

Abstract
The COVID‐19 pandemic caused by SARS‐CoV‐2 infection is spreading at an alarming rate and has created an unprecedented health emergency around the globe. There is no effective vaccine or approved drug treatment against COVID‐19 and other pathogenic coronaviruses. The development of antiviral agents is an urgent priority. Biochemical events critical to the coronavirus replication cycle provided a number of attractive targets for drug development. These include, spike protein for binding to host cell‐surface receptors, proteolytic enzymes that are essential for processing polyproteins into mature viruses, and RNA‐dependent RNA polymerase for RNA replication. There has been a lot of ground work for drug discovery and development against these targets. Also, high‐throughput screening efforts have led to the identification of diverse lead structures, including natural product‐derived molecules. This review highlights past and present drug discovery and medicinal‐chemistry approaches against SARS‐CoV, MERS‐CoV and COVID‐19 targets. The review hopes to stimulate further research and will be a useful guide to the development of effective therapies against COVID‐19 and other pathogenic coronaviruses.
Keywords: coronavirus, Covid-19, drug discovery, protease inhibitors, SARS-CoV
Effective antiviral treatment? The review highlights potential drug design targets against pathogenic coronaviruses, particularly Covid‐19 as well as past and recent progress towards the development of small‐molecule drug‐like compounds including protein‐X‐ray structure‐based design of cysteine protease inhibitors. Hopefully, the present review will stimulate development of much needed effective drugs against Covid‐19.
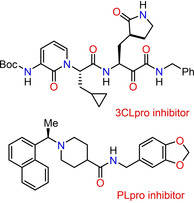
1. Introduction
Severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) originated in Wuhan in central China's Hubei Province, in December 2019.1, 2 The outbreak caused by this new coronavirus has been spreading at an alarming rate, creating a global health crisis the likes of which the world has not witnessed in over a century. SARS‐CoV‐2 has spread to over 170 countries around the globe and has adversely affected over 3.4 million individuals with more than 246,000 deaths as of May 3, 2020. The virus was initially designated as the 2019 novel coronavirus, or, 2019‐nCoV. The International Committee on Taxonomy of Viruses designated the virus as SARS‐CoV‐2.3 Subsequently, the World Health Organization (WHO) named the disease caused by the SARS‐CoV‐2 as Coronavirus disease‐2019 or COVID‐19.4 On March 11, 2020, the WHO officially declared the COVID‐19 outbreak to be a pandemic. The main form of SARS‐CoV‐2 transmission is human‐to‐human spread from respiratory droplets through sneezing, coughing, or close contacts between individuals producing aerosol concentrations. The infection ranges in severity from asymptomatic to serious fatal disease. The most common symptoms include fever, headache, non‐productive cough, dyspnea, and fatigue. Patients with severe disease develop viral pneumonia, acute respiratory distress, and hypoxia, requiring intubation and mechanical ventilation.5, 6 Thus far, there is no approved antiviral medication for the treatment of COVID‐19 patients. As the world struggles to come to grips with the uncertainty of this current global pandemic, it is very important to promote serious drug discovery efforts towards efficacious and broad‐spectrum antiviral agents against these highly pathogenic coronaviruses.
Coronaviruses (Coronaviridae) were discovered in 1960.7, 8 They are a family of positive‐sense, single‐stranded RNA viruses diversely prevalent in humans and wildlife. There are now seven known coronaviruses that cause disease in humans that include, HCoV‐229E, HCoV‐OC43, HCoV‐NL63, HCoV‐HKU1, severe acute respiratory syndrome coronavirus (SARS‐CoV) and Middle East respiratory virus coronavirus (MERS‐CoV), and now SARS‐CoV‐2.9, 10 The first four CoVs cause mild and self‐limiting disease. However, the last three coronaviruses are highly pathogenic, leading to communicable outbreak causing fatal respiratory diseases. SARS‐CoV was first reported in Guangdong Province, China, in November 2002.11, 12 It then spread to other Asian countries, North America, and Europe. The SARS‐CoV outbreak was promptly contained, although it affected more than 8000 individuals and resulted in around 774 deaths, registering 10 % mortality rate.13, 14 The MERS‐CoV was first detected in Saudi Arabia in 2012.15, 16 The MERS‐CoV outbreak was responsible for 2494 infections and led to 858 deaths, registering 35 % fatalities. The SARS‐CoV‐2 outbreak leading to COVID‐19 has grown to become the most serious public health emergency. There exists no known treatment for coronavirus infections. Attempts to re‐purpose historic antimalarial drugs or existing antivirals have yet to show efficacy. Therefore, development of new and effective broad‐spectrum antivirals against current SARS‐CoV‐2 and future outbreaks of pathogenic coronaviruses is an urgent priority. It is imperative that multiple drugs will likely be essential to tackle this pandemic. In this review, we highlight the potential drug development targets, protein X‐ray structure‐based design, lead generation and recent medicinal chemistry efforts toward the evolution of drug‐like small molecules.
2. Drug Design Targets
Although the scientific community is currently empty‐handed in terms of treatments, the availability of the virus RNA genome sequence (GenBank ID: MN908947.3) represents a valuable starting point for the identification of effective treatments. Most importantly, SARS‐CoV‐2 features 82 % similarity with SARS‐CoV (GenBank ID: NC_004718.3) with a 90 % resemblance in various essential enzymes.17, 18, 19, 20, 21
SARS‐CoV‐2 features a spike protein in charge of binding its host cell‐surface receptor, namely the angiotensin‐converting enzyme 2 (ACE2).22 Upon cell entry, viral RNA attaches to the host ribosome in order to produce two polyproteins that are essential for the production of new mature virions.23 The proteolytic cleavage of these two polyproteins is carried out by the coronavirus main proteinase (3CLpro) and the papain‐like protease (PLpro).24 Moreover, all CoVs feature an RNA‐dependent RNA polymerase (RdRp), responsible for replicating the RNA genome.25 All those proteins can represent potential targets in order to tackle SARS‐CoV‐2.21
The potential danger associated with animal reservoirs for the virus and the chance of re‐emergence of epidemic/pandemic CoV‐associated infections prompt robust research efforts in order to identify effective antiviral agents. In this context, the medicinal chemistry efforts performed towards novel therapeutic options for both SARS‐CoV and MERS‐CoV could be of great help to identify potential treatments for SARS‐CoV‐2. Among them, the development of broad‐spectrum antivirals targeting the major viral proteases, shared by all coronaviruses, could represent a very promising strategy in order to generate powerful and versatile therapeutic options against these potentially fatal respiratory illnesses.
3. Coronavirus Proteases
Coronaviruses have the largest known RNA genomes that are approximately 30kb in length, and they use a unique replication strategy. They encode two overlapping open‐reading frames that translate into two polyproteins, pp1a and pp1ab (Figure 1). These polyproteins are further processed to generate four structural proteins and 16 nonstructural proteins (nsps).26 The replicase gene encoding the 16 nsps occupies the majority of the genome, approximately 20 kb, whereas the structural and other accessory proteins account for the remaining 10 kb.27 The CoV replicase polyprotein is processed by two cysteine proteases, the papain‐like protease (PLpro) and a chymotrypsin‐like protease (3CLpro), also known as the Main protease (Mpro).27, 28 The proteolytic processing of the 16 nsps by PLpro and 3CLpro is essential for virus replication and maturation because the nsps are involved in downstream binding and replication events including the formation of the replicase complex, which is essential for viral replication and transcription of the genome.29, 30 Being that both of these proteases are essential for CoV replication, they are attractive targets for antiviral therapies such as small‐molecule inhibitors.31, 32, 33 The X‐ray structures of both 3CLpro (PDB ID: 6W63) and PLpro (PDB ID: 6W9C) from SARS‐CoV‐2 (COVID‐19) are now available for structure‐based drug design (Figure 1).
Figure 1.
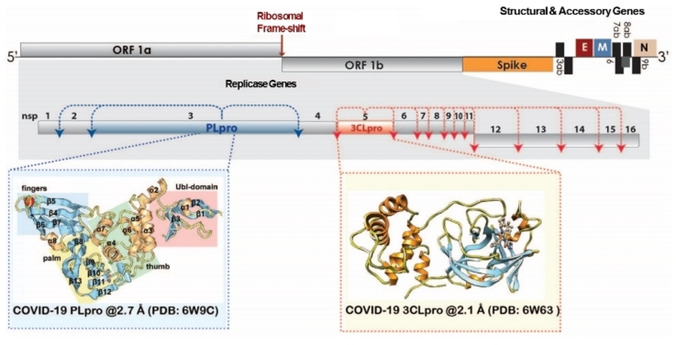
Genome organization of SARS‐CoV‐2, highlighting open reading frames (ORF 1a and ORF 1b) nonstructural, structural and accessory proteins along with both proteases and their corresponding cleavage sites. The 16 nsps are numbered and are shown shaded in gray with the exceptions of PLpro (blue) and 3CLpro (red). The structural and accessory proteins are shown and individual ribbon diagrams for each protease are also shown in individually colored boxes PLpro (blue dashed box) and 3CLpro (red dashed box). The PDB codes for 3CLpro and PLpro from SARS‐CoV‐2 (COVID‐19) are also shown.
SARS‐CoV‐2 and SARS‐CoV share a very high (96 %) sequence identity for their 3CLpro. 3CLpro, encoded in nsp5, cleaves the polyprotein at eleven sites, releasing nsps 4 to 16. Specific recognition sites in which 3CLpro cleave generate a number of other functional viral proteins, including the RNA‐dependent RNA polymerase, RNA binding proteins, exoribonuclease, helicase, and methyltransferase.34 Like PLpro, 3CLpro is also a cysteine‐protease but is active as a homodimer and utilizes a catalytic dyad (Cys‐His) instead of a triad (Cys‐His‐Asp).35 The 3CLpro enzyme must cleave itself out of the polyprotein using its own proteolytic activity in order to release the mature protease.36
Encoded in the large nsp3 protein is the PLpro domain which is a cysteine‐protease involved in processing the replicase polyprotein at the N terminus of pp1a, releasing nsp1‐nsp3.37 Not only is PLpro involved in processing the viral polyprotein, but it also is involved in removing cellular substrates like ubiquitin (Ub), termed deubiquitylation (DUB), and interferon‐stimulated gene product 15 (ISG15), termed deISGylation, from host cell proteins. Ub and ISG15 are both involved in signaling pathways recognized by the host which further stimulates the innate and anti‐viral response.28, 38 The DUB and deISGYlating activities assists in evasion of the host‐innate immune system. PLpro from SARS‐CoV‐2 and SARS‐CoV, unlike 3CLpro, share about 83 % sequence identity, with amino acid composition variations involving the whole surface of the protease, although the three secondary structure components forming the active site do not substantially vary in the two PLpro proteins.39 Therefore, inhibitors developed for the SARS‐CoV PLpro might also work for the SARS‐CoV‐2 PLpro.
4. CoV Protease Inhibitors
Since the SARS outbreak in 2003, and later on with the MERS outbreak in 2012, a large part of medicinal chemistry efforts against SARS‐CoV and MERS‐CoV was devoted to the identification of small peptides, peptidomimetics and small molecules as inhibitors of 3CLpro and PLpro. CoV proteases are appealing targets for the development of antiviral drugs able to reduce viral replication and pathogenicity. Although only a small part of the developed compounds displayed nM affinity towards the protease targets, they represent a good starting point for further optimization and a useful tool for combination antiviral therapies.40, 41, 42, 43 Moreover, the structure‐activity relationships (SAR) studies performed on the developed compounds are extremely useful and could be employed for driving design strategies of novel protease inhibitors against the SARS‐CoV‐2, to be used alone or in combination.
4.1. Biochemical assays for 3CLpro and PLpro proteases for drug discovery
Over the years, a series of biochemical assays have been developed for determining IC50 and K i values for 3CLpro and PLpro/PLP2 proteases. The kinetic properties of 3CLpro are significantly influenced by the construct and assay conditions used. Particularly important is the observation that SARS‐CoV 3CLpro is only functional as a dimer in solution.44, 45, 46 The activity of 3CLpro is significantly reduced when non‐native sequences or affinity‐tags are added to the N or C termini of the enzyme, or when the enzyme used in assays is at concentrations below the equilibrium dissociation constant of the 3CLpro dimer. Therefore, these differences need to be considered when kinetic properties and parameters between different compounds are compared. It is particularly important to implement standardized approaches for meaningful comparison of activity of different compounds.47
In the case of SARS‐CoV 3CLpro, a number of colorimetric and fluorescent substrates have been utilized and each has their advantages.47 FRET assays are typically more sensitive and offer an advantage for continuous monitoring of proteolytic activity over time. A variety of substrates, based on the 3CLpro recognition sequence have been used in the FRET‐based assays.48, 49, 50, 51
Ultimately, a FRET‐based substrate, HiLyte Fluor488TMESATLQ↓SGLRKAK‐QXL520 TM‐NH2 (Anaspec), designed by Mesecar has emerged as having the best properties for routine measurement of coronavirus 3CLpro activity and determination of IC50 and K i values. This substrate is based on a canonical cleavage sequence of SARS‐CoV 3CLpro and it utilizes a very bright HiLyte Fluor488 TM fluorescence group and efficient QXL520 TM quenching group. This was used as a substrate for 3CLpro from a number of coronaviruses including, SARS, MERS, feline infectious peritonitis virus (FIPV), mouse hepatitis virus (MHV), porcine epidemic diarrhea virus (PEDV), infectious bronchitis virus (IBV) and most recently SARS‐CoV‐2.32, 52, 53, 54, 55 Furthermore, the substrate was used for High Troughput Screening (HTS).56 The assay can be readily adaptable to 96, 384 and 1536‐well plates and requires a simple assay buffer, typically 50 mM HEPES pH=7.50, 2 mM DTT, 0.1 mg/mL BSA, and 0.01 % Triton X‐100. The BSA is included to prevent non‐specific binding of 3CLpro to the plastic of the microtiter plate, and the 0.01 % Triton X‐100 is included to help prevent non‐specific protein aggregation caused by some compounds.
The enzyme inhibitory activity of the active esters against SARS‐CoV‐3CLpro is determined using the full‐length, authentic version of the enzyme in a FRET‐based, microplate assay as described by Grum‐Tokars and co‐workers.47 The enhanced fluorescence due to cleavage of this substrate by the protease is monitored and IC50 values for inhibitors are determined by measuring the rates of reaction with increasing inhibitor concentrations.47, 57 MALDI‐TOF analysis is employed to confirm that 3CLpro is covalently modified. The mass shift is compared for the enzyme incubated with the compound and the untreated enzyme.58
For performing drug discovery studies on SARS and MERS coronavirus PLpro, we reported that a simple peptide substrate, Z‐RLRGG‐AMC, which contains the highly fluorescent 7‐amino‐4‐methyl coumarin (AMC) group that is efficiently quenched by the amide bond formed between its amino group and the carboxyl group of the glycine residue, works well for routine assays31, 59, 60, 61, 62 and for HTS.63 This substrate mimics the C terminus of the ubiquitin‐like modifiers Ubiquitin and ISG15 which are also recognized by coronavirus PLpro and PLP2 and cleaved from host‐cell protein substrates. Ubiquitin‐AMC and ISG15‐AMC are also recognized and efficiently hydrolyzed by PLpro and PLP2 but the cost of these commercial substrates is prohibitive for most labs when performing HTS. The K M value for Z‐RLRGG‐AMC is significantly high for most coronavirus PLpro and PLP2 enzymes tested to date which makes it ideal for equating the IC50 and Ki values for competitive inhibitors. Similar to 3CLpro assays, PLpro/PLP2 assays utilize a simple assay buffer typically composed of 50 mM HEPES, pH=7.5, 150 mM NaCl, 2.5 mM DTT 0.1 mg/mL BSA and 0.01 % Triton X‐100. The latter two assay components are added for the same reason as for 3CLpro.
4.2. CoV 3CLpro inhibitors
The structure and activity of SARS‐CoV 3CLpro has been unveiled and prompted the design of 3CLpro inhibitors as novel antivirals.34, 64 SARS‐CoV 3CLpro features three domains: I (comprising residues 8–101), II (comprising residues 102–184), and III (comprising residues 201–301). The active site region spans domains I and II, which are β‐barrel domains, while domain III displays an α‐helical organization. Cys145 of the active site behaves as the nucleophile, while His41 functions as the general acid base. The disclosure of the X‐ray structure of the porcine transmissible gastroenteritis coronavirus protease (TGEV 3CLpro), a related enzyme, in complex with a substrate‐analogue, namely the hexapeptidyl chloromethyl ketone (CMK) inhibitor 1 (Cbz‐Val‐Asn‐Ser‐Thr‐Leu‐Gln‐CMK, Figure 2) represents the first step towards a structure‐based design of novel 3CLpro inhibitors.65
Figure 2.
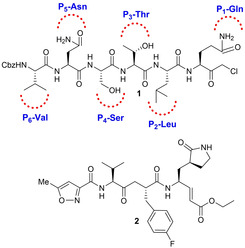
Structure of compounds 1 and 2.
The inhibitor was designed taking into account the P6 and P1 residues of the N‐terminal autoprocessing site of TGEV 3CLpro, while the SARS‐CoV 3CLpro and HCoV‐229E 3CLpro counterparts are represented by Thr‐Ser‐Ala‐Val‐Leu and Tyr‐Gly‐Thr‐Leu‐Gln, respectively. A similar binding mode for this inhibitor is superimposable to that observed for the human rhinovirus 3 C protease (3Cpro).17, 66 A further advance was represented by the discovery that compound AG7088 (2, Figure 2), a peptidomimetic prototypic inhibitor of human rhinovirus 3Cpro adopts an orientation comparable to that of inhibitor 1 in the binding‐site of TGEV 3CLpro.17, 64 Therefore, compounds 1 and 2 became reference compounds for the design of the first SARS‐CoV 3CLpro inhibitors.67
4.2.1. Peptidomimetic covalent CoV 3CLpro inhibitors
In 2005, Ghosh and collaborators reported the design and synthesis of two novel analogues of AG7088 (2), namely compounds 3 and 4 (Figure 3).55 From computational studies on SARS‐CoV 3CLpro structure, it appeared that the P2‐p‐fluorophenylmethyl moiety could be too large to properly fit the S2 pocket, therefore benzyl and prenyl groups were selected as suitable replacements.35 The terminal P1/P1’‐α,β‐unsaturated ester moiety was instead designed to function as a Michael acceptor and covalently bind Cys145. While compound 2 was essentially inactive against SARS‐CoV in cell culture and antiviral assay,68 analogues 3 and 4 were effective against SARS‐CoV 3CLpro (k inact values of 0.014 and 0.045 min−1, respectively) and in cell‐based assays (IC50 values of 45 and 70 μM, respectively). Also, they were devoid of toxic effects up to 100 μM. Compound 4 was also co‐crystallized in complex with SARS‐CoV 3CLpro to a resolution of 1.89 Å, thus unveiling key interactions with residues His164 and Glu166. Also, the P4‐oxazole group appeared to nicely fill the S4‐hydrophobic pocket (Figure 4).
Figure 3.
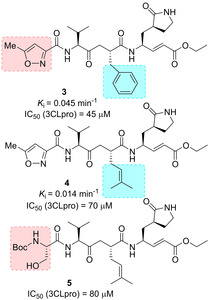
Structure of SARS‐CoV 3CLpro inhibitors 3–5.
Figure 4.
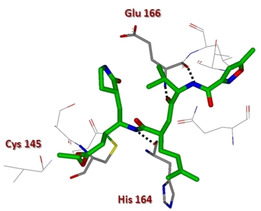
X‐ray crystal structure of compound 4 in the SARS‐CoV 3CLpro active site (PDB ID: 2ALV).
Ghosh and collaborators then speculated that the replacement of P4‐oxazole with a Boc‐Serine as the P4‐ligand would help establish additional hydrogen‐bonding interactions in the protease active site. Structure‐based incorporation of P4‐Boc‐Ser as in inhibitor 5 (Figures 4 and 5) preserved inhibitory potency (IC50=80 μM) although not substantially improving antiviral activity (IC50=75 μM).32
Figure 5.
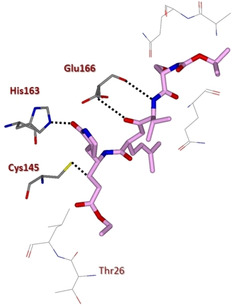
X‐ray crystal structure of compound 5 in the SARS‐CoV 3CLpro active site (PDB ID: 2QIQ).
Yang and co‐workers disclosed a potent SARS‐CoV 3CLpro inhibitor with antiviral activities against SARS‐CoV and human coronavirus (HCoV) 229E 3CL protease. Inhibitor 6 (TG‐0205221, Figure 6) displayed remarkable potency (K i=53 nM) and promising activity against viral replication with a reduction of the viral titer by 4.7 log (at 5 μM) for SARS‐CoV and 5.2 log (at 1.25 μM) for HCoV 229E. The X‐ray structure of 6 in complex with SARS 3CLpro at 1.93 Å resolution revealed an unsymmetrical binding mode with most of the hydrogen bond interactions established in proximity of the P1 site and the aldehyde moiety, while the majority of the hydrophobic interactions take place in the P2, P3, and P4 sites. Cys145 attacks the aldehyde carbonyl group thus forming a covalent C−S bond.69
Figure 6.
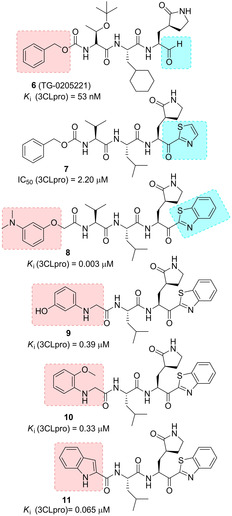
Structure of SARS‐CoV 3CLpro inhibitors 6–11.
In 2009, Hayashi and co‐workers developed a series of trifluoromethyl, benzothiazolyl and thiazolyl ketone‐containing peptidomimetic compounds as SARS‐CoV 3CLpro inhibitors. In particular, compound 7 was disclosed as a SARS‐CoV 3CLpro inhibitor (IC50=2.20 μM), containing a P1‐pyrrolidone and a thiazolyl ketone warhead. Docking studies coupled to molecular dynamics simulations highlighted key hydrogen bonding interaction with the amide backbone of Gly143, Ser144 and Cys 145 of the oxoanion generated from the attack of Cys145 to the thiazolyl ketone portion. The nitrogen of the thiazole ring was also involved in hydrogen bonding contacts with His41.70
A few years later, the same authors, based on docking studies on compound 7, embarked on an optimization process involving key modifications at P1’ and P4 portions. The m‐N,N′‐dimethylamino derivative 8, characterized by a benzothiazole warhead, exhibited the most potent inhibitory activity (K i=3.1 nM). Computational studies indicated a tighter binding of the benzothiazole unit in the S1’‐pocket with respect to the thiazole moiety.71
The same authors developed a series of dipeptide‐type inhibitors with lower molecular weight in which the P3 valine unit of the previous tripeptidomimetic compounds was replaced with different functionalities. These studies identified the N‐arylglycyl as optimum P3 moiety able to establish a hydrogen bond interaction with the backbone of Glu166 in SARS 3CLpro. Moreover, leucine and benzothiazole were identified as suitable P2 and P1′ moieties. Accordingly, compounds 9 and 10 displayed the best inhibitory potencies against 3CLpro (K i values of 0.39 and 0.33 μM, respectively).72 Further studies led to the identification of a rigid indole‐2‐carbonyl unit as one of the best P3 moieties, thus providing inhibitor 11 (K i=0.065 μM).73
In 2017, Kumar and co‐workers developed novel peptidomimetic aldehyde‐based compounds as potent and membrane permeable MERS‐CoV 3CLpro and SARS‐CoV 3CLPro dual inhibitors. Among them, compound 12 (Figure 7) stood out (SARS‐CoV 3CLPro, IC50=0.2 μM, MERS‐CoV 3CLPro, IC50=1.7 μM) In MERS‐CoV‐infected cells, 12 showed antiviral activity (EC50=0.6 μM), by reducing the viral protein production and decreasing secretion of infectious viral particles. The compound was also able to suppress other human and feline α‐ and β‐CoVs, thus unveiling its potential as broad‐spectrum antiviral agent.74 Very recently, Liu and co‐workers described the structure‐based design of capped dipeptide α‐ketoamides as broad‐spectrum antiviral agents targeting the main protease of α or β‐CoVs and the 3 C protease of enteroviruses.
Figure 7.
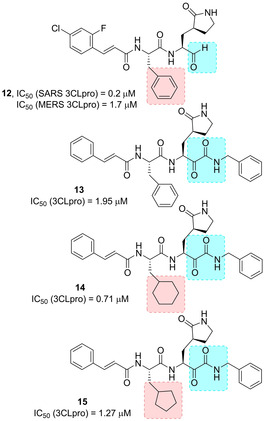
Structure of broad‐spectrum inhibitors 12–15.
The X‐ray structures of six inhibitors in complex with three different proteases were solved. The α‐ketoamide warhead was identified as ideal, with respect to previously employed Michael acceptors and aldehydes. The ketoamide moiety is able to provide two hydrogen bonding acceptors, such as the α‐keto and the amide group oxygens, differently from other warheads featuring only one hydrogen bonding acceptor. Starting from the information provided from complex of lead compound 13 (Figures 7 and 8, IC50 SARS‐CoV 3CLpro=1.95 μM) with the Mpro of HCoV NL63 (an α‐CoV protease) and SARS‐CoV (β‐CoV) as well as the 3Cpro of Coxsackievirus B3 (enterovirus proteases), a systematic study was embarked in order to modify size and flexibility of P2 ligands, in order to obtain compounds able to adapt to target proteases’ S2 pockets. The compounds which better performed were derivatives 14 and 15, bearing a P2 cyclohexylmethyl and cyclopentylmethyl moiety, respectively.75
Figure 8.
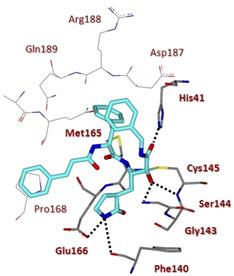
X‐ray crystal structure of compound 13 in the SARS‐CoV 3CLpro active site (PDB ID: 5N19).
Inhibitor 14 showed potent antiviral activity against MERS‐CoV when tested in Huh7 cells (EC50=400 pM), while the activity strongly lowered when Vero cells were used as the host system (EC50=5 μM). The authors speculated that, due to the strong similarity between SARS‐CoV and SARS‐CoV‐2, the new compound is likely to inhibit this new virus as well.75 Based on inhibitor 14, Zhang and co‐workers very recently developed optimized compounds as SARS‐CoV‐2 3CLpro inhibitors.76
A series of modifications were performed on inhibitor 14 in order to improve half‐life and decrease its binding to plasma proteins. In particular, in inhibitor 16 (Figure 9) the P3‐P2 amide bond was masked by incorporating it in a pyridone ring, while the terminal cinnamoyl moiety was replaced by the less hydrophobic tert‐butoxycarbonyl group. The X‐ray structure of SARS‐CoV‐2 3CLpro at 1.75 Å resolution was also solved, showing high similarity (96 %) with SARS‐CoV 3CLpro. This structure was employed to perform docking studies with 16. In order to increase inhibitory potency towards SARS‐CoV‐2, the P2 cyclohexyl moiety, which conferred the broad‐spectrum profile, was sacrificed in favor of a less hindering cyclopropyl ring. Resulting compound 17 was able to effectively inhibit SARS‐CoV‐2 3CLpro (IC50=0.67 μM), but also SARS‐CoV and MERS‐CoV 3CLpro (IC50 values of 0.90 μM and 0.58 μM, respectively). In a SARS‐CoV replicon, compound 17 inhibited RNA replication (EC50=1.75 μM). In SARS‐CoV‐2 infected Calu3 cells, an EC50 of 4–5 μM was registered. Moreover, two X‐ray crystal structures of 17‐bound SARS‐CoV‐2 3CLpro, at 1.95 and 2.20 Å resolution were solved. The compound was also profiled in terms of half‐life and lung tropism, demonstrating a promising profile.
Figure 9.
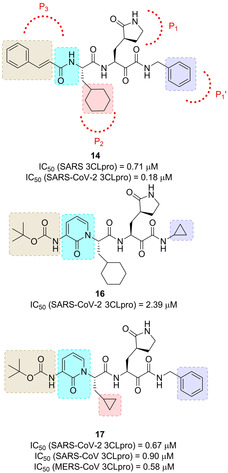
Structure of α‐ketoamide SARS‐CoV‐2 3CLpro inhibitors 16 and 17.
The substrate‐like aza‐peptide epoxide 18 (Figure 10) is representative of another class of 3CLpro covalent inhibitors.77 Although designed for clan CD cysteine peptidases,78 this class of inhibitors is also promising for SARS‐CoV 3CLpro. The (S,S) diastereomer of inhibitor 18 displayed the best inhibitory activity [k inact/K i= 1900 (±400) M−1s−1]. The X‐ray structure of 18 in complex with SARS 3CLpro highlighted the covalent bond between the Cys145 sulfur atom and the epoxide C3 (Figure 11). Computational analysis on the four diastereomers allowed the requirement for a (S,S) configuration of the epoxide to be explained.
Figure 10.
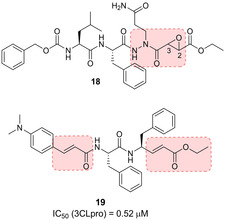
Structure of SARS‐CoV 3CLpro inhibitors 18 and 19.
Figure 11.
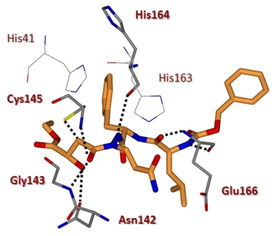
X‐ray crystal structure of compound 18 in the SARS‐CoV 3CLpro active site (PDB ID: 2A5K).
A series of tripeptide α,β‐unsaturated esters and ketomethylene isosteres as covalent SARS‐CoV 3CL protease inhibitors was also disclosed.79 Compound 19 (Figure 10) displayed the best inhibitory potency (IC50=0.52 μM) and interesting activity in cell‐based settings (EC50=0.18 μM). Molecular modelling highlighted crucial hydrogen bonding interaction with the main chain Glu166 and the side chain Gln189.
4.2.2. Small‐molecule covalent CoV 3CLpro inhibitors
Inhibitors containing peptidomimetic scaffolds sometimes lack adequate potency, especially in terms of antiviral activity, which make them unsuitable for drug‐development. Wong and co‐workers reported a series of benzotriazole esters, such as compounds 20–23 (Figure 12) as nonpeptide inhibitors of SARS‐CoV 3CLpro. Among them, compound 23, was identified as the best performing inhibitor of 3CLpro (IC50=0.2 μM; K i=7.5 nM); however, it did not exhibit any antiviral activity.80 The inhibition mechanism for 23 was confirmed via electrospray ionization mass spectrometry and involved the irreversible acylation of Cys145.
Figure 12.
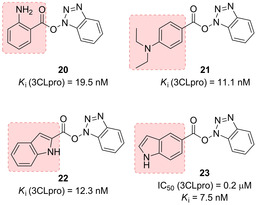
Benzotriazole esters 20–23 as covalent SARS‐CoV 3CLpro inhibitors.
Zhang and collaborators reported a series of halopyridinyl esters as very potent SARS 3CLpro inhibitors (Figure 13). Replacement of thiophene ring (compound 24) with a 2‐furanyl (compounds 25, 26) or a 2‐indolyl moiety (compound 27) led to a tenfold increase in potency. Mass spectrometry studies revealed that compound 23 formed a covalent bond with the enzyme.81, 82, 83 Inhibitor 25 exhibited good enzyme inhibitory activity (IC50=60 nM) against 3CLpro, although antiviral data were not provided for this compound.
Figure 13.
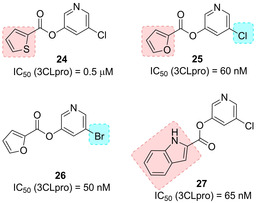
Halopyridinyl esters 24–27 as covalent SARS‐CoV 3CLpro inhibitors.
Ghosh and co‐workers reported a series of 5‐chloropyridinyl indolecarboxylates as potent 3CLpro inhibitors (compounds 28–33, Figure 14). The position of the ester functionality revealed to be a critical parameter for inhibitors’ potency. Among the 5‐substituted indole derivatives (30–32), compound 32, bearing the unsubstituted indole ring performed better than the N‐acetyl and the N‐p‐toluenesulfonyl derivatives. Derivative 33, bearing the carboxylate group at the 4‐position, displayed excellent inhibitory potency (IC50=30 nM) and the best antiviral potency of the series (EC50=6.9 μM). MALDI‐TOF studies confirmed that 3CLpro was covalently modified by 33.58
Figure 14.
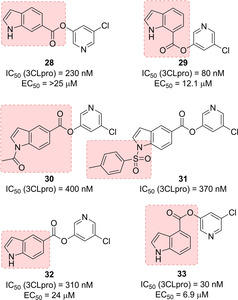
Covalent indole‐based SARS‐CoV 3CLpro inhibitors 28–33.
4.2.3. Noncovalent CoV 3CLpro inhibitors
Although the development of covalent drugs is being reconsidered in the last decade, drugs acting through covalent modifications of the target may likely be associated to off‐target liability and consequent potentially toxic effects. Additional challenges seem to be associated to the development of cysteine protease inhibitors.84 Therefore, many research efforts have been devoted to the search on novel noncovalent inhibitors for 3CLpro inhibitors.
In 2004, a series of noncovalent inhibitors of SARS‐CoV 3CLpro was disclosed, bearing keto‐glutamine‐based warheads with the phthalhydrazido group at the α‐position linked to the tripeptide Ac‐Val‐Thr‐Leu (such as in inhibitors 34–37, Figure 15) for SARS‐CoV 3CLpro. The β and β′ amino group contiguous to the keto group and intramolecular hydrogen bonding increase the electrophilicity of the carbonyl group, which becomes prone to form a hemithioacetal with the sulfur of Cys145.85
Figure 15.
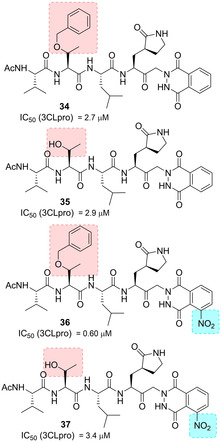
Structure of noncovalent SARS‐CoV 3CLpro inhibitors 34–37.
A series of anilides were also developed as SARS‐CoV 3CLpro inhibitors. Among derivatives 38–41 (Figure 16), compound 38 displayed the best inhibitory potency against SARS‐CoV 3CLpro (K i=0.03 μM) and behaved as a competitive noncovalent inhibitor. Computational analysis showed crucial interactions of dimethylamino, chloro and nitro groups with SARS‐CoV 3CLpro. Compound 38 also showed selectivity towards SARS‐CoV 3CLpro, displaying significantly lower activity against trypsin, chymotrypsin and papain (IC50 values of 110, 200 and 220 μM, respectively).86
Figure 16.
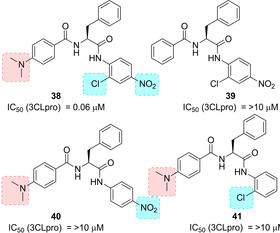
Structure of noncovalent SARS‐CoV 3CLpro inhibitors 38–41.
In 2011, Akaji and co‐workers developed a series of SARS 3CLpro inhibitors containing a terminal aldehyde. An initial lead sequence, namely Ac‐Ser‐Ala‐Val‐Leu‐His‐H (42, IC50=5.7 μM, Figure 17) was employed for further optimization. Systematic modification driven by X‐ray crystallographic analyses on the SARS 3CLpro led to compounds 43 and 44 and finally to the generation of the potent truncated inhibitor 45 (IC50=98 nM). The binding of inhibitor 45 was confirmed by X‐ray analysis, which revealed significant interaction in the protease active site (Figure 18). In particular, the nitrogen atom of the P1 imidazole formed a hydrogen bond with the imidazole nitrogen of His163, and the P2 cyclohexyl group nicely fitted into the large S2 pocket formed by His41, Met49, Met165, and Asp187. Most importantly, coupling information from X‐ray analysis and preincubation assays, it was possible to establish that 45 behaves as a competitive inhibitor and no stable covalent bonds are established with the protease.87
Figure 17.
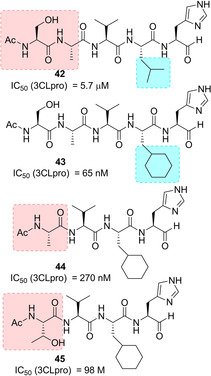
Structure of noncovalent SARS‐CoV 3CLpro inhibitors 42–45.
Figure 18.
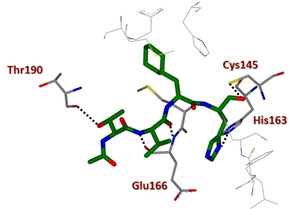
X‐ray crystal structure of compound 45 in the SARS‐CoV 3CLpro active site (PDB ID: 3ATW).
In 2015, the same authors focused on hydrophobic interactions at the cyclohexyl side‐chain of compound 45 in order to design novel inhibitors with reduced peptide features. The authors selected the decahydroisoquinolin scaffold as the suitable option for connecting the P2 cyclohexyl group of 45 to the α‐nitrogen atom of the main‐chain (Figure 19).
Figure 19.
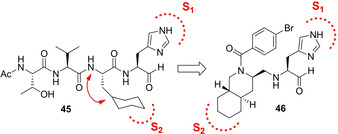
Design of decahydroisoquinolyl‐based scaffold for 3CLpro inhibitors.
Compounds 46–49 (Figure 20) were designed in order to investigate the role of the stereochemistry of the decahydroisoquinolin ring as well as different substitutions at the benzamide moiety. The X‐ray structure of 3CLpro in complex with inhibitor 46 (IC50=63 μM) revealed that the decahydroisoquinolin scaffold nicely fits into the S2 pocket, while imidazole ring occupied S1 pocket (Figure 21). These interactions were able to correctly position the terminal aldehyde moiety tightly into the active site.88
Figure 20.
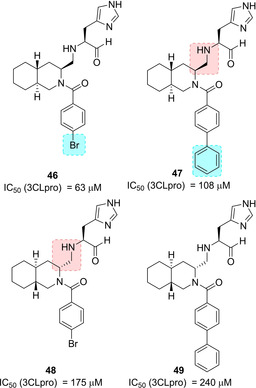
Decahydroisoquinolyl‐based SARS‐CoV 3CL pro inhibitors 46–49.
Figure 21.
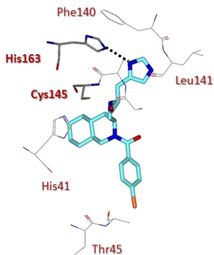
X‐ray crystal structure of compound 46 in the SARS‐CoV 3CLpro active site (PDB ID: 4TWW).
Very recently, the same authors investigated the potential of an octahydroisochromene scaffold as a novel hydrophobic core to interact with the S2 pocket of the protease. All the possible diastereomers (compounds 50–53, Figure 22) were assayed and the results suggested that a specific configuration of the octahydroisochromene scaffold, namely the (1S,3S) as in compound 51, could guarantee the correct positioning of the P1 imidazole and the aldehyde moiety within protease active site. Moreover, the n‐butyl chain at 1‐position of the fused‐ring system was also found to be crucial for hydrophobic interactions.89
Figure 22.
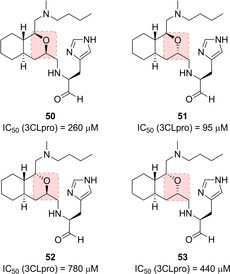
Octahydroisochromene‐based SARS‐CoV 3CLpro inhibitors 50–53.
Wong and co‐workers used computational approaches in order to optimize the SARS 3CL pro inhibitor TL‐3 (54, K i=0.6 μM, Figure 23) previously identified through screening approaches.90 Compounds 55 and 56 were the more effective competitive inhibitors (K i values of 0.073 and 0.34 μM, respectively). Compound 55 is also very selective for the 3CLpro displaying no inhibition against HIV protease. Computational studies on diol 56 in complex with the 3CLpro highlighted that both NHs of the indole rings establish hydrogen bonding interaction with the side chains of Asn142 and His41.91
Figure 23.
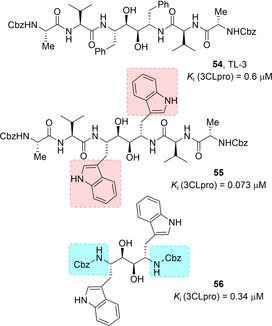
Structures of SARS‐CoV 3CLpro inhibitors 54–56.
Due to inhibitory activity of previously developed isatin derivatives on rhinovirus 3Cpro,92 a series of isatin‐based compounds were developed as SARS‐CoV 3CLpro inhibitors, due to significant similarities in the active sites of the two proteases. The compounds belonging to this series inhibited SARS‐CoV 3CLpro with IC50 in the micromolar range. Inhibitors 57 and 58 (Figure 24) displayed the best inhibitory potencies (IC50 values of 0.98 and 0.95 μM, respectively).93 Docking studies showed that the two compounds nicely fit into the active site of SARS‐CoV 3CLpro, with both carbonyl groups on isatin involved in hydrogen bonding interaction with the NH groups on Gly143, Ser144, Cys145 and the His41 side chain.
Figure 24.
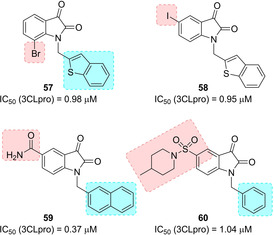
Structures of isatin‐based SARS‐CoV 3CLpro inhibitors 57–60.
Zhou and co‐workers developed a series of N‐substituted 5‐carboxamide‐isatin inhibitors and compared their activities with 5‐halogen‐substituted compounds of the previous series. Carboxamide group could serve to occupy the position of the Gln side chain and establish crucial hydrogen bonds with Phe140 and His163. Compound 59 was identified as the best SARS CoV 3CLpro inhibitor of the series (IC50=0.37 μM). MALDI‐TOFF mass spectrometry studies indicated that the inhibitor did not form a covalent bond with the protease. Compound 59 may be used as a broad‐spectrum antiviral since it also inhibited HRV‐14 3 C protease.94
In 2014 Liu and co‐workers investigated the replacement of a carboxamide group with a variety of substituted sulfonamide moieties in order to improve the inhibitory activity against SARS CoV 3CLpro. Compound 60, with the 5‐sulfonyl isatin bearing a six‐membered ring and with a pendant N‐1 benzyl ring, displayed the best inhibitory potency (IC50=1.04 μM).95
A cluster of serine residues (namely, Ser139, Ser144, Ser147) was detected in the catalytic site of SARS‐CoV 3CLpro, which supported the development of aryl boronic acid derivatives displaying IC50 values in the low micromolar range. Compound 61 (Figure 25), bearing anilide linkages, was found to be the best performing compound (IC50=0.04 μM).48 Dipeptidyl fluoromethyl ketones were also disclosed as SARS‐CoV 3CLpro inhibitors. Their activity was also assessed in cell‐based settings using infected Vero and CaCo‐2 cultures. Compound 62 was identified as the most potent SARS‐CoV inhibitor (EC50=2.5 μM). The compound also showed low toxicity.96
Figure 25.
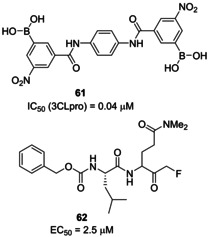
Structures of SARS‐CoV 3CLpro inhibitors 61 and 62.
A series of pyrazolone derivatives were proposed as SARS‐CoV 3CLpro inhibitors by Ramajayam and co‐workers (Figure 26). Compounds 63 and 64 (IC50=5.5 and 6.8 μM, respectively) were identified as the best performing derivatives of the series and they also behaved as moderate inhibitors against CVB3 3Cpro (IC50=20.8 and 22.4 μM, respectively), while 65 displayed the best activity against CVB3 3Cpro (IC50=9.6 μM). Therefore, these inhibitors could be potentially employed as anticoronaviral and anti‐picornaviral agents.97 The same authors also disclosed a series of 2‐(benzylthio)‐6‐oxo‐4‐phenyl‐1,6‐dihydropyrimidines as SARS‐CoV 3CLpro inhibitors (66–68, Figure 26). Compound 68 displayed the higher inhibition potency against SARS‐CoV 3CLpro (IC50=6.1 μM). The developed compounds are devoid of cytotoxicity.98
Figure 26.
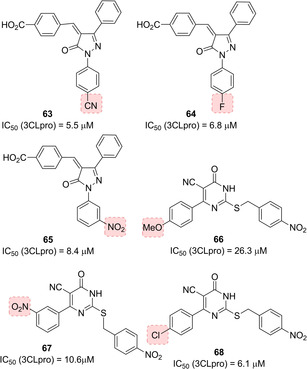
Structures of pyrazolone‐ and dihydropyrimidine‐based SARS‐CoV 3CLpro inhibitors 63–68.
4.2.4. CoV 3CLpro inhibitors from screening and natural sources
A broad variety of CoV 3CLpro inhibitors have been increasingly disclosed from screening activities on small molecule/peptidomimetics and natural compounds. In 2004, a library of 50,240 structurally diverse small molecules was screened. From this research, 104 compounds were identified displaying anti‐SARS‐CoV activity. Compound 69 (Figure 27) was found to behave as an inhibitor of SARS‐CoV 3CLpro (IC50=2.5 μM). The compound also performed well in the Vero cell‐based SARS‐CoV plaque reduction assay (EC50=7 μM).99 In the same year, another research group screened over 50 000 drug‐like molecules, using a quenched FRET assay. The 572 hits deriving from the screening underwent additional virtual and experimental filtering, which finally identified five small‐molecule inhibitors of SARS‐CoV 3CLpro (IC50=0.5–7 μM). Compounds 70 and 71 showed promising potential as SARS‐CoV 3CLpro inhibitors (IC50 values of 4.3 and 7.0 μM, respectively) with significant selectivity over four proteases (HAV 3Cpro, NS3pro, chymotrypsin and papain).81
Figure 27.
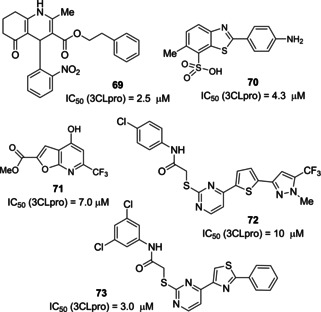
SARS‐CoV 3CLpro inhibitors 69–73 derived from screening activities.
In 2006, Tsai and co‐workers employed structure‐based virtual screening coupled to analogue search in order to identify novel SARS‐CoV 3CLpro inhibitors. More than 59,000 compounds were docked and, among them, a set of 93 derivatives underwent inhibition assay against SARS‐CoV 3CLpro. 21 compounds showed inhibition and, interestingly, three of them showed common substructures. A subsequent search for analogues with common substructure on commercial databases allowed the identification of 25 compounds endowed with inhibitory potencies (IC50 values ranging from 3 to 1000 μM). The two best performing derivatives are compounds 72 and 73, (Figure 27, IC50 values of 10 and 3 μM, respectively). The 28 compounds deriving from these two parallel activities were then analyzed through 3D‐QSAR studies in order to derive a pharmacophore model.100 A structural‐based virtual screening was also performed by Mukherjee and co‐workers in order to identify novel non‐peptide inhibitors of SARS‐3CLpro. Two inhibitors, namely 74 (PJ07) and 75 (PJ169; Figure 28, IC50 values of 18.2 and 17.2 μM, respectively), identified through this screening approach, were able to mimic the interactions of the peptide substrate with the active site of SARS‐3CLpro, with full occupancy of S1′, S1, and S2 pockets.101 In 2011, Kim and collaborators performed a structure‐based virtual screening involving 308,307 compounds. Fifty‐three compounds were shortlisted to undergo in vitro assay for inhibition of 3CLpro expressed by Escherichia coli. The IC50 of the seven best performing compounds ranged from 38.38 to 101 μM. Among them, compounds 76 and 77 behave as competitive inhibitors of 3CLpro (K i values of 9.93 and 9.11 μM, respectively). An in‐depth docking analysis for compound 76 revealed H‐bonding interaction with Phe140, Gly143, Cy145, and Glu166 residues of the S1 site of SARS‐CoV 3CLpro. The nitrophenyl moiety is involved in key H‐bond interaction with Cys145 and Gly143 as well as hydrophobic contacts with His41 and Cys145.102
Figure 28.
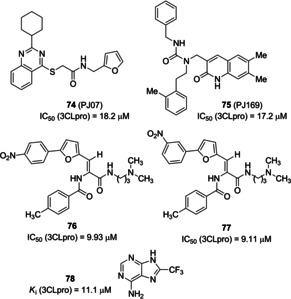
SARS‐CoV 3CLpro inhibitors 74–78 derived from screening activities.
In 2014, Johnson and co‐workers used a combination of virtual screening and HTS to search for novel inhibitors of SARS‐CoV 3CLpro. 621,000 compounds from the ZINC library were screened by integrating docking and pharmacophore‐based protocols. Subsequently, a fluorescence‐based enzymatic HTS assay was implemented for 41 000 shortlisted compounds from the previous procedure. Initial HTS was also complemented with a surface plasmon resonance (SPR)‐based assay. Inhibitor 78 (Figure 28, K i=11.1 μM) was identified from these protocols exhibiting mixed‐type inhibition. The compound was also assayed against three other cysteine proteases (SARS‐CoV PLpro and UCH−L1), a serine protease (Hepatitis C Virus NS3/4 A) and two non‐proteolytic enzymes (Bacillus anthracis dihydroorotase and Streptococcus pneumoniae PurC). Compound 78 was only able to inhibit two SARS cysteine proteases, 3CLpro and PLpro, thus displaying excellent selectivity.103
Yang and co‐workers evaluated in cell‐based assays (Vero E6 cells) 221 phytocompounds against SARS‐CoV activities. A series of terpenoids and lignoids (22 compounds in total) inhibited 50 % of Vero E6 cell proliferation. Among them, betulinic acid (79) and savinin (80; Figure 29) behaved as competitive inhibitors of SARS‐CoV 3CLpro (K i values of 8.2 and 9.1 μM, respectively).104
Figure 29.
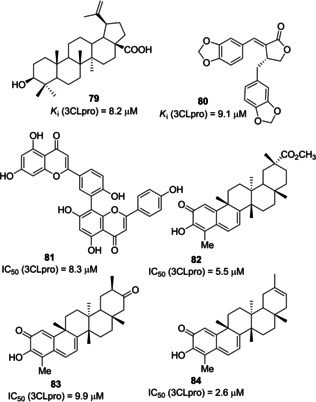
SARS‐CoV 3CLpro inhibitors 79–84 derived from natural sources.
In 2010, Lee and collaborators selected Torreya nucifera, an Asiatic medicinal plant, as a botanical source of SARS‐CoV 3CLpro inhibitors. The ethanolic extract displayed a SARS‐CoV 3CLpro inhibitory activity of 62 % at 100 mg/mL. Fractionation and subsequent FRET analysis led to the identification of the biflavone amentoflavone 81 (IC50=8.3 μM) as a promising 3CLpro inhibitor. Molecular docking helped rationalize the results deriving from enzymatic assays.105
The same authors isolated various quinone‐methide triterpenes, among them pristimerin (82), tingenone (83) and and iguesterin (84) from Triterygium regelii. The compounds were evaluated against SARS‐CoV 3CLpro showing potent inhibitory activities (IC50 values of 5.5, 9.9, and 2.6 μM, respectively). All the compounds behaved as competitive inhibitors.106
A high‐throughput screening of the NIH molecular libraries sample collection (∼293 000 compounds) led to the identification of novel SARS 3CLpro inhibitors. This novel dipeptide class of noncovalent inhibitors is represented by initial 3‐pyridyl‐based hit 85 (Figure 30). A Ugi multicomponent reaction allowed easy SAR exploration with identification of two compounds, 86 and 87 active against SARS‐CoV 3CLpro (IC50 values 4.89 and 28.1 μM, respectively). The X‐ray crystal structure of (R)‐86 (ML188) bound to SARS‐CoV 3CLpro was determined and it highlighted a similar binding orientation to that of known covalent peptidomimetic inhibitors (Figure 31). In particular the R enantiomer was shown to occupy the S3–S1′ subpockets of SARS‐CoV 3CLpro. Moreover, chiral supercritical fluid chromatography led to pure enantiomers (R)‐86 (ML188) and (S)‐86. Whereas the R enantiomer displayed inhibitory activity (IC50=1.5 μM) against SARS‐CoV 3CLpro, its S counterpart was inactive.
Figure 30.
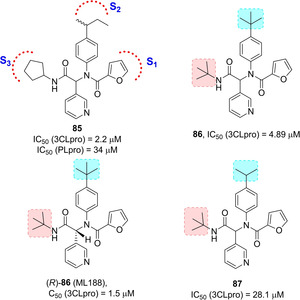
Hit compound 85 and optimized derivatives 86 and 87.
Figure 31.
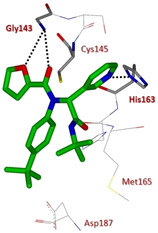
X‐ray crystal structure of compound 86 in the SARS‐CoV 3CLpro active site (PDB ID: 3 V3 M).
The SAR study around P1′showed that compounds bearing imidazole (88) and 5‐chlorofuran (89; Figure 32) exhibited equipotent profile to 86 (IC50 values of 6.0 and 5.2 μM, respectively). Exploration around P1 ligands showed that the only suitable replacement for 3‐pyridyl ring was represented by pyridazine (90) and pyrazine (91) showing IC50 values of 10 and 5.5 μM, respectively.56
Figure 32.
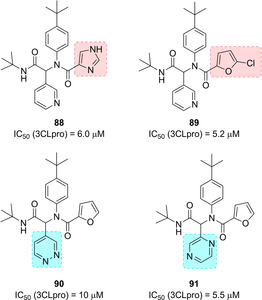
Structures of optimized SARS‐CoV 3CLpro inhibitors 88–91.
The same authors continued their quest for potent SARS‐CoV 3CLpro inhibitors focusing on a series of benzotriazoles from MLPCN screening, which allowed the identification of hit compound 92 (Figure 33). The X‐ray crystal structure of SARS‐CoV 3CLpro in complex with 92 showed the binding of the compound into an induced‐fit binding site generated by a rearrangement of the Gln189 and Met49 residue side chains (Figure 34).
Figure 33.
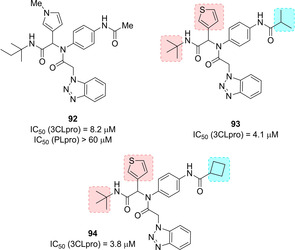
Hit compound 92 and optimized derivatives 93 and 94.
Figure 34.
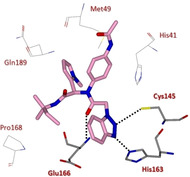
X‐ray crystal structure of compound 92 in the SARS‐CoV 3CLpro active site (PDB ID: 4MDS).
Replacement of the N‐methyl pyrrole moiety with a thiophene ring and substitutions on the acetamide moiety (P2–P1′ region) led to i‐propyl amide 93 and cyclobutylamide 94 (Figure 33) with improved inhibitory profile (IC50 values of 4.1 and 3.8 μM, respectively).
Moreover, in order to reduce the molecular weight, the authors also performed a P3‐truncation. Among the truncated amides (compounds 95–98, Figure 35), compound 95 characterized by a cyclopropyl amide moiety was identified (ML300, IC50=4.1 μM) and was employed as a chemical probe for further optimization, which led to inhibitor 98, the first sub‐micromolar inhibitor (IC50=0.051 μM).107
Figure 35.
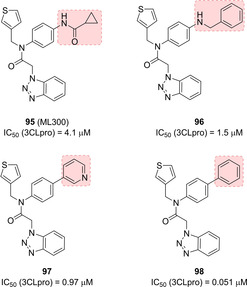
Structures of optimized SARS‐CoV 3CLpro inhibitors 95–98.
4.3. SARS‐CoV PLpro inhibitors
While many structural and mechanistic studies have been performed on SARS‐CoV 3CLpro which resulted in potent inhibitors,35, 64, 77, 81, 108, 109 the structure‐based design approaches against PLpro enzyme from SARS‐CoV or other coronaviruses were limited for a long time due to lack of structural information. SARS‐CoV PLpro is considered a comparably druggable target to 3CLpro since both are crucial for viral replication. The X‐ray structure of the catalytic domain (35‐kDa, residues 1541–1855) of PLpro of the SARS‐CoV nsp3 polyprotein domain was determined for the first time in 2006 at 1.85 Å resolution.28, 63 The SARS‐CoV nsp3 multi‐domain protein plays a crucial role in the generation of virus replication complexes, through insertion into host membranes and interactions with nsp4 and nps6.110, 111 nsp3 contains various domains most likely conserved in all CoV.112 The N‐terminal region of the nsp3 is strongly conserved among CoVs.
This region contains a ubiquitin‐like (Ubl) globular fold and a glutamic acid rich acidic‐domain (AC domain).113 Next to the AC domain is located the macro domain (or X domain) a key player in the synthesis of viral subgenomic RNAs.114, 115 The AC domain is followed by SARS unique domain (SUD), which lacks counterparts in other coronaviruses.116 SUD domain is followed by a second Ubl domain and the catalytically active PLpro domain processing the nsp1/2, nsp2/3 and nsp3/4 cleavage sites.117, 118 The nsp3 multi‐domain displayed a high frequency of point mutations, which may produce pleiotropic effects in SARS‐CoV and other CoVs pathogenicity.65, 119 The active site of SARS‐CoV PLpro features a catalytic triad, formed by Cys112−His273−Asp287. Cys112 acts as the nucleophile, His273 behaves as a general acid‐base, and Asp287 favors the His alignment, thus promoting Cys112 deprotonation. The various available X‐ray structures of SARS‐CoV PLpro highlighted a flexible substrate‐binding loop, containing Tyr269 residue, which can assume multiple conformations. This loop is in an open conformation in the unliganded SARS‐CoV PLpro, while it is found in the closed form upon inhibitor binding.63 Similar to other cysteine proteases, SARS‐CoV PLpro reacts with electrophilic warheads, via the nucleophilic attack of the thiolate on the electrophilic carbon of the warhead functionality, thus generating a covalently modified enzyme. In 2014, the first X‐ray crystal structure of SARS‐CoV PLpro covalently modified with an electrophilic warhead was solved.39
The crystal structure of MERS‐CoV PLpro was determined both alone or as a covalent complex with Ub (PLproUb).120, 121 The structure encompasses a C‐terminal catalytic domain and an N‐terminal Ubl domain, similar to SARS‐CoV.122 Two Ubl domains within nsp3 multi‐domain protein are named as Ubl1 and Ubl2. Recent studies unveiled that removing the Ubl2 domain from MERS PLpro does not affect its capability to process the viral polyprotein. The X‐ray structure of MERS PLpro‐ΔUbl2 was solved at 1.9 Å resolution and matched to PLpro featuring the N‐terminal Ubl2 domain, showing that the catalytic core of MERS‐CoV is stable and highly active even without its Ubl2 domain. Therefore, PLpro‐ΔUbl2 may be employed for structure‐based inhibitor design.59
Ghosh and co‐workers reported for the first time naphthalene‐based inhibitors against SARS‐CoV PLpro deriving from a HTS activity on 50 080 compounds.31 Two compounds featuring a naphthylmethylamine moiety, namely 99 (Figure 36) and 107 (Figure 39, below) were identified (IC50 values of 20.1 μM and 59 μM, respectively). The R enantiomer of compound 99, displayed the best inhibitory potency (IC50=8.7 μM). A preliminary SAR study on this compound highlighted that the ortho‐methyl group on the benzene ring was critical for activity, and that decoration of the naphthyl ring on the 1‐position rather than the 2‐position was strongly favored. Moreover, insertion of an amino group at the meta‐position with respect to the amide functionality increased inhibitory potency on PLpro by 4‐fold. Compound 100 displayed significantly improved potency with respect to initial hits (IC50=0.6 μM). This inhibitor also showed the ability to inhibit SARS‐CoV viral replication in Vero cells (EC50=14.5 μM), acting as a noncovalent, competitive inhibitor of SARS‐CoV PLpro (K i=0.49 μM).62
Figure 36.
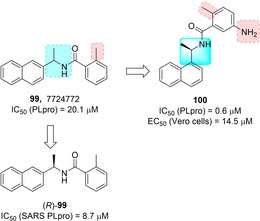
Hit compound 99 and optimized derivative 100.
Figure 39.
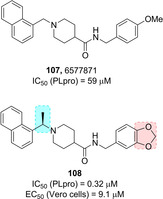
Hit compound 107 and optimized derivative 108.
The X‐ray structure of SARS‐CoV PLpro bound 100 was solved at 2.5 Å resolution (Figure 37). This structure highlights extensive interaction with the S3 and S4 subsites of SARS‐CoV PLpro.31 The amide moiety establishes two hydrogen bonds with the Asp165 and Gln270 residues, while the remaining interactions are mainly hydrophobic. The naphthyl ring forms hydrophobic contacts with the phenyl rings of Tyr265 and Tyr269 and with the side chains of Pro248 and Pro249. The (R)‐methyl group nicely inserts into a polar cavity, close to Tyr265 and Thr302, while the ortho‐methyl group points toward the floor of the cavity. The amino‐substituent of the benzene ring behaves as hydrogen‐bond acceptor.
Figure 37.
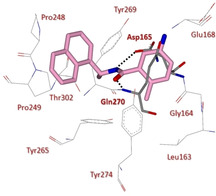
X‐ray crystal structure of compound 100 in the SARS‐CoV PLpro active site (PDB ID: 3E9S)
Based on the X‐ray crystal structure of inhibitor 100‐bound to SARS‐CoV PLpro, a drug design template was generated. A series of focused modification were performed in order to assess the importance of the amide NH and the α‐methyl group and the effect of substitution on the benzamide ring (compounds 101–106, Figure 38). This structure‐based scaffold morphing led to the development of inhibitor 105 (IC50=0.46 μM; antiviral EC50=6 μM) and its methylamine derivative 106, with a moderate enzyme inhibitory potency and the best SARS antiviral activity in the series (IC50=1.3 μM; EC50=5.2 μM). Computational studies also allowed the generation of a predictive 3D‐QSAR model for the generation of optimized SARS‐CoV PLpro inhibitors.
Figure 38.
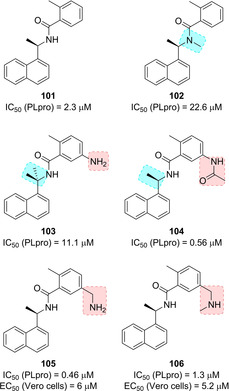
Structures of PLpro inhibitors 101–106.
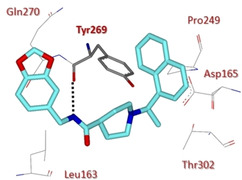
The other hit deriving from HTS, namely 107 (Figure 39), features a 1‐substituted naphthalene ring and behaves as a competitive, noncovalent inhibitor of SARS‐CoV PLpro.31 SAR studies were embarked in order to evaluate the outcome of focused substitutions. Again, the 1‐naphthyl substitution was favorite with respect to the 2‐naphthyl functionalization. In line with the previous series, a methyl group at the benzylic‐naphthyl position increased inhibitory potency, although with less marked stereochemical preference. A series of methoxy‐substituted and benzodioxole derivatives were also developed. Among them, inhibitor 108, displayed excellent inhibitory potency against SARS‐CoV PLpro (IC50=0.32 μM) and the ability to inhibit SARS‐CoV viral replication in Vero cells (EC50=9.1 μM), acting as potent, noncovalent, competitive inhibitor of PLpro.61 X‐ray studies on the co‐crystal structure of 108 in complex with SARS‐CoV PLpro at 2.63 Å resolution reveal that the inhibitor binds similarly to 100, with the naphthylmethylamine moieties in the same position and orientation (Figure 40). The piperidine moiety of 108 is positioned analogously to the carboxamide nitrogen of inhibitor 100, while the portion containing the amide and benzodioxole occupies a binding cleft not utilized by 100.
Figure 40.
X‐ray crystal structure of compound 108 in the SARS‐CoV PLpro active site (PDB ID: 3MJ5).
The amide nitrogen of 108 forms an additional hydrogen bonding interaction with the carbonyl oxygen belonging to the backbone of Tyr269, which may explain its improved potency with respect to inhibitor 100.
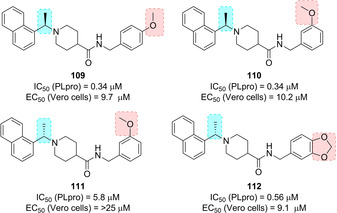
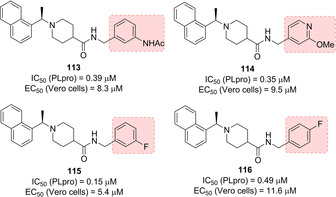
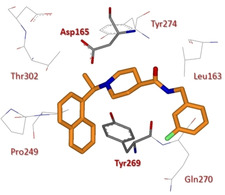
SAR studies investigated the role of the stereochemistry at the α‐methyl groups as well as substitution at the terminal aryl moiety (compounds 109–112, Figure 41). Interestingly, the (S)‐Me inhibitor 112 displays nearly equivalent enzymatic and antiviral potencies to its R enantiomer (IC50=0.56 μM; EC50=9.1 μM). Further optimization process of compounds 100 and 108 started by investigating the steric and electronic requirements of the benzylic‐naphthyl position, benzylic position, decoration and bioisosteric replacement of the benzyl and naphthyl portions, and the outdistancing between aromatic rings. Substituents placed at the benzylic position did not provide an increase in inhibitory potency, while substituents on the benzene ring strongly influence compounds’ potency. In particular, the meta‐ (115, Figure 42) and para‐fluoro substituted (116) and the meta‐acetamido‐substituted (113) derivatives displayed the best inhibitory profile (for 115, IC50=0.15 μM and EC50=5.4 μM; for 116, IC50=0.49 μM and EC50=11.6 μM; for 113, IC50=0.39 μM and EC50=8.3 μM). The increase of the distance between the amide moiety and benzene was found to be detrimental for PLpro inhibition. All the bioisosteric replacements attempted on both the naphthyl and the benzene rings did not improve inhibitor's potency, except for derivative 114, with a potency comparable to 108 (IC50=0.35 μM and EC50=9.5 μM). Inhibitor 115 was co‐crystallized with PLpro and its X‐ray structure solved at 2.1 Å resolution (Figure 43).60 Similar to 100 and 108, the compound was found to closely bind S3 and S4 subsites in proximity of the active site adopting their same orientation. An induced‐fit mechanism, involving a closed conformation of the BL2 loop, with the key contribution of Tyr269 residue, was unveiled thus explaining the strongly interaction of inhibitors. In particular, a key hydrogen bonding interaction between the backbone carbonyl of Tyr269 and the amide nitrogen is formed. The naphthyl‐rings identically bind to the hydrophobic pocket (Pro248, Pro249, Tyr265, Tyr269, and Thr302) as described for 100 and 108. Due to the superior resolution of the X‐ray structures of 115, three conserved water molecules were unveiled in the cavity filled by the (R)‐methyl substituent which strongly reduce the available space for larger groups. The metabolic stability of these compounds was evaluated in mouse liver microsomes. While 115 was found to be promptly metabolized, inhibitors 113 and 114 displayed improved metabolic stability with respect to initial hit 108.60
Figure 41.
Structures of PLpro inhibitors 109–112.
Figure 42.
Structures of PLpro inhibitors 113–116.
Figure 43.
X‐ray crystal structure of compound 115 in the SARS‐CoV PLpro active site (PDB ID: 4OW0).
In 2008, Chou and co‐workers, upon development of a FRET assay for the screening of PLpro inhibitors in a high‐throughput setting, screened a library of 960 compounds as SARS‐CoV PLpro inhibitors. Two thiopurine derivatives, namely compounds 117 (6MP) and 118 (6TG; Figure 44, previously used as anticancer agents), displayed micromolar inhibitory potency against PLpro (IC50 values of 21.6 and 5 μM, respectively). The thiocarbonyl moiety was found to play a key role and to covalently bind the active site Cys residue. However, the toxicity liabilities associated with the compounds limit their use as anti‐SARS‐CoV drugs.40, 123 In 2015, Cheng and collaborators unveiled that 117 and 118 are able to inhibit also MERS‐CoV PLpro (IC50 values of 26.9 and 24.4 μM, respectively).124
Figure 44.
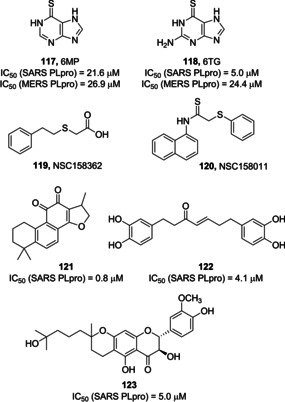
SARS‐CoV and MERS‐CoV PLpro inhibitors 117–123.
In 2011, Frieman and co‐workers screened 2000 compounds from the NIH Diversity Set library with a yeast‐based assay to identify novel SARS‐CoV PLpro inhibitors. The hits deriving from this primary assay were then investigated against SARS‐CoV replication and inhibition of PLpro function in cells. Compound 119 (NSC158362) was identified and was found able to inhibit SARS‐CoV replication devoid of cytotoxic effects, although not specifically inhibiting PLpro. A second derivative, 120 (NSC158011) was instead able of inhibiting PLpro in cell‐based assays, although with no potential of blocking SARS‐CoV replication.125
A series of natural compounds have also been identified as PLpro inhibitors. In 2012, Park and co‐workers assayed seven different tanshinones against SARS‐CoV PLpro through a continuous fluorometric assay. Since SARS‐CoV PLpro inhibition was proportional to the pre‐incubation time, a slow‐binding mechanism was hypothesized involving an isomerization of the enzyme‐inhibitor complex and covalent inhibition. The most potent tanshinone derivative as SARS‐CoV PLpro inhibitor is compound 121 (IC50=0.8 μM).126 The same authors were also able to identify a series of diarylheptanoid inhibitors of SARS‐CoV PLpro. An α,β‐unsaturated carbonyl group was found to be a key functionality for the inhibitory activity, thus supporting a covalent inhibition mode. Compound 122 was identified as the most potent diarylheptanoid SARS‐CoV PLpro inhibitor (IC50=4.1 μM).127 In 2013, Cho and co‐workers identified five novel geranylated flavonoids as SARS‐CoV PLpro inhibitors. The most active inhibitor was the geranylated flavonoid 123 (Figure 44, IC50=5.0 μM).128
In 2015, Johnson and co‐workers screened 25 000 as SARS‐CoV and MERS‐CoV PLpro inhibitors. Compound 78 (Figure 28) was identified as a small‐molecule noncovalent dual inhibitor (SARS‐CoV PLpro, IC50=10.9 μM; MERS‐CoV PLpro, IC50=6.2 μM). Kinetics and competition SPR studies ascertained that compound 78 behaves as a competitive inhibitor of MERS‐CoV PLpro, while acting as an allosteric inhibitor on SARS‐CoV PLpro. These results were explained hypothesizing a different inhibitor recognition specificity between MERS‐CoV PLpro and SARS‐CoV PLpro. The major contribution to inhibitor selectivity and behavior could be ascribed to the structural differences of the BL2 loop. Moreover, compound 78 showed selectivity for SARS‐CoV and MERS‐CoV PLpro over two human homologues, ubiquitin C‐terminal hydrolases 1 and 3 (hUCH−L1 and hUCH−L3).129
5. Anti SARS‐CoV and MERS‐CoV Compounds from Repurposing Approaches
An extensive repositioning activity of approved drugs has been embarked since the first SARS‐CoV outbreak in 2003 and later on with MERS‐CoV outbreak. This approach displays the significant advantage of involving compounds directly transferrable to clinical use or eligible for accelerated approval. In 2003, Kaletra, composed by the anti‐HIV protease inhibitors lopinavir and ritonavir, showed some antiviral activity against SARS‐CoV.130 This study prompted investigation of lopinavir, ritonavir, niclosamide, promazine, and two other HIV inhibitors, PNU and UC2 as SARS‐CoV 3CLpro, demonstrating their potential for further optimization.131, 132
A database containing 8000 approved drugs disclosed cinanserin (SQ 10 643), a serotonin antagonist, as a potential inhibitor of SARS‐CoV 3CLpro. Following in vitro assessment confirmed that cinanserin and its hydrochloride salt inhibit SARS‐CoV 3CLpro (IC50=4.92 and 5.05 μM, respectively).133 A virtual docking screening identified forty compounds upon post‐dock screening filters, including pharmacophore model and “drug‐like” filters. Calmidazolium (C3930), a calmodulin antagonist, was found to noncovalently inhibit SARS‐CoV 3CLpro (K i=61 μM).134 Additionally, three natural components of tea, namely tanic acid, 3‐isotheaflavin‐3‐gallate and theaflavin‐3,3′‐digallate demonstrated inhibitory activity against SARS‐CoV 3CLpro (IC50 values <10 μM).44
Beyond behaving as 3CLpro inhibitors many approved drugs displayed anti CoV activities with miscellaneous or unknown mechanisms.
Glycyrrhizin was able to inhibit SARS‐CoV replication after virus absorption in Vero cells (EC50=300 mg/L), most likely blocking viral entry.135 Some derivatives were even more potent against SARS‐CoV replication (EC50 values ranging from 5 μM to 50 μM), although with high cytotoxicity.136 In line with antiviral activity shown by nitric oxide (NO),137 the NO donor, S ‐nitroso‐N‐acetylpenicillamine was able to dose‐dependently block SARS‐CoV replication, possibly acting in the early steps of infection.138 HIV protease inhibitor nelfinavir,139 antihelminthic drug niclosamide 140 and the antimalarial chloroquine 141 all inhibited SARS‐CoV replication (EC50=0.048 μM, EC50=1–3 μM, and IC50=8.8 μM, respectively). Chloroquine, has also shown inhibitory effects against SARS‐CoV‐2 in Vero E6 cells (EC50=1.13 μM) and is currently in an open‐label trial (ChiCTR2000029609).142
The antiviral agent ribavirin, was employed to treat SARS patients, but it did not block viral growth at concentrations achievable in human serum.143 Interferon (IFN)‐α also displayed in vitro inhibitory activity starting at concentrations of 1000 IU/mL.144 However, the combination of ribavirin and IFN‐β led to a synergistic effect against SARS‐CoV replication.145 Trials involving anti‐HCV combination of a pegylated IFN plus ribavirin were also initiated with SARS‐CoV‐2 (ChiCTR2000029387).146
As infection from SARS‐CoV relies on the endosomal cleavage of a protein located on viral surface, the antiarrhythmic drug amiodarone underwent in vitro testing on infected Vero cells. Amiodarone was able to affect the life‐cycle of SARS‐CoV in a concentration‐dependent manner. Moreover it was found that i) the drug was able to associate with cell membranes and accumulate in acidic organelles; ii) the diethylamino‐β‐ethoxy moiety is crucial for uptake; iii) trypsin cleavage of the viral spike protein before infection, responsible for virus entry, does not influence amiodarone antiviral potency.147 In 2014, Dyall and co‐workers screened a library of 290 compounds for their antiviral potential against MERS‐CoV and SARS‐CoV. 27 compounds, belonging to 13 different drug classes, showed antiviral activity against both MERS‐CoV and SARS‐CoV. Among them both chlorpromazine hydrochloride and triflupromazine hydrochloride strongly inhibited MERS‐CoV and SARS‐CoV (EC50 values ranging from 5.76 μM to 12.9 μM, respectively). Gemcitabine also inhibited MERS‐CoV and SARS‐CoV with micromolar (EC50 values of 1.2 μM and 4.9 μM, respectively) displaying low toxicity. Moreover, two kinase inhibitors, namely imatinib mesylate and dasatinib, inhibited MERS‐CoV and SARS‐CoV activities (EC50 values ranging from 2.1 to 17.6 μM, respectively).148 In 2016, Coleman and co‐workers demonstrated that imatinib exerts its anti‐CoV activity at the early stages of infection, by blocking the fusion step of the virions at the endosomal membrane. Abelson tyrosine‐protein kinase 2 (Abl2), essential for effective SARS‐CoV and MERS‐CoV replication in vitro, was identified as the imatinib target.149
6. Potential Therapeutic Strategies for SARS‐CoV‐2
SARS‐CoV‐2 is a single‐stranded RNA β‐coronavirus. As above described for SARS‐CoV and MERS‐CoV, its genome is able to encode nonstructural proteins (such as 3CLpro, PLpro, helicase, and RNA‐dependent RNA polymerase (RdRp)), as well as structural (spike protein, nucleocapsid (N) matrix (M) and envelope (E)) and accessory proteins. While the nonstructural proteins are crucial for CoVs life cycle, the spike protein is necessary during viral entry for virus–host cell interaction.150 Regarding the four enzymes, their genomic sequences from SARS‐CoV‐2 clearly highlight the high sequence similarity and the high conservation of drug‐binding pockets with respect to their SARS and MERS counterparts, thus supporting their role as valuable therapeutic targets for SARS‐CoV‐2.21 Insights into the development of therapeutics against coronavirus diseases by targeting N protein have also been disclosed over years and could represent a suitable strategy to be pursued also for SARS‐CoV‐2.151 Moreover, repurposing inhibitors designed for MERS‐CoV and SARS‐CoV enzymes could represent a reasonable starting point for identifying novel therapeutic options for SARS‐CoV‐2.
We have already described in detail in paragraph 4 the work from Zhang and co‐workers which allowed the identification of very potent α‐ketoamide based 3CLpro inhibitors as promising strategy to target SARS‐CoV‐2.76
Similar to SARS‐CoV, very recently it was shown that the SARS‐CoV‐2 uses the angiotensin‐converting enzyme 2 (ACE2) as a receptor for cell entry.2, 152 The fusion of the viral and cell membranes occurs upon binding of the spike protein on the virion with ACE2. Fusions allows viral RNA replication inside the host cell and generate new virions able to infect other cells.153 The CoV spike glycoprotein is a crucial target for the development of vaccines, antibodies, and diagnostics. Very recently, a cryo‐EM structure of the SARS‐CoV‐2 spike trimer in the prefusion conformation has been solved at 3.5 Å‐resolution by Wrapp and co‐workers. The protein trimer displays only one of the three receptor‐binding domains (RBDs) in a receptor‐accessible conformation.154 The authors also demonstrated that SARS‐CoV‐2 spike protein binds ACE2 with higher affinity with respect to SARS‐CoV spike counterpart. Also, many monoclonal antibodies specifically developed for SARS‐CoV RBD (e. g., m396, CR3014) do not effectively bind SARS‐CoV‐2 spike protein. This disclosed structure should then be crucial to rapidly develop novel therapeutics and diagnostics for SARS‐CoV‐2.154
Very recently, a SARS‐CoV‐specific human monoclonal antibody, namely CR3022, was found to potently bind SARS‐CoV‐2 RBD (K D=6.3 nM). Interestingly, CR3022 epitope does not overlap with the ACE2 binding site within SARS‐CoV‐2 RBD. This antibody should then be exploited as an effective therapeutic option, either alone or in combination with other antibodies.155
A series of molecules emerged over years acting as viral entry inhibitors. Inhibitor 124 (Figure 45) was identified through a screening protocol performed on 50,240 compounds. It was able to block pseudovirus entry and SARS‐CoV plaque formation (EC50 values of 3 and 1.6 μM, respectively).99 Another screening of small molecules deriving from Chinese herbal medicine was performed, thus unveiling the potential of luteolin (125) and tetra‐O‐galloyl‐β‐d‐glucose (126, TGG) as viral entry inhibitors (EC50 values of 4.5 and 10.6 μM, respectively).156 Upon binding with ACE2, SARS‐CoV is incorporated in a vesicle in order to enter the cell. Cathepsin enzymes exert their action in the acid medium which characterizes the vesicle, facilitating fusion of viral and the vesicle membrane. In particular, the study from Simmons and co‐workers in 2005 highlighted compound 127 (MDL28170), a cathepsin L inhibitor, as a novel therapeutic option for targeting SARS‐CoV entry (EC50=100 nM).157
Figure 45.
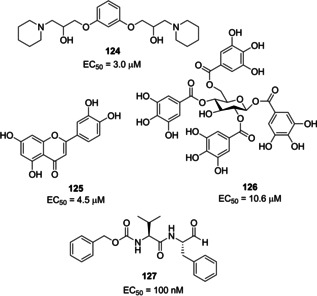
Viral entry inhibitors 124–127.
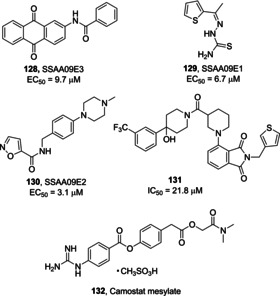
In 2013, Adedeji and co‐workers screened a chemical library of around 14,000 drug‐like compounds for blocking SARS‐CoV entry. This study unveiled compounds, among them 128–130 (Figure 46), acting by at least three different mechanisms. Compound 130 (SSAA09E2) inhibits SARS‐CoV by blocking the interaction between ACE2 and SARS spike protein RBD; compound 129 (SSAA09E1) acts as a cathepsin L inhibitor; compound 128 (SSAA09E3) blocks viral entry by inhibiting fusion of the viral membrane with the host cell membrane. Dose‐response experiments for the three compounds were performed to assess SARS/HIV pseudotyped virus entry inhibition (128, EC50=9.7 μM; 129, EC50=6.7 μM; 130, EC50=3.1 μM).158
Figure 46.
Viral entry inhibitors 128–132.
Kandell and co‐workers performed a virtual screening on 1.56 million compounds, thus engaging the best performing hits in cell‐cell fusion and MERS‐CoV plaques inhibition assays. The activity led to the identification of three promising derivatives. Among them, compound 131 (IC50=21.8 μM), at 10 μM concentration, was able to produce 74.5 % reduction in MERS‐CoV plaques formation, with low cell toxicity (HEK293 and Vero cells), and favorable predicted drug‐like parameters.159
Very recently, it was also demonstrated that SARS‐CoV‐2 uses the host serine protease TMPRSS2 for spike protein priming. The serine protease inhibitor camostat mesylate (132, Figure 46), able to inhibit TMPRSS2 activity,42, 160 was found to block viral entry and reduce infection of Calu‐3 lung cells by SARS‐CoV‐2. This compound is approved in Japan for the treatment of chronic pancreatitis‐associated pain and might constitute a novel treatment option for SARS‐CoV‐2 outbreak.161
RdRp is another key enzyme in CoVs life cycle. As such, it became a suitable target for a variety of RNA viruses, such as hepatitis C and Zika viruses.162, 163 Another promising example is provided from a recent study showing that remdesivir (133, RDV, Figure 47), which interferes with the viral polymerase, and IFNβ have superior antiviral activity to lopinavir and ritonavir (currently involved in human trials for MERS infection in Saudi Arabia). In murine experiments, RDV showed prophylactic and therapeutic potential ameliorating the pulmonary function and reducing viral loads in lungs.164 Since active site of RdRp is highly conserved, its utility as a target might be extended also to SARS‐CoV‐2. In this context, Elfiky recently performed a sequence analysis, followed by docking studies in order to model SARS‐CoV‐2 RdRp. The study supports the potential of Sofosbuvir (134), IDX‐184 (135), Ribavirin (136), and RDV against SARS‐CoV‐2 RdRp.165
Figure 47.
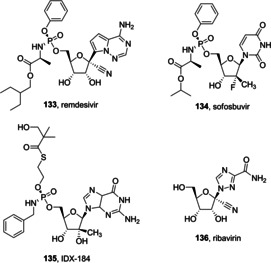
RdRp inhibitors 133–136.
The antimalarial agent chloroquine (137, Figure 48) demonstrated significant inhibition in the spread of SARS‐CoV by interfering with ACE2 in Vero E6 cell lines.166 It was shown that chloroquine can act at both entry and post‐entry stages during SARS‐CoV‐2 infection in Vero E6 cells. Also, it shows immune‐modulating activity, thus enhancing its antiviral potency in vivo.142 Recent clinical trials conducted in China demonstrated acceptable safety in COVID‐19 patients and efficacy in reducing exacerbation of pneumonia, and shortening the disease course.167 The actual clinical benefit of chloroquine in Covid‐19 patients however has not been conclusive.
Figure 48.
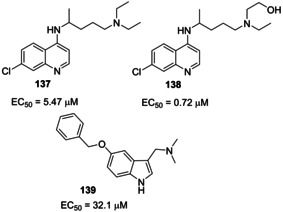
Structure of chloroquine (137), hydroxychloroquine (138) and compound 139.
Hydroxychloroquine (138), a less toxic aminoquinoline, has an N‐hydroxyethyl side chain which makes it more soluble than chloroquine.168 Similar to chloroquine, hydroxychloroquine possesses antiviral effects. In addition, hydroxychloroquine exerts a modulating effect on activated immune cells, downregulating the expression of Toll‐like receptors (TLRs) and TLR‐mediated signal transduction, and reducing the production of interleukin‐6.169 Hydroxychloroquine (EC50=0.72 μM) was found to be more potent than chloroquine (EC50=5.47 μM) to inhibit SARS‐CoV‐2 in vitro.170 Although, the clinical efficacy of hydroxychloroquine in COVID‐19 patients has not yet been established.
Lin and co‐workers recently disclosed a novel approach based on protein‐protein interactions (PPIs). The authors exploited a conserved hydrophobic cavity at the interface of the dimeric structure of the N‐terminal domain of the MERS‐CoV nucleocapsid protein (MERS‐CoV N‐NTD) to perform a structure‐based screening of small molecules.
Compound 139 (5‐benzyloxygramine) was identified through this approach, acting as a potent stabilizer of MERS CoV N protein and displaying antiviral profile in Vero E6 cells infected with MERS‐CoV (EC50=32.1 μM). X‐ray studies were also performed on the identified compounds. Compound 139 blocks MERS‐CoV activity most likely prompting abnormal aggregation of the N protein within the cell.171 Targeting non‐native PPIs may represent a valuable approach to be possibly extended to SARS‐CoV‐2.
7. Conclusion
The Covid‐19 pandemic caused by highly transmissible SARS‐CoV‐2 has become a major public health crisis in the world, today. The pandemic is spreading at an alarming rate overwhelming existing healthcare system, and causing inordinate fatalities, particularly to elderly and immunocompromised patients around the globe. Covid‐19 has induced huge economic damages with shortage and uncertainty worldwide. At present, there is no vaccine or approved antiviral medication available for the treatment of Covid‐19. In our current pandemic situation, effective antiviral treatment could have made a significant impact on reducing morbidity and mortality. Antivirals could also be used as cheap prophylaxis. Thus, it is essential to proceed with very serious efforts for the development of effective, broad‐spectrum antivirals against Covid‐19. Past and present research efforts on the coronavirus replication cycle provided a number of significant biochemical targets for drug development. As the SARS‐CoV‐2 genome has over 80 % similarity to SARS‐CoV, previous work on the development of antivirals against SARS is very beneficial and timely. Much ground work has been laid in terms of small‐molecule lead generation, identification and limited medicinal chemistry optimization of lead structures. This review outlines various important protein targets for drug development and highlights principles and strategies for drug design along with a host of small‐molecule lead structures. We hope that this review will stimulate drug design and discovery efforts toward the development of broad‐spectrum antivirals against COVID‐19 and future pathogenic coronaviruses.
Abbreviations
- ACE2
angiotensin‐converting enzyme 2
- AMC
7‐amino‐4‐methyl coumarin
- Boc
tert‐butyloxycarbonyl
- BSA
bovine serum albumin
- 3CLpro
coronavirus main proteinase
- CMK
chloromethyl ketone
- CoVs
coronaviruses
- 3Cpro
3 C protease
- FIPV
feline infectious peritonitis virus
- FRET
fluorescence resonance energy transfer
- HTS
high‐throughput screening
- IBV
infectious bronchitis virus
- IFN
interferon
- IRF3
interferon regulatory factor 3
- MERS‐CoV
Middle East respiratory virus coronavirus
- MHV
mouse hepatitis virus
- NIH
National Institutes of Health
- PEDV
porcine epidemic diarrhea virus
- PLpro
papain‐like protease
- PPI
protein‐protein interaction
- QSAR
quantitative structure‐activity relationships
- RBDs
receptor‐binding domains
- SAR
structure‐activity relationships
- SARS‐CoV
severe acute respiratory syndrome coronavirus
- SARS‐Cov‐2
severe acute respiratory syndrome coronavirus 2
- SPR
surface plasmon resonance
- SUD
SARS Unique Domain
- RdRp
RNA‐dependent RNA polymerase
- TGEV 3CLpro
transmissible gastroenteritis coronavirus protease
- TGG
tetra‐O‐galloyl‐β‐d‐glucose
- TLRs
Toll‐like receptors
- Ubl
ubiquitin‐like
- hUCH−L1
ubiquitin C‐terminal hydrolase 1
- hUCH−L3
ubiquitin C‐terminal hydrolase 3
- WHO
World Health Organization
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Arun K. Ghosh obtained his PhD in 1985 at the University of Pittsburgh. He pursued postdoctoral research at Harvard University (1985–1988). He was a research fellow at Merck Research Laboratories. In 1994, he joined the chemistry faculty at the University of Illinois‐Chicago as an assistant Professor and became full Professor in 1998. In 2005, he moved to Purdue University where he is currently the Ian P. Rothwell Distinguished Professor in the Department of Chemistry and Department of Medicinal Chemistry and Molecular Pharmacology. His interests include bioactive natural products, developing tools and strategies for protein structure‐based molecular design and drug discovery.

Biographical Information
Margherita Brindisi received her PhD in Pharmaceutical Sciences from the University of Siena in 2008. She was a postdoctoral research fellow in Professor Ghosh's research group 2010 and again in 2016. She worked on protease inhibitors for the treatment of HIV/AIDS and Alzheimer's disease. She also worked at the University of Siena on developing treatments for cancer, viral and parasitic diseases, and brain disorders. In April 2019, she became an Assistant Professor at the Department of Pharmacy at University of Naples Federico II, where she is working on the application of sustainable methodologies to her drug discovery projects.

Biographical Information
Dana Shahabi obtained her B.S. in applied chemistry in 2007 at the University of Shahreza, Iran, her M.S. degree in organic chemistry from Yazd University, Iran, in 2010, and her Ph.D. from Isfahan University of Technology in 2017 on developing methods for heterocyclic compounds by using green catalysts and computational chemistry. In 2019, she joined Professor Ghosh's group at Purdue University as a postdoctoral research fellow. Her current research focuses on the design, synthesis and biological evaluation of next‐generation anti‐HIV agents and drug design for severe acute respiratory syndrome (SARS) coronavirus and Covid‐19.

Biographical Information
Mackenzie E. Chapman obtained her Bachelor of Science and Arts from Purdue University in 2018. In Professor Mesecar's lab, she is studying the structure and function of coronaviral papain‐like proteases (PLPs) encoded in nonstructural protein 3 (nsp3). She is using X‐ray crystallography to perform detailed structural analyses for comparison of PLPs from α and β coronaviruses, and enzyme kinetics and biochemistry to understand substrate and inhibitor selectivity of PLPs. Her structural information is being used to design small‐molecule drugs and attenuated vaccines to treat coronavirus infections in humans (SARS‐CoV‐2, SARS‐CoV, MERS and NL63) and in animals (PEDV and FIPV).

Biographical Information
Andrew D. Mesecar received his Ph.D. in Biochemistry from the University of Notre Dame in 1995, then pursued post‐doctoral research at UC Berkeley under Prof. Daniel E. Koshland, Jr. He joined the University of Illinois‐Chicago as an assistant professor in 1999, becoming a full Professor in 2008. In 2010, he was recruited to Purdue as the Walther Professor of Structural Biology and Deputy Director of the Purdue Center for Cancer Research. He is interested in the structure and function of medically important enzymes involved in cancer, coronavirus infections and Alzheimer's disease, and using structure‐based approaches to design therapeutics against these enzymes.

Acknowledgements
Financial support of this research in part by the National Institutes of Health (AI150466, A.K.G.) and Purdue University is gratefully acknowledged. This work was also supported in part by the National Institute of Allergy and Infectious Disease of the National Institutes of Health under contract HHSN272201700060 C to A.D.M. and A.K.G. and award number R01AI085089 to A.D.M. M.E.C. is supported by an NIH/NIGMS T32 Training Grant for Structural Biology and Biophysics (#GM132024). We also acknowledge MIUR Grant Dipartimento di Eccellenza (Department of Excellence, M.B.) 2018–2022 to the Department of Pharmacy, University of Naples Federico II.
A. K. Ghosh, M. Brindisi, D. Shahabi, M. E. Chapman, A. D. Mesecar, ChemMedChem 2020, 15, 907.
References
- 1. Wu F., Zhao S., Yu B., Chen Y M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., Zhang Y. Z., Nature 2020, 579, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou P., Yang X. L., Wang X. G., Hu B., Zhang L., Zhang W., Si H. R., Zhu Y., Li B., Huang C. L., Chen H. D., Chen J., Luo Y., Guo H., Jiang R. D., Liu M. Q., Chen Y., Shen X. R., Wang X., Zheng X. S., Zhao K., Chen Q. J., Deng F., Liu L. L., Yan B., Zhan F. X., Wang Y. Y., Xiao G. F., Shi Z. L., Nature 2020, 579, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu Y., Ho W., Huang Y., Jin D. Y., Li S., Liu S. L., Liu X., Qiu J., Sang Y., Wang Q., Yuen K. Y., Zheng Z. M., Lancet 2020, 395, 949–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coronavirus disease, COVID pandemic. Accessed: March 20, 2020: http://www.who.int/emergencies/diseases/novel-coronavirus-2019.
- 5.Coronavirus disease COVID pandemic, 2020: situation summary. Accessed: March 20, 2020: https://www.cdc.gov/coronaviru/2019-ncov/cases-updates/summary.html.
- 6. Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B., Lancet 2020, 395, 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kahn J. S., McIntosh K., Pediatr. Infect. Dis. J. 2005, 24, S223–227, discussion S226. [DOI] [PubMed] [Google Scholar]
- 8. Sexton N. R., Smith E. C., Blanc H., Vignuzzi M., Peersen O. B., Denison M. R., J. Virol. 2016, 90, 7415–7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cui J., Li F., Shi Z. L., Nat. Rev. Microbiol. 2019, 17, 181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zumla A., Hui D. S., Perlman S., Lancet 2015, 386, 995–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Peiris J. S., Guan Y., Yuen K. Y., Nat. Med. 2004, 10, S88-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pyrc K., Berkhout B., van der Hoek L., Expert. Rev. Anti. Infect. Ther. 2007, 5, 245–253. [DOI] [PubMed] [Google Scholar]
- 13. Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H. R., Becker S., Rabenau H., Panning M., Kolesnikova L., Fouchier R. A., Berger A., Burguiere A. M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J. C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H. D., Osterhaus A. D., Schmitz H., Doerr H. W., N. Engl. J. Med. 2003, 348, 1967–1976. [DOI] [PubMed] [Google Scholar]
- 14. Christian M. D., Poutanen S. M., Loutfy M. R., Muller M. P., Low D. E., Clin. Infect Dis. 2004, 38, 1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Memish Z. A., Zumla A. I., Al-Hakeem R. F., Al-Rabeeah A. A., Stephens G. M., N. Engl. J. Med. 2013, 368, 2487–2494. [DOI] [PubMed] [Google Scholar]
- 16. Rahman A., Sarkar A., Am. J. Public. Health. 2019, 109, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hilgenfeld R., Peiris M., Antiviral. Res. 2013, 100, 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eckerle I., Muller M. A., Kallies S., Gotthardt D. N., Drosten C., Virol. J. 2013, 10, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Butler D., Nature 2015, 522, 139–140. [DOI] [PubMed] [Google Scholar]
- 20.WHO Director-General's opening remarks at the media briefing on COVID-19. Accessed: March 14, 2020: https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-mission-briefing-on-covid-2019–2013-march-2020.
- 21. Morse J. S., Lalonde T., Xu S., Liu W. R., ChemBioChem 2020, 21, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dong N., Yang X., Ye L., Chen K., Wai-Chi Chan E., Yang M., Chen S., bioRxiv 2020, DOI:10.1101/2020.1101.1120.913368. [Google Scholar]
- 23. Baranov P. V., Henderson C. M., Anderson C. B., Gesteland R. F., Atkins J. F., Howard M. T., Virology 2005, 332, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ziebuhr J., Snijder E. J., Gorbalenya A. E., J. Gen. Virol. 2000, 81, 853–879. [DOI] [PubMed] [Google Scholar]
- 25. Xu X., Liu Y., Weiss S., Arnold E., Sarafianos S. G., Ding J., Nucl. Acids. Res. 2003, 31, 7117–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Forni D., Cagliani R., Clerici M., Sironi M., Trends Microbiol. 2017, 25, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.“Coronaviruses: An Overview of Their Replication and Pathog.enesis”, A. R. Fehr, S. Perlman in Coronaviruses, Vol. 1282, Springer, New York, 2015. [DOI] [PMC free article] [PubMed]
- 28. Ratia K., Saikatendu K. S., Santarsiero B. D., Barretto N., Baker S. C., Stevens R. C., Mesecar A. D., Proc. Natl. Acad. Sci. USA 2006, 103, 5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham R. L., Sparks J. S., Eckerle L. D., Sims A. C., Denison M. R., Virus Res. 2008, 133, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanjanahaluethai A., Chen Z., Jukneliene D., Baker S. C., Virology 2007, 361, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B. S., Johnson M. E., Baker S. C., Ghosh A. K., Mesecar A. D., Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ghosh A. K., Xi K., Grum-Tokars V., Xu X., Ratia K., Fu W., Houser K. V., Baker S. C., Johnson M. E., Mesecar A. D., Bioorg. Med. Chem. Lett. 2007, 17, 5876–5880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Deng X., StJohn S. E., Osswald H. L., O′Brien A., Banach B. S., Sleeman K., Ghosh A. K., Mesecar A. D., Baker S. C., J. Virol. 2014, 88, 11886–11898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thiel V., Ivanov K. A., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., Snijder E. J., Rabenau H., Doerr H. W., Gorbalenya A. E., Ziebuhr J., J. Gen. Virol. 2003, 84, 2305–2315. [DOI] [PubMed] [Google Scholar]
- 35. Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H., Gao G. F., Anand K., Bartlam M., Hilgenfeld R., Rao Z., Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muramatsu T., Takemoto C., Kim Y. T., Wang H., Nishii W., Terada T., Shirouzu M., Yokoyama S., Proc. Natl. Acad. Sci. USA 2016, 113, 12997–13002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ratia K., Mesecar A., O'Brien A., Baker S. C. in Handbook of Proteolytic Enzymes Elsevier, 2013, pp. 2195–2199. [Google Scholar]
- 38. Baez-Santos Y. M., Mielech A. M., Deng X., Baker S., Mesecar A. D., J. Virol. 2014, 88, 12511–12527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ratia K., Kilianski A., Baez-Santos Y. M., Baker S. C., Mesecar A., PLoS Pathog. 2014, 10, e1004113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen X., Chou C. Y., Chang G. G., Antivir. Chem. Chemother. 2009, 19, 151–156. [DOI] [PubMed] [Google Scholar]
- 41. Park J. Y., Ko J. A., Kim D. W., Kim Y. M., Kwon H. J., Jeong H. J., Kim C. Y., Park K. H., Lee W. S., Ryu Y. B., J. Enzyme Inhib. Med. Chem. 2016, 31, 23–30. [DOI] [PubMed] [Google Scholar]
- 42. Zhou Y., Vedantham P., Lu K., Agudelo J., R. Carrion, Jr. , Nunneley J. W., Barnard D., Pohlmann S., McKerrow J. H., Renslo A. R., Simmons G., Antiviral. Res. 2015, 116, 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kumar V., Jung Y. S., Liang P. H., Expert. Opin. Ther. Pat. 2013, 23, 1337–1348. [DOI] [PubMed] [Google Scholar]
- 44. Chen C. N., Lin C. P., Huang K. K., Chen W. C., Hsieh H. P., Liang P. H., Hsu J. T., Evid. Based Complement Alternat. Med. 2005, 2, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fan K., Wei P., Feng Q., Chen S., Huang C., Ma L., Lai B., Pei J., Liu Y., Chen J., Lai L., J. Biol. Chem. 2004, 279, 1637–1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shi J., Wei Z., Song J., J. Biol. Chem. 2004, 279, 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grum-Tokars V., Ratia K., Begaye A., Baker S. C., Mesecar A. D., Virus Res. 2008, 133, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bacha U., Barrila J., Velazquez-Campoy A., Leavitt S. A., Freire E., Biochemistry 2004, 43, 4906–4912. [DOI] [PubMed] [Google Scholar]
- 49. Graziano V., McGrath W. J., DeGruccio A. M., Dunn J. J., Mangel W. F., FEBS Lett. 2006, 580, 2577–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kao R. Y., To A. P., Ng L. W., Tsui W. H., Lee T. S., Tsoi H. W., Yuen K. Y., FEBS Lett. 2004, 576, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kuo C. J., Chi Y. H., Hsu J. T., Liang P. H., Biochem. Biophys. Res. Commun. 2004, 318, 862–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. St John S. E., Therkelsen M. D., Nyalapatla P. R., Osswald H. L., Ghosh A. K., Mesecar A. D., Bioorg. Med. Chem. Lett. 2015, 25, 5072–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. St John S. E., Tomar S., Stauffer S. R., Mesecar A. D., Bioorg. Med. Chem. 2015, 23, 6036–6048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tomar S., Johnston M. L., St John S. E., Osswald H. L., Nyalapatla P. R., Paul L. N., Ghosh A. K., Denison M. R., Mesecar A. D., J. Biol. Chem. 2015, 290, 19403–19422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ghosh A. K., Xi K., Ratia K., Santarsiero B. D., Fu W., Harcourt B. H., Rota P. A., Baker S. C., Johnson M. E., Mesecar A. D., J. Med. Chem. 2005, 48, 6767–6771. [DOI] [PubMed] [Google Scholar]
- 56. Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S. A., Chase P., Eggler A., Dawson E. S., Baez-Santos Y. M., Tomar S., Mielech A. M., Baker S. C., Lindsley C. W., Hodder P., Mesecar A., Stauffer S. R., J. Med. Chem. 2013, 56, 534–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhu L., George S., Schmidt M. F., Al-Gharabli S. I., Rademann J., Hilgenfeld R., Antiviral. Res. 2011, 92, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghosh A. K., Gong G., Grum-Tokars V., Mulhearn D. C., Baker S. C., Coughlin M., Prabhakar B. S., Sleeman K., Johnson M. E., Mesecar A. D., Bioorg. Med. Chem. Lett. 2008, 18, 5684–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Clasman J. R., Baez-Santos Y. M., Mettelman R. C., O′Brien A., Baker S. C., Mesecar A. D., Sci. Rep. 2017, 7, 40292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baez-Santos Y. M., Barraza S. J., Wilson M. W., Agius M. P., Mielech A. M., Davis N. M., Baker S. C., Larsen S. D., Mesecar A. D., J. Med. Chem. 2014, 57, 2393–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ghosh A. K., Takayama J., Rao K. V., Ratia K., Chaudhuri R., Mulhearn D. C., Lee H., Nichols D. B., Baliji S., Baker S. C., Johnson M. E., Mesecar A. D., J. Med. Chem. 2010, 53, 4968–4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ghosh A. K., Takayama J., Aubin Y., Ratia K., Chaudhuri R., Baez Y., Sleeman K., Coughlin M., Nichols D. B., Mulhearn D. C., Prabhakar B. S., Baker S. C., Johnson M. E., Mesecar A. D., J. Med. Chem. 2009, 52, 5228–5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Baez-Santos Y. M., St John S. E., Mesecar A. D., Antiviral. Res. 2015, 115, 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., Hilgenfeld R., Science 2003, 300, 1763–1767. [DOI] [PubMed] [Google Scholar]
- 65. Snijder E. J., Bredenbeek P. J., Dobbe J. C., Thiel V., Ziebuhr J., Poon L. L., Guan Y., Rozanov M., Spaan W. J., Gorbalenya A. E., J. Mol. Biol. 2003, 331, 991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anand K., Palm G. J., Mesters J. R., Siddell S. G., Ziebuhr J., Hilgenfeld R., EMBO J. 2002, 21, 3213–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ghosh A. K., Xi K., Johnson M. E., Baker S. C., Mesecar A. D., Annu. Rep. Med. Chem. 2007, 41, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Seipelt J., Guarne A., Bergmann E., James M., Sommergruber W., Fita I., Skern T., Virus Res. 1999, 62, 159–168. [DOI] [PubMed] [Google Scholar]
- 69. Yang S., Chen S. J., Hsu M. F., Wu J. D., Tseng C. T., Liu Y. F., Chen H. C., Kuo C. W., Wu C. S., Chang L. W., Chen W. C., Liao S. Y., Chang T. Y., Hung H. H., Shr H. L., Liu C. Y., Huang Y. A., Chang L. Y., Hsu J. C., Peters C. J., Wang A. H., Hsu M. C., J. Med. Chem. 2006, 49, 4971–4980. [DOI] [PubMed] [Google Scholar]
- 70. Regnier T., Sarma D., Hidaka K., Bacha U., Freire E., Hayashi Y., Kiso Y., Bioorg. Med. Chem. Lett. 2009, 19, 2722–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Konno S., Thanigaimalai P., Yamamoto T., Nakada K., Kakiuchi R., Takayama K., Yamazaki Y., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S. E., Freire E., Hayashi Y., Bioorg. Med. Chem. 2013, 21, 412–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Kiso Y., Kawasaki Y., Chen S. E., Naser-Tavakolian A., Schon A., Freire E., Hayashi Y., Eur J. Med. Chem. 2013, 65, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thanigaimalai P., Konno S., Yamamoto T., Koiwai Y., Taguchi A., Takayama K., Yakushiji F., Akaji K., Chen S. E., Naser-Tavakolian A., Schon A., Freire E., Hayashi Y., Eur J. Med. Chem. 2013, 68, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kumar V., Shin J. S., Shie J. J., Ku K. B., Kim C., Go Y. Y., Huang K. F., Kim M., Liang P. H., Antiviral. Res. 2017, 141, 101–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., Snijder E. J., Liu H., Hilgenfeld R., J. Med. Chem. 2020, DOI: 10.1021/acs.jmedchem.1029b01828.. [DOI] [PubMed] [Google Scholar]
- 76. Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R., Science 2020, 368, 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lee T. W., Cherney M. M., Huitema C., Liu J., James K. E., Powers J. C., Eltis L. D., James M. N., J. Mol. Biol. 2005, 353, 1137–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Asgian J. L., James K. E., Li Z. Z., Carter W., Barrett A. J., Mikolajczyk J., Salvesen G. S., Powers J. C., J. Med. Chem. 2002, 45, 4958–4960. [DOI] [PubMed] [Google Scholar]
- 79. Shie J. J., Fang J. M., Kuo T. H., Kuo C. J., Liang P. H., Huang H. J., Wu Y. T., Jan J. T., Cheng Y. S., Wong C. H., Bioorg. Med. Chem. 2005, 13, 5240–5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wu C. Y., King K. Y., Kuo C. J., Fang J. M., Wu Y. T., Ho M. Y., Liao C. L., Shie J. J., Liang P. H., Wong C. H., Chem. Biol. 2006, 13, 261–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., Brown E. D., Chem. Biol. 2004, 11, 1445–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang J., Pettersson H. I., Huitema C., Niu C., Yin J., James M. N., Eltis L. D., Vederas J. C., J. Med. Chem. 2007, 50, 1850–1864. [DOI] [PubMed] [Google Scholar]
- 83. Niu C., Yin J., Zhang J., Vederas J. C., James M. N., Bioorg. Med. Chem. 2008, 16, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Turk B., Nat. Rev. Drug. Discov. 2006, 5, 785–799. [DOI] [PubMed] [Google Scholar]
- 85. Jain R. P., Pettersson H. I., Zhang J., Aull K. D., Fortin P. D., Huitema C., Eltis L. D., Parrish J. C., James M. N., Wishart D. S., Vederas J. C., J. Med. Chem. 2004, 47, 6113–6116. [DOI] [PubMed] [Google Scholar]
- 86. Shie J. J., Fang J. M., Kuo C. J., Kuo T. H., Liang P. H., Huang H. J., Yang W. B., Lin C. H., Chen J. L., Wu Y. T., Wong C. H., J. Med. Chem. 2005, 48, 4469–4473. [DOI] [PubMed] [Google Scholar]
- 87. Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A., J. Med. Chem. 2011, 54, 7962–7973. [DOI] [PubMed] [Google Scholar]
- 88. Shimamoto Y., Hattori Y., Kobayashi K., Teruya K., Sanjoh A., Nakagawa A., Yamashita E., Akaji K., Bioorg. Med. Chem. 2015, 23, 876–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Yoshizawa S. I., Hattori Y., Kobayashi K., Akaji K., Bioorg. Med. Chem. 2020, 28, 115273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Wu C. Y., Jan J. T., Ma S. H., Kuo C. J., Juan H. F., Cheng Y. S., Hsu H. H., Huang H. C., Wu D., Brik A., Liang F. S., Liu R. S., Fang J. M., Chen S. T., Liang P. H., Wong C. H., Proc. Natl. Acad. Sci. USA 2004, 101, 10012–10017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Shao Y. M., Yang W. B., Peng H. P., Hsu M. F., Tsai K. C., Kuo T. H., Wang A. H., Liang P. H., Lin C. H., Yang A. S., Wong C. H., ChemBioChem 2007, 8, 1654–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Webber S. E., Tikhe J., Worland S. T., Fuhrman S. A., Hendrickson T. F., Matthews D. A., Love R. A., Patick A. K., Meador J. W., Ferre R. A., Brown E. L., DeLisle D. M., Ford C. E., Binford S. L., J. Med. Chem. 1996, 39, 5072–5082. [DOI] [PubMed] [Google Scholar]
- 93. Chen L. R., Wang Y. C., Lin Y. W., Chou S. Y., Chen S. F., Liu L. T., Wu Y. T., Kuo C. J., Chen T. S., Juang S. H., Bioorg. Med. Chem. Lett. 2005, 15, 3058–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhou L., Liu Y., Zhang W., Wei P., Huang C., Pei J., Yuan Y., Lai L., J. Med. Chem. 2006, 49, 3440–3443. [DOI] [PubMed] [Google Scholar]
- 95. Liu W., Zhu H. M., Niu G. J., Shi E. Z., Chen J., Sun B., Chen W. Q., Zhou H. G., Yang C., Bioorg. Med. Chem. 2014, 22, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Zhang H. Z., Zhang H., Kemnitzer W., Tseng B., J. Cinatl Jr. , Michaelis M., Doerr H. W., Cai S. X., J. Med. Chem. 2006, 49, 1198–1201. [DOI] [PubMed] [Google Scholar]
- 97. Ramajayam R., Tan K. P., Liu H. G., Liang P. H., Bioorg. Med. Chem. 2010, 18, 7849–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ramajayam R., Tan K. P., Liu H. G., Liang P. H., Bioorg. Med. Chem. Lett. 2010, 20, 3569–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kao R. Y., Tsui W. H., Lee T. S., Tanner J. A., Watt R. M., Huang J. D., Hu L., Chen G., Chen Z., Zhang L., He T., Chan K. H., Tse H., To A. P., Ng L. W., Wong B. C., Tsoi H. W., Yang D., Ho D. D., Yuen K. Y., Chem. Biol. 2004, 11, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Tsai K. C., Chen S. Y., Liang P. H., Lu I. L., Mahindroo N., Hsieh H. P., Chao Y. S., Liu L., Liu D., Lien W., Lin T. H., Wu S. Y., J. Med. Chem. 2006, 49, 3485–3495. [DOI] [PubMed] [Google Scholar]
- 101. Mukherjee P., Desai P., Ross L., White E. L., Avery M. A., Bioorg. Med. Chem. 2008, 16, 4138–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nguyen T. T., Ryu H. J., Lee S. H., Hwang S., Breton V., Rhee J. H., Kim D., Bioorg. Med. Chem. Lett. 2011, 21, 3088–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Lee H., Mittal A., Patel K., Gatuz J. L., Truong L., Torres J., Mulhearn D. C., Johnson M. E., Bioorg. Med. Chem. 2014, 22, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Wen C. C., Kuo Y. H., Jan J. T., Liang P. H., Wang S. Y., Liu H. G., Lee C. K., Chang S. T., Kuo C. J., Lee S. S., Hou C. C., Hsiao P. W., Chien S. C., Shyur L. F., Yang N. S., J. Med. Chem. 2007, 50, 4087–4095. [DOI] [PubMed] [Google Scholar]
- 105. Ryu Y. B., Jeong H. J., Kim J. H., Kim Y. M., Park J. Y., Kim D., Nguyen T. T., Park S. J., Chang J. S., Park K. H., Rho M. C., Lee W. S., Bioorg. Med. Chem. 2010, 18, 7940–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ryu Y. B., Park S. J., Kim Y. M., Lee J. Y., Seo W. D., Chang J. S., Park K. H., Rho M. C., Lee W. S., Bioorg. Med. Chem. Lett. 2010, 20, 1873–1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Turlington M., Chun A., Tomar S., Eggler A., Grum-Tokars V., Jacobs J., Daniels J. S., Dawson E., Saldanha A., Chase P., Baez-Santos Y. M., Lindsley C. W., Hodder P., Mesecar A. D., Stauffer S. R., Bioorg. Med. Chem. Lett. 2013, 23, 6172–6177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S. H., J. Med. Chem. 2016, 59, 6595–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ramajayam R., Tan K. P., Liang P. H., Biochem. Soc. Trans. 2011, 39, 1371–1375. [DOI] [PubMed] [Google Scholar]
- 110. Imbert I., Snijder E. J., Dimitrova M., Guillemot J. C., Lecine P., Canard B., Virus Res. 2008, 133, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Angelini M. M., Akhlaghpour M., Neuman B. W., Buchmeier M. J., mBio 2013, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Neuman B. W., Joseph J. S., Saikatendu K. S., Serrano P., Chatterjee A., Johnson M. A., Liao L., Klaus J. P., J. R. Yates, 3rd , Wuthrich K., Stevens R. C., Buchmeier M. J., Kuhn P., J. Virol. 2008, 82, 5279–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Serrano P., Johnson M. A., Almeida M. S., Horst R., Herrmann T., Joseph J. S., Neuman B. W., Subramanian V., Saikatendu K. S., Buchmeier M. J., Stevens R. C., Kuhn P., Wuthrich K., J. Virol. 2007, 81, 12049–12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Egloff M. P., Malet H., Putics A., Heinonen M., Dutartre H., Frangeul A., Gruez A., Campanacci V., Cambillau C., Ziebuhr J., Ahola T., Canard B., J. Virol. 2006, 80, 8493–8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Saikatendu K. S., Joseph J. S., Subramanian V., Clayton T., Griffith M., Moy K., Velasquez J., Neuman B. W., Buchmeier M. J., Stevens R. C., Kuhn P., Structure 2005, 13, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Tan J., Kusov Y., Mutschall D., Tech S., Nagarajan K., Hilgenfeld R., Schmidt C. L., Biochem. Biophys. Res. Commun. 2007, 364, 877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Barretto N., Jukneliene D., Ratia K., Chen Z., Mesecar A. D., Baker S. C., Adv. Exp. Med. Biol. 2006, 581, 37–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Harcourt B. H., Jukneliene D., Kanjanahaluethai A., Bechill J., Severson K. M., Smith C. M., Rota P. A., Baker S. C., J. Virol. 2004, 78, 13600–13612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Chouljenko V. N., Lin X. Q., Storz J., Kousoulas K. G., Gorbalenya A. E., J. Gen. Virol. 2001, 82, 2927–2933. [DOI] [PubMed] [Google Scholar]
- 120. Lei J., Mesters J. R., Drosten C., Anemuller S., Ma Q., Hilgenfeld R., Antiviral. Res. 2014, 109, 72–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bailey-Elkin B. A., Knaap R. C., Johnson G. G., Dalebout T. J., Ninaber D. K., van Kasteren P. B., Bredenbeek P. J., Snijder E. J., Kikkert M., Mark B. L., J. Biol. Chem. 2014, 289, 34667–34682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Faesen A. C., Luna-Vargas M. P., Sixma T. K., Biochem Soc Trans 2012, 40, 539–545. [DOI] [PubMed] [Google Scholar]
- 123. Chou C. Y., Chien C. H., Han Y. S., Prebanda M. T., Hsieh H. P., Turk B., Chang G. G., Chen X., Biochem Pharmacol 2008, 75, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Cheng K. W., Cheng S. C., Chen W. Y., Lin M. H., Chuang S. J., Cheng I. H., Sun C. Y., Chou C. Y., Antiviral. Res. 2015, 115, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Frieman M., Basu D., Matthews K., Taylor J., Jones G., Pickles R., Baric R., Engel D. A., PLoS One 2011, 6, e28479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Park J. Y., Kim J. H., Kim Y. M., Jeong H. J., Kim D. W., Park K. H., Kwon H. J., Park S. J., Lee W. S., Ryu Y. B., Bioorg. Med. Chem. 2012, 20, 5928–5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Park J. Y., Jeong H. J., Kim J. H., Kim Y. M., Park S. J., Kim D., Park K. H., Lee W. S., Ryu Y. B., Biol Pharm Bull 2012, 35, 2036–2042. [DOI] [PubMed] [Google Scholar]
- 128. Cho J. K., Curtis-Long M. J., Lee K. H., Kim D. W., Ryu H. W., Yuk H. J., Park K. H., Bioorg. Med. Chem. 2013, 21, 3051–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Lee H., Lei H., Santarsiero B. D., Gatuz J. L., Cao S., Rice A. J., Patel K., Szypulinski M. Z., Ojeda I., Ghosh A. K., Johnson M. E., ACS Chem. Biol. 2015, 10, 1456–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Vastag B., JAMA 2003, 290, 1695–1696. [DOI] [PubMed] [Google Scholar]
- 131. Zhang X. W., Yap Y. L., Bioorg. Med. Chem. 2004, 12, 2517–2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Rajnarayanan R. V., Dakshanamurthy S., Pattabiraman N., Biochem. Biophys. Res. Commun. 2004, 321, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chen L., Gui C., Luo X., Yang Q., Gunther S., Scandella E., Drosten C., Bai D., He X., Ludewig B., Chen J., Luo H., Yang Y., Zou J., Thiel V., Chen K., Shen J., Shen X., Jiang H., J. Virol. 2005, 79, 7095–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Liu Z., Huang C., Fan K., Wei P., Chen H., Liu S., Pei J., Shi L., Li B., Yang K., Liu Y., Lai L., J Chem Inf Model 2005, 45, 10–17. [DOI] [PubMed] [Google Scholar]
- 135. Cinatl J., Morgenstern B., Bauer G., Chandra P., Rabenau H., Doerr H. W., Lancet 2003, 361, 2045–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Hoever G., Baltina L., Michaelis M., Kondratenko R., Tolstikov G. A., Doerr H. W., J. Cinatl, Jr. , J. Med. Chem. 2005, 48, 1256–1259. [DOI] [PubMed] [Google Scholar]
- 137. Lane T. E., Paoletti A. D., Buchmeier M. J., J. Virol. 1997, 71, 2202–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Akerstrom S., Mousavi-Jazi M., Klingstrom J., Leijon M., Lundkvist A., Mirazimi A., J. Virol. 2005, 79, 1966–1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Yamamoto N., Yang R., Yoshinaka Y., Amari S., Nakano T., Cinatl J., Rabenau H., Doerr H. W., Hunsmann G., Otaka A., Tamamura H., Fujii N., Biochem. Biophys. Res. Commun. 2004, 318, 719–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Wu C. J., Jan J. T., Chen C. M., Hsieh H. P., Hwang D. R., Liu H. W., Liu C. Y., Huang H. W., Chen S. C., Hong C. F., Lin R. K., Chao Y. S., Hsu J. T., Antimicrob. Agents. Chemother. 2004, 48, 2693–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Keyaerts E., Vijgen L., Maes P., Neyts J., Van Ranst M., Biochem. Biophys. Res. Commun. 2004, 323, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G., Cell. Res. 2020, 30, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Koren G., King S., Knowles S., Phillips E., CMAJ 2003, 168, 1289–1292. [PMC free article] [PubMed] [Google Scholar]
- 144. Stroher U., DiCaro A., Li Y., Strong J. E., Aoki F., Plummer F., Jones S. M., Feldmann H., J. Infect. Dis. 2004, 189, 1164–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Morgenstern B., Michaelis M., Baer P. C., Doerr H. W., J. Cinatl, Jr. , Biochem. Biophys. Res. Commun. 2005, 326, 905–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Li G., De Clercq E., Nat. Rev. Drug. Discov. 2020, 19, 149–150. [DOI] [PubMed] [Google Scholar]
- 147. Stadler K., Ha H. R., Ciminale V., Spirli C., Saletti G., Schiavon M., Bruttomesso D., Bigler L., Follath F., Pettenazzo A., Baritussio A., Am. J. Respir. Cell. Mol. Biol. 2008, 39, 142–149. [DOI] [PubMed] [Google Scholar]
- 148. Dyall J., Coleman C. M., Hart B. J., Venkataraman T., Holbrook M. R., Kindrachuk J., Johnson R. F., G. G. Olinger, Jr. , Jahrling P. B., Laidlaw M., Johansen L. M., Lear-Rooney C. M., Glass P. J., Hensley L. E., Frieman M. B., Antimicrob. Agents. Chemother. 2014, 58, 4885–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Coleman C. M., Sisk J. M., Mingo R. M., Nelson E. A., White J. M., Frieman M. B., J. Virol. 2016, 90, 8924–8933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zumla A., Chan J. F., Azhar E. I., Hui D. S., Yuen K. Y., Nat. Rev. Drug. Discov. 2016, 15, 327–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Chang C. K., Lo S. C., Wang Y. S., Hou M. H., Drug. Discov. Today 2016, 21, 562–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Li W., Moore M. J., Vasilieva N., Sui J., Wong S. K., Berne M. A., Somasundaran M., Sullivan J. L., Luzuriaga K., Greenough T. C., Choe H., Farzan M., Nature 2003, 426, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kruse R. L., F1000Res 2020, 9, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Wrapp D., Wang N., Corbett K. S., Goldsmith J. A., Hsieh C. L., Abiona O., Graham B. S., McLellan J. S., Science 2020, 367, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Tian X., Li C., Huang A., Xia S., Lu S., Shi Z., Lu L., Jiang S., Yang Z., Wu Y., Ying T., Emerg. Microbes. Infect. 2020, 9, 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G., Zhang H., Luo H., Zhu L., Jiang P., Chen L., Shen Y., Luo M., Zuo G., Hu J., Duan D., Nie Y., Shi X., Wang W., Han Y., Li T., Liu Y., Ding M., Deng H., Xu X., J. Virol. 2004, 78, 11334–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Simmons G., Gosalia D. N., Rennekamp A. J., Reeves J. D., Diamond S. L., Bates P., Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Adedeji A. O., Severson W., Jonsson C., Singh K., Weiss S. R., Sarafianos S. G., J. Virol. 2013, 87, 8017–8028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kandeel M., Yamamoto M., Al-Taher A., Watanabe A., Oh-Hashi K., Park B. K., Kwon H. J., Inoue J. I., Al-Nazawi M., Biomol. Ther. (Seoul) 2020, doi: 10.4062/biomother.2019.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Kawase M., Shirato K., van der Hoek L., Taguchi F., Matsuyama S., J. Virol. 2012, 86, 6537–6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., Schiergens T. S., Herrler G., Wu N. H., Nitsche A., Muller M. A., Drosten C., Pohlmann S., Cell 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Elfiky A. A., J. Med. Virol. 2016, 88, 2044–2051. [DOI] [PubMed] [Google Scholar]
- 163. Elfiky A. A., Ismail A., Life Sci. 2019, 238, 116958. [DOI] [PubMed] [Google Scholar]
- 164. Sheahan T. P., Sims A. C., Leist S. R., Schafer A., Won J., Brown A. J., Montgomery S. A., Hogg A., Babusis D., Clarke M. O., Spahn J. E., Bauer L., Sellers S., Porter D., Feng J. Y., Cihlar T., Jordan R., Denison M. R., Baric R. S., Nat. Commun. 2020, 11, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Elfiky A. A., Life Sci. 2020, 248, 117477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Vincent M. J., Bergeron E., Benjannet S., Erickson B. R., Rollin P. E., Ksiazek T. G., Seidah N. G., Nichol S. T., Virol. J. 2005, 2, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Gao J., Tian Z., Yang X., Biosci. Trends 2020, 14, 72–73. [DOI] [PubMed] [Google Scholar]
- 168. Sahraei Z., Shabani M., Shokouhi S., Saffaei A., Int. J. Antimicrob. Agents. 2020, 105945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jorge A. M., Melles R. B., Zhang Y., Lu N., Rai S. K., Young L. H., Costenbader K. H., Ramsey-Goldman R., Lim S. S., Esdaile J. M., Clarke A. E., Urowitz M. B., Askanase A., Aranow C., Petri M., Choi H., Arthritis. Res. Ther. 2018, 20, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D., Clin. Infect Dis. 2020, DOI: 10.1093/cid/ciaa1237.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Lin S. M., Lin S. C., Hsu J. N., Chang C. K., Chien C. M., Wang Y. S., Wu H. Y., Jeng U. S., Kehn-Hall K., Hou M. H., J. Med. Chem. 2020, 63, 3131–3141. [DOI] [PubMed] [Google Scholar]