

Abstract
BRD4, a member of the bromodomain and extra-terminal domain (BET) protein family, plays a role in the organization of super-enhancers and transcriptional activation of oncogenes in cancer and is recognized as a promising target for cancer therapy. microRNAs (miRNAs), endogenous small noncoding RNAs, cause mRNA degradation or inhibit protein translation of their target genes by binding to complementary sequences. miRNA mimics simultaneously targeting several tumor-promoting genes and BRD4 may be useful as therapeutic agents of tumor-suppressive miRNAs (TS-miRs) for cancer therapy. To investigate TS-miRs for the development of miRNA-based cancer therapeutics, we performed function-based screening in 10 cancer cell lines with a library containing 2,565 human miRNA mimics. Consequently, miR-1293, miR-876-3p, and miR-6571-5p were identified as TS-miRs targeting BRD4 in this screening. Notably, miR-1293 also suppressed DNA repair pathways by directly suppressing the DNA repair genes APEX1 (apurinic-apyrimidinic endonuclease 1), RPA1 (replication protein A1), and POLD4 (DNA polymerase delta 4, accessory subunit). Concurrent suppression of BRD4 and these DNA repair genes synergistically inhibited tumor cell growth in vitro. Furthermore, administration of miR-1293 suppressed in vivo tumor growth in a xenograft mouse model. These results suggest that miR-1293 is a candidate for the development of miRNA-based cancer therapeutics.
Keywords: miR-1293, BRD4, DNA repair, cancer, nucleic acid therapeutics
Graphical Abstract
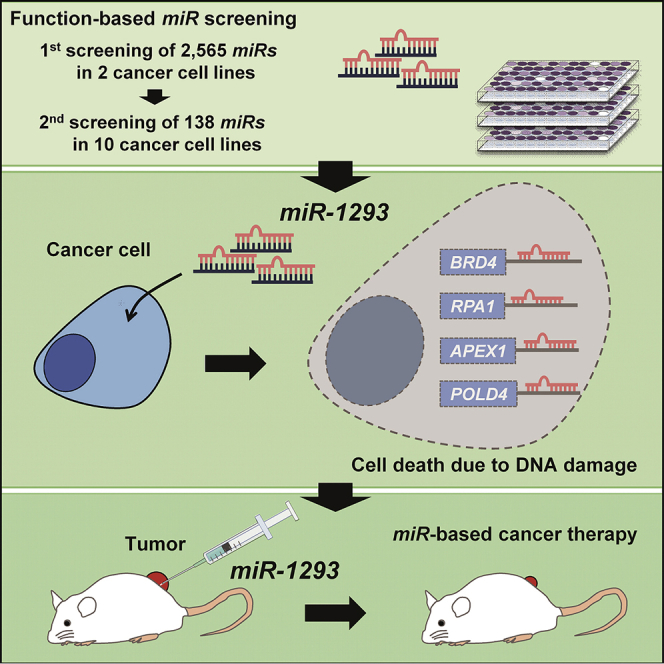
Takagawa and colleagues identified miR-1293 as a candidate for miRNA-based cancer therapeutics from the screening of 2,565 human miRNA mimics. miR-1293 targeted concurrently BRD4 and DNA repair genes, resulting in strong suppression of cancer cell growth by DNA damage accumulation. Local administration of miR-1293 suppressed tumor cell growth in vivo.
Introduction
BRD4, a member of the bromodomain and extra-terminal domain (BET) protein family, facilitates transcriptional activation as an epigenetic reader through two bromodomains.1 BRD4 is widely acknowledged in cancer for its role in the organization of super-enhancers and transcriptional regulation of oncogenes.2 Pharmacological inhibition of BRD4 by BET inhibitors (BETi) has shown potent efficacy in preclinical studies against many types of cancers, such as acute myeloid leukemia,3 multiple myeloma,4 triple negative breast cancer,5 melanoma,6 pancreatic cancer,7 and prostate cancer.8 For this reason, BETi or the recently developed proteolysis targeting chimera (PROTAC) that degrades BRD4 is considered one of the most promising agents for the treatment of cancers.9, 10, 11 However, a first-in-class BETi, birabresib (OTX015), has shown little but not sufficient therapeutic benefit in advanced solid tumors for use as a single agent in a clinical trial.12 Recent studies have shown that the combination of BETi with several pathway inhibitors clearly enhanced antitumor activity in preclinical models.10,11
microRNAs (miRNAs), endogenous small noncoding RNAs of 20–25 nucleotides, mediate posttranscriptional gene silencing by binding to complementary mRNA sequences of their target genes.13 Because a single miRNA can simultaneously regulate numerous target genes, miRNA mimics targeting several tumor-promoting genes may be useful as therapeutic agents for cancer therapy. From this viewpoint, we have previously identified several tumor-suppressive miRNAs (TS-miRs), including miR-3140, using miRNA libraries containing up to 1,090 miRNAs.14 We showed that miR-3140 inhibited tumor growth by concurrently targeting BRD4 and the cell proliferation pathway (EGFR and CDK2). Nucleic acid therapeutics, including miRNA formulations, are now regarded as a next-generation treatment for cancer.15,16
Here, to identify candidates for the development of miRNA-based cancer therapeutics, we conducted miRNA screening using a miRNA library containing 2,565 miRNAs based on the inhibitory effects of in vitro tumor cell growth. We identified miR-1293 as a TS-miR targeting BRD4. We further determined that the tumor-suppressive function of miR-1293 was mediated by the combination of downregulation of BRD4 and genes associated with the DNA repair pathway. The therapeutic effects of miR-1293 administration were evaluated using a xenograft mouse model.
Results
miR-1293 Was Identified as a TS-miR by a Function-Based miRNA Library Screening
To identify TS-miRs, we performed function-based miRNA screening using a library containing 2,565 miRNA mimics in HCT116 p53+/+ (HCT116+/+) and HCT116 p53−/− (HCT116−/−) cells. A flow chart of the strategy and summary of the results are shown in Figure 1A. 72 h after transfection of each miRNA in this library, the relative cell number was assessed using a relative ratio normalized to the number of cells transfected with negative control miRNA (miR-NC). Figure 1B shows the results of the 1st screening in HCT116+/+ and HCT116−/− cells. A total of 630 miRNAs and 141 miRNAs markedly suppressed cell proliferation (relative cell number < 0.3) in HCT116+/+ and HCT116−/− cells, respectively. Since p53-inactivated cells were more resistant to cytotoxic agents than p53 wild-type (WT) cells (Figure S1), as previously reported,17 we extracted the 138 common miRNAs that suppressed cell proliferation in both cell lines regardless of the p53 status (Table S1). We then performed a 2nd screening using these common 138 miRNAs in 10 cancer cell lines (HCT116+/+, HCT116−/− [colon cancer], SW48 [colon cancer], HT29 [colon cancer], HOC313 [oral cancer], HSC-2 [oral cancer], AGS [gastric cancer], A549 [lung cancer], KYSE150 [esophageal cancer], and MIAPaCa2 [pancreatic cancer]). As shown in Figure 1C and 7 miRNAs, miR-1293, miR-876-3p, miR-4438, miR-6751-5p, miR-634, miR-3140-3p, and miR-92a-2-5p suppressed cell proliferation in all 10 cancer cells (relative cell number < 0.6). Among them, miR-876-3p, miR-634, and miR-3140-3p were previously reported as TS-miRs by our group and others.14,18,19 Overall, miR-1293, miR-4438, miR-6751-5p, and miR-92a-2-5p were identified as TS-miRs.
Figure 1.

miR-1293 Was Identified as a Tumor-Suppressive miRNA (TS-miR) by Function-Based miRNA Library Screening
(A) Flow chart of the study to identify TS-miRNAs. (B) Results of function-based miRNA library 1st screening in HCT116+/+ (p53 WT) cells and HCT116−/− (p53 null) cells, using the mirVana miR mimic Library v21 consisting of 2,565 mature human miRNA mimics. Relative cell viability was evaluated by CV staining using a relative ratio normalized to the cell survival rate of cells transfected with negative control miRNA (miR-NC). Common 138 miRNAs that suppressed cell proliferation in both cell lines (red dots). (C) 7 common miRNAs that remarkably inhibited cell growth in all 10 cancer cell lines in the second screening. HCT116+/+, SW48, AGS, and A549 cells harbor WT p53, whereas the others do not.
miR-1293 Suppressed the Expression of BRD4 by Directly Targeting Its 3′ UTR
We have previously shown that miR-3140-3p, one of 7 TS-miRs identified in this screening, exerts tumor-suppressive effects by targeting BRD4, EGFR, and CDK2 simultaneously.14 A recent study revealed that targeting BRD4 and other pathway inhibitors has synergistic antitumor activity.11,20,21 Thus, we explored TS-miRs directly targeting BRD4. First, we determined whether those 7 TS-miRs downregulate the expression of BRD4 in all 10 cell lines. As shown in Figures 2A and S2A, the expression of BRD4 was decreased in all 10 cell lines transfected with miR-1293, miR-876-3p, miR-6751-5p, miR-3140-3p, and miR-92a-2-5p, although the expression of BRD4 was not decreased in HCT116−/−, MIAPaCa2, and AGS cells transfected with miR-634, and in HT29 and AGS cells transfected with miR-4438. Knockdown of BRD4 using small interfering RNA (siRNA) targeting BRD4 (si-BRD4) reduced cell proliferation in all 10 cell lines (Figure 2B; Figures S2B and S2C), suggesting that downregulation of BRD4 is one of the tumor-suppressive mechanisms of these TS-miRs except for miR-634 and miR-4438.
Figure 2.

miR-1293 Directly Targets the 3′ UTR of BRD4
(A) Western blot analysis of BRD4 in HCT116+/+, HCT116−/−, and HOC313 cells 48 h after transfection with 10 nmol/L miR-NC or the indicated 7 miRNAs. (B) Knocking down BRD4 suppressed in vitro cell proliferation in HCT116+/+, HCT116−/−, and HOC313 cells. Western blot analysis (right) and cell growth assay (left) in each cell line after transfection with 20 nmol/L negative control siRNA (si-NC) or siRNA targeting BRD4 (si-BRD4). ∗p < 0.05. (C–E) Luciferase reporter assay. HOC313 cells were cotransfected with pmirGLO dual-luciferase vectors containing wild-type (WT) BRD4-3′ UTR or mutant variants of BRD4-3′ UTR and miR-NC, miR-1293 (C), miR-876-3p (D), or miR-6751-5p (E). Top, putative binding sequences of each miRNA within the 3′ UTR of BRD4 and mutant sequences are indicated. Bottom, the results of the luciferase assay; ∗p < 0.05.
We next examined whether each of the 7 miRNAs could directly bind to the 3′ UTR of BRD4. Five miRNAs except for miR-634 and miR-3140-3p were predicted to bind to the 3′ UTR of BRD4 according to the TargetScan program (http://www.targetscan.org). Luciferase assays, using reporter plasmid vectors containing WT or mutant (Mt) seed sequences of the 3′ UTR of BRD4 mRNA were performed in HOC313 cells (Figures 2C–2E; Figures S3A–S3C). The luciferase activity of the WT vector was decreased compared with that of the empty vector (EV) in miR-1293-, miR-876-3p-, and miR-6751-5p-transfected cells and was completely restored with the Mt vector, suggesting that miR-1293, miR-876-3p, and miR-6751-5p can directly bind to the 3′ UTR of BRD4. On the other hand, miR-4438 and miR-92a-2-5p could not bind the 3′ UTR or coding sequence (CDS) of BRD4 (Figures S3B and S3C). Collectively, these data suggested that miR-1293, miR-876-3p, and miR-6751-5p are TS-miRs targeting BRD4.
miR-1293 Inhibited In Vitro Tumor Cell Growth in Various Cancer Cell Lines
We next focused on a detailed analysis of miR-1293 because miR-1293 is a TS-miR that inhibited in vitro tumor cell growth most efficiently in the 2nd screening. To validate the growth-suppressive effect of miR-1293, we evaluated cell proliferation in vitro after transfections of various cell lines with miR-1293 or miR-NC. In agreement with the results of the 2nd screening, overexpression of miR-1293 significantly inhibited tumor cell proliferation in these cell lines (Figures 3A and 3B; Figure S4A). Annexin V and propidium iodide (PI) staining showed that the apoptotic cell population was increased in miR-1293-transfected cells compared with miR-NC-transfected cells (Figure 3C; Figure S4B). Western blotting showed increased expression of cleaved PARP (cl. PARP) in miR-1293-transfected cells (Figure 3D; Figure S4C). Taken together, the results demonstrate that miR-1293 inhibited in vitro tumor cell growth by inducing apoptosis.
Figure 3.

Overexpression of miR-1293 Induced Apoptosis in Cancer Cells In Vitro
(A) In vitro cell growth assay in various cancer cell types. Cells were transfected with 10 nmol/L miR-NC or miR-1293. Cell growth rate was assessed with the CV staining assay using a relative ratio compared with cell growth on day 0. Error bar, SD for triplicate experiments. ∗p < 0.05. (B) Phase contrast images of HCT116+/+, HCT116−/−, and HOC313 cells transfected with 10 nmol/L miR-NC or miR-1293. Images were obtained 48 h after transfection. (C) The percentage of apoptotic cells in HCT116+/+, HCT116−/−, and HOC313 cells transfected with miR-NC or miR-1293. Cells were double stained with Annexin V and propidium iodide (PI) 48 h after transfection and analyzed by flow cytometry. The percentage of cells indicates late apoptotic cells (Annexin V+/ PI+). Error bar, SD for triplicate experiments. ∗p < 0.05. (D) Western blot analysis of cleaved PARP (cl. PARP) in HCT116+/+, HCT116−/−, and HOC313 cells 48 h after transfection with 10 nmol/L miR-NC or miR-1293.
miR-1293 Directly Targeted APEX1, RPA1, and POLD4 by Binding Their 3′ UTRs
To explore target genes of miR-1293 other than BRD4, we performed gene expression array analysis in 5 cell lines, HCT116+/+, A549, KYSE150, HOC313, and HSC-2, after transfection of miR-1293 or miR-NC. As shown in a Venn diagram (Figure 4A), 1,248 genes were commonly downregulated (fold change > 1.5) by transfection of miR-1293 in 4 or 5 out of 5 cell lines. The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of these 1248 genes using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/) revealed that miR-1293 significantly suppressed several genes related to DNA repair pathways, such as base excision repair, nucleotide excision repair, mismatch repair, and homologous recombination (HR) (Figure 4A; Figure S5A). We then examined whether genes associated with the DNA repair pathway might be direct targets of miR-1293. Among these targets, APEX1 (apurinic-apyrimidinic endonuclease 1), RPA1 (replication protein A1), and POLD4 (DNA polymerase delta 4, accessory subunit) were decreased at protein levels in miR-1293-transfected HCT116+/+, HCT116−/−, HOC313, KYSE150, A549, and HSC-2 cells compared with those in miR-NC-transfectants (Figure 4B; Figure S6A). The expression of ɣH2AX, a marker of DNA damage, was also increased in miR-1293-transfected cells, especially in HCT116−/− and A549 cells (Figure 4B; Figure S6B). Luciferase assays using reporter plasmid vectors with WT or Mt seed sequences of the 3′ UTRs of APEX1, RPA1, and POLD4 showed that in miR-1293-transfected cells, the luciferase activity was significantly decreased in the WT vector compared with the EV, whereas it was completely restored in the Mt vector (Figure 4C). The luciferase activity of the WT vector containing the predicted seed sequences in the 3′ UTR or CDS of RAD51, XRCC3, BLM, POLE2, PARP1, EXO1, or MSH6 was not decreased in miR-1293-transfectants (Figure S5B). The protein levels of CUL4B, RFC2, and RFC3, which were also listed in the present KEGG pathway enrichment analysis, were not reduced appreciably in miR-1293-transfected cells (Figure S5C). Overall, miR-1293 directly suppressed the expression of APEX1, RPA1, and POLD4 by binding the 3′ UTRs of these genes.
Figure 4.

miR-1293 Downregulated the Expression of APEX1, RPA1, and POLD4 by Directly Targeting Their 3′ UTRs
(A) Identification of downregulated genes and pathways after miR-1293 transfection. Left, a Venn diagram showing 1,248 genes that were commonly downregulated (fold change > 1.5) by transfection of miR-1293 in at least 4 out of 5 cell lines. Right, the table shows the results of the pathway analysis of the 1,248 commonly downregulated genes using DAVID Bioinformatics Resources 6.8. Fisher’s exact test was used for genes selection. p < 0.05 were considered significant. (B) Western blot analysis of APEX1, RPA1, POLD4, and ɣH2AX in HCT116+/+, HCT116−/−, HOC313, KYSE150, A549, and HSC-2 cells 48 h after transfection with 10 nmol/L miR-NC or miR-1293. (C) Luciferase reporter assays. HOC313 cells were cotransfected with pmirGLO dual-luciferase vectors containing the wild-type (WT) 3′ UTRs of APEX1, RPA1, and POLD4 or mutant (Mt) variants of these genes and miR-NC or miR-1293. Top, putative binding site of miR-1293 within the 3′ UTR of each gene and mutant sequences. Bottom, the results of the luciferase assay; ∗p < 0.05.
miR-1293 Suppressed In Vitro Tumor Cell Growth by Concurrent Downregulation of BRD4, APEX1, RPA1, and POLD4
We next examined the effects of concurrent downregulation of BRD4, APEX1, RPA1, and POLD4 using siRNA targeting each gene. As shown in Figures 5A–5D, knockdown of BRD4 in combination with knockdown of RPA1 or POLD4 led to a more pronounced suppression of in vitro cell growth with increased expression of cl. PARP in HCT116−/− cells, although these synergistic effects of combination of si-BRD4 and si-APEX1 were not significant. Similar synergistic effects on cell growth were also observed in HOC313 cells, although those were not significant in HCT116+/+ cells (Figure S7A). Collectively, these results suggested that concurrent downregulation of BRD4, RPA1, and POLD4 may contribute to the miR-1293-mediated growth-suppressive effects.
Figure 5.

miR-1293 Inhibited DNA Damage Repair by Downregulating BRD4 and DNA Repair Genes Synergistically In Vitro
(A–D) Evaluation of the effect of si-BRD4 and si-APEX1, si-RPA1, or si-POLD4. Western blot analysis (A, si-BRD4 and si-APEX1; B, si-BRD4 and si-RPA1; C, si-BRD4 and si-POLD4) and cell growth assay (D) in indicated cell lines after transfection with siRNA (si-NC [40 nmol/L]; si-BRD4, si-APEX1, si-RPA1, and si-POLD4 [each 20 nmol/L]). 120 h after transfection with the siRNAs, the cell growth rate was assessed with the CV staining assay using a relative ratio compared with that of si-NC-transfected cells. Error bar, SD for triplicate experiments. ∗p < 0.05. (E) Immunofluorescence analysis of HCT116−/− cells that were double labeled with anti-γH2AX (red) and DAPI (blue; nuclei). Cells were transfected with 10 nmol/L miR-NC, miR-1293, si-NC, or the indicated siRNAs for 6 h following treatment with neocarzinostatin (200 ng/mL) for 30 min. (F) The percentage of ɣH2AX-positive cells in (E). ∗p < 0.05. (G) Homologous recombination (HR) repair analysis. U2OS DR-GFP cells were transfected with si-NC, the indicated siRNAs, miR-NC, or miR-1293, 6 h after transfection with the I-Sce1 endonuclease expression vector for 48 h. The HR efficiency of cells cotransfected with si-BRD4 and miR-1293 was compared with that of cells cotransfected with si-NC and miR-NC based on the percentage of GFP+ cells detected by flow cytometry. Error bar, SD for triplicate experiments. ∗p < 0.05. (H) Schematic models for the mechanism by which miR-1293 suppresses the DNA repair pathway and tumor growth.
Next, to examine the effects of miR-1293 on DNA repair, the accumulation and clearance of DNA damage were evaluated in miR-1293- and miR-NC-transfected HCT116−/− and HOC313 cells following neocarzinostatin (NCS) treatment. We observed a significantly delayed clearance of γH2AX foci, a marker of unrepaired DNA damage, in miR-1293-transfected cells compared miR-NC-transfected cells (Figures 5E and 5F; Figure S7B). These results were partially reproduced by the combined knockdown of APEX1, RPA1, POLD4, and BRD4. We also evaluated the frequency of HR, a major pathway of double-stranded DNA break repair, using the DR-GFP reporter in U2OS cells as previously reported.22 The HR efficiency of U2OS DR-GFP cells transfected with miR-1293 was significantly decreased (Figure 5G). Knockdown of BRD4, APEX1, RPA1, and POLD4 also suppressed the HR efficiency in HCT116−/− cells. Collectively, these results demonstrated that miR-1293 inhibited tumor cell growth and the DNA repair pathway by concomitant suppression of BRD4, APEX1, RPA1, and POLD4 (Figure 5H).
miR-1293 Suppressed In Vivo Tumor Growth in a Xenograft Mouse Model
The in vivo therapeutic effect of miR-1293 was investigated using a xenograft mouse model of HCT116−/− cells. miR-NC or miR-1293 was administered into the subcutaneous space around tumors 5 times (3, 7, 10, 14, and 17 days after the injection of cells; Figure 6A). There were no obvious adverse consequences, such as body weight loss or skin damage around the administration site of miRNAs (Figure S8A). As a result, tumors treated with miR-1293 at 19 days after the injection of HCT116−/− cells were significantly smaller than tumors treated with miR-NC (Figures 6B–6D). qRT-PCR and in situ hybridization showed that the expression of miR-1293 was significantly higher in the miR-1293-treated tumors compared to the miR-NC-treated tumors, suggesting that miR-1293 was actually delivered into the cancer cells of the miR-1293-treated tumor (Figure 6E; Figure S8B). Immunohistochemical staining showed that the expression levels of BRD4, APEX1, RPA1, and POLD4 were decreased in the resected tumors treated with miR-1293 (Figure 6F). Similarly, administration of miR-1293 inhibited tumor growth in nude mice that were subcutaneously inoculated with SAS cells, as well as in vitro (Figures S8C–S8H and S9A–S9M). Taken together, these findings suggest that the administration of miR-1293 might be effective for tumor growth suppression in vivo.
Figure 6.

Therapeutic Effects of miR-1293 on Tumor Growth In Vivo
(A) The experimental schedule for miR-1293 treatment in nude mice. Tumors were formed by subcutaneous injection of HCT116−/− cells in nude mice. miR-NC or miR-1293 was administered subcutaneously around the tumors for a total of 5 times (3, 7, 10, 14, and 17 days after the injection of cells). (B) Representative images of tumor-bearing nude mice and resected tumors at 19 days after the injection of HCT116−/− cells. Scale bar, 10 mm. (C) Tumor growth curves of xenograft mouse models treated with miR-NC or miR-1293 (n = 10, each). Tumor volume was calculated using the following formula: (shortest diameter) 2 × (longest diameter) × 0.5. Error bar, SD for 10 mice; ∗p < 0.05, ∗∗p < 0.01. (D) Weights of the resected tumors. Tumor weights are shown as a boxplot. Error bar, SD for 10 mice; ∗p < 0.05. (E) Expression analysis of miR-1293 in the resected tumors. The expression level of miR-1293 was measured by qRT-PCR. Each experiment was performed in duplicate. The relative ratio was normalized to the expression of RNU6B. Error bar, SD for 10 mice; ∗∗p < 0.01. (F) Representative images (upper) and evaluation (lower) of immunohistochemical staining for BRD4, APEX1, RPA1, and POLD4 in resected tumors. The density per field was defined using the single point selection tool on the Nikon NIS Elements Br 4.0 software, and the results were normalized to the values of tumors treated with miR-NC (see the Materials and Methods section). Scale bar, 50 μm. ∗∗p < 0.01.
Discussion
Here, we identified miR-1293, miR-876-3p, and miR-6751-5p as TS-miRs targeting BRD4 by function-based screening of 2,565 miRNAs. We showed that miR-1293 suppressed DNA repair pathways by concurrently targeting BRD4, APEX1, RPA1, and POLD4, thereby inhibiting tumor cell growth. Administration of miR-1293 suppressed in vivo tumor growth in a xenograft mouse model.
Previous studies, including ours, have reported the results of function-based miRNA library screening in cancer cells. For example, we previously screened the growth-suppressive effects of 1,090 human miRNAs in pancreatic cancer cells,14 and Zhou et al.23 examined a total of 858 human miRNAs in KRAS mutant cancer cells. Since 2,654 mature human miRNAs are currently registered in miRBase v22 (http://www.mirbase.org/), the tumor-suppressive functions of numerous human miRNAs have not yet been investigated. Thus, we examined a total of 2,565 miRNAs, which covered ~96% of the registered human miRNAs, on the basis of their growth-inhibitory effects in cancer cells. In agreement with past reports including ours, the results of the 1st screening included several known TS-miRs, such as miR-1298-3p, miR-491-5p, miR-634, and miR-3140-3p (Table S1).14,19,23,24 Furthermore, the 2nd screening identified 7 miRNAs as the most growth-inhibitory miRNAs in all 10 cancer cell lines tested.
Since BRD4 regulates the expression of genes associated with cancer cell growth, BRD4-targeting therapy such as BETi or PROTAC that degrade BRD4 was reported to exhibit potent efficacy against many types of cancer.2, 3, 4, 5, 6, 7, 8,10 Thus, TS-miRs targeting BRD4 are a reasonable strategy for the development of miRNA-based therapeutics. Our results showed that miR-1293, miR-876-3p, and miR-6751-5p are TS-miRs targeting BRD4. Since recent studies have shown that the combined inhibition of BRD4 and other signaling pathways markedly enhanced antitumor activity,10,11 these TS-miRs, similar to miR-3140-3p,14 may suppress BRD4 and other oncogenic targets concomitantly.
According to the TCGA database, the expression of miR-1293, miR-876, and miR-6751 are extremely low in comparison to the expression of the representative miRNA miR-34a in both tumor tissues and non-tumor tissues of colon, esophagus, head and neck, lung, pancreas, and stomach, although the expression of miR-1293 is slightly elevated in esophagus squamous cell carcinoma (ESCC) and head and neck squamous cell carcinoma (HNSCC) (Figure S10A). The expression of miR-1293 was not correlated with overall survival in ESCC, and HNSCC (Figure S10B). On the other hand, the expression of miR-1293 was reported to be decreased in the serum of patients with colon cancer25 and to be increased in the tissues of oral cancer.26 Further investigation of the clinical relevance of miR-1293 is required.
Moreover, we showed that miR-1293 directly suppressed the DNA repair genes APEX1, RPA1, and POLD4. APEX1, a key enzyme for the repair of single-strand DNA breaks, is frequently overexpressed in solid tumors.27, 28, 29 RPA1, one of the main eukaryotic single-stranded DNA-binding proteins, plays an important role in DNA damage repair by recognizing DNA damage. Overexpression of RPA1 has been reported to be associated with poor clinical outcomes in several cancers.30, 31, 32 POLD4 not only plays a critical role in DNA replication and repair,33 but also promotes tumor cell growth.34 Since cancer cells depend more on DNA repair than normal cells because of oncogene-induced replication stress,35 targeting DNA repair genes such as APEX1, RPA1, and POLD4 may be rational for cancer therapy.
Recent studies revealed that BRD4 promotes DNA damage repair, including the non-homologous end-joining recombination pathway and HR pathway,20,36,37 and consequently, inhibition of the DNA repair pathway along with BRD4 suppression is synthetically lethal in many types of cancer cells.20,36,37 Consistent with these reports, we showed that concurrent downregulation of BRD4, APEX1, RPA1, and POLD4 synergistically suppressed DNA repair and inhibited tumor cell growth in vitro. Since these effects were similar to those of miR-1293, the tumor-suppressive effects of miR-1293 are thought to be mediated by the concurrent targeting of BRD4, APEX1, RPA1, and POLD4. The combination of miR-1293 with DNA-damaging agents or radiation therapy may be effective for cancer therapy.
Finally, we showed that local administration of miR-1293 suppressed in vivo tumor growth without obvious adverse events. These results indicated the potential of miR-1293 for miRNA-based therapy. For future studies, to translate these findings into the clinical, several challenges need to be overcome that include chemical modification of the nucleic acids for improved stability, development of a drug delivery system, and evaluation of the effects of miR-1293 on normal cells.15 In conclusion, miR-1293 suppresses tumor cell growth by concurrently targeting BRD4 and DNA repair genes. miR-1293 may be a promising candidate for the development of miRNA-based cancer therapeutics.
Materials and Methods
Cell Culture
The human colon cancer cell line HCT116 (HCT116+/+) and its derived isogenic p53 null cell line (HCT116−/−) were kindly provided by Dr. Bert Vogelstein (The Johns Hopkins University). HCT116+/+, HCT116−/−, HT29, SW48, MIAPaCa2, HSC2, HOC313, and A549 cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS). KYSE150 cells, a gift from Dr. Y. Shimada (Toyama University),38 and AGS cells were maintained in RPMI 1640 medium containing 10% FBS. SAS cells were maintained in Dulbecco’s modified Eagle’s medium with Ham’s F12 medium containing 10% FBS. All cell lines were maintained at 37°C and 5% CO2. All experiments were carried out in accordance with the approved guidelines and regulations (G2019-013A).
Reagents
Olaparib (AZD2281, Ku-0059436; S1060) was purchased from Selleck Chemicals (Houston, TX), puromycin was obtained from Invivogen (San Diego, CA), neocarzinostatin (N9162) was purchased from Sigma-Aldrich (St. Louis, MO), and doxorubicin hydrochloride was obtained from WAKO Pure Chemical Industries (Tokyo, Japan). These reagents were used for the treatment of cultured cells in vitro at the indicated concentrations.
Function-Based miRNA Screening
In the 1st screening, 6,000 HCT116+/+ and HCT116−/− cells were seeded in 96-well plates. After 24 h, each cell line was transfected in duplicate with each of the 2,565 double-stranded RNAs (dsRNA) from the mirVana miR mimic Library v21 (Thermo Fisher Scientific, Waltham, MA) or a negative control miRNA (miR-NC) using an RNA concentration of 10 nmol/L. After 72 h, viable cell numbers were assessed by the crystal violet (CV) staining assay. The results were normalized to the numbers of cells transfected with miR-NC. In the 2nd screening, HCT116+/+, HCT116−/− (colon cancer), SW48 (colon cancer), HT29 (colon cancer), HOC313 (oral cancer), HSC-2 (oral cancer), AGS (gastric cancer), A549 (lung cancer), KYSE150 (esophageal cancer), and MIAPaCa2 (pancreatic cancer) cells were used. 24 h after cell seeding, each cell line was transfected in duplicate with each of the 138 dsRNAs selected from the 1st screening or a miR-NC using an RNA concentration of 10 nmol/L. After 72 h, viable cell numbers were assessed as described above.
Transfection of miRNAs and siRNAs
The dsRNAs mimicking mature human miR-1293 (MC13698), miR-876-3p (MC12886), miR-4438 (MC22634), miR-6751-5p (MC27194), miR-634 (MC11538), miR-3140-3p (MC17496), and miR-92a-2-5p (MC12524) and a nonspecific control miRNA (negative control #1) were purchased from Thermo Fisher Scientific. The SMARTpool siRNA for BRD4 (M-004937-02), APEX1 (M-010237-01), RPA1 (M-015749-01), POLD4 (M-014013-01), and nonspecific control siRNAs were obtained from GE Healthcare (Buckinghamshire, UK). Each SMARTpool siRNA contains 4 siRNA duplexes designed to target different mRNA sequences of the same gene. miRNAs and siRNAs were transfected individually into cells at the indicated concentrations using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions.
In Vitro Cell Growth Assay
Cell survival was evaluated by the CV staining assay. Cells were washed in PBS and fixed with 0.1% CV in 10% formaldehyde in PBS for 5 minutes. After the excess CV solution was discarded, stained cells were completely air-dried, and then lysed with a 2% SDS solution with shaking for 1 h. Optical density (OD) absorbance was measured at 560 nm using a microplate reader (ARVOmx; PerkinElmer), and the percent absorbance of every well was determined. The OD absorbance values of cells in control wells were arbitrarily set at 100% to determine the percentage of viable cells.
Apoptosis Assay
48 h after transfection with 10 nmol/L miRNA (miR-NC or miR-1293), HCT116+/+, HCT116−/−, and HOC313 cells were double stained with Annexin V-FITC and PI using a MEBCYTO Apoptosis kit (MBL, Nagoya), according to the manufacturer’s instructions. Cells were analyzed by the Accuri C6 Flow Cytometer (BD Biosciences).
Gene Expression Array Analysis
Gene expression array analysis was carried out as previously described.19 Briefly, HCT116+/+, A549, KYSE150, HOC313, and HSC-2 cells were transfected with 10 nmol/L miRNA (miR-NC or miR-1293). Total RNA was extracted 48 h after transfection. For gene expression analysis, the Agilent 8 × 60 K array was used according to the manufacturer’s instructions (Agilent Technologies). The data were analyzed by GeneSpring software (Agilent Technologies, Japan).
Western Blotting
Western blotting was performed as previously described.19 The following primary antibodies were used for western blotting: antibodies against BRD4 (#13440), cleaved PARP (#9541), phospho-histone H2AX (Ser139) (#9718), and RPA70 (#2267) were purchased from Cell Signaling Technology; the antibody against APEX1 (10203-1-AP) was purchased from Proteintech (Chicago, IL); anti-POLD4 antibody (MBS9609765) was obtained from MyBioSource.com (San Diego, CA, USA); and anti-β-actin (A5441) and anti-Vinculin (V9131) were obtained from Sigma-Aldrich (Tokyo, Japan).
Luciferase Activity Assay
The luciferase activity assay was carried out as previously described.19 Luciferase reporter plasmids were constructed by inserting the 3′ UTRs of BRD4, APEX1, RPA1, and POLD4 downstream of the luciferase gene in the pmirGLO Dual-Luciferase miRNA Target Expression Vector (Promega, Madison, WI). All site-specific mutations were generated by the KOD -Plus- Mutagenesis Kit (TOYOBO, Osaka, Japan). Luciferase reporter plasmids or the control plasmid (pmirGLO) were transfected into HOC313 cells using Lipofectamine 2000 (Thermo Fisher Scientific), and 10 nmol/L of the indicated miRNA mimic was also transfected 6 h later. After 2 days, firefly and Renilla luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega), and relative luciferase activity was calculated by normalizing the firefly luciferase reading to its corresponding internal Renilla luciferase control.
Plasmid Construction and Transfection
The BRD4 expression vector was previously generated.14 The BRD4 expression vector was transfected into MIAPaCa2 cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Immunofluorescence Analysis
Immunofluorescence was carried out as described previously.39 For immunostaining of phospho-histone H2AX, cells were fixed with 3.7% formaldehyde, permeabilized with 0.1% Triton X-100, blocked with 3% bovine serum albumin for 30 min, and then incubated with primary antibodies (rabbit anti-phospho-histone H2AX, 1:400 dilution) for 12 h at 4°C. Bound antibodies were visualized using Alexa Fluor 555-conjugated anti-rabbit immunoglobulin G (IgG) antibody (both 1:500 dilution; Life Technologies), and coverslips were mounted in VECTASHIELD containing DAPI (Vector). Images were obtained by confocal fluorescence microscopy (Nikon).
In Vivo Tumor Growth and miRNA Administration
In vivo miRNA administration was performed as previously described.19 6-week-old female BALB/c nude mice were purchased from Oriental Bio Service, Japan. Briefly, a total of 2.0 × 106 cells in 100 μL of PBS were subcutaneously injected into the dorsal side of the mice. After tumor formation on day 3, a mixture of 1 nmol dsRNA (miR-NC or miR-1293) and 100 μL AteloGene (KOKEN, Tokyo, Japan) was administered around the tumor. miRNAs were administered on days 3, 7, 10, 14, and 17, and, on day 19 after cell injection, mice were sacrificed and tumors were resected. Tumor volume was calculated using the following formula: (shortest diameter)2 × (longest diameter) × 0.5. All experimental protocols conducted on the mice were approved by the Tokyo Medical and Dental University Animal Care and Use Committee (A2019-101A).
Immunohistochemistry
Immunohistochemistry was performed as previously described.14 The resected tumors from the xenograft mouse model were fixed in 10% formaldehyde in PBS for 24 h and stored in 70% ethanol and then embedded in paraffin. The following primary antibodies were used for immunohistochemistry. The antibody against BRD4 (HPA061646, 1:500) was purchased from Atlas Antibodies (Stockholm, Sweden), and the anti-RPA70 (ab79398, 1:100) antibody was purchased from Abcam. The anti-APEX1 (10203-1-AP, 1:100) antibody was purchased from Proteintech. The anti-POLD4 antibody (MBS9609765, 1:100) was purchased from MyBioSource.com. For measurement of the density per field, each of three images of sections from three tumors treated with miR-NC or miR-1293 were manually captured, and a threshold value was defined for each image using the single point selection tool on the Nikon NIS Elements Br 4.0 software. The sum density was calculated, and the results were normalized to the density values of tumors treated with miR-NC.
qRT-PCR
Total RNA was extracted using TRIsure reagent (BIOLINE, London, UK) according to the manufacturer’s instructions. For miRNA detection, total RNA was reverse transcribed using the TaqMan Reverse Transcription Kit followed by qRT-PCR performed using Custom TaqMan miRNA assays (Applied Biosystems, Foster City, CA). The miRNA expression level was normalized to that of the internal control RNU6B. The following primers were used for the TaqMan assay (Thermo Fisher Scientific): human miR-1293 (002905) and RNU6B (001093).
HR Repair Analysis
The pHPRT-DRGFP vector and the plasmid expressing I-SceI (pCBASce) were gifts from Maria Jasin (Addgene plasmid # 26476; http://www.addgene.org/26476/; RRID: Addgene_26476) (Addgene plasmid # 26477; http://www.addgene.org/26477/; RRID: Addgene_26477).22,40 U2OS cells were transfected with pHPRT-DRGFP. Puromycin was added 24 h later at 2.5 μg/mL. After puromycin selection, colonies were isolated and analyzed by RT-PCR. U2OS DR-GFP cells contain a single copy of the HR repair reporter substrate DR-GFP, which contains two nonfunctional GFP open reading frames, including one GFP-coding sequence that is separated by a recognition site for the I-SceI endonuclease. The expression of I-SceI leads to the formation of a DSB in the I-SceI GFP allele, which can be repaired by HR using the nearby GFP sequence lacking the N- and C-termini, thereby producing functional GFP that can be detected by flow cytometry. Cells were transfected with miR-1293 or miR-NC for 6–8 h and subsequently transfected with pCBASceI for 48 h. Then, GFP+ cells were analyzed by the Accuri Flow Cytometer.
In Situ Hybridization (ISH) Analysis for the Detection of miRNAs
The ISH assay was performed using miRCURY LNA microRNA ISH Optimization Kit (Exiqon/QIAGEN, Vedbaek, Denmark), as previously described.41 Briefly, formalin-fixed and paraffin-embedded (FFPE) tissue sections were incubated with proteinase-K (Exiqon/QIAGEN) for 10 min at 37°C after deparaffinization. Subsequently, the sections were hybridized with digoxigenin (DIG)-labeled miR-1293 probes (Exiqon/QIAGEN) for 16 h at 60°C and probed with a specific anti-DIG antibody (Sigma) directly conjugated to alkaline phosphatase (AP) (Roche, Basel, Switzerland). Nuclei were counterstained with nuclear fast red (Vector Laboratories, Burlingame, CA).
Public Datasets
To explore the generality of the miRNA expression among colon cancer, ESCC, HNSCC, lung adenocarcinoma, lung squamous cell carcinoma, pancreatic ductal adenocarcinoma, and stomach adenocarcinoma, we examined the public datasets from TCGA (https://cancergenome.nih.gov) retrieved on July 24, 2017. The correlation between prognosis and expression of miR-1293 in ESCC and HNSCC was analyzed by the Kaplan-Meier Plotter (http://kmplot.com/analysis/), a database that integrates gene expression data and clinical data.42
Statistical Analysis
Differences between groups were determined with Student’s t test, one-way ANOVA with Bonferroni adjustment, Fisher’s exact test, Mann-Whitney U test, or log-rank test as appropriate. All statistical analyses were carried out with R software. p values < 0.05 were considered significant.
Author Contributions
Y.T. and Y.G. were involved in the research design, performed the experiments, analyzed the data, and wrote the manuscript. T.M. was involved in the research design, performed the experiments, and analyzed the data. K.T. contributed to public database analysis. J. Inoue was involved in the research design. H.H. supervised the study. J. Inazawa was involved in the research design, wrote the manuscript, and supervised the study.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank Ayako Takahashi and Rumi Mori for technical assistance. This work was supported by KAKENHI(18H02688 and 19K07709) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) and partially supported by the Project for Cancer Research and Therapeutic Evolution (P-CREATE) from Japan Agency for Medical Research and Development, AMED.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.04.001.
Contributor Information
Yasuyuki Gen, Email: ygen.cgen@mri.tmd.ac.jp.
Johji Inazawa, Email: johinaz.cgen@mri.tmd.ac.jp.
Supplemental Information
References
- 1.Belkina A.C., Denis G.V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer. 2012;12:465–477. doi: 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fujisawa T., Filippakopoulos P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017;18:246–262. doi: 10.1038/nrm.2016.143. [DOI] [PubMed] [Google Scholar]
- 3.Zuber J., Shi J., Wang E., Rappaport A.R., Herrmann H., Sison E.A., Magoon D., Qi J., Blatt K., Wunderlich M. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delmore J.E., Issa G.C., Lemieux M.E., Rahl P.B., Shi J., Jacobs H.M., Kastritis E., Gilpatrick T., Paranal R.M., Qi J. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shu S., Lin C.Y., He H.H., Witwicki R.M., Tabassum D.P., Roberts J.M., Janiszewska M., Huh S.J., Liang Y., Ryan J. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–417. doi: 10.1038/nature16508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Segura M.F., Fontanals-Cirera B., Gaziel-Sovran A., Guijarro M.V., Hanniford D., Zhang G., González-Gomez P., Morante M., Jubierre L., Zhang W. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73:6264–6276. doi: 10.1158/0008-5472.CAN-13-0122-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahai V., Kumar K., Knab L.M., Chow C.R., Raza S.S., Bentrem D.J., Ebine K., Munshi H.G. BET bromodomain inhibitors block growth of pancreatic cancer cells in three-dimensional collagen. Mol. Cancer Ther. 2014;13:1907–1917. doi: 10.1158/1535-7163.MCT-13-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Civenni G., Bosotti R., Timpanaro A., Vàzquez R., Merulla J., Pandit S., Rossi S., Albino D., Allegrini S., Mitra A. Epigenetic Control of Mitochondrial Fission Enables Self-Renewal of Stem-like Tumor Cells in Human Prostate Cancer. Cell Metab. 2019;30:303–318. doi: 10.1016/j.cmet.2019.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Donati B., Lorenzini E., Ciarrocchi A. BRD4 and Cancer: going beyond transcriptional regulation. Mol. Cancer. 2018;17:164. doi: 10.1186/s12943-018-0915-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cochran A.G., Conery A.R., Sims R.J., 3rd Bromodomains: a new target class for drug development. Nat. Rev. Drug Discov. 2019;18:609–628. doi: 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- 11.Stathis A., Bertoni F. BET proteins as targets for anticancer treatment. Cancer Discov. 2018;8:24–36. doi: 10.1158/2159-8290.CD-17-0605. [DOI] [PubMed] [Google Scholar]
- 12.Lewin J., Soria J.C., Stathis A., Delord J.P., Peters S., Awada A., Aftimos P.G., Bekradda M., Rezai K., Zeng Z. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J. Clin. Oncol. 2018;36:3007–3014. doi: 10.1200/JCO.2018.78.2292. [DOI] [PubMed] [Google Scholar]
- 13.Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tonouchi E., Gen Y., Muramatsu T., Hiramoto H., Tanimoto K., Inoue J., Inazawa J. miR-3140 suppresses tumor cell growth by targeting BRD4 via its coding sequence and downregulates the BRD4-NUT fusion oncoprotein. Sci. Rep. 2018;8:4482. doi: 10.1038/s41598-018-22767-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Setten R.L., Rossi J.J., Han S.P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019;18:421–446. doi: 10.1038/s41573-019-0017-4. [DOI] [PubMed] [Google Scholar]
- 16.Anthiya S., Griveau A., Loussouarn C., Baril P., Garnett M., Issartel J.P., Garcion E. MicroRNA-Based Drugs for Brain Tumors. Trends Cancer. 2018;4:222–238. doi: 10.1016/j.trecan.2017.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Vazquez A., Bond E.E., Levine A.J., Bond G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 18.Yang F., Zhao W.J., Jia C.L., Li X.K., Wang Q., Chen Z.L., Jiang Q. MicroRNA-876-3p functions as a tumor suppressor gene and correlates with cell metastasis in pancreatic adenocarcinoma via targeting JAG2. Am. J. Cancer Res. 2018;8:636–649. [PMC free article] [PubMed] [Google Scholar]
- 19.Fujiwara N., Inoue J., Kawano T., Tanimoto K., Kozaki K., Inazawa J. miR-634 activates the mitochondrial apoptosis pathway and enhances chemotherapy-induced cytotoxicity. Cancer Res. 2015;75:3890–3901. doi: 10.1158/0008-5472.CAN-15-0257. [DOI] [PubMed] [Google Scholar]
- 20.Sun C., Yin J., Fang Y., Chen J., Jeong K.J., Chen X., Vellano C.P., Ju Z., Zhao W., Zhang D. BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell. 2018;33:401–416. doi: 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stratikopoulos E.E., Dendy M., Szabolcs M., Khaykin A.J., Lefebvre C., Zhou M.M., Parsons R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell. 2015;27:837–851. doi: 10.1016/j.ccell.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce A.J., Hu P., Han M., Ellis N., Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev. 2001;15:3237–3242. doi: 10.1101/gad.946401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou Y., Dang J., Chang K.Y., Yau E., Aza-Blanc P., Moscat J., Rana T.M. MIR-1298 inhibits mutant KRAS-driven tumor growth by repressing FAK and LAMB3. Cancer Res. 2016;76:5777–5787. doi: 10.1158/0008-5472.CAN-15-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X., Liu Y., Granberg K.J., Wang Q., Moore L.M., Ji P., Gumin J., Sulman E.P., Calin G.A., Haapasalo H. Two mature products of MIR-491 coordinate to suppress key cancer hallmarks in glioblastoma. Oncogene. 2015;34:1619–1628. doi: 10.1038/onc.2014.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y., Li M., Ding Y., Fan Z., Zhang J., Zhang H., Jiang B., Zhu Y. Serum MicroRNA profile in patients with colon adenomas or cancer. BMC Med. Genomics. 2017;10:23. doi: 10.1186/s12920-017-0260-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chattopadhyay E., Singh R., Ray A., Roy R., De Sarkar N., Paul R.R., Pal M., Aich R., Roy B. Expression deregulation of mir31 and CXCL12 in two types of oral precancers and cancer: importance in progression of precancer and cancer. Sci. Rep. 2016;6:32735. doi: 10.1038/srep32735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bobola M.S., Blank A., Berger M.S., Stevens B.A., Silber J.R. Apurinic/apyrimidinic endonuclease activity is elevated in human adult gliomas. Clin. Cancer Res. 2001;7:3510–3518. [PubMed] [Google Scholar]
- 28.Kelley M.R., Cheng L., Foster R., Tritt R., Jiang J., Broshears J., Koch M. Elevated and altered expression of the multifunctional DNA base excision repair and redox enzyme Ape1/ref-1 in prostate cancer. Clin. Cancer Res. 2001;7:824–830. [PubMed] [Google Scholar]
- 29.Moore D.H., Michael H., Tritt R., Parsons S.H., Kelley M.R. Alterations in the expression of the DNA repair/redox enzyme APE/ref-1 in epithelial ovarian cancers. Clin. Cancer Res. 2000;6:602–609. [PubMed] [Google Scholar]
- 30.Dahai Y., Sanyuan S., Hong L., Di Z., Chong Z. A relationship between replication protein A and occurrence and prognosis of esophageal carcinoma. Cell Biochem. Biophys. 2013;67:175–180. doi: 10.1007/s12013-013-9530-y. [DOI] [PubMed] [Google Scholar]
- 31.Hoxha M., Fabris S., Agnelli L., Bollati V., Cutrona G., Matis S., Recchia A.G., Gentile M., Cortelezzi A., Morabito F. Relevance of telomere/telomerase system impairment in early stage chronic lymphocytic leukemia. Genes Chromosomes Cancer. 2014;53:612–621. doi: 10.1002/gcc.22171. [DOI] [PubMed] [Google Scholar]
- 32.Li S., Xu K., Gu D., He L., Xie L., Chen Z., Fan Z., Zhu L., Du M., Chu H. Genetic variants in RPA1 associated with the response to oxaliplatin-based chemotherapy in colorectal cancer. J. Gastroenterol. 2019;54:939–949. doi: 10.1007/s00535-019-01571-z. [DOI] [PubMed] [Google Scholar]
- 33.Zhang S., Chao H.H., Wang X., Zhang Z., Lee E.Y.C., Lee M.Y.W.T. Loss of the p12 subunit of DNA polymerase delta leads to a defect in HR and sensitization to PARP inhibitors. DNA Repair (Amst.) 2019;73:64–70. doi: 10.1016/j.dnarep.2018.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang Q.M., Akashi T., Masuda Y., Kamiya K., Takahashi T., Suzuki M. Roles of POLD4, smallest subunit of DNA polymerase δ, in nuclear structures and genomic stability of human cells. Biochem. Biophys. Res. Commun. 2010;391:542–546. doi: 10.1016/j.bbrc.2009.11.094. [DOI] [PubMed] [Google Scholar]
- 35.Gaillard H., García-Muse T., Aguilera A. Replication stress and cancer. Nat. Rev. Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- 36.Takashima Y., Kikuchi E., Kikuchi J., Suzuki M., Kikuchi H., Maeda M., Shoji T., Furuta M., Kinoshita I., Dosaka-Akita H. Bromodomain and extraterminal domain inhibition synergizes with WEE1-inhibitor AZD1775 effect by impairing nonhomologous end joining and enhancing DNA damage in nonsmall cell lung cancer. Int. J. Cancer. 2019;1:1–12. doi: 10.1002/ijc.32515. [DOI] [PubMed] [Google Scholar]
- 37.Stanlie A., Yousif A.S., Akiyama H., Honjo T., Begum N.A. Chromatin reader Brd4 functions in Ig class switching as a repair complex adaptor of nonhomologous end-joining. Mol. Cell. 2014;55:97–110. doi: 10.1016/j.molcel.2014.05.018. [DOI] [PubMed] [Google Scholar]
- 38.Shimada Y., Imamura M., Wagata T., Yamaguchi N., Tobe T. Characterization of 21 newly established esophageal cancer cell lines. Cancer. 1992;69:277–284. doi: 10.1002/1097-0142(19920115)69:2<277::aid-cncr2820690202>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 39.Gen Y., Yasui K., Kitaichi T., Iwai N., Terasaki K., Dohi O., Hashimoto H., Fukui H., Inada Y., Fukui A. ASPP2 suppresses invasion and TGF-β1-induced epithelial-mesenchymal transition by inhibiting Smad7 degradation mediated by E3 ubiquitin ligase ITCH in gastric cancer. Cancer Lett. 2017;398:52–61. doi: 10.1016/j.canlet.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 40.Richardson C., Moynahan M.E., Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gokita K., Inoue J., Ishihara H., Kojima K., Inazawa J. Therapeutic Potential of LNP-Mediated Delivery of miR-634 for Cancer Therapy. Mol. Ther. Nucleic Acids. 2020;19:330–338. doi: 10.1016/j.omtn.2019.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nagy Á., Lánczky A., Menyhárt O., Győrffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018;8:9227. doi: 10.1038/s41598-018-27521-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.