D.R., Cohen P. The selectivity of protein kinase inhibitors: a further update. Biochem. J. 2007;408(3):297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webber J.L., Tooze S.A. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 2010;29(1):27–40. doi: 10.1038/emboj.2009.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Menon M.B., Dhamija S., Kotlyarov A., Gaestel M. The problem of pyridinyl imidazole class inhibitors of MAPK14/p38α and MAPK11/p38β in autophagy research. Autophagy. 2015;11(8):1425–1427. doi: 10.1080/15548627.2015.1059562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiacchiera F., Simone C. Inhibition of p38α unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy. 2009;5(7):1030–1033. doi: 10.4161/auto.5.7.9252. [DOI] [PubMed] [Google Scholar]
- 38.Comes F., Matrone A., Lastella P., Nico B., Susca F., Bagnulo R., Ingravallo G., Modica S., Sasso G.L., Moschetta A. A novel cell type-specific role of p38α in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ. 2007;14(4):693. doi: 10.1038/sj.cdd.4402076. [DOI] [PubMed] [Google Scholar]
- 39.Simone C. Signal-dependent control of autophagy and cell death in colorectal cancer cell: the role of the p38 pathway. Autophagy. 2007;3(5):468–471. doi: 10.4161/auto.4319. [DOI] [PubMed] [Google Scholar]
- 40.Menon M.B., Kotlyarov A., Gaestel M. SB202190-induced cell type-specific vacuole formation and defective autophagy do not depend on p38 MAP kinase inhibition. PloS One. 2011;6(8) doi: 10.1371/journal.pone.0023054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Song J.-X., Sun Y.-R., Peluso I., Zeng Y., Yu X., Lu J.-H., Xu Z., Wang M.-Z., Liu L.-F., Huang Y.-Y. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy. 2016;12(8):1372–1389. doi: 10.1080/15548627.2016.1179404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim J.-W., Li M.-H., Jang J.-H., Na H.-K., Song N.-Y., Lee C., Johnson J.A., Surh Y.-J. 15-Deoxy-Δ12, 14-prostaglandin J2 rescues PC12 cells from H2O2-induced apoptosis through Nrf2-mediated upregulation of heme oxygenase-1: potential roles of Akt and ERK1/2. Biochem. Pharmacol. 2008;76(11):1577–1589. doi: 10.1016/j.bcp.2008.08.007. [DOI] [PubMed] [Google Scholar]
- 43.Tong B.C.-K., Lee C.S.-K., Cheng W.-H., Lai K.-O., Foskett J.K., Cheung K.-H. Familial Alzheimer's disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci. Signal. 2016;9(444) doi: 10.1126/scisignal.aaf1371. ra89-ra89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edelstein A.D., Tsuchida M.A., Amodaj N., Pinkard H., Vale R.D., Stuurman N. Advanced methods of microscope control using μManager software. J. Biol. Methods. 2014;1(2) doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Medina D.L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., Settembre C., Wang W., Gao Q., Xu H., Sandri M., Rizzuto R., De Matteis M.A., Ballabio A. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015;17(3):288–299. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou J., Tan S.-H., Nicolas V., Bauvy C., Yang N.-D., Zhang J., Xue Y., Codogno P., Shen H.-M. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res. 2013;23(4):508. doi: 10.1038/cr.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klionsky D.J., Abdelmohsen K., Abe A., Abedin M.J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C.M., Adams P.D., Adeli K. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimura S., Noda T., Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 49.Enslen H., Raingeaud J., Davis R.J. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 1998;273(3):1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- 50.Martina J.A., Puertollano R. Protein phosphatase 2A stimulates activation of TFEB and TFE3 transcription factors in response to oxidative stress. J. Biol. Chem. 2018;293(32):12525–12534. doi: 10.1074/jbc.RA118.003471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu J., Farmer J.D., Jr., Lane W.S., Friedman J., Weissman I., Schreiber S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66(4):807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- 52.Høyer-Hansen M., Bastholm L., Szyniarowski P., Campanella M., Szabadkai G., Farkas T., Bianchi K., Fehrenbacher N., Elling F., Rizzuto R. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-β, and Bcl-2. Mol. Cell. 2007;25(2):193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 53.Crabtree G.R. Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 2001;276(4):2313–2316. doi: 10.1074/jbc.R000024200. [DOI] [PubMed] [Google Scholar]
- 54.Billman G.E. Intracellular calcium chelator, BAPTA-AM, prevents cocaine-induced ventricular fibrillation. Am. J. Physiol. Heart Circ. Physiol. 1993;265(5):H1529–H1535. doi: 10.1152/ajpheart.1993.265.5.H1529. [DOI] [PubMed] [Google Scholar]
- 55.Wang C., Niederstrasser H., Douglas P.M., Lin R., Jaramillo J., Li Y., Olswald N.W., Zhou A., McMillan E.A., Mendiratta S. Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 2017;8(1):2270. doi: 10.1038/s41467-017-02332-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hirosawa M., Nakahara M., Otosaka R., Imoto A., Okazaki T., Takahashi S. The p38 pathway inhibitor SB202190 activates MEK/MAPK to stimulate the growth of leukemia cells. Leuk. Res. 2009;33(5):693–699. doi: 10.1016/j.leukres.2008.09.028. [DOI] [PubMed] [Google Scholar]
- 57.Chiacchiera F., Matrone A., Ferrari E., Ingravallo G., Sasso G.L., Murzilli S., Petruzzelli M., Salvatore L., Moschetta A., Simone C. p38α blockade inhibits colorectal cancer growth in vivo by inducing a switch from HIF1α-to FoxO-dependent transcription. Cell Death Differ. 2009;16(9):1203. doi: 10.1038/cdd.2009.36. [DOI] [PubMed] [Google Scholar]
- 58.Zhang J., Wang J., Zhou Z., Park J.-E., Wang L., Wu S., Sun X., Lu L., Wang T., Lin Q. Importance of TFEB acetylation in control of its transcriptional activity and lysosomal function in response to histone deacetylase inhibitors. Autophagy. 2018;14(6):1043–1059. doi: 10.1080/15548627.2018.1447290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sha Y., Rao L., Settembre C., Ballabio A., Eissa N.T. STUB1 regulates TFEB‐induced autophagy–lysosome pathway. EMBO J. 2017;36(17):2544–2552. doi: 10.15252/embj.201796699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tong B.C.-K., Wu A.J., Li M., Cheung K.-H. BBA)-Molecular Cell Research; 2018. Calcium signaling in Alzheimer's disease & therapies, Biochimica et Biophysica Acta. [DOI] [PubMed] [Google Scholar]
- 61.Görlach A., Bertram K., Hudecova S., Krizanova O. Calcium and ROS: a mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.La Rovere R.M., Roest G., Bultynck G., Parys J.B. Intracellular Ca2+ signaling and Ca2+ microdomains in the control of cell survival, apoptosis and autophagy. Cell Calcium. 2016;60(2):74–87. doi: 10.1016/j.ceca.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 63.Yorimitsu T., Klionsky D.J. Endoplasmic reticulum stress: a new pathway to induce autophagy. Autophagy. 2007;3(2):160–162. doi: 10.4161/auto.3653. [DOI] [PubMed] [Google Scholar]
- 64.Mathew R., Karantza-Wadsworth V., White E. Role of autophagy in cancer. Nat. Rev. Canc. 2007;7(12):961. doi: 10.1038/nrc2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pastore N., Brady O.A., Diab H.I., Martina J.A., Sun L., Huynh T., Lim J.-A., Zare H., Raben N., Ballabio A. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy. 2016;12(8):1240–1258. doi: 10.1080/15548627.2016.1179405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.He Y., Xu Y., Zhang C., Gao X., Dykema K.J., Martin K.R., Ke J., Hudson E.A., Khoo S.K., Resau J.H. Identification of a lysosomal pathway that modulates glucocorticoid signaling and the inflammatory response. Sci. Signal. 2011;4(180) doi: 10.1126/scisignal.2001450. ra44-ra44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cadwell K. Crosstalk between autophagy and inflammatory signalling pathways: balancing defence and homeostasis. Nat. Rev. Immunol. 2016;16(11):661. doi: 10.1038/nri.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Martini-Stoica H., Xu Y., Ballabio A., Zheng H. The autophagy–lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39(4):221–234. doi: 10.1016/j.tins.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Menzies F.M., Fleming A., Rubinsztein D.C. Compromised autophagy and neurodegenerative diseases. Nat. Rev. Neurosci. 2015;16(6):345. doi: 10.1038/nrn3961. [DOI] [PubMed] [Google Scholar]
- 70.Yang C., Cai C.-Z., Song J.-X., Tan J.-Q., Durairajan S.S.K., Iyaswamy A., Wu M.-Y., Chen L.-L., Yue Z., Li M. NRBF2 is involved in the autophagic degradation process of APP-CTFs in Alzheimer disease models. Autophagy. 2017;13(12):2028–2040. doi: 10.1080/15548627.2017.1379633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.