

Abstract
Coronavirus disease 2019 emerged as the first example of “Disease X”, a hypothetical disease of humans caused by an unknown infectious agent that was named as novel coronavirus and subsequently designated as severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). The origin of the outbreak at the animal market in Wuhan, China implies it as a case of zoonotic spillover. The study was designed to understand evolution of Betacoronaviruses and in particular diversification of SARS‐CoV‐2 using RNA dependent RNA polymerase (RdRp) gene, a stable genetic marker. Phylogenetic and population stratification analyses were carried out using maximum likelihood and Bayesian methods, respectively. Molecular phylogeny using RdRp showed that SARS‐CoV‐2 isolates cluster together. Bat‐CoV isolate RaTG13 and Pangolin‐CoVs are observed to branch off prior to SARS‐CoV‐2 cluster. While SARS‐CoV form a single cluster, Bat‐CoVs form multiple clusters. Population‐based analyses revealed that both SARS‐CoV‐2 and SARS‐CoV form separate clusters with no admixture. Bat‐CoVs were found to have single and mixed ancestry and clustered as four sub‐populations. Population‐based analyses of Betacoronaviruses using RdRp revealed that SARS‐CoV‐2 is a homogeneous population. SARS‐CoV‐2 appears to have evolved from Bat‐CoV isolate RaTG13, which diversified from a common ancestor from which Pangolin‐CoVs have also evolved. The admixed Bat‐CoV sub‐populations indicate that bats serve as reservoirs harboring virus ensembles that are responsible for zoonotic spillovers such as SARS‐CoV and SARS‐CoV‐2. The extent of admixed isolates of Bat‐CoVs observed in population diversification studies underline the need for periodic surveillance of bats and other animal reservoirs for potential spillovers as a measure towards preparedness for emergence of zoonosis.
Keywords: bat coronavirus, COVID‐19, disease‐X, evolution, pandemic, population genetics, SARS‐CoV‐2, virus bioinformatics
Research Highlights
SARS‐CoV‐2 shares a common ancestor with Bat‐CoV (isolate RaTG13) and Pangolin‐CoVs.
Analysis based on RdRp gene revealed that SARS‐CoV‐2 form a distinct homogeneous population.
Bat‐CoVs are found to be admixed ensembles indicative of their potential for zoonotic spillovers.
Our study recommends use of RdRp gene for survelliance to track emerging RNA viruses.
1. INTRODUCTION
An infectious disease of unknown origin and etiology (Disease X) was reported for the first time in China during December 2019 and was named as coronavirus disease 2019 (COVID‐19) due to presentation of symptoms similar to severe acute respiratory syndrome coronavirus (SARS‐CoV). The causative agent was subsequently identified and referred to as an unknown novel virus of animal origin based on the epicenter of the outbreak at the animal market in Wuhan, China. 1 The virus, believed to have acquired ability to infect human, was named as 2019‐nCoV (also referred as nCoV‐19) by the World Health Organisation and as SARS‐CoV‐2 by the International Committee on Taxonomy of Viruses (ICTV) with a referral to the genus Betacoronavirus of the family Coronaviridae. 2
With the emergence of COVID‐19 in China, 3 the disease has spread globally and has been declared as a pandemic. To tackle the disease and to characterize the virus, global scientific community has generated large data sets across domains. Multidisciplinary teams are undertaking data analysis from the perspective of both, basic and applied research with objectives to decipher origin and evolution of the virus as well as to embark on development of diagnostics, drugs and vaccines.
Members of the family Coronaviridae include viruses capable of infecting mammals, birds, and fishes 4 and are known to be the largest positive sense single‐stranded RNA viruses. Genus Betacoronavirus of family Coronaviridae have been responsible for three major outbreaks in the past two decades that include SARS epidemic in 2002 to 2003, 5 Middle East respiratory syndrome (MERS) epidemic in 2012, 6 and the present SARS‐CoV‐2 outbreak in 2019. 7 In all the above cases, bats have been implicated to play a role of a reservoir along with intermediate hosts like palm‐civets in the case of SARS 8 and dromedary camels in the case of MERS, 9 prior to host‐spill over to humans. In case of SARS‐CoV‐2, role of pangolin as an intermediate host has been suggested on the basis of analysis of virome of Malayan pangolins. 10 , 11
Phylogenetic analyses of Betacoronavirus members revealed that MERS‐CoV, Human‐CoV HKU1, Human‐CoV OC43, Bat‐CoVs (with Pipistrellus and Tylonycteris as hosts), and Mus musculus MHV‐1 form a distinct sister clade due to evolutionary divergence that is also reflected as variations in their genomic sequences. 10
Due to efforts of various research labs across China and elsewhere, more than 170 genomes have been sequenced and made available for researchers globally. 12 Genome length of SARS‐CoV‐2 is ~29 kb and encodes multiple Open Reading Frames (ORFs). The 5’ end of the genome contains replicase gene that constitutes 2/3rd of the genome and contains two overlapping ORFs viz, 1a and 1b. Cleavage by proteinases results in multiple protein products including RNA dependent RNA polymerase (RdRp). 4 The 3’ end of the genome encodes structural (spike, envelope, membrane, and nucleocapsid) and accessory genes. 13
RdRp gene has been found to be useful for evolutionary analysis of global RNA virome due to its universal presence in all RNA viruses. 14 , 15 , 16 Therefore, in this study, we have used RdRp gene to analyse the population structure of members of genus Betacoronavirus. Our group has designed workflows for population stratification studies of RNA viruses using the complete genome data to identify the factors shaping the genetic diversity of individual viruses infecting human such as Rhinoviruses, Dengue viruses, and Measles viruses. 17 , 18 Similar population diversification studies have been carried out for the members of the genus Begomovirus that are known to infect plants. 19
An attempt has been made to delineate genetic diversity of SARS‐CoV‐2 in comparison with a few Betacoronaviruses using phylogeny and population structure analysis of RdRp gene. It is envisaged that population‐scale diversity studies based on a stable genetic marker would augment phylogenetic analysis and provide an insight into evolution and origin of emerging viral zoonoses in general and that of SARS‐CoV‐2 in particular.
2. MATERIALS AND METHODS
2.1. Data curation
RdRp gene sequences of SARS‐CoV‐2 and Pangolin‐CoV isolates were extracted from the genomes deposited in GISAID database 12 using BLASTn 20 with RefSeq: NC_045512.2 as a query. Low resolution sequence data have been excluded. RdRp gene sequences belonging to members of genus Betacoronavirus were retrieved using online BLASTn search against nonredundant nucleotide database 21 (as of 18 February 2020), and filtered using % overlap of more than 90% and e‐value cutoff (e < 0.0001).
We have used the nomenclature SARS‐CoV (for SARS and SARS‐related and SARS‐like Coronaviruses), Pangolin‐CoV (for Betacoronavirus isolates derived from pangolin), Bat‐CoV (for Betacoronavirus isolates derived from bats). The total number of sequences in the RdRp data set are 307 (Table S1) which belong to viruses SARS‐CoV‐2 (91), Pangolin‐CoV (5), SARS‐CoV (125), Bat‐CoV (23), and SARS‐like‐Bat‐CoV (63). MERS‐CoV was not included in this analysis as it did not meet the cutoff criteria of overlap and identity used for data curation.
2.2. Multiple sequence alignment
Multiple sequence alignments (MSA) of nucleotide sequence of RdRp gene were carried out using MAFFT. 22 RDP4 23 was used to detect recombination events.
2.3. Phylogenetic analysis
Phylogenetic tree of RdRp gene data set was built using maximum likelihood (ML) method available in IQ‐TREE 24 with 1000 bootstrap replicates. iTOL (URL: https://itol.embl.de/) was used for visualization of phylogenetic trees.
2.4. Population stratification studies
Parsimonious sites extracted from the MSA of RdRp gene were used as input to study population structure of Betacoronaviruses. STRUCTURE tool 25 and the protocol described by Waman et al 2014 17 was used for fine‐level clustering of isolates belonging to genus Betacoronavirus using RdRp gene. Linkage equilibrium was estimated using LIAN 26 with p value 10−4 for 10 000 replicates and DnaSP tools. 27 Linkage equilibrium is measured in terms of standardised index of association (IS A); mean value of the difference between observed and expected haplotype frequency which is normalized by either maximum or minimum possible value of this difference (|Dʹ|); squared value of the difference between observed and expected haplotype frequency normalized by variance of allele frequency (r 2). Admixture and linkage models were used starting with burn‐in of 100 000 to 175 000 and Markov Chain Monte Carlo (MCMC) run of 100 000 to 175 000 steps each. An isolate was said to have admixture if its score for minor membership is ≥0.05. Number of clusters representing sub‐populations (k) was varied from 1 to 10 with ten replicates each. Optimal k was chosen using the STRUCTURE harvester. 28
3. RESULTS
3.1. RdRp analysis
MSA of RdRp nucleotide sequences revealed 99.78% identity amongst SARS‐CoV‐2 (91 isolates, as of 18 February 2020) and 55.66% identity amongst all Betacoronavirus members used in this study (SARS‐CoV, Pangolin‐CoV, Bat‐CoV, SARS‐like Bat‐CoV, SARS‐CoV, and SARS‐related‐CoV, totaling 307 isolates). RdRp gene was found to be devoid of recombination. The number of parsimonious sites in RdRp gene were found to be 784 out of 2796 bases. Linkage disequilibrium calculated in terms of IS A, |Dʹ|, and r 2 was found to be 0.22, 0.775 and 0.144, respectively.
Phylogenetic tree derived using ML method and TIM2 + F + I + G4 substitution model based on Bayesian information criterion score, revealed 7 branches representing clusters (Figure 1C). All the SARS‐CoV‐2 viruses formed an independent cluster. RaTG13 is the closest virus isolate that appears to branch off before the SARS‐CoV‐2 cluster, followed by the cluster of Pangolin‐CoVs. SARS‐like‐Bat‐CoV isolated from Kenya (GenBank: KY352407) remains independent. Overall, SARS‐like‐Bat‐CoVs and Bat‐CoVs form four clusters, based on host species from which the Bat‐CoVs are isolated. As expected, SARS‐CoVs formed a cluster that is distant from SARS‐CoV‐2 viruses.
Figure 1.
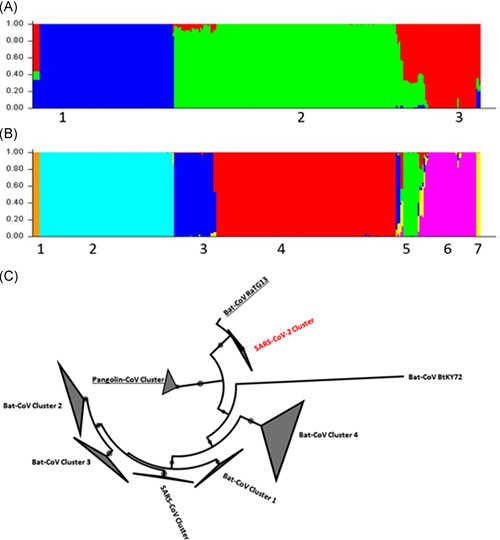
Fine level clustering of members of Betacoronavirus based on RdRp gene. A, Population stratification at optimal peak k = 3 wherein the labels 1, 2, and 3 represent SARS‐CoV‐2, SARS‐CoV, and Bat‐CoV, respectively. B, Population stratification at minor peak k = 7 wherein the labels 1,2,3,4,5,6,7 represent Pangolin‐CoV, SARS‐CoV‐2, Bat‐CoV‐Cluster 1, SARS‐CoV, Bat‐CoV‐Cluster 2, Bat‐CoV‐Cluster 3, and Bat‐CoV‐Cluster 4, respectively. C, Phylogenetic tree of Betacoronavirus members (SARS‐CoV, SARS‐CoV‐2, Pangolin CoV, and Bat‐CoV) derived using RdRp gene employing maximum‐likelihood method. The black filled circles on the nodes denote bootstrap support more than 70%. The seven clusters observed using population stratification studies (Figure 1A, B) are mapped onto the phylogenetic tree. RdRp, RNA dependent RNA polymerase; SARS‐CoV, severe acute respiratory syndrome coronavirus
3.2. Population stratification
Population genetics analysis of RdRp gene sequences revealed an optimal peak at k = 3 (Figure 1A) corresponding to the three groups of Betacoronavirus viz, SARS‐CoV, Bat‐CoV, and SARS‐CoV‐2 viruses. The SARS‐CoV‐2 isolates clustered together as a homogeneous group (membership score of 1). Similarly, SARS‐CoV clustered as a distinct homogeneous group (membership score of 1). It was interesting to observe that the isolate RaTG13, despite being a Bat‐CoV showed major membership (0.86) to SARS‐CoV‐2 cluster and only a minor membership (0.102) to Bat‐CoV cluster, and thereby substantiates that it is the closest Bat‐CoV to SARS‐CoV‐2 as was also observed in the phylogenetic tree (Figure 1C). Pangolin‐CoV isolates exhibited mixed ancestry (Figure 1A) with major membership to Bat‐CoV (membership score 0.56) and minor memberships to SARS‐CoV‐2 (membership score 0.343) as well as SARS‐CoV (membership score 0.09). Bat‐CoV members show unique pattern of ancestry wherein a few isolates are genetically homogeneous (membership score of 1) and a few others show mixed ancestry to SARS‐CoV (Figure 1A). Bat‐CoV isolates with host as Rhinolophus monoceros, R. thomasi, R. pearsonii and a few members with R. sinicus as host clustered together as a homogeneous group (membership score of 1). Bat‐CoV HKU isolates also clustered with this group with a membership score of 1. Bat‐CoV with R. ferrumequinum as host showed major membership to Bat‐CoV cluster (membership score 0.668) and minor membership to SARS‐CoV cluster (membership score 0.329). Three Bat‐CoV isolates displayed admixture with major membership to Bat‐CoV cluster and minor membership to both SARS‐CoV and SARS‐CoV‐2 clusters, which is also evident in the branching pattern observed in the phylogenetic tree. These include BtKY72 (isolated from Kenya in 2007; GenBank: KY352407); BtCoV/BM48‐31/BGR/2008 (isolated from Bulgaria, in 2008; GenBank: GU190215 with R. blasii as host), Zhejiang 2013 (isolated from China in 2013, GenBank: KF636752 with Hipposideros pratti as host).
The same population stratification study also revealed a minor peak at k = 7 (Figure 1B) in which the admixed members of Bat‐CoV (identified at k = 3) were observed to be further stratified based on their host species whereas Pangolin‐CoV isolates clustered independently. In all, there are four Bat‐CoV clusters observed. Members with R. sinicus, R. rex, Miniopterus schreibersii, and Aselliscus stoliczkanus as host clustered together with membership score of more than 0.9 (designated as Bat‐CoV cluster 1). This cluster also included an isolate each with R. affinis as host (GenBank: MK211376) and R. ferrumequinum as host (GenBank: KY417145). It is interesting to note that all other isolates with R. ferrumequinum as host clustered separately (designated as Bat‐CoV cluster 2). Three isolates namely, BtKY72 (isolated from Kenya in 2007; GenBank: KY352407); BtCoV/BM48‐31/BGR/2008 (isolated from Bulgaria, in 2008; GenBank: GU190215 with R. blasii as host), Zhejiang2013 (isolated from China in 2013, GenBank: KF636752 with Hipposideros pratti as host) clustered together (designated as Bat‐CoV cluster 3). Isolates with R. monoceros, R. thomasi, R. pearsonii, and a few members with R. sinicus as host along with Bat‐CoV HKU isolates clustered together with membership score of more than 0.9 (designated as Bat‐CoV cluster 4). Isolate Rp/Shaanxi2011 with R. pusillus as host (GenBank: JX993987) was found to be admixed with major membership to Bat‐CoV cluster 2 and minor membership to other three clusters of Bat‐CoVs. Isolates BtCoV/279/2005 and Rm1 with R. macrotis as host (GenBank: DQ648857 and DQ412043) were found to be admixed with membership to all four clusters of Bat‐CoVs.
4. DISCUSSION
From genomic sequences to 3‐dimensional structures, experiments in the domains of virology, molecular biology, biochemistry, biotechnology, and immunology have been performed for SARS‐CoV‐2 in a short span of 2 months. The data are analyzed simultaneously and concurrently by the multi‐disciplinary teams studying evolutionary biology, statistics, computational biology, and bioinformatics with objectives to limit spread of SARS‐CoV‐2 and to combat COVID‐19. This resulted in sequencing of more than 170 genomes in a span of ~60 days that are being tracked by resources such as Nextstrain (URL: https://nextstrain.org/) and Virological.org (URL: virological.org) in real time. With availability of data, studies on molecular evolution and characterization were performed to gain an insight into evolution and epidemiology of SARS‐CoV‐2. The data were also used gainfully to propose hypothesis regarding progenitor and intermediate hosts. It has now been accepted that bats are reservoirs and pangolins are intermediate host of SARS‐CoV‐2. 10 , 11 , 29
RdRp gene was chosen as it is the common gene essential for replication of all RNA viruses 14 and the phylogenetic tree generated using RdRp closely approximates whole genome phylogenetic analyses of Betacoronaviruses (data not shown), an observation also reported by Zhang et al 2020. 10 Therefore, a study using phylogenetic and population genetics analyses was designed based on RdRp gene and data curation was done using stringent criteria based on alignment coverage and sequence similarity. Relaxation of cut‐off criteria led to inclusion of MERS‐CoVs in our analysis. However, it not only adversely effected quality of the alignment due to sequence divergence and presence of indels resulting in doubling of parsimoniously informative sites and thereby impacted evolutionary model building studies. 15 Hence, relaxation of cut‐off criteria to include MERS‐CoV was ruled out and the analysis was carried out using Betacoronaviruses such as SARS‐CoV, Bat‐CoV, Pangolin‐CoV, and SARS‐CoV‐2.
These analyses revealed that SARS‐CoV‐2 isolates emerge as an independent homogeneous cluster with one of the Bat‐SARS‐isolate (RaTG13) as the closest isolate followed by a group of Pangolin‐CoVs. SARS‐CoV isolates form a distinct homogeneous cluster. The analysis of membership scores based on genetic similarity indicate that both, SARS‐CoV and SARS‐CoV‐2 are independent viruses. The Bat‐CoVs serve as interim linking the two independent clusters of SARS‐CoV and SARS‐CoV‐2. Population of Bat‐CoVs appear to be an ensemble of four sub‐populations having single as well as mixed ancestry, as evident from fine level clustering observed (at k = 7; Figure 1B) and their placement in the multiple branches of the phylogenetic tree (Figure 1C). Thus, bats serve as reservoirs of several viruses that appear to evolve independently and therefore have equal opportunity to jump hosts. 30 , 31 Our studies quantitatively substantiate hypothesis that the Bat‐SARS‐isolate (RaTG13) as one of the progenitor 32 of SARS‐CoV‐2 with Pangolin‐CoVs 10 , 11 , 29 that share a common ancestor along with the possibility of any other intermediate host that is not known so far.
Moderate value of LD (0.22) was observed for Betacoronaviruses which could be attributed to the fact that multiple viruses (SARS CoV, Bat‐CoV, Pangolin‐CoV, and SARS‐CoV‐2) infecting diverse hosts (human, bats, and pangolins) and possibly transmitted by several intermediate hosts have been analysed together. In view of this, the system was equilibrated and sampled for MCMC runs of 175 000 iterations allowing for it to capture variations. Additional MCMC runs with 200 000 and 225 000 iterations showed that the population structure sampled at k = 7 remains the same except that the SARS‐CoV cluster diversifies into two sub‐populations, SARS‐CoVs and SARS‐related‐CoVs as expected.
Analyses of 140 complete genomes of SARS‐CoV‐2 revealed the presence of only 50+ parsimonious sites (data not shown) indicating that the SARS‐CoV‐2 isolates characterized (as of 18 February 2020) represent a single spill over event. This hints at absence of further diversification of SARS‐CoV‐2 into lineages that was reported recently. 33
5. CONCLUSIONS
The outcome of population analysis of Betacoronaviruses using RdRp indicates that SARS‐CoV‐2 evolved as a homogeneous population distinct from both, SARS‐CoV as well as Bat‐CoVs. Both, the Bat‐CoV (RaTG13) and Pangolin‐CoV share a common ancestor with SARS‐CoV‐2. The Bat‐CoVs, however, appear to be emerging viral ensembles based on their mixed ancestry thereby hinting at their role and potential for zoonotic spillovers in future. The outcome of our study recommends use of RdRp gene for surveillance to track emerging RNA viruses (that include positive‐sense, negative‐sense, single‐stranded, and double‐stranded). SARS‐CoV‐2 pandemic exemplified need for surveillance and data‐driven infrastructural preparedness to combat zoonotic perils to meet the sustainable development goal (UN SDG 3: health for all) that recognizes interconnections between people‐animals‐plants and their shared environments.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
AUTHOR CONTRIBUTION
SMK, MK, and SL were involved in data curation, analysis, and writing‐original draft preparation. MMK was involved in formal analysis and writing‐original draft preparation. UKK contributed to conceptualization, methodology and writing‐review and editing.
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors would like to profoundly thank the global scientific community involved in data generation, curation, and dissemination. In particular, GISAID for hosting genomes and providing access to the global scientific community. We would like to acknowledge the Department of Biotechnology, Government of India for the Centre of Excellence in Bioinformatics grant.
Kasibhatla SM, Kinikar M, Limaye S, Kale MM, Kulkarni‐Kale U. Understanding evolution of SARS‐CoV‐2: A perspective from analysis of genetic diversity of RdRp gene. J Med Virol. 2020;92:1932–1937. 10.1002/jmv.25909
Sunitha M. Kasibhatla, Meenal Kinikar, and Sanket Limaye contributed equally to this study and are co‐first authors.
REFERENCES
- 1. Wu F, Zhao S, Yu B, et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265‐269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses . The species severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5:536‐544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese center for disease control and prevention. JAMA. 2020;323:1239‐1242. 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 4. de Groot RJ, Baker SC, Baric R, et al. Family coronaviridae. In: King AMQ, Adam MJ, Carstens EB, Lefkowitz EJ, eds. Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses. London, UK: Academic Press, Ltd; 2011:806‐828. [Google Scholar]
- 5. Drosten C, Günther S, Preiser W, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348:1967‐1976. [DOI] [PubMed] [Google Scholar]
- 6. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367:1814‐1820. [DOI] [PubMed] [Google Scholar]
- 7. Chan JFW, Kok KH, Zhu Z, et al. Genomic characterization of the 2019 novel human‐pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect. 2020;9:221‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song HD, Tu CC, Zhang GW, et al. Cross‐host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc Natl Acad Sci USA. 2005;102(7):2430‐2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Alagaili AN, Briese T, Mishra N, et al. Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. mBio. 2014;5. 10.1128/mBio.00884-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang T, Wu Q, Zhang Z. Probable pangolin origin of SARS‐CoV‐2 associated with the COVID‐19 outbreak. Curr Biol. 2020;30:1346‐1351. 10.1016/j.cub.2020.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lam TT, Shum MH, Zhu HC, et al. Identifying SARS‐CoV‐2 related coronaviruses in Malayan pangolins. Nature. 2020. 10.1038/s41586-020-2169-0 [published online ahead of print March 26, 2020]. [DOI] [PubMed] [Google Scholar]
- 12. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data ‐ from vision to reality. Euro Surveill. 2017;22(13):30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fehr AR, Perlman S. Coronaviruses: an overview of their replication and pathogenesis. In: Maier H, Bickerton E, Britton P, eds. Coronaviruses. Methods in Molecular Biology. Vol 1282. New York, NY: Humana Press; 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wolf YI, Kazlauskas D, Iranzo J, et al. Origins and evolution of the global RNA virome. mBio. 2018;9. 10.1128/mBio.02329-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Holmes EC, Duchêne S. Can sequence phylogenies safely infer the origin of the global virome? mBio. 2019;10. 10.1128/mBio.00289-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wolf YI, Kazlauskas D, Iranzo J, et al. Reply to Holmes and Duchêne, "can sequence phylogenies safely infer the origin of the global virome?": deep phylogenetic analysis of RNA viruses is highly challenging but not meaningless. mBio. 2019;10. 10.1128/mBio.00542-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waman VP, Kolekar PS, Kale MM, Kulkarni‐Kale U. Population structure and evolution of Rhinoviruses. PLoS One. 2014;9(2):e88981. 10.1371/journal.pone.0088981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vaidya SR, Kasibhatla SM, Bhattad DR, et al. Characterization of diversity of measles viruses in India: genomic sequencing and comparative genomics studies. J Infect. 2020;80(3):301‐309. [DOI] [PubMed] [Google Scholar]
- 19. Prasanna H, Sinha DP, Verma A, et al. The population genomics of begomoviruses: global scale population structure and gene flow. Virol J. 2010;7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Altschul S, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25(17):3389‐3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sayers EW, Cavanaugh M, Clark K, Ostell J, Pruitt KD, Karsch‐Mizrachi I. GenBank. Nucleic Acids Res. 2020;48(D1):D84‐D86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katoh K, Rozewicki J, Yamada KD. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 2017;20(4):1160‐1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015;1(1):vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W‐IQ‐TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016;44:W232‐W235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Linkage analysis. Bioinformatics. 2000;16(9):847‐848. [DOI] [PubMed] [Google Scholar]
- 27. Rozas J, Ferrer‐Mata A, Sánchez‐DelBarrio JC, et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol Biol Evol. 2017;34(12):3299‐3302. [DOI] [PubMed] [Google Scholar]
- 28. Dent EA, vonHoldt BM. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour. 2012;4(2):359‐361. [Google Scholar]
- 29. Li X, Zai J, Zhao Q, et al. Evolutionary history, potential intermediate animal host, and cross‐species analyses of SARS‐CoV‐2. J Med Virol. 2020;92:602‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fan Y, Zhao K, Shi ZL, Zhou P. Bat coronaviruses in China. Viruses. 2019;11(3):210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng VC, Lau SK, Woo PC, Yuen KY. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin Microbiol Rev. 2007;20(4):660‐694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou P, Yang XL, Wang XG, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS‐CoV‐2. Natl Sci Rev. 2020. 10.1093/nsr/nwaa036 [published online ahead of print March 3, 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information