

Abstract
Pre‐clinical responses to fast‐moving infectious disease outbreaks heavily depend on choosing the best isolates for animal models that inform diagnostics, vaccines and treatments. Current approaches are driven by practical considerations (e.g. first available virus isolate) rather than a detailed analysis of the characteristics of the virus strain chosen, which can lead to animal models that are not representative of the circulating or emerging clusters. Here, we suggest a combination of epidemiological, experimental and bioinformatic considerations when choosing virus strains for animal model generation. We discuss the currently chosen SARS‐CoV‐2 strains for international coronavirus disease (COVID‐19) models in the context of their phylogeny as well as in a novel alignment‐free bioinformatic approach. Unlike phylogenetic trees, which focus on individual shared mutations, this new approach assesses genome‐wide co‐developing functionalities and hence offers a more fluid view of the ‘cloud of variances’ that RNA viruses are prone to accumulate. This joint approach concludes that while the current animal models cover the existing viral strains adequately, there is substantial evolutionary activity that is likely not considered by the current models. Based on insights from the non‐discrete alignment‐free approach and experimental observations, we suggest isolates for future animal models.
Keywords: alignment‐free phylogeny, bioinformatics, COVID‐19, genomics, PHEIC, viral evolution
1. INTRODUCTION
The world is witnessing increasing instances of emerging and re‐emerging diseases caused by viruses. For instance, there have been six ‘Public Health Emergency of International Concern’ (PHEIC) declarations by the WHO since 2009, viz. H1N1 (swine flu), Polio, West Africa Ebola, Zika, and the ongoing Kivu Ebola and SARS‐CoV‐2 coronavirus outbreaks (Eurosurveillance Editorial Team, 2019, 2020); two of these viruses (H1N1 and SARS‐CoV2) have resulted in pandemics within 10 years (WHO, 2020).
The SARS outbreak of 2002–2004, the MERS outbreaks since 2012 and the current COVID‐19 outbreak since 2019 demonstrate the potential of coronaviruses, especially bat‐derived betacoronaviruses (Zhou et al., 2020), to cause PHEICs, with COVID‐19 having escalated to a global pandemic. Viruses in a new host (humans) have the potential to evolve rapidly and present quasispecies diversity (Eigen, McCaskill, & Schuster, 1988), which is a hallmark of RNA viruses that exist as a ‘cloud of variants’ due to low fidelity, high polymorphism and viral polymerases lacking the capability to correct errors (Drew, 2011; Wilke, Wang, Ofria, Lenski, & Adami, 2001). As a result, most variants are a random accumulation of errors, useful for tracing aetiology, but typically without substantial functional change (Grubaugh, Petrone, & Holmes, 2020). Unlike most other RNA viruses, coronaviruses express a 3′‐to‐5′ exoribonuclease that enables the high‐fidelity replication of their relatively large 26–32 kb ssRNA(+) genome (Minskaia et al., 2006;Snijder et al., 2003). Coronaviruses have a moderate mutation rate (0.80–2.38 × 10–3 nucleotide substitutions per site per year for the SARS‐CoV genome; Zhao et al., 2004) allowing a wider evolutionary space to be explored more deliberately. This can complicate the outbreak response in terms of rapid development and evaluation of diagnostics, vaccines, antivirals and antibody therapies as many diverse strains with unknown functional differences exist (Figure 1).
FIGURE 1.
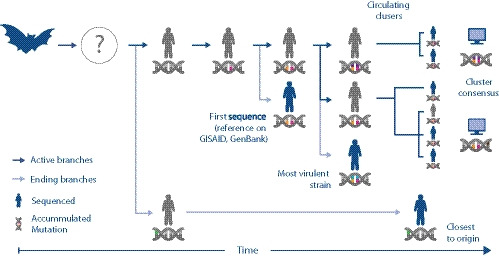
Illustration of coronavirus spread while it accumulates mutations. The dark blue arrows represent the main volume of transmissions, while the nucleic acid symbol illustrates mutations acquired by the different viral strains as they enter humans from a primary/reservoir host (represented by the bat symbol) through an intermediate host (which is yet to be identified for SARS‐CoV‐2). The first human SARS‐CoV‐2 isolate sequenced (with orange and pink mutation) may not have been the original strain that first infected humans (grey). It is possible that a strain sequenced later (green) may be genetically closer to the original strain. In this scenario, the original strain has not been captured through sequencing at all. It also shows that there may be two currently circulating strains (orange‐pink‐purple and orange‐pink‐brown), which in turn might be different from the most virulent one (orange‐pink‐blue). In the absence of clinical data correlated with SARS‐CoV‐2 genome isolates, bioinformatic analysis (represented by the computer symbol) can identify clusters and consensus sequences to investigate the genetic diversity of the emerging SARS‐CoV‐2 strains
This is particularly exacerbated by increased movement of people (enabled by global air travel), animals and goods spreading new viruses across the world's population and exposing them to huge variations in environment, demographics, age structure, socio‐economic status, co‐morbidities and equitable access to health care. The sheer number of these inter‐connected influencing factors often makes an unfolding situation hard to comprehend fully and challenges the traditional virology and public health disciplines by rendering them less effective in coping with the spread of the virus.
Bioinformatic approaches may be able to better inform epidemiology and responses to trans‐boundary viruses, by synthesizing complex information more effectively and systematically. Enabled by the advances in genomic sequencing technology (e.g. Oxford Nanopore, Illumina) and the willingness to share the sequenced information in the public domain (e.g. GenBank, GISAID), bioinformatic approaches developed in the human domain have enabled comparative analysis of the emerging genomic diversity of a virus as it spreads throughout a host population.
For the ongoing COVID‐19 outbreak, as illustrated in Figure 1, applying in silico approaches can rapidly provide answers to questions like: Is the first sequenced SARS‐CoV‐2 genome (often called the ‘reference genome’ in GISAID, GenBank, etc.) closest to the original or ‘true reference’ strain which entered humans (which may not have been sequenced and/or which may still be circulating with minimal mutations)? Combined with epidemiological or experimental evidence, bioinformatics can help address questions like ‘Are there one or multiple circulating strains and are they different to the most virulent ones’ (if yes can we identify the molecular basis)? Finally, can synthetic consensus sequences be used to represent the genetic diversity of individual isolates in larger circulating clusters?
Answers to these questions are not just of academic interest; they can help inform outbreak response and development and evaluation of diagnostics, vaccines and countermeasures. For instance, outbreak response efforts including vaccine evaluation should focus on the most prevalent circulating strain (lest we may end up rejecting acceptable candidates by raising the efficacy threshold too high), but it would be desirable to evaluate therapeutics against the most virulent strain. However, with the field not yet using bioinformatic tools to their fullest potential, current data collection practices are not capturing the information necessary for advanced analysis. For example, pathogen sequences derived from patients are typically not annotated with deidentified disease progression and other clinically relevant information, and this in turn hampers the identification of the most virulent strain or associating pathogen properties with genomic modifications.
Here, we use bioinformatic analysis to identify emerging trends among the SARS‐CoV‐2 isolates. From this, we sought to identify the most representative strains for animal models and pre‐clinical research, in particular, which strains among the emerging Australian isolates are good choices for animal model development and which ones are not representative.
2. MATERIALS AND METHODS
2.1. Sequencing of two Australian isolates
Two Australian isolates (BetaCoV/Australia/VIC02/2020 and BetaCoV/Australia/SA01/2020) were sequenced with the MiSeq platform (Illumina, Inc). In brief, RNA was purified from each isolate using a Direct‐zol RNA Miniprep kit (Zymo Research). Purified RNA was reverse transcribed using a TaqMan Reverse Transcription kit (Applied Biosystems) with random octamers linked to a specific primer sequence, followed by second‐strand cDNA synthesis using Klenow DNA Polymerase I (Promega). Complementary DNA was further amplified (using primers specific to the sequences added to the random octamers used for reverse transcription) with a KAPA HiFi HotStart kit (Roche). Resulting DNA was purified using a DNA Clean and Concentrator kit (Zymo Research). Fragmentation and dual‐index library preparation was conducted using a Nextera XT DNA Library Preparation Kit (Illumina, Inc), and denatured libraries were sequenced using a 300‐cycle MiSeq Reagent kit v2 (Illumina, Inc). Sequencing reads were trimmed for quality and mapped to the published reference sequence (BetaCoV/Wuhan‐Hu‐1/2019; GenBank accession number NC_045512.2) using Geneious 11.1.4. Consensus genome sequences for the isolates were generated for analysis.
2.2. GISAID pre‐processing and alignment
All available viral sequences were downloaded from GISAID (on 05/03/2020, see Appendix S3 (Elbe, & Buckland‐Merrett, 2017)), filtering for complete sequences of human origin (187 genomes in total). Low‐quality sequences (defined as sequences with an N content greater than 1%) were filtered out leaving 178 strains in total. We also included one recently reported viral sequence from the European Virus Archive global (Ref‐SKU: 026V‐03883) and the two Australian sequences in the full data set.
This data set of 181 sequences was aligned against each other using Muscle (v3.8.31) (Madeira et al., 2019). Based on the alignments, we could see significant variation at the ends of the sequences (Figure S1), likely due to sequencing artefacts and errors preventing the full viral genomes from being sequenced. To reduce the impact of potential sequencing errors, we wrote a custom perl script to trim the sequences such that only positions with 95% coverage were retained. This trimming entailed removing the first 101 and last 72 bp from the alignments.
Once trimmed and converting all ‘U’ to ‘T’, we identified 54 sequences that were identical. To limit the effect of duplicate sequences on subsequent analysis, we collapsed these into a single entry. These collapsed sequences are summarized in Table S1.
To validate the methodology, we have included sequences from other coronaviruses. We chose a mixture of controls as follows: 7 SARS sequences of human origin (GenBank accession numbers AY274119.3, AY291451.1, AY502923.1, AY502932.1, AY559083.1, AY559084.1 and AY559087.1), 10 SARS sequences of bat origin (accession numbers KY417142.1 to KY417152.1), 4 MERS sequences of human origin (accession numbers KJ477102.1, KT006149.2, KT026453.1 and KT029139.1) and 2 MERS sequences of bat origin (accession numbers MG596802.1 and MG596803.1).
2.3. Phylogenetic tree
The maximum likelihood phylogenetic tree was generated from the above alignments using RAxML‐NG (Kozlov, Darriba, Flouri, Morel, & Stamatakis, 2019). The evolutionary model used was a general time reversable model with gamma‐distributed rate heterogeneity and invariant sites (GTR + G + I). We use this mode since it is the most general model and has been proposed to produce equal trees to optimally selected models (Abadi, Azouri, Pupko, & Mayrose, 2019). The tree was visualized using iTOL (Letunic & Bork, 2019) as a midpoint rooted tree and shows the likely evolutionary relationships between the sampled strains.
2.4. K‐mer method
Every organism and potentially isolate can have a unique genomic signature based on the composition of their genomic sequence. To quantify this signature, we determined the K‐mer frequency. Counting of all possible strings of length k in the sequence of the virus has emerged as an alternative to phylogenetic trees in other disciplines (Sims, Jun, Wu, & Kim, 2009). The conceptual distance between all isolates can then be visualized by running a principal component analysis (PCA) over all genomic signatures to reduce this high‐dimensional K‐mer frequency vector into a two‐dimensional space (Jolliffe & Cadima, 2016). Please see Appendix S1 for more details of this method.
Custom scripts were used to calculate the K‐mer frequency for each sequence using a k of 10 (Sims et al., 2009). K‐mers containing ambiguous bases (i.e. N's) were removed. We then calculated the relative proportion of each K‐mer, resulting in a frequency vector. We used the PCA implementation of Python scikit‐learn to reduce the genomic signatures containing 1,048,576 10‐mer proportions into a vector containing two principal components. Finally, custom scripts were used to compare the genomic signatures for all aforementioned coronavirus sequences.
3. RESULTS AND DISCUSSION
3.1. Phylogeny reveals three current clusters
The evolutionary structure of the 181 isolates as determined by the phylogenetic tree reveals three major clusters (C1‐C3) (Figure 2). The C1 cluster mainly represents Wuhan and isolates captured early on; C2 and C3 contain later isolates, such as Sydney/3, Australia/VIC01 and France/IDF0372 in C2 and Australia/NSW01, Australia/QLD01‐3, Australia/VIC02 and USA/WA1 in C3. The three clusters are separated by distinct mutations (Table 1) but contain a substantial number of other unique mutations, which we define as diversity within the cluster. These individual mutations are outside of the established hotspots of diversity (Wang et al., 2020) as shown in Figure S4, which are predominantly of concern for PCR primer design rather than aetiology. There may also be three additional clusters emerging (C4‐6) with C4 capturing the suspected community spread from Lombardy (‘Narrative: Genomic analysis of COVID‐19 spread’, n.d.), C5 regionally mixed (Asia and North Amerika) and C6 from Australia and Asia, notably Australia/NSW05‐7 (see fully annotated tree in Figure S5).
FIGURE 2.
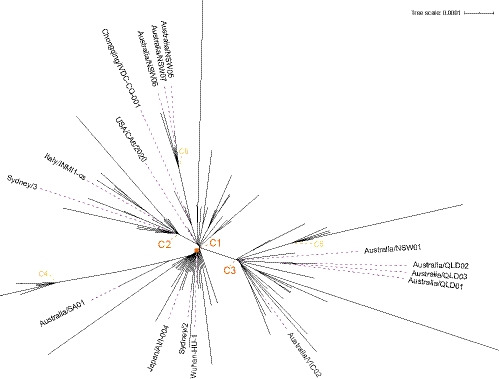
Phylogenetic tree highlighting isolates of interest with branch points of the six clusters labelled to indicate mature (orange) and emerging (yellow) disease clusters (full list of identical sequences for these branch points are in Table S1 and complete image in Figure S5)
TABLE 1.
Mutations characterizing phylogenetic clusters
| Cluster | Mutations in common | Diversity within cluster |
|---|---|---|
| C1 (Wuhan‐hu‐1) | Reference strain | 107 unique mutations |
| C2 (Vic01, France/IDF0372, Sydney/3) | G26144T | 31 unique mutations |
| C3 (Australia/NSW01, USA/WA1) | C8782T, T28144C | 68 unique mutations |
| C4 | C241T, C3037T, A23403G, C14408T, GGG28881AAC, | 9 unique mutations |
| C5 | C8782T, T28144C, C24034T, T26729C, G28077C | 7 unique mutations |
| C6 | G11083T, G1397A, T28688C, G29742T | 10 unique mutations |
Positions are relative to trimmed alignments (see Methods for more details). The following sequences were excluded from the analysis: Beijing/IVDC‐BJ‐005, Shenzhen/SZTH‐001, Shenzhen/SZTH‐004 and Wuhan/HBCDC‐HB‐04 because their high number of mutations is likely due to sequencing errors. Cluster diversity of C1 includes the diversity of cluster C4 and C6, and cluster diversity of C3 includes the cluster diversity of C5.
This finding is different to Tang et al. (2020), who postulate two clusters (S and L). Their analysis was only on 103 GISAID isolates and includes betacoronaviruses from bats, which roots the tree differently and merges C1 and C2. However, as the aetiology is not fully demonstrated, especially with the intermediate vector yet unknown, artificially rooting the tree by introducing a distant relative may bias the results.
Irrespective of the root placement, both trees allow the assessment of individual isolates that are not part of major branches by being genetically divergent offshoots. For example, Sydney/2 appears to be an off‐shoot from Wuhan‐Hu‐1. Upon further inspection of the Sydney/2 strand, we discovered a 41‐bp deletion, which overlaps the infectious bronchitis virus' (IBV) motif at the 3′ end (Goebel, Taylor, & Masters, 2004). Inspecting other isolates, Australia/VIC01 has also a 10‐bp deletion within the genomic location of the 41‐bp deletion (Figure S3). Despite this similarity, Australia/VIC01 was placed into C2 and Sydney/2 in C1 since Australia/VIC01 contains the G26144T mutation indicative of C2 which Sydney/2 lacks (See Table 2 for a list of referenced isolates).
TABLE 2.
Hamming Distance Mutation analysis (trimmed) relative to Wuhan‐HU‐1
| Strain | Shorthand | Condensed/Uncondensed (in core) | Core identical sequences | Interest |
|---|---|---|---|---|
| BetaCoV/USA/WA1/2020|EPI_ISL_404895 | USA/WA1 | 5/24 (3/3) | 4 | Animal model |
| BetaCoV/Germany/BavPat1/2020|EPI_ISL_406862 | BavPat1 | 7/124 (3/3) | 2 | Animal model |
|
Human 2019‐nCoV Human 2019‐nCoV 026V‐03883 |
7/71 (3/3) | 2 | Animal model | |
|
BetaCoV/Australia/VIC01/2020|EPI_ISL_406844 |
Vic01 | 4/13 (4/13) | Animal model | |
|
BetaCoV/Canada/ON‐VIDO‐01/2020|EPI_ISL_413015 |
Canada/ON‐VIDO‐01 | 8/27 (5/13) | Animal model, deletion | |
|
BetaCoV_France_IDF0372_2020_C2 |
France/IDF0372 | 4/31 (2/2) | 4 | Animal model |
| BetaCoV/Sydney/2/2020|EPI_ISL_408976 | Syd02 | 7/164 (3/43) | Deletion | |
| BetaCoV/Sydney/3/2020|EPI_ISL_408977 | Syd03 | 5/122 (1/1) | ||
| BetaCoV/USA/CA6/2020|EPI_ISL_410044 | USA/CA6 | 4/45 (24/2) | Deletion | |
| BetaCoV/Japan/AI/I‐004/2020|EPI_ISL_407084 | Japan/AI/I‐004 | 6/57 (26/3) | Deletion | |
| BetaCoV/Australia/NSW01/2020|EPI_ISL_407893 | NSW01 | 6/123 (2/2) | 5 | |
| BetaCoV/Chongqing/IVDC‐CQ‐001/2020|EPI_ISL_408481 | Chongqing/IVDC‐CQ‐001 | 3/22 (1/1) | 4 | Potential recombination with Sydney/3 |
| BetaCoV/Italy/INMI1‐cs/2020|EPI_ISL_410546 | Italy/INMI1‐cs | 5/39 (3/3) | 2 | Potential recombination result between Chongqing/1VDC‐CQ‐001 and Sydney/3 |
A more systematic sequence analysis revealed that many isolates have deletions in their core genome (Table S2). While these deletions appear to be specific to each isolate (Figure S4), their effect on virus structure or function might be pronounced. For example, Australia/VIC01 and Sydney/2 may have the IBV motif disrupted with implications for replication, while USA/CA6 and Japan/AI‐004 may have disruption in the non‐structural protein 1 (nsp1), with implications for host gene expression (Figure S3).
While the full impact of these genomic variations can only be confirmed through functional genomics experiments, coronaviruses' proof‐reading ability likely lifts them above random accumulation of errors. As such, having a methodology able to take deletions into consideration when calculating genomic distance is desirable.
3.2. Alignment‐free phylogeny captures evolutionary distances
Aiming to overcome the limitations of phylogenetic tree approaches, we also investigated whether an alignment‐free approach can be used to understand how the genomic content of different SARS‐CoV‐2 isolates changes over time and with respect to each other by also taking deletions into account. We therefore calculated the frequency of all possible 10‐mers across each viral genome followed by principal component analysis (PCA) to reduce the high‐dimensional K‐mer vector space into a two‐dimensional image for visual comparison.
We first demonstrate the methods' ability to faithfully separate distant coronavirus strains by comparing all SARS‐CoV‐2 against 17 SARS and 6 MERS isolates. As shown in Figure 3 (inset), this alignment‐free approach separates the isolates into their three respective clusters of SARS‐CoV‐2, SARS and MERS. This indicates that the genetic distance between the different isolates of the same coronavirus strain is relatively small, while there are substantial differences between the different coronavirus virus strains. Predictably, we separated MERS isolates into two subclusters, reflecting their different host origins (human and bat).
FIGURE 3.
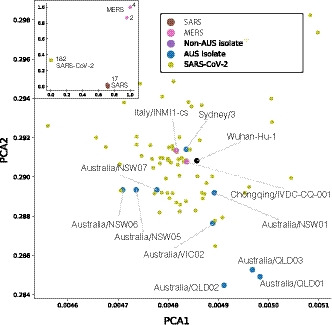
PCA plots showing the genomic signatures of different coronavirus sequences. Each point represents the genomic signature for an isolate. Inset Comparison of genomic signatures across different strains of coronavirus. Numbers correspond to the number of isolates at each location. Overall, the genomic signatures for isolates of different coronavirus strains were relatively far apart. Main image Zoomed in PCA plot of the cluster of SARS‐CoV‐2 isolates, showing the overall genomic signatures of the different strains
To further investigate how the genomic content of different SARS‐CoV‐2 viruses relates to each other, we re‐ran the PCA analysis on just the SARS‐CoV‐2 sequences. Here, the genomic signatures of isolates that are likely to be closely related cluster together (e.g. Australia/QLD01, Australia/QLD03 samples from close family relations), while strains separated by time are far apart (e.g. Wuhan‐Hu‐1, collection date 31/12/2019, and Australia/NSW05, collection date 28/2/2020).
Of the Australian isolates, Sydney/3 and Australia/NSW01 were the closest to Wuhan‐Hu‐1, which is consistent with the phylogenetic results and reflects the fact that these sequences have only mutational changes in their core sequences compared to Wuhan‐Hu‐1 (Table S2). However, this alignment‐free method positions isolates with deletions (viz. Australia/VIC01 and Canada/ON‐VIDO‐01) further away from Wuhan‐Hu‐1 than in the phylogenetic tree, demonstrating the ability of the K‐mer method to represent deletions accurately (see Supplemental Table 7).
Of the 181 isolates, we found that Singapore/4, Taiwan/NTU01, Finland/1, USA/IL1 and Shenzhen/SZTH‐001 were among the furthest from Wuhan‐Hu‐1 (data not shown). For both Singapore/4 and Taiwan/NTU01, this is due to these being shorter than the core sequences, so the K‐mer fingerprint accurately reflects the missing sequences. Meanwhile, USA/IL1 and Finland/1 contained several ambiguous bases effectively shortening the length of similar sequence, so the method correctly places them further from the other isolates.
While the K‐mer approach is not as suggestive as phylogenetic trees with respect to visualizing the potential transmission route (e.g. Lombardy), it may more accurately reflect the fluidity of changes (‘cloud of variants’) and capture recombination events (Graham & Baric, 2010). An example is Italy/INMI1, which has mutations in common with both Sydney/3 (C1 cluster, G26144T) and Chongqing/IVDC‐CQ‐001 (C3 cluster, G11083T) making it impossible to definitively place it in the discrete phylogenetic tree (it was placed in the C2 cluster, Figure 2), while the PCA plot shows the fluid evolution placing it between the Italian and Australian isolate (Figure 3).
More generally, phylogenetic analysis is based on the presence of shared mutations; for example, two strains which share most SNPs are likely to be closely related and therefore exist on neighbouring branches in the phylogenetic tree. In comparison, an alignment‐free method (such as K‐mer signatures) is more concerned with global similarities and difference, for example changes averaged across the whole genome rather than at specific locations. This can be informative about high‐level similarities between genomes, for example evolution of distinct genomic islands with common functions or recombination events. While this does not create visually clear clades, it offers a more holistic representation of pair‐wise distances between all isolates. Together with clinical information, this could hence help determine the most virulent strains (Figure 1).
3.3. How representative are the currently chosen isolates for pre‐clinical models?
Currently, the process of determining which isolate to use for animal models is less informed than it could be due to the lack of shared genomic information and readily available bioinformatic methodologies, especially towards the beginning of an outbreak. For example, while China published the genomic sequence of SARS‐CoV‐2 (Zhou et al., 2020), patient samples or virus isolates were not made available, and with reverse genetics of a large RNA virus taking time (Thao et al., 2020), countries had to wait for imported cases. Australia's Doherty Institute was the first in the world to isolate the virus (Australia/VIC01) and made it available for pre‐clinical animal models to the authors (CSIRO, 2020), Public Health England, etc. This practical consideration dictated the initial choice, however, with more isolates to choose from subsequently a more informed approach can be taken (Figure 1). Two questions are pertinent: Is VIC01 an appropriate strain to continue with further development and characterization of the animal model? If not, what options are more representative and appropriate?
While Nextstrain ('Narrative: Genomic analysis of COVID‐19 spread', n.d.) is a powerful aid in visualizing the available strains in real time, it currently relies on phylogeny only and thus may be hampered in its conclusions. In this section, we offer a static view of the alignment‐free approach for the currently used strains for animal model research (that we know of) and interpret their likely representativeness with respect to future evolution of the virus.
According to the phylogenetic tree shown in Figure 4a, the main clusters are represented by the current animal models. That is, C1 is represented by Germany/BavPat1 and Human 2019‐nCoV (whose core sequence is identical), though they seem to be half‐way to the C4 cluster (suspected Lombardy cluster). C2 is represented by Australia/VIC01 and France/IDF0372, and C3 is represented by USA/WA1. Note that Scotland/CVR01 was removed due to its high N content (2.3%). However, with phylogenetic methods focusing on the presence of shared mutations rather than the overall genomic similarity (e.g. shared evolved functionality), the assumption that current animal models adequately cover the evolutionary space of the actively circulating virus might be misleading as the PCA plot indicates (Figure 4b). Here, the top‐left and bottom‐right areas seem to be underrepresented.
FIGURE 4.
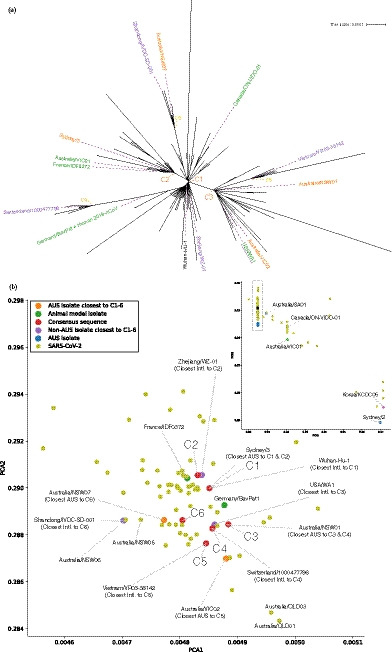
Identification of potential viral strains for animal models. Phylogenetic methods (a) show that current animal models (highlighted in green) cover the major clusters (C1‐3) but may not capture the emerging clusters. A K‐mer based analysis (b) is able to suggest alternative strains that cover all emerging clusters (C4‐6). The inset shows the wider region with the main image extent marked by a rectangle
To join the strain‐etiology of phylogenetic approaches with the more fluid distance measure of alignment‐free methods, we created the consensus sequences of the major and emerging clusters (Appendix S2) and re‐ran the PCA analysis to find more robust and future relevant isolates (Figure 4b). Of the existing animal models, only USA/WA1 is close to a consensus (C3); all other clusters have different representative isolates, viz. C1 directly overlaps Wuhan‐HU‐1, C2 is nearest to Zhejiang/WZ‐01, C3 directly overlaps Australia/NSW01, C4 directly overlaps Switzerland/1000477796, C5 directly overlaps Vietnam/VR03‐38142, and C6 is nearest to Australia/NSW07.
The central location of Germany/BavPat1 and France/IDF0372 may reflect a broad representation across multiple clusters, in contrast to Canada/ON‐VIDO‐01 and Australia/VIC01, which are located further away from the SARS‐CoV‐2 centre marked by the rectangle in Figure 4b inset. In this ‘outer’ view, the dominant driver for placing isolates away from the centre is ‘missing’ bases either due to deletions, missing sequences at the tails or ambiguous bases. The vertical lines hence cluster isolates with a similar number of ‘missing’ bases; for example, Australia/VIC01 and Canada/ON‐VIDO‐01 have 10‐bp deletions, while Korea/KCDC05 and Australia/Sydney02 have a 40‐bp shorter sequence and 42‐bp deletion, respectively.
Among the Australian strains, the genomic distance analysis marks Australia/Sydney2 as a less optimal target for animal models, compared to, say Australia/VIC01, Australia/VIC02 and Australia/SA01 (in no particular order). In this context, USA/WA1 (available through BEI) could also be a good choice due to its central location, likely ability to represent especially the newly emerging clusters, and comparability of animal models with non‐human primate studies in the United States which have chosen this on the basis of being readily available. Clinical observations (e.g. severity of symptoms and mortality if applicable) and experimental observations (e.g. growth and titre) can refine the choice of isolate for animal model development, with further refinement based on observations from animal challenge studies such as shedding of the virus, associated histopathology and clinical signs if applicable.
It is clear that at this early stage of the pandemic, our initial questions around the number of circulating strains and their virulence cannot be answered with the currently available epidemiological and clinical data even when applying sophisticated computational analysis tools. However, we have demonstrated that creating synthetic consensus sequences can be used to demarcate the evolutionary space already claimed by the virus. While SARS‐CoV‐2 does have a proof‐reading exoribonuclease domain in nsp14, its genetic drift remains a point of uncertainty with respect to the long‐lasting efficacy of a vaccine candidates currently being developed.
In conclusion, joining bioinformatics, epidemiological and experimental results can help inform animal model choice for efficient pre‐clinical responses (Callaway, 2020). Moving away from purely practical considerations towards a more deliberate approach that assesses current and future characteristics of isolate choices will lead to a better coverage of actively circulating strains (Figure 1). With more sharing of isolates internationally and information collected about patient‐deidentified details of case severity and outcome, more sophisticated machine learning approaches can be generated to assist in triaging and treatment choices. Additional information, such as co‐morbidities, socio‐economic and smoking status, may also help in anticipating public health demand. Furthermore, releasing the full high‐throughput sequencing data sets rather than the consensus sequences would allow a more detailed exploration of the existing quasispecies to further improve isolate selection.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ETHICAL APPROVAL
Ethical Statement is not applicable.
Supporting information
Appendix S1
Appendix S2
Appendix S3
ACKNOWLEDGEMENTS
The two Australian isolates not yet in GISAID were kindly provided by Dr Julian Druce from the Victorian Infectious Disease Reference Laboratory. SSV acknowledges the Coalition for Epidemic Preparedness Innovations (CEPI) ‘High Containment Animal Studies to Support Product Development Program' for funding in partnership with the Commonwealth Scientific and Industrial Research Organisation (CSIRO). Images were designed with elements from FreePik from www.flaticon.com under the ‘Attribution' licence. DCB and SSV conceived the idea and wrote the paper, with inputs from APT, LOW, DR, CH and AJM. DR did the mutation rate analysis. APT performed the principal component analysis (PCA) using the k‐mer method. Hamming distance was calculated by AJM and APT, who also performed the sequence alignment. LOWW and CH built the phylogenetic tree. AM, EP, ST, VS, MJN, MT, TWD and SSV were involved in iterative interpretation and re‐design of bioinformatic approaches to understand the implications of quasispecies diversity on corona virology and the development and evaluation of diagnostics, vaccines and countermeasures. The authors would like to acknowledge the colleagues at AAHL and H&B business units and Chris Hammang for designing the cover art.
Bauer DC, Tay AP, Wilson LOW, et al. Supporting pandemic response using genomics and bioinformatics: A case study on the emergent SARS‐CoV‐2 outbreak. Transbound Emerg Dis. 2020;67:1453–1462. 10.1111/tbed.13588
Denis C. Bauer and Aidan P. Tay contributed equally to this work.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available in GISAID at https://www.gisaid.org/.
REFERENCES
- Abadi, S. , Azouri, D. , Pupko, T. , & Mayrose, I. (2019). Model selection may not be a mandatory step for phylogeny reconstruction. Nature Communications, 10(1), 934. 10.1038/s41467-019-08822-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway, E. (2020). Labs rush to study coronavirus in transgenic animals – Some are in short supply. Nature, 579(7798), 183. 10.1038/d41586-020-00698-x [DOI] [PubMed] [Google Scholar]
- CSIRO (2020). Working against the new coronavirus. March 5, 2020, Retrieved from https://www.csiro.au/en/Research/Health/Infectious‐dieases‐coronavirus/coronavirus [Google Scholar]
- Drew, T. W. (2011). The emergence and evolution of swine viral diseases: To what extend have husbandry systems and global trade contributed to their distribution and diversity? Revue Scientifique et Technique de l’OIE, 30(1), 95–106. 10.20506/rst.30.1.2020 [DOI] [PubMed] [Google Scholar]
- Eigen, M. , McCaskill, J. , & Schuster, P. (1988). Molecular quasi‐species. The Journal of Physical Chemistry, 92(24), 6881–6891. 10.1021/j100335a010 [DOI] [Google Scholar]
- Elbe, S. , & Buckland‐Merrett, G. (2017). Data, disease and diplomacy: GISAID's innovative contribution to global health. Global Challenges, 1, 33–46. 10.1002/gch2.1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurosurveillance Editorial Team . (2019). Ebola public health emergency of international concern, democratic republic of the congo, 2019. Euro Surveillance, 24(29), 190718e. 10.2807/1560-7917.ES.2019.24.29.190718e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eurosurveillance Editorial Team (2020). Note from the editors: World Health Organization declares novel coronavirus (2019‐nCoV) sixth public health emergency of international concern. Euro Surveillance, 25(5), 200131e. 10.2807/1560-7917.ES.2020.25.5.200131e [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebel, S. J. , Taylor, J. , & Masters, P. S. (2004). The 3’ cis‐acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. Journal of Virology, 78(14), 7846–7851. 10.1128/JVI.78.14.7846-7851.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham, R. L. , & Baric, R. S. (2010). Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross‐species transmission. Journal of Virology, 84(7), 3134–3146. 10.1128/JVI.01394-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubaugh, N. D. , Petrone, M. E. , & Holmes, E. C. (2020). We shouldn't worry when a virus mutates during disease outbreaks. Nature Microbiology, 10.1038/s41564-020-0690-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe, I. T. , & Cadima, J. (2016). Principal component analysis: A review and recent developments. Philosophical Transactions. Series A, Mathematical, Physical, and Engineering Sciences, 374(2065), 20150202. 10.1098/rsta.2015.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlov, A. M. , Darriba, D. , Flouri, T. , Morel, B. , & Stamatakis, A. (2019). RAxML‐NG: A fast, scalable and user‐friendly tool for maximum likelihood phylogenetic inference. Bioinformatics, 35(21), 4453–4455. 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2019). Interactive tree of life (iTOL) v4: Recent updates and new developments. Nucleic Acids Research, 47(W1), W256–W259. 10.1093/nar/gkz239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira, F. , Park, Y. M. , Lee, J. , Buso, N. , Gur, T. , Madhusoodanan, N. , … Lopez, R. (2019). The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Research, 47(W1), W636–W641. 10.1093/nar/gkz268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minskaia, E. , Hertzig, T. , Gorbalenya, A. E. , Campanacci, V. , Cambillau, C. , Canard, B. , & Ziebuhr, J. (2006). Discovery of an RNA virus 3’‐>5’ exoribonuclease that is critically involved in coronavirus RNA synthesis. Proceedings of the National Academy of Sciences of the United States of America, 103(13), 5108–5113. 10.1073/pnas.0508200103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narrative: Genomic analysis of COVID‐19 spread . (n.d.). March 5, 2020, Retrieved from https://nextstrain.org/narratives/ncov/sit‐rep/2020‐03‐04
- Sims, G. E. , Jun, S.‐R. , Wu, G. A. , & Kim, S.‐H. (2009). Alignment‐free genome comparison with feature frequency profiles (FFP) and optimal resolutions. Proceedings of the National Academy of Sciences of the United States of America, 106(8), 2677–2682. 10.1073/pnas.0813249106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder, E. J. , Bredenbeek, P. J. , Dobbe, J. C. , Thiel, V. , Ziebuhr, J. , Poon, L. L. M. , … Gorbalenya, A. E. (2003). Unique and conserved features of genome and proteome of SARS‐coronavirus, an early split‐off from the coronavirus group 2 lineage. Journal of Molecular Biology, 331(5), 991–1004. 10.1016/s0022-2836(03)00865-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, X. , Wu, C. , Li, X. , Song, Y. , Yao, X. , Wu, X. , … Lu, J. (2020). On the origin and continuing evolution of SARS‐CoV‐2. National Science Review. 10.1093/nsr/nwaa036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thao, T. T. N. , Labroussaa, F. , Ebert, N. , V’kovski, P. , Stalder, H. , Portmann, J. … Thiel, V. (2020). Rapid reconstruction of SARS‐CoV‐2 using a synthetic genomics platform. bioRxiv. 10.1101/2020.02.21.959817 [DOI] [PubMed] [Google Scholar]
- Wang, C. , Liu, Z. , Chen, Z. , Huang, X. , Xu, M. , He, T. , & Zhang, Z. (2020). The establishment of reference sequence for SARS‐CoV‐2 and variation analysis. Journal of Medical Virology, 92(6), 667–674. 10.1002/jmv.25762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO (2020). WHO Director‐General's opening remarks at the media briefing on COVID‐19. March 11, 2020, Retrieved from https://www.who.int/dg/speeches/detail/who‐director‐general‐s‐opening‐remarksat‐the‐media‐briefing‐on‐covid‐19–‐11‐march‐2020.
- Wilke, C. O. , Wang, J. L. , Ofria, C. , Lenski, R. E. , & Adami, C. (2001). Evolution of digital organisms at high mutation rates leads to survival of the flattest. Nature, 412(6844), 331–333. 10.1038/35085569 [DOI] [PubMed] [Google Scholar]
- Zhao, Z. , Li, H. , Wu, X. , Zhong, Y. , Zhang, K. , Zhang, Y.‐P. , … Fu, Y.‐X. (2004). Moderate mutation rate in the SARS coronavirus genome and its implications. BMC Evolutionary Biology, 4, 21. 10.1186/1471-2148-4-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, P. , Yang, X.‐L. , Wang, X.‐G. , Hu, B. , Zhang, L. , Zhang, W. , … Shi, Z.‐L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Appendix S2
Appendix S3
Data Availability Statement
The data that support the findings of this study are available in GISAID at https://www.gisaid.org/.