

Abstract
Currently, most antioxidants do not show any favorable clinical outcomes in reducing myocardial ischemia-reperfusion (I/R) injury, suggesting an urgent need for exploring a new regulator of redox homeostasis in I/R hearts. Here, using heart-specific transgenic (TG) and knockdown (KD) mouse models, tumor susceptibility gene 101 (Tsg101) is defined as a novel cardiac-protector against I/R-triggered oxidative stress. RNA sequencing and bioinformatics data surprisingly reveal that most upregulated genes in Tsg101-TG hearts are transcribed by Nrf2. Accordingly, pharmacological inhibition of Nrf2 offsets Tsg101-elicited cardio-protection. Mechanistically, Tsg101 interacts with SQSTM1/p62 through its PRR domain, and promotes p62 aggregation, leading to recruitment of Keap1 for degradation by autophagosomes and release of Nrf2 to the nucleus. Furthermore, knockout of p62 abrogates Tsg101-induced cardio-protective effects during I/R. Hence, our findings uncover a previously unrecognized role of Tsg101 in the regulation of p62/Keap1/Nrf2 signaling cascades and provide a new strategy for the treatment of ischemic heart disease.
Keywords: Oxidative stress, Myocardial ischemia-reperfusion, Nrf2, p62 aggregation, Cardiac autophagy, Tsg101
Graphical abstract
Highlights
-
•
Most antioxidants are disappointing in clinically reducing cardiac oxidative damage.
-
•
Cardiac Tsg101 expression is altered during development and upon oxidative stress.
-
•
Tsg101 is a novel cardiac-protector against ischemia/reperfusion injury.
-
•
Tsg101 is new positive regulator of Nrf2 signaling pathway.
-
•
Tsg101 promotes p62 aggregation and autophagic degradation of Keap1.
1. Introduction
Myocardial infarction remains a leading cause of mortality in the world. Currently, effective therapy for ischemic heart disease is the immediate reperfusion through thrombolytic agents or primary percutaneous coronary intervention [1]. However, reperfusion causes massive generation of reactive oxygen species (ROS) by the myocardium itself or by infiltrating inflammatory cells, leading to extensive cardiomyocyte death (apoptosis, necrosis and necroptosis) in ischemic hearts [[2], [3], [4]]. Hence, over the past 5 decades, tremendous effort has been made to test whether various antioxidants (i.e., vitamins C/E, N-acetylcysteine, and SODs) can scavenge free radicals or target the sources of ROS in the setting of myocardial ischemia/reperfusion (I/R). Unfortunately, clinical trials showed that most antioxidants did not have any favorable outcomes in reducing myocardial reperfusion injury [5,6], suggesting the incomplete knowledge about endogenous regulatory mechanisms underlying I/R-triggered oxidative stress. Therefore, it will be of urgent need and significance to define the role of any new cardiac endogenous molecules in the regulation of redox homeostasis during I/R.
Tumor susceptibility gene 101 (Tsg101) was originally identified as a tumor suppressor, as it was mutated or deleted in human breast cancer patient samples [7]. Later on, multiple studies elucidated that Tsg101 was not a tumor suppressor but an oncogene and plays an important role in tumor progression through its regulation of cell cycle mediators, i.e., p53, p21 and MDM2 [[8], [9], [10]]. Recent evidence further demonstrates that Tsg101 is actually a critical member of the endosomal sorting complexes required for transport (ESCRT) [11]. As a key component of the ESCRT complex I, Tsg101 enables ubiquitination and sorting of endosomal cargo into multivesicular bodies (MVB) and late endosomes to be targeted for degradation by lysosomes [11]. Notably, Tsg101 is essential for viruses (i.e. HIV and Ebola) to reproduce and release from cells. For example, Tsg101 could interact with HIV-1 Gag protein through the PTAP domain to regulate assembly of HIV in cells [12,13]. More interestingly, our recent work shows that Tsg101 is also highly expressed in animal hearts, and involved in exercise-induced physiological cardiac hypertrophy as well as endotoxin-triggered cardiac dysfunction [14,15]. Nevertheless, the functional role of Tsg101 in myocardial I/R-caused oxidative damage remains totally unknown.
At present, the Kelch-like ECH-associated protein 1 (Keap1)-Nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is a well-characterized cellular defense mechanism against oxidative stress [16,17]. Under quiescent conditions, the transcriptional factor Nrf2, bound to its partner, Keap1, is constitutively degraded through the ubiquitin-proteasome pathway [16,17]. However, under stress conditions, the specific cysteine residues on Keap1 required for binding to Nrf2 are modified, leading to release and translocation of Nrf2 to the nucleus for initiating transcription of genes regulated by the antioxidant response elements (ARE) [16,17]. Of more interest, SQSTM1/p62 (referred to as p62 hereafter), a selective autophagy adaptor, has been recently identified as the key regulator of the Keap1-Nrf2 pathway [18,19]. For example, several studies have shown that aggregation of p62 enhances its interaction with Keap1, which sequesters Keap1 to be degraded by autophagosomes and consequently, activates translocation of Nrf2 to the nucleus [18,19]. Nonetheless, how to regulate p62-dependent sequestration of Keap1 in the heart has never been investigated. Thus, it will be very significant to determine whether Tsg101 modulates the p62/Keap1/Nrf2 axis in favor of protection against I/R-induced cardiac injury.
In this study, we first determined cardiac expression levels of Tsg101 during development and upon oxidative stress. Then, cardiac-specific overexpression and knockdown mouse models were utilized to examine possible roles of Tsg101 in response to both ex vivo and in vivo myocardial ischemia/reperfusion (I/R) injury. In addition, we performed RNA-seq, bioinformatics assay, and a series of molecular/biochemical analyses to dissect the mechanisms underlying Tsg101-elicited effects. Lastly, using both pharmacological and genetic inhibition approaches, we further validated the functional roles of Tsg101 in mouse hearts upon ischemia/reperfusion injury and cultured cardiomyocytes in response to oxidative stress as well as the associated mechanisms.
2. Materials and methods
2.1. Animal models
All mice used in this study were maintained and bred in the Division of Laboratory Animal Resources at the University of Cincinnati Medical Center. All animal protocols conformed to the Guidelines for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences, and approved by the University of Cincinnati Animal Care and Use Committee. Tsg101-transgenic (TG) mice (FVB/N background) were generated by constructing a DNA vector in which a 1.176-Kb cDNA fragment of murine Tsg101 gene was inserted to the downstream of the cardiac-specific α-myosin heavy chain promoter (α-MHCp) in the Transgenic Animal and Genome Editing Core at the Cincinnati Children's Hospital Center [14]. Mice having the floxed Tsg101 allele (Tsg101 fl/fl) under the 129/SvJ genetic background were provided by Dr. Kay-Uwe Wagner (Wayne State University) [20]. Mice expressing an inducible Cre recombinase transgene driven by the αMHC promoter (αMHC-MerCre-Mer) were purchased from the Jackson Laboratory (Stock No: 005657, C57/BL6 background). Tsg101fl/fl mice were crossed with αMHC-MerCreMer mice to obtain heterozygotes (Cre+/Tsg101flox/wt, B6/129 background), which were injected i.p. with tamoxifen (Sigma, 30 μg/g, 3 consecutive days) at 8-week old to induce knockdown of Tsg101 in the heart. Cre−/Tsg101flox/wt littermates were used as control mice (B6/129 background). All biochemical and physiological experiments were performed in mice at one week after the last injection. P62 global knockout mice (C57/BL6 background) were provided by Dr. Tetsuro Ishii (University of Tsukuba, Tsukuba, Japan) [21]. Tsg101-TG/p62−/− (FVB/B6 background) were obtained by crossing Tsg101-TG mice with p62-knockout mice. All primers used for genotyping are included in the Supplemental Table S1.
2.2. Myocardial global no-flow I/R ex vivo
Ischemia/reperfusion (I/R) of mouse hearts ex vivo by the Langendorff method was described previously [22]. In brief, hearts were dissected from mice after anesthesia, immediately cannulated and then hung to the Langendorff system. Hearts were perfused with a phosphate-free Krebs-Henseleit buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, 0.003 mM Na2EDTA, 11 mM Glucose pH 7.4) equilibrated at 37 °C with 5% CO2/95% O2. After a 10-min equilibration period, mouse hearts underwent no-flow global ischemia for 30 min, 45 min or 60 min, followed by 60-min reperfusion. During the reperfusion, the parameters of left ventricular maximum rate of contraction (+dP/dt), and minimum rate of relaxation (–dP/dt) were recorded with a DigiMed Heart performance Analyzer (Micro-Med).
2.3. Ex vivo I/R-induced cardiac damage analysis
Total lactate dehydrogenase (LDH) in the Langendroff reperfusion buffer released from I/R hearts were determined by using an in vitro Toxicology Assay Kit (Sigma) and expressed as units per ml of wet heart weight (U/ml/g). The perfused buffer in the first 10 min were collected and then mixed with the Lactate Dehydrogenase Assay Mixture complex. After 30-min incubation, the absorbance at the 490 nm and background absorbance at 690 nm were measured in a microplate reader (EZ Read 400, Bio-Tek Instruments). Relative LDH value was determined by subtracting absorbance value at 690 nm from absorbance value at 490 nm.
I/R-induced cardiac apoptosis was assessed by TUNEL staining and Caspase-3 activity [4]. For the TUNEL staining, hearts were fixed in 4% paraformaldehyde, embedded in paraffin, cut into 5-μm thickness sections and treated per the instruction of the DeadEnd Fluorometric TUNEL kit (Promega), followed by staining with an anti-actin (α-Sarcomeric) antibody (Sigma-Aldrich) and Alexa Fluor-568 goat anti mouse secondary antibody. The nuclei were stained with DAPI. TUNEL-positive (green) nuclei located in cardiomyocytes were counted from 5 randomly chosen microscope fields (400X) of the mid-ventricular section and were expressed as a percentage of total nuclei from the same field. Caspase-3 activity was determined in cardiac lysates (200 μg) using Caspase-3/CPP32 Fluorometric Assay kit (BioVision). Results were standardized to the sham group for comparison of the fold change in caspase-3 activity.
2.4. Myocardial I/R in vivo and infarct size analysis
In vivo myocardial I/R surgery was performed as previously described [4]. Briefly, male adult mice (10–12 week old) were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (5 mg/kg), then orally intubated, and connected to a mouse ventilator (Model 845, Harvard Apparatus). Subsequently, these mice underwent the occlusion of the left anterior descending coronary artery (LAD) by ligation with 6-0 silk sutures around fine PE-10 tubing. The suture was then tied and tightened for 30min or 1 h, followed by varying periods of reperfusion. Sham-operated mice were subjected to the same surgical procedures, except that the suture was passed under the LAD but not tied. To measure myocardial infarction size, Evans blue (0.2–0.5 ml of a 2% solution; Sigma-Aldrich) was injected into the aorta to demarcate the non-ischemic myocardium, as previous described [4]. The heart was rapidly excised, immediately frozen in −80 °C fridge for 4 h and sliced into 5 slices which were subsequently incubated in 1% 2,3,5-triphenyltetrazolium chloride (Sigma-Aldrich) for 30 min at 37 °C to distinguish the ischemia and infarct myocardium within area at risk (AAR). And then the slices were fixed in 10% neutral formaldehyde buffer and were digitally photographed 12 h later. The infarct size (IS), area at risk (AAR), and non-ischemic left ventricle (LV) were assessed with image analysis software. The ratio of AAR/LV and IS/AAR were calculated.
2.5. Assessment of cardiac function using echocardiography
Transthoracic echocardiography was performed by one high-resolution micro-imaging system (Visual Sonics Vevo 2100) with a 40-MHz transducer, as previously described [23]. Mice were anesthetized with 1.5% isoflurane. Hearts were imaged in 2D long-axis view at the level of the greatest left ventricular (LV) diameter and also the mid-ventricular short axis. LV internal diameters end-diastole and end-systole (LVIDd, LVIDs), Interventricular septal end diastole and end systole (IVSd, IVSs), Left ventricular posterior wall end diastole and end systole (LVPWd, LVPWs) were measured from M-mode recordings. LVEF and LVFS were calculated as: EF% = [(LVIDd) 3 - (LVIDs) 3/(LVIDd) 3 × 100]; FS% = [(LVIDd - LVIDs)/LVDId × 100].
2.6. Measurement of ROS levels in the heart
Dihydroethidium (DHE) dye (Thermo Fisher) was reconstituted in DMSO and diluted with PBS to a concentration of 5 μM before use. Fresh samples of the left ventricular myocardium after ex vivo ischemia/reperfusion were sliced and incubated with the diluted DHE at 37 °C for 10 min in a dark room, followed by washing with PBS for 3 times. The fluorescence intensity was examined under a confocal LSM 710 (Carl Zeiss Microimaging, Jena, Germany). In addition, ROS levels were determined in heart homogenates by the fluorescence indicator DCFH, as described previously [24]. Images were taken under fluorescence microscopes (Olympus, Japan) and analyzed using ImageJ software. Three myocardial sections from each group were analyzed. For quantification, three randomly selected visual fields were analyzed in each section.
2.7. Isolation and culture of adult animal cardiomyocytes
Cardiomyocytes were isolated from adult rats (4–5 week old, male, Sprague Dawley from Charles River Laboratory) or adult mice (2-month old, male, FVB/N) as previously described [23,24]. Briefly, the heart was excised quickly after animals were anesthetized. Then the aorta was cannulated and mounted on a Langendorff perfusion apparatus and perfused with Ca-free solution (113 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 12 mM NaHCO3, 10 mM KHCO3, 10 mM HEPES, 30 mM taurine, 10 mM BDM, and 5.5 mM glucose, pH 7.4) at 37 °C for 4 min. The perfusion buffer was replaced with the same solution containing Liberase™ TH (Thermolysin High) Research Grade (0.25 mg/ml, Roche) and CaCl2 (50 μM), and continued for 8–15 min until the heart became flaccid. After removing the atria, the ventricular tissue was teased to small pieces using forceps and gently triturated with a transfer pipette until clumps disappear. The cell suspension was filtered through a 240 μm mesh screen and cardiomyocytes were allowed to settle by gravity for 15 min. The adult animal cardiomyocytes were cultured on laminin (10 μg/ml, Life technologies)-coated plates with AW medium (Cellutron life technologies) with 10% FBS, followed by treatment with H2O2 (200 μM) for 1 h. Then, cell survival was determined using a MTS analysis kit (Promega).
2.8. Isolation of neonatal animal cardiomyocytes and infection/transfection with recombinant adenoviruses and siRNAs
Neonatal cardiomyocytes were isolated from ventricles of 1–3 day old neonatal Sprague Dawley rats, or mice (FVB/N, 1–3 day old) using a neonatal cardiomyocyte isolation kit (Worthington Biochemical, LK003300). Per the manufacturer's instructions, cell pellets were obtained after enzyme digestion and centrifugation. Cells were plated and incubated in cell culture incubator. 2 h later, adherent fibroblasts were discarded and non-adherent cardiomyocytes were re-suspended and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FBS, and 1% penicillin/streptomycin at 37 °C and 5% CO2. Subsequently, cells were allowed to adhere for 48 h, neonatal rat cardiomyocytes (NRCMs) were infected with recombinant adenovirus vectors (Ad.GFP-Tsg101 or Ad. GFP) for 24 h, and transfected with 50 nM p62-siRNA (Sigma, SASI_Rn01_00098873 and SASI_Rn01_00098874) or control siRNA (Sigma) for 48 h, using the lipofectamine 3000 reagent (Thermo-Fisher) according to the manufacturer's instructions. Subsequently, these NRCMs were treated with H2O2 (500 μM) for 2 h to measure ROS levels, or for 6 h to determine cell survival, as described above.
2.9. Immunofluorescence staining
Neonatal rat cardiomyocytes (NRCMs) were cultured on coverslips, fixed in 4% PFA (Sigma-Aldrich), permeabilized with 0.3% Triton X-100 in PBS and then blocked with 5% bovine serum albumin (BSA) for 30 min at room temperature. Coverslips were incubated overnight at 4 °C with the following antibodies: p62 (Sigma, P0067, 1:500), Keap1 (Proteintech, 10503-2-AP, 1:200), LC3A/B (Cell Signaling Technology, 4108S, 1:500), EEA1 (Cell Signaling Technology, 2411S, 1:500), CD63 (Santa Cruz, sc-15363, 1:250). After being washed with PBS for 3 times, coverslips were incubated with fluorescence conjugated secondary antibodies for 60 min at room temperature (Thermo Fisher Scientific). DAPI (Invitrogen) was used for nuclear staining. Cells were imaged with a confocal LSM 710 (Carl Zeiss Microimaging, Jena, Germany). Images were analyzed with Image J software.
2.10. Preparation of total, soluble and insoluble proteins, subcellular fractions, Western-blotting and co-immunoprecipitation analysis
Total protein samples were extracted from cells or animal hearts, which were homogenized in NP-40 lysis buffer (Invitrogen, FNN0021) containing 1 mM PMSF and protease inhibitor cocktail (Roche). For soluble protein extraction, mouse hearts or cells were homogenized in lysis buffer consisting of 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM Na2EDTA, 1 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail (Roche), and were lysed on ice for 20 min during which the sample was shaken 15s every 5 min. Samples were centrifuged for 15 min at 12000 rpm in 4 °C and the supernatant was used as the soluble protein. For insoluble protein extraction, after centrifugation for the soluble protein (above), the pellet was washed twice with the same buffer and then used as the detergent-insoluble fraction. Such fractions were dissolved in SDS sample buffer [20 mmol/L Tris (pH6.8), 150 mM NaCl, 1%SDS, 1 mM Na2EDTA, 1 mM EDTA, 1 mM PMSF, and protease inhibitor cocktail (Roche)]. Samples were centrifuged 15 min at 12000 rpm in 4 °C and the supernatant was used as the insoluble protein.
Nuclear and cytosol extracts were prepared using the Nuclear Extraction Kit (Abcam, ab113474), according to the manufacturer's instructions. Subsequently, these cellular fractions underwent Western-blotting assays (below). Lamin B1 was used as nuclear fraction marker, and β-Tubulin or GAPDH was used as loading control for cytosol protein.
Western-blotting analysis was performed as described previously [25]. In brief, protein samples (30–100 μg) were loaded to 10–12% SDS-PAGE and then transferred to 0.2 or 0.45 μm nitrocellulose blotting membrane (GE Healthcare Life Science). The membranes were blocked with 5% non-fat milk in TBS for 1 h and subsequently incubated at 4 °C overnight with primary antibodies against Tsg101 (Santa Cruz, sc-7964, 1:500), p62 (Sigma, P0067, 1:500), Keap1 (Proteintech, 10503-2-AP, 1:500), Nrf2 (Cell Signaling, 12721s, 1:1000), Lamin B1 (Cell Signaling, 13435S, 1:500), β-Tubulin (Abcam, ab6046, 1:1000), LC3A/B (Cell Signaling, 4108S, 1:500), GAPDH (Cell Signaling, 2118S, 1:2000). After overnight incubation, membranes were incubated in corresponding HRP-conjugated secondary antibody for 1 h. Finally, the specific bands were detected by super ECL reagent (Pierce, Rockford, IL). The corresponding bands were quantified by densitometry and the intensity of the GAPDH band was used as a loading control for comparison between samples.
For co-immunoprecipitation (co-IP) assays, mouse heart tissues or cells were homogenized and lysed in IP lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, 1 mM Na2EDTA, 1 mM EDTA, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride, complete protease inhibitor cocktail (Roche)]. After determination of protein concentration, 500 μg protein of each sample was incubated with 1–2 μg of corresponding primary antibodies overnight at 4 °C on a rotary wheel, followed by incubation with Protein G Agarose Beads (Cell Signaling) for 4 h. The beads were then separated by centrifugation, and washed with ice-cold lysis buffer for 5 times. The immuno-precipitates were eluted in same volume of 2x SDS loading buffer by boiling at 95 °C for 5 min. The eluted proteins were subjected to SDS-PAGE for Western-blotting analysis.
2.11. RNA-sequencing, bio-informatics and RT-PCR analysis
Total RNAs from heart samples or NRCMs were isolated using Trizol reagent (Invitrogen), and purified by the miRNeasy Mini kit (Qiagen). The quality and concentration of RNA was measured by the NanoDrop 2000 system (Thermo Fisher). RNA-sequencing analysis was performed using total RNAs isolated from hearts of Tsg101-TG and WT mice (2-month old, male) by the Genomics, Epigenomics and Sequencing Core (GESC) at the University of Cincinnati. To analyze differential gene expression, sequence reads were aligned to the mouse genome by using standard pipeline, which was performed by The Laboratory for Statistical Genomics and Systems Biology at the University of Cincinnati. Specifically, sequence alignment was done by using Tophat [26] and differential expression analysis was done by using edgeR [27]. 48 genes with adjusted p-value < 0.01 and fold change >3 were chosen to perform Promoter binding motif analysis using the Homer Known Motif Enrichment software. RT-PCR was performed using the miScript PCR Starter kit (Qiagen) to generate complementary DNA and the SYBR Green ER qPCR SuperMix (Invitrogen, Carlsbad, CA). Each reaction was performed in duplicate, and relative fold expression of every target gene expression normalized to GAPDH levels was calculated by the 2−ΔΔCt method. All primer sequences for RT-PCR are listed in Supplemental Table S2.
2.12. Measurement of autophagic flux in vivo and in vitro
The in vivo autophagic flux analysis was performed as described previously [28]. In brief, mice were i.p. injected with chloroquine (CQ, 50 mg/kg; Thermo-Fisher, AC45524500), an inhibitor for autophagosome-lysosome fusion, and PBS-injected mice as controls. Six hours after injection, mouse hearts were excised, minced and homogenized for Western-blotting analysis of the LC3A/B levels. The in vitro autophagy flux analysis was conducted in neonatal rat cardiomyocytes which were transfected with mRFP-GFP-LC3 (Addgene plasmid # 21074) and Myc-Tsg101 or myc-vector (Origene, MR206104, PS100001) for 48 h. Then, these transfected cells were treated with or without BafA1 (100 nM) for 4 h and fixed with 4% paraformaldehyde (PFA). The yellow dots represent combined signals of GFP with mRFP in autophagosomes, whereas the red dots that do not overlap with GFP fluorescence signal stand for autolysosomes. The number of autophagosomes and autolysosomes were quantified in each cell (20–30 cells per group) under a confocal fluorescence microscope.
2.13. Tsg101-p62 interaction domain analysis
DYKDDDDK (Flag)-tagged human p62 and its truncations, M1 (amino acids 1–102), M2 (amino acids 103–440), M3 (amino acids 103–343), M4 (amino acids 103–440, D321–390) and M5 (amino acids 103–320), were generously provided by Dr. Zhuo-Wei Hu (Peking Union Medical College), as described previously [29]. mCherry-Tsg101 was a gift from James Hurley (Addgene plasmid #21505; http://n2t.net/addgene:21505). The different truncations of human Tsg101-myc were generously offered by Dr. Tzu-Hao Cheng (National Yang-Ming University), as described previously [30]. Tsg101-p62 interaction domain analysis was performed in HEK293T cells by co-transfection of Flag-p62 with respective truncated Tsg101 plasmids, or mCherry-Tsg101 with respective truncated p62 plasmids for 48 h. Then cells were collected and lysed for Western-blotting and co-IP analysis, as described above. For domain function analysis, HEK293 cells were seeded at 1 × 105 in 6-well plates and transfected with Tsg101 truncates for 48 h. Subsequently, cells were treated with 500 μM H2O2 for 6 h, after which cells were washed 3 times with PBS. Cells were then incubated with 2 μM DHE in serum-free medium at 37 °C for 15 min, washed once with serum-free medium. Images were taken under fluorescence microscopes for determination of ROS levels. In parallel, cells were seeded at 1 × 104 in 96-well plates and transfected with Tsg101 truncates for 48 h. Then cells were treated with 500 μM H2O2 for 12 h, followed by the addition of MTS reagent (20 μl) to determine cell survival, as described above.
2.14. Statistics
Data are presented as means ± SD. Analysis of variance (ANOVA) was conducted first across all investigated groups in measurements of myocardial function during ex vivo I/R in which we performed the Shapiro-Wilk test for normality, and there was no evidence of deviation from normality for all variables. Then post hoc pairwise tests were performed with assessment of statistical significance after Bonferroni correction of p values. P < 0.05 was considered statistically significant when Student's t-test was used for two-group comparisons.
3. Results
3.1. Cardiac Tsg101 expression is altered during development and upon oxidative stress
We first determined the Tsg101 expression pattern during cardiac development in mice. Western-blotting analyses showed that Tsg101 was highly expressed in neonatal hearts, but gradually decreased significantly from postnatal day 7 into adulthood (Fig. 1A). Similarly, Tsg101 protein levels in isolated adult mouse cardiomyocytes were significantly lower in comparison to neonatal cardiomyocytes (Fig. 1B). Given that neonatal cardiomyocytes are more resistant to oxidative stress than adult cardiomyocytes [31], these results suggest that Tsg101 may be critical for the heart in response to ischemic conditions. Indeed, we observed that the expression of Tsg101 was greatly reduced by ~50% and ~75% in the ex vivo I/R hearts (IR: 45min/1 h and 1h/1 h, respectively), compared to sham samples (Fig. 1C). However, protein levels of Tsg101 were significantly elevated in the in vivo I/R hearts (Fig. 1D). These inconsistent data suggest that the in vivo upregulation of Tsg101 may be compensatory for the heart to maintain its function and animal survival during I/R. To exclude such in vivo compensatory effects caused by neurohumoral factors and inflammatory components, we utilized the in vitro cultured adult rat cardiomyocytes, a well-controlled system, which underwent H2O2 treatment for 1 h. We observed that exposure of cardiomyocytes to H2O2 caused a remarkable down-regulation of Tsg101 expression, compared to PBS-treated cells (Fig. 1E). Put together, these data indicate that reduction of Tsg101 may be responsible for the sensitivity of cardiomyocytes to oxidative damage, and the in vivo transient elevation of Tsg101 may provide compensatory protection against I/R-induced cardiac injury.
Fig. 1.

Cardiac Tsg101 expression is altered during development and upon oxidative stress. (A) Representative immunoblots and quantitative analyses showing protein levels of Tsg101 in cardiac lysates extracted from postnatal day (P) 1 (P1), P7, P21 and adult (2-month old, male) mice (FVB/N). *, p < 0.05 vs. P1. (B) Representative immunoblots and quantitative analyses showing protein levels of Tsg101 in cardiomyocytes isolated from neonates (P1) and adult (2-month old, male) mice (FVB/N). *, p < 0.05 vs. neonates. (C) Representative immunoblots and quantitative analysis showing protein levels of Tsg101 in 2-month old mouse (FVB/N, male) hearts subjected to ex vivo 30-min, 45-min or 1-h global ischemia followed by 1-h reperfusion. *, p < 0.05 vs. pre-I/R samples. (D) Representative immunoblots and quantitative analyses revealing protein levels of Tsg101 in 2-month old mouse (FVB/N, male) hearts subjected to in vivo 30-min or 1-h ischemia followed by 4-h reperfusion. *, p < 0.05 vs. Sham-operated samples. (e) Representative immunoblots and quantitative analysis showing protein levels of Tsg101 in adult Sprague Dawley rat (4–5 week old, male) cardiomyocytes treated with PBS or H2O2 (100 μM) for 1 h *, p < 0.05 vs. PBS. GAPDH or α-Actin was used as a loading control.
3.2. Overexpression of Tsg101 improves the post-ischemic recovery of cardiac function
To explore the possible role of Tsg101 in I/R-induced cardiac damage, we next generated a heart-specific Tsg101-transgenic (TG) mouse model in which Tsg101 was driven by a cardiac-specific promoter, α-MHC promoter (Fig. 2A). Western blotting showed that expression levels of Tsg101 were increased by ~4.5-fold in TG-hearts rather than other tissues, compared to WT controls (Fig. 2B/C). Importantly, TG-hearts exhibited better functional recovery than WT-hearts during I/R, as measured by the rates of contraction (+dP/dt) and relaxation (-dP/dt) (Fig. 2D/E). In addition, the release of lactate dehydrogenase (LDH), a biochemical marker for myocyte necrosis, was greatly reduced in TG-hearts during reperfusion, compared to WT samples (Fig. 2F). Remarkably, the degree of I/R-triggered apoptotic death, as evaluated by TUNEL staining, was lower in TG-cardiomyocytes than WTs (Fig. 2G/H). Caspase-3 activity was also significantly decreased in TG hearts upon I/R, compared to WT controls (Fig. 2I). Together, these data indicate that elevation of Tsg101 improves cardiac function recovery and reduces cardiomyocyte death (necrosis and apoptosis) during ex vivo I/R.
Fig. 2.

Tsg101-transgenic (TG) hearts exhibit improved recovery of cardiac function after I/R injury. (A) Schematic diagram showing the Tsg101-TG construct. (B) Representative Western blots and (C) quantification analyses showing the Tsg101 expression in heart, liver, spleen, lung, and kidney of WT- and TG-mice (FVB/N, male, 10-12-week old). (D/E) Tsg101-overexpressing hearts (TG) showed better contractile function recovery after 45-min global ischemia followed by 1-h reperfusion, compared to wild-type (WT) hearts. n = 6 for WT, n = 6 for TG. *, p < 0.05 vs. WT. (F) Total LDH in the coronary effluent collected during the first 10 min of reperfusion from WT- and TG-hearts. *, p < 0.05 vs. WT. Representative images (G) and quantitative results (H) of the TUNEL staining in WT- and TG-hearts after ex vivo I/R (45min/2 h). Green, TUNEL-positive nuclei; Red, α-actinin; blue, DAPI. *, p < 0.05 vs. WT. (I) Caspase-3 activity in the WT- and TG-hearts subjected to ex vivo I/R (45min/1 h). *, p < 0.05 vs. WT. (J) Representative in vivo myocardial infarction images (white/gray: infarct size; red: area at risk; and blue: non-ischemic area) and (K) quantitative results of infarction size and (L) the ratio of risk region to left ventricular (LV) area. *, p < 0.05 vs. WTs. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To further determine the consequence of increased Tsg101 expression under in vivo I/R conditions, we subjected WT and TG hearts to in vivo 1-h myocardial ischemia, via coronary artery occlusion, followed by 24-h reperfusion. TG-mice exhibited smaller infarction size than WTs (p < 0.01) (Fig. 2J/K), whereas the region at risk was not significantly different between these two groups (Fig. 2L). In addition, TG-mice exhibited better cardiac function than WT mice at 24 h after I/R (Supplemental Table S3A). Moreover, TG mice showed less cardiac fibrosis and better myocardial function than WT mice at one month after in vivo I/R (Supplement Fig. S1 and Table S3B). Collectively, these in vivo data further suggest that overexpression of Tsg101 could elicit cardio-protective effects on I/R-induced injury.
3.3. Tsg101 positively regulates the Nrf2 signaling pathway
To dissect potential mechanisms underlying the Tsg101-induced cardio-protective effects on I/R injury, we performed RNA sequencing (RNA-seq) analysis in Tsg101-TG hearts and WT-hearts. RNA-seq results identified over 1700 differentially expressed genes in TG-hearts compared to WT controls (Fig. 3A/B, GEO accession number GSE126653). Interestingly, the top 20 up-regulated genes in TG hearts are well-known targets of transcription factor Nrf2 (Supplement Fig. S2A). We further conducted Promoter Binding Motif Search analysis for 48 genes that were upregulated by 3-fold in Tsg101-hearts, and screened their upstream promoter sequences from either −400 to 100 or from −2000 to 500 around the transcription start site (TSS) (Supplement Fig. S2B/C). The Homer Known Motif Enrichment analysis results showed that Nrf2 is the top-ranked transcriptional factor that binds to promoters of these 48 genes (Fig. 3C). We then selected 7 genes (Gsta1, Gstp1, Gstm1, Nqo1, Gclc, Fth1 and p62) for validation using qRT-PCR. Consistent with RNA-seq data, mRNA levels of these Nrf2-controlled genes were significantly elevated in TG hearts, compared to WTs (Fig. 3D). However, mRNA levels of Nrf2 were unchanged in Tsg101 TG-hearts (Fig. 3D). Considering that Nrf2 activates its target gene expression as a transcriptional factor only in the nucleus [16,17], we next sought to determine the nuclear translocation of Nrf2 in Tsg101-TG hearts. As expected, we did observe that Nrf2 contents in the nuclear fraction was significantly higher in TGs than in WTs, whereas cytosolic Nrf2 levels were similar between these two groups, but total protein levels of Nrf2 were greatly increased by 2-fold in TG hearts, relative to WTs (Fig. 3E/F). Hence, these results suggest that elevation of Tsg101 could affect cardiac Nrf2 activity and its translocation but not its mRNA levels.
Fig. 3.

Tsg101 enhances the Nrf2 signaling pathway in the heart. (A) Heat map and (B) Volcano plot from RNA-seq analysis, comparing heart genes from 12-week-old WT and TG mice (n = 3 per group, male). Gene changes were considered significant if FDR <0.1 and fold-change were normalized to log2 expression levels. (C) Promoter Binding Motif analysis showing Nrf2 was the top ranked transcriptional factor in TG hearts compared with WT hearts. (D) mRNA levels of Nrf2-target genes (Gsta1, Gstp1, Nqo1, Gclc, Fth1, p62) in WT- and TG-mouse hearts. *, p < 0.05 vs. WT. (E) Representative Western blots and (F) quantification analysis showing the expression levels of Tsg101, total Nrf2, nuclear Nrf2 and cytosolic Nrf2 in WT- and TG-hearts. *, p < 0.05 vs. WT; n.s. stands for no significance. Lamin B1 was used as a loading control for nuclear protein, β-Tubulin was used as a loading control for cytosol protein, and GAPDH was used as a loading control for total protein.
3.4. Blockade of Nrf2 activation with ML385 dampens Tsg101-mediated protective effects against I/R injury
We then asked whether Tsg101-elicited cardio-protective effects were dependent on the activation of Nrf2. To clarify this issue, we pre-treated mice with ML385 for 3 times (30 μg/g i.p. injection daily), followed by a series of ex vivo and in vivo experiments (Fig. 4A). ML385 is a newly identified chemical compound that specifically binds to the Neh1 domain of Nrf2 and inhibits its downstream target gene expression, as well as reduces its own expression (Note: Nrf2 can autoregulate its own transcription) [32]. Hearts were collected from mice at 24 h after the last injection and underwent the validation analysis for ML385-mediated inhibition of Nrf2 signaling. Western-blotting results confirmed that pre-treatment of both WT and TG mice with ML385 markedly attenuated both total and nuclear levels of Nrf2, compared to buffer-treated controls (Fig. 4B/C). Consistently, the expression levels of Nrf2-downstream genes (i.e., Gsta1, Gstm1 Gstp1 and Nqo1) were significantly reduced in the ML385-treated mouse hearts, compared to buffer-controls (Fig. 4D). Ex vivo I/R analysis showed that Tsg101-mediated beneficial effects on cardiac function recovery were greatly offset by pre-treatment with ML385 (Fig. 4E/F). In addition, reduction of LDH release and caspase-3 activity in Tsg101-overexpressing hearts were dramatically inhibited by ML385 pre-treatment, relative to buffer-controls (Fig. 4G/H). Furthermore, reduction of ROS levels in TG hearts upon ex vivo I/R was also significantly suppressed after injection of ML385 (Fig. 4I/J). We also subjected ML385-treated mice and control mice to in vivo 1-h myocardial ischemia, via coronary artery occlusion, followed by 24-h reperfusion. While the infarction size was significantly smaller in buffer-treated TG hearts than WT-controls, such reduction was not exhibited in ML385-treated TG hearts, compared to ML385-injected WT controls (Fig. 4K/L). The region at risk showed no differences among these groups (Fig. 4M). Taken together, these data demonstrate that Tsg101-induced protective effects against cardiac I/R injury are dependent on the activation of Nrf2 signaling pathway.
Fig. 4.

Blockade of Nrf2 activation by ML385 suppresses Tsg101-mediated cardio-protective effects. (A) Schematic diagram depicting the experimental protocol for intraperitoneal (i.p.) injection of ML385 daily for 3 times, 24 h after the last injection, heart samples were collected for a series of experiments. (B) Representative Western-blots and (C) quantitative analysis validating the inhibitory effects of ML385 on the nuclear levels of Nrf2 in WT- and TG-hearts, and (D) RT-PCR analysis for Nrf2-target gene expression in mouse hearts with or without ML385-injection; *, p < 0.05 vs. WT-controls, n = 4. Cardiac functional recovery assessed by (E) LV dp/dt (max) and (F) LV dp/dt (min) in ML385-treated WT- and TG-hearts during ex vivo I/R (45min/1 h). n = 5 for each group. *, p < 0.05 vs. WT-controls. (G) Total LDH levels in coronary effluent collected during the first 10 min of reperfusion. *, p < 0.05 vs. WT-controls. (H) Caspase-3 activity in the WT- and TG-hearts, pretreated with or without ML385, upon ex vivo I/R (45min/1 h). *, p < 0.05 vs. WT-controls. (I) Representative images and (J) quantification analysis of Dihydroethidium (DHE) staining in heart sections from WT- and TG-hearts, pretreated with or without ML385, upon ex vivo I/R (45min/5min). *, p < 0.05. (K) Representative infarction images of mouse hearts upon in vivo I/R (1h/24 h) and (L) quantitative results of infarction size and (M) the ratio of risk region to left ventricular (LV) area. *, p < 0.05 vs. WT-controls. n.s. stands for no significance.
3.5. Knockdown of Tsg101 aggravates I/R-triggered cardiac dysfunction
Next, we examined the effects of Tsg101-deficiency on cardiac functional recovery during ex vivo I/R, using an inducible cardiac-specific knockdown (KD) mouse model (Fig. 5A). It is worthy to note that mice with complete knockout of Tsg101 are lethal [8]. Western blotting analysis revealed that Tsg101 expression was reduced by ~47% in KD-hearts (Fig. 5B). Tsg101-KD and corresponding control (Cre−/Tsg101flox/wt) hearts were mounted onto the Langerdorff apparatus and stabilized for 10 min with perfusion buffer, followed by 30-min no-flow ischemia and 1-h reperfusion. During reperfusion, KD-hearts exhibited significantly depressed functional recovery compared to controls, evidenced by the decreased rates of contraction (+dP/dt) and relaxation (-dP/dt), relative to controls (Fig. 5C/D). Levels of LDH released from ischemic/reperfused KD hearts were 2.8-fold higher than Controls (Fig. 5E). Similarly, the degree of I/R-induced cardiac apoptosis, determined by Caspase-3 activity, was significantly increased in KD group, compared to controls (Fig. 5F). Collectively, these results indicate that Tsg101-deficiency inhibits cardiac contractile function recovery during I/R and exaggerates I/R-triggered myocyte damage.
Fig. 5.

Knockdown of Tsg101 aggravates I/R-triggered cardiac dysfunction and ROS production. (A) Schematic diagram depicting the generation of inducible cardiac-specific Tsg101 knockdown (Tsg101-KD) mice (B6/129). (B) Representative Western-blots and quantification analyses showing Tsg101 protein levels in control (Ctrl: Cre−/Tsg101flox/wt littermates) and Tsg101-KD mouse hearts. *, p < 0.05 vs. Ctrl. (C/D) Tsg101-KD hearts demonstrated worse contractile function recovery during ex vivo I/R (30min/1 h), compared to controls. n = 6 for each group. *, p < 0.05 vs. Ctrl. (E) Total LDH levels in coronary effluent collected during the first 10 min of reperfusion. *, p < 0.05 vs. Ctrl. (F) Caspase-3 activity in Ctrl- and KD-hearts subjected to ex vivo I/R (30min/1 h), *, p < 0.05 vs. Ctrl. (G) Representative Western-blots and (H) quantification analysis showing the expression levels of Tsg101, total Nrf2, nuclear Nrf2 and cytosolic Nrf2 in pre-I/R and post-I/R hearts. *, p < 0.05 vs. Ctrl, n = 4. GAPDH was used as a loading control for total and cytosolic proteins, and Lamin B1 was used as a loading control for nuclear proteins. (I) mRNA levels of Nrf2 target genes in post-I/R hearts. *, p < 0.05 vs. Ctrl. (J) Representative images and (K) quantification analysis of DHE staining in sections from Ctrl- and KD-hearts subjected to ex vivo I/R (30min/5min). *, p < 0.05 vs. Ctrl-I/R. (L) ROS levels, measured by DCFH2M fluorescence intensity, in Ctrl- and KD-hearts subjected to ex vivo I/R (30min/5min). *, p < 0.05 vs. Ctrl-I/R. (M) Representative infarction images of mouse hearts upon in vivo I/R (30min/24 h) and (N) quantitative results of infarction size and (O) the ratio of risk region to left ventricular (LV) area. *, p < 0.05 vs. Ctrl.
To elucidate whether the detrimental effects of Tsg101-knockdown were associated with the inhibition of Nrf2 signaling cascades, we measured both total and nuclear levels of Nrf2 in mouse hearts under pre- and post-I/R conditions, and observed that both total and nuclear Nrf2 levels were significantly reduced in Tsg101-KD hearts upon pre- and post-I/R, in relation to control hearts, whereas cytosolic Nrf2 levels were similar between groups (Fig. 5G/H). We further measured mRNA levels of Nrf2 target genes (i.e., Gsta1, Gstp1, Gstm1, Nqo1, Gclc, Fth1 and p62) in post-I/R hearts, and observed that Nrf2-target genes were greatly down-regulated in KD-hearts, compared to controls (Fig. 5I). Cardiac ROS levels, assessed by DHE staining and DCFH2M, in response to ex vivo I/R (30min/5min), were significantly increased in KD-hearts, compared to WT-controls (Fig. 5J-L). Moreover, upon in vivo 30-min coronary artery occlusion (myocardial ischemia), followed by 24-h reperfusion, Tsg101-KD mouse hearts exhibited a significant larger infarction size than control hearts (Fig. 5M/N), while the region at risk did not show any difference between groups (Fig. 5O). Altogether, these results demonstrate that knockdown of Tsg101 in the heart could suppress Nrf2-mediated anti-oxidative signaling pathway and augment I/R-induced cardiac injury.
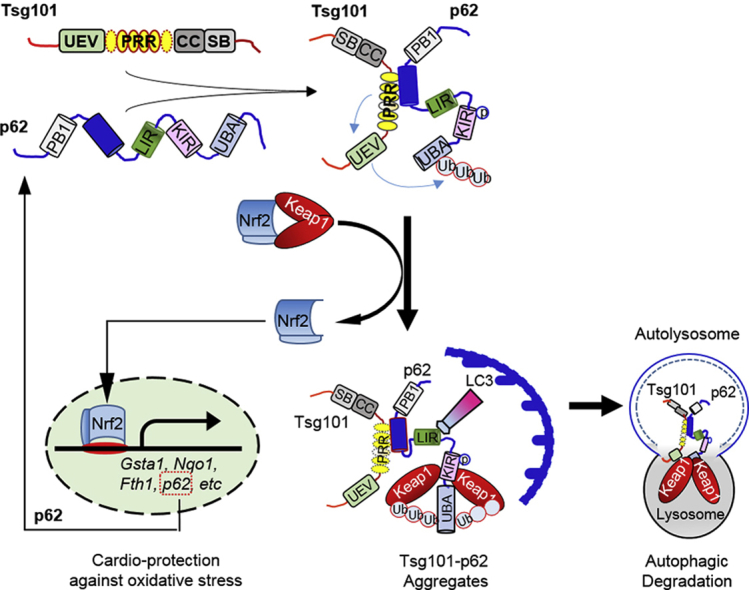
3.6. Tsg101 promotes p62 aggregation and autophagic degradation of Keap1 in the heart
We next asked how Tsg101 controlled activation of Nrf2 in the heart. To address this mechanistic question in-depth, we examined whether Tsg101 had any relationship with p62, as Tsg101 did not alter mRNA but protein levels of Nrf2 (Fig. 3D–F) and importantly, p62 is well characterized for its ability to aggregate and disrupt the inhibitory effects of Keap1 on Nrf2 [[17], [18], [19]] (Fig. 6A). In general, aggregation of p62 could recruit and sequester Keap1 to autophagosomes for degradation and consequently, Nrf2 is released freely and translocated to the nucleus where it activates transcription of anti-oxidant genes [[17], [18], [19]] (Fig. 6A). We thus first determined protein levels of Keap1, p62 and its phosphorylation in mouse hearts. Western-blotting analysis revealed that Keap1 protein levels were significantly diminished, whereas the levels of p62 and its Ser351-phosphorylation were dramatically increased in TG hearts, compared to WTs (Fig. 6B). In contrast, KD hearts showed higher levels of Keap1 and lower levels of p62 in comparison to controls (Supplement Fig. S3A/B; Note: we did not measure Ser351-p62 levels in KD-hearts, as it may be too low to be detectable). Collectively, these gain- and loss-of-function data suggest a possible connection between Tsg101 and the p62/Keap1/Nrf2 axis. Therefore, we next examined whether elevation of Tsg101 could affect the formation of p62 aggregates in the heart. We isolated soluble and insoluble fractions from heart homogenates using 1% Triton X-100 and 1% SDS lysis buffer, respectively. As shown in Fig. 6C, while p62 aggregates could be observed in the soluble fractions of both WT and TG hearts, the degree of p62 aggregation was much stronger in TGs than WTs, which was consistent with higher levels of p62 phosphorylation. More interestingly, p62 aggregates were non-visible in the insoluble fraction of WT-hearts, whereas TG hearts displayed the strong formation of p62 aggregates (Fig. 6C). In parallel, we observed that Tsg101 was also aggregated in the insoluble fractions of TG hearts (Fig. 6D). Accordingly, increased aggregation of p62 recruited more Keap1, leading to higher levels of Keap1 in the insoluble fraction of TG-hearts than WTs, which was opposite to the lower levels of Keap1 in the soluble fractions of TG hearts, relative to WTs (Fig. 6D).
Fig. 6.

Tsg101 promotes p62 aggregation and Keap1 recruitment to the Tsg101-p62 complex. (A) Diagram showing the p62-Keap1-Nrf2 signal pathway. Aggregation of p62 increases affinity of p62 to Keap1 and thereby, releasing Nrf2 from the Keap1-inhibition and translocating to the nucleus to initiate transcription of genes involved in anti-oxidant response. (B) Representative immunoblots and quantification analysis showing the expression levels of Tsg101, Keap1, p62 and S351-p62 in WT- and TG-hearts. *, p < 0.05 vs. WT. (C/D) Representative Western-blots showing aggregates of p62 and Tsg101 in the soluble and insoluble extracts from WT- and TG-hearts. n = 4 for each group. (E) Co-immunoprecipitations (Co-IP) with anti-p62 antibody and immunoblotting with Tsg101-antibody, using insoluble fractions of WT-and TG-hearts. Insoluble fractions of TG hearts were used as positive controls, and immunoprecipitates with IgG as negative control. HC stands for non-specific IgG heavy-chain bands. (F) Co-IP with Tsg101-antibody and immunoblotting with p62-antibody, using insoluble fractions of WT and TG hearts. Insoluble fractions of TG hearts were used as positive controls, and immunoprecipitates with IgG as negative control. (G) Co-IP with anti-p62 antibody and immunoblotting with Tsg101-antibody, using the homogenates from pre- and post-I/R wild-type hearts. (H) Co-IP with Tsg101-antibody and immunoblotting with p62-antibody, using the homogenates from pre- and post-I/R wild-type hearts. Immunoprecipitates with IgG as negative controls. (I) Representative images showing immunofluorescence staining with antibodies to p62- (red) and Keap1 (purple) in Ad.GFP-Tsg101 (green)-infected neonatal rat cardiomyocytes (NRCMs). Similar results were observed in three independent experiments. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To determine whether Tsg101 directly interacted with p62 in such aggregates, we performed co-immunoprecipitation (co-IP) of insoluble fractions with anti-p62 and anti-Tsg101 antibodies, respectively. As shown in Fig. 6E/F, Tsg101 strongly interacted with p62 and promoted its aggregation. To further clarify whether such interaction of Tsg101 with p62 existed in WT-hearts under basal and I/R conditions, we conducted co-IP for total lysates collected from ex vivo pre- and post-I/R hearts. As shown in Fig. 6G/H, there was an interaction between endogenous Tsg101 and p62, which was impaired upon I/R stress. In addition, immunofluorescence staining of cardiomyocytes also confirmed that p62 co-localized with Keap1 and Tsg101 (Fig. 6I).
Considering that p62 aggregates could recruit Keap1 and activate autophagic degradation of Keap1 [33], we then examined whether Tsg101 activated cardiac autophagy. As shown in Fig. 7A/B, the expression levels of autophagic flux marker, LC3A/B-II, were greatly increased in Tsg101-TG hearts, compared to WTs. What's more, we injected CQ (chloroquine, 50 mg/kg, i.p.), an inhibitor of autophagic flux, into WT and TG mice. Six hours later, hearts were collected for Western-blotting analysis of LC3A/B. While the LC3A/B-II protein was significantly accumulated in both WT- and TG-hearts upon CQ treatment, the degree of such accumulation was more enhanced in CQ-treated TGs than WT controls (Fig. 7C/D). Accordingly, autophagy-mediated degradation of Keap1 was suppressed in CQ-treated hearts, as evidenced by higher levers of Keap1 in the CQ-samples than PBS-treated controls (Fig. 7C/E). These data suggest that elevation of Tsg101 in the heart could activate autophagic flux. Utilizing the in vitro mRFP-GFP-LC3 reporter system, we also observed that transfection of cardiomyocytes with Tsg101-myc caused an increase in the red florescent dots of mRFP-GFP-LC3 (note: red fluorescence dot indicates fusion of autophagosomes with lysosomes), compared to controls (Fig. 7F/G). In addition, such red florescent dots were remarkably diminished by treatment of bafilomycin A1 (BafA1), an inhibitor of autolysosome formation (Fig. 7F/G). Additionally, immuno-staining results showed that Tsg101 co-localized with autophagosomes (LC3) and late endosomes (CD63) but not early endosomes (EEA1) (Supplement Fig. S4). Put together, these data suggest that Tsg101 interacts directly with p62 and enhances its ubiquitination and aggregation and consequently, activates autophagic degradation of Keap1.
Fig. 7.

Tsg101 promotes autophagic flux in the heart. (A) Representative immunoblots and (B) quantification analysis showing the expression levels of Tsg101 and LC3A/B-II in WT- and TG-hearts. *, p < 0.05 vs. WTs, n = 4. (C) Representative immunoblots and (D/E) quantification analysis showing the protein levels of Tsg101, LC3A/B-II and Keap1 in WT- and TG-mouse hearts treated with or without chloroquine (CQ, 50 mg/kg) for 6 h *, p < 0.05, n = 4. (F) Representative images and (G) quantitative analysis of neonatal rat cardiomyocytes (NRCMs) transfected with the mRFP-GFP-LC3 reporter plasmid and treated with either DMSO (0.001%) or Bafilomycin A1 (BafA1, 100 nM, 4 h). *, p < 0.05, n = 3 litters of neonatal Sprague Dawley rats (1–3 days old) and a total of 20–30 cells per group. Those yellow dots represent combined signals of GFP with mRFP in autophagosomes, whereas those red dots (indicated as arrows in merged images) that do not overlap with GFP fluorescence signal stand for autolysosomes. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.7. Tsg101 interacts with p62 through its PRR domain
We next dissected how Tsg101 interacted with p62. Tsg101 gene is highly conserved between human and mouse [34]. Both protein structures are similar and contain four known structural motifs: the N-terminal UEV domain, followed by a proline-rich region (PRR), a coiled coil (CC) region, and a C-terminal α–helical/steadiness box (SB) domain (Fig. 8A) [34]. To map the specific region of Tsg101 that binds to p62, we co-transfected the truncated variants of Myc-tagged human Tsg101 plasmids with the Flag-tagged human p62 plasmid in HEK-293T cells (Fig. 8A). Western-blotting analysis confirmed the expression of these transfected plasmids in 293T cells (Fig. 8B). Co-IP assays showed that those variants containing PRR domain (plasmids #2, 3, 6 and 7) could bind to p62 (Fig. 8C/D). To further determine the effects of PRR deletion on Tsg101-induced anti-oxidative stress, we transfected HEK-293 cells with empty vector, Myc-full length Tsg101, and Myc-delta PRR (#4 Myc-UEV+#8 Myc-CC/SB), respectively. 48 h later, these transfected cells were treated with H2O2 (500 μM) for 12 h, and then ROS levels and cell survival were measured. As shown in Fig. 8E/F, overexpression of Tsg101 significantly reduced ROS levels, compared to empty vector-controls; however, such reduction of ROS levels was greatly suppressed by the deletion of PRR domain. Accordingly, cell survival analysis results revealed that deletion of PRR domain did not provide any protection against H2O2-triggered cell death, while full-length Tsg101-transfected cells exhibited better survival than empty vector-controls (Fig. 8G). Collectively, these results described above demonstrate that Tsg101 interacts with p62 through its PRR domain, and disruption of such interaction by the deletion of PRR domain offsets Tsg101-elicited cellular protective effects against oxidative stress injury.
Fig. 8.

Tsg101 interacts with p62 through the PRR domain. (A) Schematic diagram of plasmid structures for Tsg101 full-length and its truncated mutations. (B) Representative immunoblots showing the expression of co-transfected Tsg101 mutants and Flag-p62 in HEK-293T cells. (C) Co-immunoprecipitations (Co-IP) with anti-Flag antibody, and immunoblotting with anti-Myc antibody in HEK-293T cells co-transfected with indicated Tsg101 mutants and Flag-p62. Similar results were observed in three independent experiments. HC stands for non-specific IgG heavy-chain signal bands. (D) Co-IP with anti-Myc antibody, and immunoblotting with anti-Flag antibody in HEK-293T cells co-transfected with indicated Tsg101 mutants and Flag-p62 plasmid. Similar results were observed in three independent experiments. (E) Representative images and (F) quantification analysis of DHE staining in HEK-293T cells transfected with Tsg101-full length (FL) and its PRR-deletion plasmids. *, p < 0.05. Similar results were observed in three independent experiments. (G) Cell survival rate was determined by MTS incorporation in HEK-293T cells transfected with Tsg101-FL and its PRR-deletion plasmids, followed by treatment with 500 μM H2O2 for 12 h *, p < 0.05. Similar results were observed in three independent experiments. (H) A diagram showing the structures of p62-FL and its truncated mutations. (I) Representative immunoblots showing the expression of indicated Flag-p62 mutants and mCherry-Tsg101 in HEK-293T cells. (J) Co-IP with mCherry antibody and immunoblotting with Flag antibody in HEK-293T cells co-transfected with indicated Flag-p62 mutants and mCherry-Tsg101. Similar results were observed in three independent experiments. HC stands for non-specific IgG heavy-chain bands. (K) Co-IP with anti-Flag antibody, and immunoblotting with mCherry antibody in HEK-293T cells co-transfected with indicated Flag-p62 mutants and mCherry-Tsg101. Similar results were observed in three independent experiments. HC stands for non-specific IgG heavy-chain bands.
To further map which domain of p62 interacts with Tsg101, various Flag-tagged deletion mutants of p62 (Fig. 8H) were transfected into HEK293, together with mCherry-tagged Tsg101 (Fig. 8I). Co-IP results showed that deletions of PB1 (M2-M5), KIR-UBA (M3), LIR (M4), and LIR-KIR-UBA(M5) did not affect the interaction with Tsg101, whereas the region lacking AA103-440 (M1) disrupted such interaction (Fig. 8J/K). These data suggest that Tsg101 does not interact with the regions of PB1, LIR, KIR and UBA, but with the region of AA103-320. Given that: 1) the PB1 domain is responsible for homo-oligomerization or hetero-oligomerization, 2) the LIR domain (LC3-interacting region) and the C-terminal ubiquitin-associated domain (UBA) involve in the sequestration of ubiquitinated substrates into the autophagosome, and 3) the KIR domain is Keap1-interacting region [35], Tsg101 binding to the AA103-320 region therefore would not interfere with the formation of p62 aggregates and its sequestration of Keap1 for autophagic degradation, which is consistent with our data presented in Fig. 6, Fig. 7.
3.8. Ablation of p62 offsets Tsg101-elicited protective effects on cardiac I/R injury
Now that Tsg101 interacts with p62, we then asked whether p62 was required for Tsg101-induced cardio-protection against I/R injury. To this end, we generated a new mouse model in which p62 was ablated in Tsg101-TG hearts by crossing global p62-KO mice with Tsg101-TG mice (hereafter referred to as TG/KO) (Fig. 9A). Western blotting results confirmed that Tsg101 was overexpressed whereas p62 was knocked out in the heart of this cross mouse model under pre- and post-I/R conditions, but I/R caused significant reductions in protein levels of both Tsg101 and p62 (Fig. 9B/C). Notably, p62 levels remained higher in TG hearts than WTs upon I/R (Fig. 9B/C). Furthermore, we observed that protein levels of Keap1 were greatly increased whereas Nrf2 levels were reduced in both p62-KO and TG/KO hearts under pre-I/R conditions, and such alterations remained in post-I/R hearts (Fig. 9B/C). Accordingly, Tsg101-mediated activation of autophagy, measured by the protein levels of LC3A/B-II was remarkably attenuated by the knockout of p62 in mouse hearts upon both pre- and post-I/R conditions (Fig. 9B/C). qRT-PCR analysis also confirmed that Tsg101-induced elevation of Gsta1, Gstm1, and Nqo1 mRNA levels were completely suppressed by the ablation of p62 (Supplement Figs. S5A–C). Next, we subjected hearts of WT, Tsg101-TG, p62-KO and TG/KO mice to the Langendorff system for ex vivo I/R (45min/1 h). Tsg101-TG hearts showed better cardiac function recovery during reperfusion, but such beneficial effects were greatly attenuated when p62 was absent in Tsg101-TG hearts (Fig. 9D/E). Similarly, we observed that I/R-caused cardiomyocyte cell death (LDH release and Caspase-3 activity, Fig. 9F/G) and ROS production (Fig. 9H/I) were significantly reduced in Tsg101-TG hearts, but not in TG/KO hearts, compared to WTs. The in vivo I/R (1h/24 h) results also showed that the infarction size was smaller in Tsg101-TG hearts, but not in p62-KO or in TG/KO-hearts, compared to WTs (Fig. 9J/K). Notably, the region at risk did not exhibit any differences among these groups (Fig. 9L). Collectively, these data demonstrate that Tsg101-induced cardio-protective effects against I/R injury are dependent on p62.
Fig. 9.

Ablation of p62 offsets Tsg101-elicited protective effects on cardiac I/R injury. (A) A diagram depicting the generation of the cross mouse model (Tsg101-TG/p62-KO, FVB/B6 background) in which Tsg101 was overexpressed and p62 was ablated in the mouse hearts. (B) Representative Western-blots and (C) quantification analysis showing the expression levels of Tsg101, Keap1, p62, Nrf2 and LC3A/B in the indicated mouse hearts upon pre-I/R and post-I/R. *, p < 0.05; n = 4 for each group. Cardiac functional recovery assessed by (D) LV dp/dt (max) and (E) LV dp/dt (min) in WT, TG, p62-KO and TG/KO hearts during I/R. n = 5 for each group, *, p < 0.05 vs. WTs. (H) Total LDH levels in the coronary effluent collected during the first 10 min of reperfusion. *, p < 0.05 vs. WTs. (G) Caspase-3 activity determined in WT, TG, p62-KO and TG/KO hearts subjected to ex vivo I/R (45min/1 h). *, p < 0.05 vs. WTs. (H) Representative images and (I) quantification analysis of DHE staining in heart sections from WT, TG, p62-KO and TG/KO hearts subjected to ex vivo I/R (45min/5min). *, p < 0.05 vs. WTs. (J) Representative infarction images of mouse hearts upon in vivo I/R (1h/24 h) and (K) quantitative results of infarction size and (L) the ratio of risk region to left ventricular (LV) area. *, p < 0.05 vs. WTs. n.s. stands for no significance.
3.9. Knockdown of p62 in Tsg101-overexpressing cardiomyocytes dampens its protective effects against oxidative stress
One could argue that p62-KO mouse model is not cardiac-specific, and different mouse strains may contribute to physiological differences in the heart. To address this concern, we therefore performed in vitro studies using cultured neonatal rat cardiomyocytes (NRCMs), a well-controlled experimental setting (Note: given that adult cardiomyocytes are not easy to be transfected with siRNA, we used neonatal rather than adult cardiomyocytes here). NRCMs were transfected with p62-siRNA or control-siRNA for 48 h, followed by infection with Ad.Tsg101 or Ad.GFP for 24 h. Western blotting analysis showed that protein levels of p62 were reduced by 56% in siRNA-p62-transfected cells (Fig. 10A/B). Consistent with the in vivo data presented in Fig. 9B, protein levels of Keap1 were significantly reduced in Ad.Tsg101-myocytes, but not in siRNA-p62-transfected Tsg101-cells, compared to Ad.GFP-siRNA control myocytes (Fig. 10A/B). Accordingly, Tsg101-triggered nuclear translocation of Nrf2 in cardiomyocytes was largely blocked by knockdown of p62 (Fig. 10A/C). As a consequence, H2O2-caused oxidative stress, measured by DHE staining, was greatly reduced in Ad.Tsg101-myocytes, compared to Ad.GFP-cells (Fig. 10D/E). However, such reduction was remarkably inhibited in Ad.Tsg101-cells when p62 expression was knocked down (Fig. 10D/E). Consequently, the degree of cell survival was significantly improved in Ad.Tsg101-cardiomyocytes, but not in siRNA-p62-transfected Tsg101-overexpressing cells upon H2O2 treatment, compared to Ad.GFP-siRNA controls (Fig. 10F). These in vitro data further indicate that p62 is essential for Tsg101-mediated protective effects against oxidative stress-induced cardiomyocyte damage.
Fig. 10.

Knockdown of p62 in cardiomyocytes dampens Tsg101-mediated protective effects against oxidative stress. (A) Representative Western-blots and (B/C) quantification analysis of Tsg101, p62, Keap1, cytosolic and nuclear Nrf2 in neonatal Sprague Dawley rat cardiomyocytes (NRCMs) transfected with p62-siRNA or control siRNA for 48 h, followed by infection with Ad.Tsg101 or Ad.GFP for 24 h *, p < 0.05; n = 3. GAPDH was used as a loading control for total and cytosolic proteins, and Lamin B1 was used as a loading control for nuclear proteins. (D) Representative images and (E) quantitative analysis of DHE immuno-staining in NRMCs transfected with the indicated siRNAs for 48 h and infected with the indicated adenovirus vectors for 24 h, then treated with H2O2 (500 μM, 2 h). (F) MTS analysis of cell survival in NRMCs transfected with the indicated siRNAs for 48 h and infected with the indicated adenovirus vectors for 24 h, then treated with or without H2O2 (500 μM, 6 h) *, p < 0.05, n = 4; n.s. stands for no significance. Similar results were obtained in other two independent experiments.
4. Discussion
The present study, using gain- and loss-of-function approaches, uncovered that downregulation of Tsg101 contributed significantly to cardiac injury during I/R, whereas overexpression of Tsg101 protected hearts against I/R-induced oxidative stress and cardiomyocyte death. Mechanistically, we identified that Tsg101 interacted with p62 through the PRR domain and enhanced p62 aggregation, resulting in sequestration of Keap1 to autophagosomes for degradation and release of Nrf2 into the nucleus where it activates endogenous antioxidant response. We believe that our study presented here uncovers a novel functional role for Tsg101 as a key modulator of the p62/Keap1/Nrf2 axis in the heart, which may provide a new strategy for cardio-protection against I/R injury.
Over the past decades, numerous studies have implicated Nrf2 as a master regulator of cellular antioxidant response in ischemic hearts [16,36,37]. Nrf2 belongs to a small family of transcription factors containing a unique basic-leucine-zipper (bZIP) motif [37]. Domain analysis of Nrf2 has shown that Nrf2 is composed of seven conserved Nrf2-erythroid cell-derived protein with CNC homology (Neh) domains [37]. Notably, the Neh1 domain allows Nrf2 to bind to the antioxidant-response element (ARE) sequence and activates the expression of >200 genes that are associated with a series of enzymes with antioxidant and detoxifying activity. The Neh2 domain is responsible for binding to the Nrf2 inhibitor Keap1. Under basal or unstressed conditions, Nrf2 remains in an inactive cytoplasmic form through association with the Keap1 [38]. Although it was initially thought that Nrf2 could not be detected in the cytosol under basal conditions because of the rapid degradation by proteasome, increasing evidence has shown that Nrf2 protein levels are actually kept at low levels in both cytosol and nucleus through multiple layers of mechanisms (i.e., p62 as described below, non-coding RNAs and other epigenetic modifications) [37]. With regard to the regulation of Nrf2 activity, recent studies have concentrated on its non-canonical pathway [38]. The canonical approach to activation of Nrf2 is through the oxidative and electrophilic stress-induced oxidation of cysteine residues in Keap1, thereby releasing its inhibitory effects on Nrf2 [38]. The non-canonical mode of activation of Nrf2 is through disrupting the interaction between the KIR binding domain of Keap1 with the DLG and ETGE motifs in Neh2 domain of Nrf2 [38]. Thus far, a number of molecules that disrupt such interaction have been identified including p62, p21, dipeptidyl peptidase III (DPP3) and BRCA1 [[39], [40], [41], [42]]. Among these disruptors, p62 has drawn the most attention in this field. P62 is believed to interrupt the binding of Keap1 to Nrf2 by aggregation with Keap1 for autophagic degradation [39]. Interestingly, p62 expression is also controlled by Nrf2 and thereby, forms a p62-Nrf2-p62 positive feedback loop [43]. Nonetheless, how to regulate such p62-Keap1-Nrf2 signaling cascade remains largely unknown. In the present study, we identified Tsg101 as a novel molecule to regulate p62/Keap1/Nrf2 axis through aggregation of p62, evidenced by high levels of aggregated p62 in Tsg101-transgenic hearts than WT controls (Fig. 6C). More so, we demonstrated that Tsg101 was bound to p62 and both proteins were found in the insoluble fraction of protein aggregates collected from TG hearts (Fig. 6C/D), indicating that Tsg101 may induce p62 aggregation.
Currently, it has been reported that the recruitment and attachment of ubiquitin to p62 is critical for the formation of p62 aggregates [36]. As a matter of fact, Tsg101 is an E2 ubiquitin ligase that possesses the ubiquitin E2 variant (UEV) domain [35,44]. Although the UEV domain lacks the essential cysteine residues, Tsg101 has been shown to bind ubiquitin [44]. For instance, Tsg101 is ubiquitinated by ubiquitin E3 ligase, Mahogunin Ring Finger-1 (MGRN1), which aids in the fusion of amphisomes to autolysosomes [45]. In addition, Tsg101 utilizes the UEV domain to bind and sort ubiquitinated endosomal cargo into multivesicular bodies and late endosomes [46]. Previous reports had suggested that binding to PRR domain of Tsg101 could enhance the function of the UEV domain [34,47]. For example, Tsg101 binds to fellow ESCRT protein Alix through the PRR domain and consequently, activates the UEV domain to increase ubiquitination of HIV Gag proteins for facilitating assembly and release of the virus [34,47]. Similarly, in the present study, we observed that Tsg101 was bound to p62 through the PRR domain (Fig. 8). Thus, it is conceivable that binding of Tsg101 to p62 may increase the availability of ubiquitin for p62, thereby promoting the aggregation of p62 and eventual sequestration of Keap1 to autophagosomes for degradation.
Regarding the Nrf2-inhibition experiments, we selected pharmacological inhibition with ML385 rather than Nrf2-knockout mouse model. Recent studies revealed that chronic knockout of Nrf2 adaptively upregulated eNOS expression, leading to smaller infarction size following myocardial ischemia/reperfusion injury than WT mice [48]. In addition, Ashrafian et al. also observed that Nrf2-KO mice exhibited similar infarction size to WT mice following a coronary artery ligation for 24 h [49]. Therefore, the Nrf2-KO mouse model may not be suitable for testing whether Tsg101-mediated cardio-protective effects are dependent on Nrf2. By contrast, ML385 is newly developed inhibitor that specifically binds to Neh1, the Cap 'N' Collar Basic Leucine Zipper (CNC-bZIP) domain of Nrf2, and blocks Nrf2 transcriptional activity [32]. The in vivo experimental results demonstrated that pre-injection of ML385 into mice selectively inhibited Nrf2 activation without off-target effects [32]. The present study also confirmed the efficacy of ML385 in the suppression of Nrf2 activity in mouse hearts (Fig. 4B–D).
In conclusion, our findings define a novel role of Tsg101 in the regulation of ischemia/reperfusion-induced cardiac injury. Elevation of Tsg101 expression in the heart could confer protection against oxidative stress-induced cardiac damage via the activation of p62-Keap1-Nrf2 axis. Thus, our study may provide a new avenue for the treatment of ischemic heart disease.
Data availability
All relevant data are available from the authors. RNA sequencing data can be accessed on the NCBI website using the GEO accession number GSE126653.
Author contributions
S.D., K.E. and X. W. designed, performed experiments, and analyzed the data. S.D., and K.E. wrote and edited the manuscript. Y.L., J.P., X.M. T.P., and K.H. assisted with some experiments and discussed the results. W.H. and Y.W. performed in vivo chronic cardiac ischemia/reperfusion and measurement of cardiac function. J.C. and A.J. conducted bioinformatics analysis. D.-S. J. provided human patient samples. C.W. and J.-L. G. for p62-KO analysis. G.C.F. supervised and conceptualized the study, wrote and edited the manuscript.
Declaration of competing interest
None.
Acknowledgements
This work was supported partially by National Institutes of Health (NIH) grants R01 GM-126061, GM-132149, and American Heart Association (AHA) Established Investigator Award 17EIA33400063 (G.-C. F), and AHA Pre-doctoral Fellowship (18PRE34030123 to K. E).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101453.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Ibáñez B., Heusch G., Ovize M., Van de Werf F. Evolving therapies for myocardial ischemia/reperfusion injury. J. Am. Coll. Cardiol. 2015;65:1454–1471. doi: 10.1016/j.jacc.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 2.Granger D.N., Kvietys P.R. Reperfusion injury and reactive oxygen species: the evolution of a concept. Redox Biol. 2015;6:524–551. doi: 10.1016/j.redox.2015.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou T., Chuang C.C., Zuo L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. BioMed Res. Int. 2015;2015:864946. doi: 10.1155/2015/864946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin D., Wang X., Li Y., Yang L., Wang R., Peng J., Essandoh K., Mu X., Peng T., Han Q., Yu K.J., Fan G.C. MicroRNA-223-5p and -3p cooperatively suppress necroptosis in ischemic/reperfused hearts. J. Biol. Chem. 2016;291:20247–20259. doi: 10.1074/jbc.M116.732735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braunersreuther V., Jaquet V. Reactive oxygen species in myocardial reperfusion injury: from physiopathology to therapeutic approaches. Curr. Pharmaceut. Biotechnol. 2012;13:97–114. doi: 10.2174/138920112798868782. [DOI] [PubMed] [Google Scholar]
- 6.Granger D.N., Kvietys P.R. Reperfusion therapy-What's with the obstructed, leaky and broken capillaries? Pathophysiology. 2017;24:213–228. doi: 10.1016/j.pathophys.2017.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Zhong Q., Chen C.F., Chen Y., Chen P.L., Lee W.H. Identification of cellular TSG101 protein in multiple human breast cancer cell lines. Canc. Res. 1997;57:4225–4228. [PubMed] [Google Scholar]
- 8.Carstens M.J., Krempler A., Triplett A.A., Van Lohuizen M., Wagner K.U. Cell cycle arrest and cell death are controlled by p53-dependent and p53-independent mechanisms in Tsg101-deficient cells. J. Biol. Chem. 2004;279:35984–35994. doi: 10.1074/jbc.M400408200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh H., Mammucari C., Nenci A., Cabodi S., Cohen S.N., Dotto G.P. Negative regulation of cell growth and differentiation by TSG101 through association with p21 (Cip1/WAF1) Proc. Natl. Acad. Sci. U. S. A. 2002;99:5430–5435. doi: 10.1073/pnas.082123999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li L., Liao J., Ruland J., Mak T.W., Cohen S.N. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl. Acad. Sci. U. S. A. 2001;98:1619–1624. doi: 10.1073/pnas.98.4.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slagsvold T., Pattni K., Malerød L., Stenmark H. Endosomal and non- endosomal functions of ESCRT proteins. Trends Cell Biol. 2006;16:317–326. doi: 10.1016/j.tcb.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 12.Myers E.L., Allen J.F. Tsg101, an inactive homologue of ubiquitin ligase e2, interacts specifically with human immunodeficiency virus type 2 gag polyprotein and results in increased levels of ubiquitinated gag. J. Virol. 2002;76:11226–11235. doi: 10.1128/JVI.76.22.11226-11235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sette P., Nagashima K., Piper R.C., Bouamr F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology. 2013;10:79. doi: 10.1186/1742-4690-10-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Essandoh K., Deng S., Wang X., Jiang M., Mu X., Peng J., Li Y., Peng T., Wagner K.U., Rubinstein J., Fan G.C. Tsg101 positively regulates physiologic-like cardiac hypertrophy through FIP3-mediated endosomal recycling of IGF-1R. Faseb. J. 2019;33:7451–7466. doi: 10.1096/fj.201802338RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Essandoh K., Wang X., Huang W., Deng S., Gardner G., Mu X., Li Y., Kranias E.G., Wang Y., Fan G.C. Tumor susceptibility gene 101 ameliorates endotoxin-induced cardiac dysfunction by enhancing Parkin-mediated mitophagy. J. Biol. Chem. 2019;294:18057–18068. doi: 10.1074/jbc.RA119.008925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q.M., Maltagliati A.J. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol. Genom. 2018;50:77–97. doi: 10.1152/physiolgenomics.00041.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canning P., Sorrell F.J., Bullock A.N. Structural basis of Keap1 interactions with Nrf2. Free Radic. Biol. Med. 2015;88:101–107. doi: 10.1016/j.freeradbiomed.2015.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartolini D., Dallaglio K., Torquato P., Piroddi M., Galli F. Nrf2-p62 autophagy pathway and its response to oxidative stress in hepatocellular carcinoma. Transl. Res. 2018;193:54–71. doi: 10.1016/j.trsl.2017.11.007. [DOI] [PubMed] [Google Scholar]
- 19.Kansanen E., Kuosmanen S.M., Leinonen H., Levonen A.L. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris C.R., Stanton M.J., Manthey K.C., Oh K.B., Wagner K.U. A knockout of the Tsg101 gene leads to decreased expression of ErbB receptor tyrosine kinases and induction of autophagy prior to cell death. PloS One. 2012;7 doi: 10.1371/journal.pone.0034308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okada K., Yanagawa T., Warabi E., Yamastu K., Uwayama J., Takeda K., Utsunomiya H., Yoshida H., Shoda J., Ishii T. The alpha-glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol. Res. 2009;39:490–500. doi: 10.1111/j.1872-034X.2008.00478.x. [DOI] [PubMed] [Google Scholar]
- 22.Fan G.C., Ren X., Qian J., Yuan Q., Nicolaou P., Wang Y., Jones W.K., Chu G., Kranias E.G. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–1799. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 23.Wang X., Gu H., Huang W., Peng J., Li Y., Yang L., Qin D., Essandoh K., Wang Y., Peng T., Fan G.C. Hsp20-mediated activation of exosome biogenesis in cardio-myocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes. 2016;65:3111–3128. doi: 10.2337/db15-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fan G.C., Zhou X., Wang X., Song G., Qian J., Nicolaou P., Chen G., Ren X., Kranias E.G. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res. 2008;103:1270–1279. doi: 10.1161/CIRCRESAHA.108.182832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peng J., Li Y., Wang X., Deng S., Holland J., Yates E., Chen J., Gu H., Essandoh K., Mu X., Wang B., McNamara R.K., Peng T., Jegga A.G., Liu T., Nakamura T., Huang K., Perez-Tilve D., Fan G.C. An Hsp20-FBXO4 axis regulates adipocyte function through modulating PPARγ ubiquitination. Cell Rep. 2018;23:3607–3620. doi: 10.1016/j.celrep.2018.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trapnell C., Pachter L., Salzberg S.L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu G.S., Zhu H., Cai W.F., Wang X., Jiang M., Essandoh K., Vafiadaki E., Haghighi K., Lam C.K., Gardner G., Adly G., Nicolaou P., Sanoudou D., Liang Q., Rubinstein J., Fan G.C., Kranias E.G. Regulation of BECN1-mediated autophagy by HSPB6: insights from a human HSPB6 (S10F) mutant. Autophagy. 2018;14:80–97. doi: 10.1080/15548627.2017.1392420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hua F., Li K., Yu J.J., Lv X.X., Yan J., Zhang X.W., Sun W., Lin H., Shang S., Wang F., Cui B., Mu R., Huang B., Jiang J.D., Hu Z.W. TRB3 links insulin/IGF to tumour promotion by interacting with p62 and impeding autophagic/proteasomal degradations. Nat. Commun. 2015;6:7951. doi: 10.1038/ncomms8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin Y.S., Chen Y.J., Cohen S.N., Cheng T.H. Identification of TSG101 functional domains and p21 loci required for TSG101-mediated p21 gene regulation. PloS One. 2013;8 doi: 10.1371/journal.pone.0079674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ost'ádal B., Ost'ádalová I., Skárka L., Kolár F., Kopecký J. Ischemic injury of the developing heart. Exp. Clin. Cardiol. 2002;7:93–98. [PMC free article] [PubMed] [Google Scholar]
- 32.Singh A., Venkannagari S., Oh K.H., Zhang Y.Q., Rohde J.M., Liu L., Nimmagadda S., Sudini K., Brimacombe K.R., Gajghate S., Ma J., Wang A., Xu X., Shahane S.A., Xia M., Woo J., Mensah G.A., Wang Z., Ferrer M., Gabrielson E., Li Z., Rastinejad F., Shen M., Boxer M.B., Biswal S. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016;11:3214–3225. doi: 10.1021/acschembio.6b00651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sánchez-Martín P., Komatsu M. p62/SQSTM1 - steering the cell through health and disease. J. Cell Sci. 2018;131:21. doi: 10.1242/jcs.222836. [DOI] [PubMed] [Google Scholar]
- 34.Jiang Y., Ou Y., Cheng X. Role of Tsg101 in cancer. Front. Biosci. 2013;18:279–288. doi: 10.2741/4099. [DOI] [PubMed] [Google Scholar]
- 35.Pornillos O., Alam S.L., Davis D.R., Sundquist W.I. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat. Struct. Biol. 2002;9:812–817. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- 36.Lee Y., Weihl C.C. Regulation of SQSTM1/p62 via UBA domain ubiquitination and its role in disease. Autophagy. 2017;13:1615–1616. doi: 10.1080/15548627.2017.1339845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen Y., Liu X., Shi J., Wu X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019;125:496–502. doi: 10.1016/j.ijbiomac.2018.11.190. [DOI] [PubMed] [Google Scholar]
- 38.Silva-Islas C.A., Maldonado P.D. Canonical and non-canonical mechanisms of Nrf2 activation. Pharmacol. Res. 2018;134:92–99. doi: 10.1016/j.phrs.2018.06.013. [DOI] [PubMed] [Google Scholar]
- 39.Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y.S., Ueno I., Sakamoto A., Tong K.I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 40.Chen W., Sun Z., Wang X.J., Jiang T., Huang Z., Fang D., Zhang D.D. Direct interaction between Nrf2 and p21 (Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol. Cell. 2009;34:663–673. doi: 10.1016/j.molcel.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hast B.E., Goldfarb D., Mulvaney K.M., Hast M.A., Siesser P.F., Yan F., Hayes D.N., Major M.B. Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Canc. Res. 2013;73:2199–2210. doi: 10.1158/0008-5472.CAN-12-4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gorrini C., Baniasadi P.S., Harris I.S. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013;210:1529–1544. doi: 10.1084/jem.20121337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jain A., Lamark T., Sjøttem E., Larsen K.B., Awuh J.A., Øvervatn A., McMahon M., Hayes J.D., Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sundquist W.I., Schubert H.L., Kelly B.N., Hill G.C., Holton J.M., Hill C.P. Ubiquitin recognition by the human TSG101 protein. Mol. Cell. 2004;13:783–789. doi: 10.1016/s1097-2765(04)00129-7. [DOI] [PubMed] [Google Scholar]
- 45.Majumder P., Chakrabarti O. Mahogunin regulates fusion between amphisomes/MVBs and lysosomes via ubiquitination of TSG101. Cell Death Dis. 2015;6 doi: 10.1038/cddis.2015.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu Q., Hope L.W., Brasch M., Reinhard C., Cohen S.N. TSG101 interaction with HRS mediates endosomal trafficking and receptor down-regulation. Proc. Natl. Acad. Sci. U. S. A. 2003;100:7626–7631. doi: 10.1073/pnas.0932599100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren X., Hurley J.H. Proline-rich regions and motifs in trafficking: from ESCRT interaction to viral exploitation. Traffic. 2011;12:1282–1290. doi: 10.1111/j.1600-0854.2011.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Erkens R., Suvorava T., Sutton T.R., Fernandez B.O., Mikus-Lelinska M., Barbarino F., Flögel U., Kelm M., Feelisch M., Cortese-Krott M.M. Nrf2 deficiency unmasks the significance of nitric oxide synthase activity for cardioprotection. Oxid. Med. Cell. Longev. 2018;2018 doi: 10.1155/2018/8309698. 8309698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ashrafian H., Czibik G., Bellahcene M. Fumarate is cardioprotective via activation of the Nrf2 antioxidant pathway. Cell Metabol. 2012;15:361–371. doi: 10.1016/j.cmet.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the authors. RNA sequencing data can be accessed on the NCBI website using the GEO accession number GSE126653.