

Summary
Translation efficiency contributes several orders of magnitude difference in the overall yield of exogenous gene expression in bacteria. In diverse bacteria, the translation initiation site, whose sequence is the primary determinant of the translation performance, is comprised of the start codon and the Shine–Dalgarno box located upstream. Here, we have examined how the sequence of a spacer between these main components of the translation initiation site contributes to the yield of synthesized protein. We have created a library of reporter constructs with the randomized spacer region, performed fluorescently activated cell sorting and applied next‐generation sequencing analysis (the FlowSeq protocol). As a result, we have identified sequence motifs for the spacer region between the Shine–Dalgarno box and AUG start codon that may modulate the translation efficiency in a 100‐fold range.
Understanding the principles that determine mRNA translation efficiency is of primary value for deciphering translational control of gene expression and for optimization of protein synthesis in biotechnology. In this work we combined Flowseq method with randomization of a region within the spacer between the Shine‐Dalgarno box and AUG start codon to decipher an influence of this mRNA part on translation efficiency.

Introduction
Protein synthesis in heterological expression systems, such as bacteria, is one of the major goals of biotechnology. A protein is synthesized by ribosomes following instructions encoded in the sequence of mRNA (Brenner et al., 1961; Gros et al., 1961). Non‐coding regions of mRNA, in particular the 5’‐untranslated region (5’‐UTR), contribute greatly to the efficiency of protein synthesis (Laursen et al., 2005; Brenneis and Soppa, 2009). Computational and experimental analysis of bacterial 5’‐UTRs revealed a number of features affecting translation (Chen et al., 1994; Salis et al., 2009; Salis, 2011; Espah Borujeni et al., 2014; Farasat et al., 2014). The most important and best‐known 5’‐UTR sequence element of bacterial mRNAs is the Shine–Dalgarno (SD) box complementary to the 3’‐end region of the small subunit 16S rRNA (Shine and Dalgarno, 1974). The length of the complementary region in E. coli varies between 4 and 8 nucleotides (nt) (Shultzaberger et al., 2001). The optimal distance between the SD box and the start codon for efficient gene expression is 5–9 nt (Hartz et al., 1991; Osterman et al., 2013).
Experimental analysis of 5’‐UTR sequence elements that affect protein synthesis is commonly done via monitoring the expression of a reporter gene placed downstream of a set of specific 5’‐UTR sequences (Vimberg et al., 2007). High‐throughput analysis of the translation efficiency as a function of mRNA sequence became possible with development of the FlowSeq method (Kudla et al., 2009; Goodman et al., 2013; Evfratov et al., 2017). This method is based on sorting of cells transformed by a library of reporter constructs encoding a fluorescent protein, followed by next‐generation sequencing of sorted fractions of the library. This method allowed us to identify multiple features of the 5’‐UTR influencing the translation efficiency (Evfratov et al., 2017). However, complete randomization of 20 or 30 nt 5’‐UTR regions (Evfratov et al., 2017) resulted in a library whose diversity exceeded the throughput of the FlowSeq method, precluding complete sampling of the sequence space and hence complicating in‐depth analysis of particular 5’‐UTR determinants of the translation efficiency.
Here, we have applied FlowSeq to assess the translational influence of the least studied region of the 5’‐UTR, the spacer between the SD box and the start codon.
Results and discussion
Library construction and FlowSeq analysis
To analyse the influence of the spacer region sequence on the translation efficiency, we used the plasmid encoding the red (RFP) and cerulean (CER) fluorescent proteins (Osterman et al., 2012; Osterman et al., 2013; Evfratov et al., 2017). Both fluorescent protein genes were controlled by identical T5 promoters (Fig. 1, see Fig. S1A for details). 5’‐UTRs of the rfp gene were identical in all plasmids in the library, allowing us to use RFP as an internal standard, whereas 5’‐UTRs of cer were subjected to partial randomization. Randomized inserts of four nucleotides were placed into the 8 nt spacer region between the SD box and the start codon of the 22 nt long 5’‐UTR of the cer gene (Fig. 1, see Fig. S1B for details). E. coli cells transformed by this plasmid library were sorted into six fractions according to the CER/RFP ratio measured as the ratio of fluorescence intensities at 405/530 and 561/582 nm and indicated by the inclined frames of the fractions on the real FACS plot (Fig. 1).
Fig. 1.
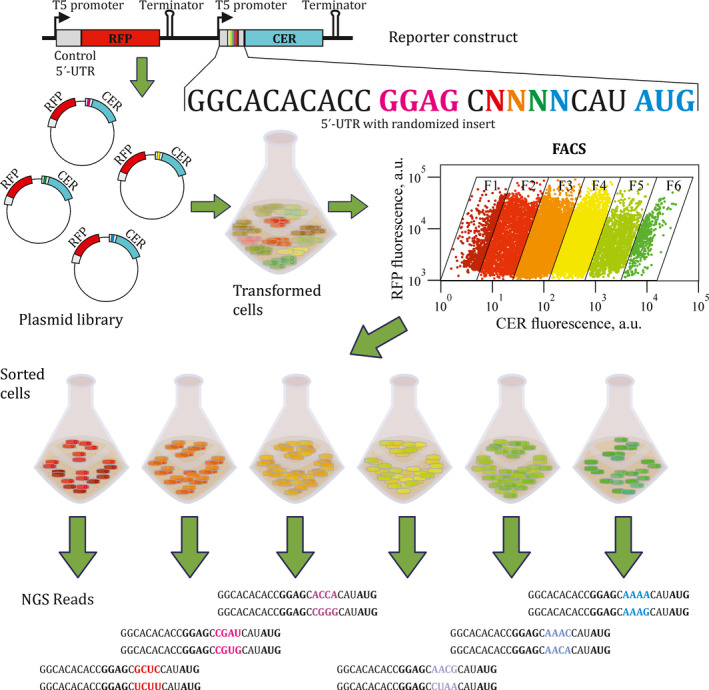
Scheme of the FlowSeq experiment. On the top, a scheme of the reporter construct library is presented. Promoters, terminators, 5’‐UTRs, RFP and CER fluorescent protein coding regions are marked. The sequence of the CER gene 5’‐UTR is shown below the scheme. Shine–Dalgarno sequence is coloured pink, start codon is coloured blue, while the 4 nt randomized fragment is rainbow‐coloured. Creation of the plasmid library, cell transformation, growth, sorting, fraction collection and next‐generation sequencing are shown schematically. The real FACS plot for the library with 4 nt randomized spacer region is presented.
Sorted cells from these fractions were collected, used for plasmid isolation and PCR amplification of the region containing the randomized 5’‐UTR part (Fig. S2). Next‐generation sequencing of the amplicons allowed us to deduce the distribution of cells carrying particular variants among the fractions separated by the CER/RFP ratio (Table S1) and hence to assign the translation efficiency value to each construct.
Sequencing of the cer 5’‐UTR regions from the sorted cells yielded 249 unique inserts out of 256 variants theoretically possible for a 4 nt randomized region (Tables S1 and S2). Unlike our FlowSeq analysis of reporter construct libraries with 5’‐UTRs containing 20 and 30 nt randomized regions (Evfratov et al., 2017) demonstrating four orders of magnitude span of translation efficiencies, the reporters containing 4 nt randomized region between the SD box and the start codon demonstrated at most a 100‐fold difference between the highest and lowest CER protein yield (Fig. S3). This efficiency range provides for moderate, yet substantial contribution of the spacer region sequence to the overall translation rate.
Taking into account a relatively narrow distribution of the observed translation efficiencies and an uneven distribution of 5’‐UTR variants among six initially obtained fractions (Fig. S3), we combined these fractions into two groups, representing high and low translation efficiencies in such a way that both groups contained (almost) equal number of sequences. These two groups were subjected to further computational analysis.
Spacer region influence on translation efficiency
The comparison of the nucleotide composition in the randomized spacer (Fig. 2A) for efficiently and poorly translated mRNAs revealed a significant difference in the nucleotide composition for all positions of the spacer (chi‐square test, P‐value < 10–6). At all four positions, adenosine appeared to be the most beneficial for the translation, while cytidines were unfavourable, in agreement with the results previously obtained for reporters carrying larger randomized regions (Evfratov et al., 2017). Our results are also in consent with previous work (Mirzadeh et al., 2015) where an influence of the spacer region positions −6 to −1 on translation efficiency was examined. Likewise, in these data, oligoadenosine tracks of 3 or 4 residues at positions −6 to −3 could be found predominantly in mRNAs possessing high expression efficiency. Many efficiently translated mRNAs in our library possess A‐rich sequences with U residue in the spacer region. For instance, mRNAs with AAAU, AAUA, AUAA, AUAU sequence variants have the mean fraction number more than five (Table S1) demonstrating one of the highest translation efficiency. Analysis of GC content of the randomized mRNA region for mRNAs possessing different translation efficiencies (Fig. S4) also suggests a positive role of AU content of the examined mRNA part on protein yield. This might reflect a positive influence of AU‐rich enhancers (Komarova et al., 2005) that may bind ribosomal protein S1 (Boni et al., 1991; Komarova et al., 2005; Duval et al., 2013; Osterman et al., 2013), although these enhancers are generally assumed to be positioned upstream of the SD sequence. Alternative explanation for the preference towards adenosines in the spacer region of efficiently translated mRNAs is enhanced interaction with the ribosome. While spacer region of mRNA is not involved in base pairing with the 16S rRNA, nucleotides −1 to −3 of the 4 nt long spacer are stacked on each other and on top of the 16S rRNA nucleotide G926 (Hussain et al., 2016). Nucleotide A1503 of the 16S rRNA contacts mRNA nucleotide −4 (relative to the first P‐site nucleotide) in several initiation complexes (Jenner et al ., 2010; Hussain et al., 2016) and nucleotide −2 in the intermediate of translocation (Zhou et al ., 2013). While little is known about the structure of initiation complexes containing longer spacer regions, such as ones used in our study, it might be hypothesized that ribosome may form some sequence‐specific, most likely stacking interactions with this mRNA region.
Fig. 2.
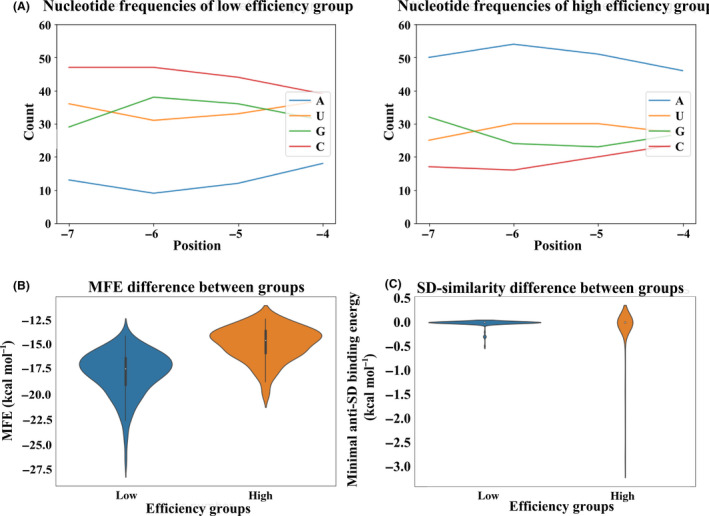
Properties of the spacer region of the efficiently and poorly translated CER mRNAs.
A. Frequency of nucleotides at specific positions of the randomized region for poorly (left panel) and efficiently (right panel) translated mRNAs. Nucleotide positions are numbered by the distance to the start codon.
B. Distributions of the minimal free energy (MFE) of the secondary structure folding in mRNA groups with different translation efficiencies.
C. Distributions of the minimal hybridization energy of the spacer region between the constant SD sequence and AUG start codon containing entire randomized mRNA region and its 1 nt upstream and 4 nt downstream flanks to the 3’‐terminal region of the 16S rRNA in mRNA groups with different translation efficiencies.
The spacer region might be a part of a secondary structure that may mask other components of the translation initiation site, such as the SD box or the start codon. Indeed, formation of a secondary structure inhibits the initiation of translation (Smit and van Duin, 1990; Osterman et al., 2012; Evfratov et al., 2017). To analyse the influence of the spacer sequence on the folding energy of the translation initiation site, and hence on the translation efficiency, we modelled secondary structures for all mRNA sequences in our data set, using a window encompassing the entire 22 nt 5’‐UTR and the first 50 nt of the coding region. These sequences were used to calculate the minimal free energy (MFE), a standard measure of the predicted secondary structure stability, with lower MFE values corresponding to more stable secondary structures (Fig. 2B). The Kolmogorov–Smirnov test demonstrated that the difference in the secondary structure stability between efficiently and poorly translated mRNAs was significant at the level 10–15. To show that this effect was not a trivial consequence of the difference in the nucleotide content, we performed permutational analysis (see Methods) and demonstrated that the difference of MFE among fractions was significantly larger than expected given the observed positional nucleotide frequencies (P‐value = 0.001, see Fig. S5).
All 5’‐UTR sequences in our data set contained a four‐nucleotide SD box, located 8 nt upstream of the AUG start codon. To check whether extension of this standard box yielding additional regions complementary to the 16S rRNA 3’‐end region could influence the efficiency of translation, we calculated the free hybridization energy of the anti‐SD sequence CACCUCCU at the 3’‐terminal region of the 16S rRNA with CNNNNCAUA 5’‐UTR part containing the entire randomized region (Fig. 2C). Overall, two observed distributions of the free hybridization energy did not differ significantly (the Kolmogorov–Smirnov test p‐value > 0.05). However, spacers from poorly translated cer mRNAs had no SD‐like patches in addition to the standard SD box, whereas several spacers from the efficiently translated set contained a sequence that could form complementary interactions with the 16S rRNA 3’‐end region, such as the ones whose randomized regions are AAGG, AGGA, GAGG, GGAG, GGGG and GGGA. This observation corroborates our earlier result (Evfratov et al., 2017) on an additive influence of multiple SD‐like sequences in the 5’‐UTR on the efficiency of translation. However, this mechanism may explain the observed high translation rate of only a limited subset of efficiently translated mRNAs.
Several tools for prediction of the translation efficiency are published for monocistronic (Salis et al., 2009; Salis, 2011; Bonde et al ., 2016) and bicistronic constructs (Mutalik et al ., 2013; Nieuwkoop et al ., 2019). Comparison of the measured translation efficiency of mRNAs in our data set with that predicted by RBS Calculator (Salis et al., 2009; Salis, 2011) (Table S1, Predicted translation efficiency) demonstrated relatively good, although not ideal correlation (r = 0.62).
The data obtained here allow us to suggest the following recommendations for the choice of spacer regions between the SD box and the start codon for bacterial expression systems. In order to boost translation and hence protein yield, it seems reasonable to use oligoadenylate or other A‐rich spacers while avoiding cytidine residues, although it cannot be ruled out that some particular mRNAs with A‐rich spacers might mask translation initiation site within the secondary structure if, e.g., coding region would be U‐rich. Our results might help to adjust the level of exogenous gene expression to a particular biotechnological need. For coexpression of genes whose products should be synthesized at a particular stoichiometry, e.g., if the proteins are subunits of a heteromultimeric complex, the expression levels might be fine‐tuned by proper selection of spacers between the SD boxes and the start codons of the designed mRNAs.
Experimental procedures
Strains and plasmids
All cloning manipulations were carried out on the Escherichia coli strain JM109. Cells were grown at 37⁰C in LB medium with 100 µg ml−1 ampicillin added if required.
Plasmid pRFPCER (Osterman et al., 2012) was used as the host vector for randomized library construction (Fig. S1), similar to our previously published protocol (Evfratov et al., 2017) presented in the supporting information (Fig. S1B). The methodology was adapted from published procedure (Oliphant et al., 1986). Briefly, oligonucleotide 5’‐ACTGCCGCGGCACACACC GGAGCNNNNCATATG‐3’ containing randomized region was self‐annealed and converted to double‐stranded form by Klenow fragment of DNA polymerase I. Double‐stranded inserts were cloned into the pRFPCER vector via SacII and NdeI recognition sites. Products of ligation were used for transformation of ultra‐competent JM109 cells (Sambrook and Russell, 2006). In parallel, the control plasmid with the same 5’‐UTR upstream the start codons of the both fluorescent protein genes and as a consequence with approximately the same ratio of CER to RFP proteins was transformed into JM109 E. coli strain and next prepared for sorting.
Sorting and sequencing
Cells transformed by the plasmid library were grown overnight at 37°C in the liquid LB medium supplemented with 100 µg ml−1 ampicillin with agitation. After washing in phosphate‐buffered saline and diluting in PBS to ca 0.004 A600, the cells were sorted by Becton Dickinson FACSAria III while simultaneously monitoring CER and RFP fluorescence intensities at 405/530 and 561/582 nm correspondingly. Six fractions with different CER/RFP fluorescence intensity ratio (log scale) were collected according to the indicated inclined frames of the fractions on the real FACS plot (Fig. 1). Inclined frames are used since they represent cells of equal CER/RFP fluorescence ratio.
The number of cells collected for each fraction was proportional to the total abundance of cells with particular CER/RFP fluorescence intensity ratio, i.e. without artificial enrichment for rare variants. The total number of the sorted cells was 104 covering several times the diversity of 4 nt randomized library.
The sorted pools were grown overnight at 37°C in the liquid LB medium supplemented with 100 µg ml−1 ampicillin with agitation and used for plasmid preparation and PCR amplification using primers 5’‐CCATCTCATCCCTGCGTGTCTCATTTGCTTTCAGGAAAATTTTTCTG‐3’ and 5’‐CCACTACGCCTCCGCTTTCCTC NNNN TCACCAGGCCGCTCTCGTCC‐3’, for the last one the region NNNN corresponds to six barcode variants for further sequencing, the CER coding region is underlined (Fig. S2). The sequencing of the amplicon library was conducted with Ion Torrent (Rothberg et al., 2011) PGM (Life Technologies) using Ion PGMTM Template OT2 200 Kit for emulsion PCR amplification and Ion Chips 314 or 318 along with the reagent kit Ion PGMTM Sequencing 200 Kit v2 following instructions of manufacturer.
Data analysis
To extract 5’‐UTR sequences from raw read data in the FASTQ format, all 50 to 100 nt long reads were used to search for regions that differed by at most two mismatches, including indels, from the GGCACACACCGGAGC and CATATGAAAGAGACGGACGAGAGCGGCCTGGTGA sequences flanking the 4 nt randomized region. Each sequence variant was searched for in all sorted fractions. The collected data were summarized as a table counting the occurrences of each sequence variant in each of the sorted fractions (Table S1). Whereas the same sequence variant is fractionally spread, each sequence variant in the experiment was assigned a mean weighted number of its fraction assuming a Gaussian distribution (the last column in Table S1). Due to strongly different numbers of variants in the fractions and a relatively narrow overall range of the translation efficiency in our experiment (Fig. S3), we divided all 5’‐UTR variants into two groups of roughly equal size. The group of low translation efficiency contains 125 out of 249 mRNA variants whose mean fraction numbers belong to the range from 1.47 to 3.07, while the group of high translation efficiency contains 124 out of 249 mRNA variants whose mean fraction numbers belong to the range from 3.08 to 5. Following analysis was conducted on these two classes.
Nucleotide frequencies were counted for each position within the 4 nt randomized regions separately for efficiently and poorly translated mRNAs. Minimal folding energies (MFE) were calculated for the region encompassing the entire 22 nt 5’‐UTR and 50 nt of the downstream cer coding region using RNAfold ver. 2.1.7 of the Vienna RNA package with default parameters (Lorenz et al., 2011). To estimate the statistical significance of the difference in MFE between efficiently and poorly translated mRNAs, while controlling for the nucleotide content, we used a Monte Carlo modelling to generate 1000 pairs of sets of shuffled sequence variants by randomly permuting nucleotides in each position, separately for both groups. The MFE calculation was performed for each randomly shuffled variant, the distribution of MFE was constructed for each set, and then the Kolmogorov–Smirnov statistic was calculated for the pairs of these modelled distributions, yielding the distribution shown in Fig. S5. The Kolmogorov–Smirnov statistic for the real pair was calculated and used to estimate the p‐value of the observed difference.
To estimate a propensity of a particular spacer region to hybridize with the 16S rRNA 3’‐terminal region, we calculated the free hybridization energy of 5’‐UTR fragments CNNNNCAUA that contained the randomized part with the anti‐SD sequence CACCUCCU using the RNAfold programme of the Vienna RNA package (Lorenz et al., 2011). The distributions of the energy of interaction with the anti‐SD sequence were constructed for both mRNAs groups.
The general sequence data processing was implemented in Python (Sanner, 1999), whereas statistical analysis and plotting were executed in R (Dessau and Pipper, 2008).
Conflict of interest
The authors declare that they have no competing interests.
Supporting information
Fig. S1. Creation of the reporter construct library. A. Scheme of the plasmid pRFPCER with used restriction sites shown. B. Cloning scheme of the randomized fragment in spacer region into pRFPCER reporter vector upstream CER fluorescent protein gene. Following designations are used: “ss oligos”, single‐stranded oligonucleotides (~ 30nt), “ds oligos”, double‐stranded oligonucleotides (~ 60nt), “cleaved oligos”, oligonucleotides treated by restriction endonucleases (~ 25 nt), “PolIk”, Klenow fragment of E.coli DNA polymerase I, “SacII”, “NdeI”, corresponding endonucleases.
Fig. S2. Electrophoresis of amplicons, containing the randomized part, used for NGS in 2% agarose gel. The plasmids isolated from the sorted cells were separated into six fractions and used for PCR amplification with primers flanking both sides of the randomized region. M is GeneRulerTM 1kb Plus DNA Ladder.
Fig. S3. Average CER and RFP fluorescence intensity and proportion of reads in each of six fractions after sorting. Fractions F1 to F6 correspond to cell pools sorted by the translation efficiency from the lowest efficiency (F1) to the highest one (F6). Bulk fluorescence intensity of cells in each fraction measured by a fluorimeter is shown as green (CER) and red (RFP) bars on the left side of the panel and starts from 103 for convenience, the fluorescence intensity scale is presented as decimal logarithm of absolute values. Proportions of individual reads in all extracted sequence variants from the NGS dataset that correspond to each fraction sorted by the translation efficiency are shown in the right panel.
Fig. S4. GC‐content analysis of the 4 nt randomized sequence in the spacer region of the efficiently and poorly translated CER mRNAs. GC‐content was plotted against the mean expression fraction (Table S1, last column) of each sequence variant (A) or the number of variants in two classes with low and high translation efficiencies (B). The more the mean fraction number the more translation efficiency can be observed for the sequence variant.
Fig. S5. Histogram of the Kolmogorov–Smirnov (KS) statistic values for mRNA groups with permuted sequences. Sequences in the two groups were shuffled 1000 times, yielding sets of permuted sequences with the same number of random variants and the same positional nucleotide frequencies. For each pair of sets the distributions of the secondary structure energy were compared using the Kolmogorov‐Smirnov statistics. The KS statistic value for real sequences in the experiment is shown by the black arrow. The calculated p‐value is 0.001.
Table S1. Properties of mRNA variants in the dataset.
Table S2. Variants of the randomized sequence fragment what are theoretically possible but have not been found among sequenced variants.
Acknowledgements
This work was supported by RSF grant 19‐14‐00043 (P.S.) with library preparation supported by RFBR grant 17‐00‐00366 (P.S.) and computational analysis supported by RSF grant 18‐14‐00358 (M.G.). Moscow State University Scientific School (O.D.) and Institute of functional genomics government support (P.S.) were used for stipends of participants.
Microbial Biotechnology (2020) 13(4), 1254–1261
Funding Information
This work was supported by RSF grant 19‐14‐00043 (P.S.) with library preparation supported by RFBR grant 17‐00‐00366 (P.S.) and computational analysis supported by RSF grant 18‐14‐00358.
References
- Bonde, M. , Pedersen, M. , Klausen, M. , Jensen, S.I. , Wulff, T. , Harrison, S. , et al (2016) Predictable tuning of protein expression in bacteria. Nat Methods 13: 233–236. [DOI] [PubMed] [Google Scholar]
- Boni, I.V. , Isaeva, D.M. , Musychenko, M.L. , and Tzareva, N.V. (1991) Ribosome‐messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res 19: 155–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneis, M. , and Soppa, J. (2009) Regulation of Translation in Haloarchaea: 5’‐ and 3’‐UTRs are essential and have to functionally interact in vivo . PLoS ONE 4: e4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, S. , Jacob, F. , and Meselson, M. (1961) An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 190: 576–581. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Bjerknes, M. , Kumar, R. , and Jay, E. (1994) Determination of the optimal aligned spacing between the Shine‐Dalgarno sequence and the translation initiation codon of Escherichia coli mRNAs. Nucleic Acids Res 22: 4953–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessau, R.B. , and Pipper, C.B. (2008) ‘R’–project for statistical computing. Ugeskr Laeger 170: 328–330. [PubMed] [Google Scholar]
- Duval, M. , Korepanov, A. , Fuchsbauer, O. , Fechter, P. , Haller, A. , Fabbretti, A. , et al (2013) Escherichia coli ribosomal protein S1 unfolds structured mRNAs onto the ribosome for active translation initiation. PLoS Biol 11: e1001731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espah Borujeni, A. , Channarasappa, A.S. , and Salis, H.M. (2014) Translation rate is controlled by coupled trade‐offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res 42: 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evfratov, S.A. , Osterman, I.A. , Komarova, E.S. , Pogorelskaya, A.M. , Rubtsova, M.P. , Zatsepin, T.S. , et al (2017) Application of sorting and next generation sequencing to study 5’‐UTR influence on translation efficiency in Escherichia coli . Nucleic Acids Res. 45: 3487–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farasat, I. , Kushwaha, M. , Collens, J. , Easterbrook, M. , Guido, M. , and Salis, H.M. (2014) Efficient search, mapping, and optimization of multi‐protein genetic systems in diverse bacteria. Mol Syst Biol 10: 731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman, D.B. , Church, G.M. , and Kosuri, S. (2013) Causes and effects of N‐terminal codon bias in bacterial genes. Science 342: 475–479. [DOI] [PubMed] [Google Scholar]
- Gros, F. , Hiatt, H. , Gilbert, W. , Kurland, C.G. , Risebrough, R.W. , and Watson, J.D. (1961) Unstable ribonucleic acid revealed by pulse labelling of Escherichia coli . Nature 190: 581–585. [DOI] [PubMed] [Google Scholar]
- Hartz, D. , McPheeters, D.S. , and Gold, L. (1991) Influence of mRNA determinants on translation initiation in Escherichia coli. J. Mol. Biol. 218: 83–97. [DOI] [PubMed] [Google Scholar]
- Hussain, T. , Llacer, J.L. , Wimberly, B.T. , Kieft, J.S. , and Ramakrishnan, V. (2016) Large‐scale movements of IF3 and tRNA during bacterial translation initiation. Cell 167: 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner, L. , Demeshkina, N. , Yusupova, G. , and Yusupov, M. (2010) Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 17: 555–560. [DOI] [PubMed] [Google Scholar]
- Komarova, A.V. , Tchufistova, L.S. , Dreyfus, M. , and Boni, I.V. (2005) AU‐rich sequences within 5' untranslated leaders enhance translation and stabilize mRNA in Escherichia coli . J Bacteriol 187: 1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudla, G. , Murray, A.W. , Tollervey, D. , and Plotkin, J.B. (2009) Coding‐sequence determinants of gene expression in Escherichia coli . Science 324: 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen, B.S. , Sorensen, H.P. , Mortensen, K.K. , and Sperling‐Petersen, H.U. (2005) Initiation of protein synthesis in bacteria. Microbiol Mol Biol Rev 69: 101–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz, R. , Bernhart, S.H. , Siederdissen, C. , Höner, Z. , Tafer, H. , Flamm, C. , et al. (2011) ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadeh, K. , Martinez, V. , Toddo, S. , Guntur, S. , Herrgard, M. , Elofsson, A. , et al (2015) Enhanced protein production in Escherichia coli by optimization of cloning scars at the vector:coding sequence junction. ACS Synth. Biol. 4: 959–965. [DOI] [PubMed] [Google Scholar]
- Mutalik, V. , Guimaraes, J. , Cambray, G. , Lam, C. , Christoffersen, M.J. , Maiet, Q.‐A. , et al (2013) Precise and reliable gene expression via standard transcription and translation initiation elements. Nat Methods 10: 354–360. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop, T. , Claassens, N.J. , and van der Oost, J. (2019) Improved protein production and codon optimization analyses in Escherichia coli by bicistronic design. Microb Biotechnol. 12: 173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant, A.R. , Nussbaum, A.L. , and Struhl, K. (1986) Cloning of random‐sequence oligodeoxynucleotides. Gene 44: 177–183. [DOI] [PubMed] [Google Scholar]
- Osterman, I.A. , Prokhorova, I.V. , Sysoev, V.O. , Boykova, Y.V , Efremenkova, O.V. , Svetlov, M.S. , et al (2012) Attenuation‐based dual‐fluorescent‐protein reporter for screening translation inhibitors. Antimicrob Agents Chemother 56: 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterman, I.A. , Evfratov, S.A. , Sergiev, P.V. , and Dontsova, O.A. (2013) Comparison of mRNA features affecting translation initiation and reinitiation. Nucleic Acids Res. 41: 474–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothberg, J.M. , Hinz, W. , Rearick, T.M. , Schultz, J. , Mileski, W. , and Davey, M. , et al (2011) An integrated semiconductor device enabling non‐optical genome sequencing. Nature 475: 348–352. [DOI] [PubMed] [Google Scholar]
- Salis, H.M. (2011) The Ribosome Binding Site Calculator. Amsterdam, Netherlands: Elsevier Inc, 498, pp. 19–42. [DOI] [PubMed] [Google Scholar]
- Salis, H.M. , Mirsky, E.A. , and Voigt, C.A. (2009) Automated design of synthetic ribosome binding sites to precisely control protein expression. Nat Biotechnol 27: 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , and Russell, D.W. (2006) The Inoue method for preparation and transformation of competent E. coli: "ultra‐competent" cells. CSH Protoc. [DOI] [PubMed] [Google Scholar]
- Sanner, M.F. (1999) Python: a programming language for software integration and development. J Mol Graph Model 17: 57–61. [PubMed] [Google Scholar]
- Shine, J. , and Dalgarno, L. (1974) The 3'‐terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc Natl Acad Sci USA 71: 1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultzaberger, R.K. , Bucheimer, R.E. , Rudd, K.E. , and Schneider, T.D. (2001) Anatomy of Escherichia coli ribosome binding sites. J Mol Biol 313: 215–228. [DOI] [PubMed] [Google Scholar]
- Smit, M.H. , and van Duin, J. (1990) Secondary structure of the ribosome binding site determines translational efficiency: a quantitative analysis. Proc Nati. Acad Sci USA 87: 7668–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vimberg, V. , Tats, A. , Remm, M. , and Tenson, T. (2007) Translation initiation region sequence preferences in Escherichia coli . BMC Mol Biol 8: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Lancaster, L. , Donohue, J.P. , and Noller, H.F. (2013) Crystal structures of EF‐G‐ribosome complexes trapped in intermediate states of translocation. Science 340: 1236086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Creation of the reporter construct library. A. Scheme of the plasmid pRFPCER with used restriction sites shown. B. Cloning scheme of the randomized fragment in spacer region into pRFPCER reporter vector upstream CER fluorescent protein gene. Following designations are used: “ss oligos”, single‐stranded oligonucleotides (~ 30nt), “ds oligos”, double‐stranded oligonucleotides (~ 60nt), “cleaved oligos”, oligonucleotides treated by restriction endonucleases (~ 25 nt), “PolIk”, Klenow fragment of E.coli DNA polymerase I, “SacII”, “NdeI”, corresponding endonucleases.
Fig. S2. Electrophoresis of amplicons, containing the randomized part, used for NGS in 2% agarose gel. The plasmids isolated from the sorted cells were separated into six fractions and used for PCR amplification with primers flanking both sides of the randomized region. M is GeneRulerTM 1kb Plus DNA Ladder.
Fig. S3. Average CER and RFP fluorescence intensity and proportion of reads in each of six fractions after sorting. Fractions F1 to F6 correspond to cell pools sorted by the translation efficiency from the lowest efficiency (F1) to the highest one (F6). Bulk fluorescence intensity of cells in each fraction measured by a fluorimeter is shown as green (CER) and red (RFP) bars on the left side of the panel and starts from 103 for convenience, the fluorescence intensity scale is presented as decimal logarithm of absolute values. Proportions of individual reads in all extracted sequence variants from the NGS dataset that correspond to each fraction sorted by the translation efficiency are shown in the right panel.
Fig. S4. GC‐content analysis of the 4 nt randomized sequence in the spacer region of the efficiently and poorly translated CER mRNAs. GC‐content was plotted against the mean expression fraction (Table S1, last column) of each sequence variant (A) or the number of variants in two classes with low and high translation efficiencies (B). The more the mean fraction number the more translation efficiency can be observed for the sequence variant.
Fig. S5. Histogram of the Kolmogorov–Smirnov (KS) statistic values for mRNA groups with permuted sequences. Sequences in the two groups were shuffled 1000 times, yielding sets of permuted sequences with the same number of random variants and the same positional nucleotide frequencies. For each pair of sets the distributions of the secondary structure energy were compared using the Kolmogorov‐Smirnov statistics. The KS statistic value for real sequences in the experiment is shown by the black arrow. The calculated p‐value is 0.001.
Table S1. Properties of mRNA variants in the dataset.
Table S2. Variants of the randomized sequence fragment what are theoretically possible but have not been found among sequenced variants.