

Summary
Pf prophages are ssDNA filamentous prophages that are prevalent among various Pseudomonas aeruginosa strains. The genomes of Pf prophages contain not only core genes encoding functions involved in phage replication, structure and assembly but also accessory genes. By studying the accessory genes in the Pf4 prophage in P. aeruginosa PAO1, we provided experimental evidence to demonstrate that PA0729 and the upstream ORF Rorf0727 near the right attachment site of Pf4 form a type II toxin/antitoxin (TA) pair. Importantly, we found that the deletion of the toxin gene PA0729 greatly increased Pf4 phage production. We thus suggest the toxin PA0729 be named PfiT for Pf4 inhibition toxin and Rorf0727 be named PfiA for PfiT antitoxin. The PfiT toxin directly binds to PfiA and functions as a corepressor of PfiA for the TA operon. The PfiAT complex exhibited autoregulation by binding to a palindrome (5′‐AATTCN5 GTTAA‐3′) overlapping the ‐35 region of the TA operon. The deletion of pfiT disrupted TA autoregulation and activated pfiA expression. Additionally, the deletion of pfiT also activated the expression of the replication initiation factor gene PA0727. Moreover, the Pf4 phage released from the pfiT deletion mutant overcame the immunity provided by the phage repressor Pf4r. Therefore, this study reveals that the TA systems in Pf prophages can regulate phage production and phage immunity, providing new insights into the function of TAs in mobile genetic elements.
PfiT/PfiA in Pf4 prophage forms a type II toxin‐antitoxin system in Pseudomonas aeruginosa. PfiT inhibits production of Pf4 phage.
Introduction
Toxin/antitoxin (TA) systems are genetic modules widely distributed in prokaryotes. TA genes usually encode a toxin that kills cells or inhibits cell growth and a cognate antitoxin that neutralizes the toxicity of the toxin. A total of six types of TA systems have been identified based on the molecular features (protein or RNA) of antitoxins and the mechanisms they used to mask the toxicity of toxins (Mruk and Kobayashi, 2014). In type II TA systems, both toxins and antitoxins are proteins, and antitoxins neutralize the toxicity of toxins by direct protein–protein interactions. Toxin and antitoxin genes are in the same operon, and the cognate toxins either work as repressors or activators of antitoxins to autoregulate the expression of the TA operon (Magnuson and Yarmolinsky, 1998; Afif et al., 2001; Overgaard et al., 2008; Winther and Gerdes, 2012; Turnbull and Gerdes, 2017). These type II TA systems are found in both chromosomes and mobile genetic elements including plasmids and prophages (Wang and Wood, 2016; Harms et al., 2018). Studies of TA systems in plasmids are more extensive than those in prophages. The studied plasmid‐encoded TA systems include the first type II TA CcdB/CcdA characterized ‘addiction’ systems on the F sex factor plasmid (Ogura and Hiraga, 1983), ParE/ParD, Hok/Doc, HigB/HigA and HicB/HicA (Lehnherr et al., 1993; Roberts et al., 1994; Hayes, 2003; Christensen‐Dalsgaard and Gerdes, 2006; Kroll et al., 2010).
Prophages and satellite prophages are some of the major horizontal gene transfer elements that are widespread among bacteria, and they constitute up to 20% of bacterial genomes. Many sequenced bacterial genomes contain multiple prophages, e.g. eighteen prophages were identified in E. coli O157 Sakai (Asadulghani et al., 2009), and nine prophages were identified in E. coli K12 MG1655 (Wang et al., 2010). Prophages confer a series of phenotypic traits to their hosts, including pathogenicity (Sweere et al., 2019), antibiotic tolerance and resistance (Wang and Wood, 2016), biofilm formation and general stress (Wang et al., 2010; Wang and Wood, 2011; Zeng et al., 2016). The genomes of most prophages not only contain genes encoding functions involved with phage replication, structure and assembly, but also contain accessory genes. For example, the well‐characterized MG1655 prophages encode type I, type II and type IV toxin/antitoxin (TA) systems. In particular, the product of toxin ralR in the rac prophage is a DNase, and the type I RalR/RalA TA pair increased cell resistance to fosfomycin (Guo et al., 2014). In addition, the type IV TA pair CbtA/CbeA in the cryptic prophage CP4‐44 has been related to resistance to norfloxacin, novobiocin and spectinomycin (Kohanski et al., 2007; Masuda et al., 2012). In Shewanella oneidensis, a type II TA pair ParESO/CopASO in the cryptic prophage CP4So stabilizes the circular prophage CP4So in host cells after its excision (Yao et al., 2018). In addition, infection of lytic phages is also inhibited by plasmid‐ or chromosomal‐encoded TA systems. The type I TA system Hok/Sok from plasmid R1 excludes T4 infection in E. coli (Pecota and Wood, 1996), and the chromosomal type II TA system MazE/MazF protects cells from P1 phage infection (Hazan and Engelberg‐Kulka, 2004). In addition, the first type III TA system, ToxN/ToxI, was found in a cryptic plasmid of the plant pathogen Pectobacterium atrosepticum that supplies cells with an ability to resist to other phages by the release of the ribonuclease toxin ToxN (Fineran et al., 2009).
Pseudomonas aeruginosa is an opportunistic pathogen found to infect plants, invertebrates and vertebrates (Palleroni, 1984) and is clinically important for chronic lung infections in cystic fibrosis (CF) patients (Lyczak et al., 2002). These P. aeruginosa strains frequently contain prophages, and prophages are important in the CF‐epidemic strains. The filamentous phage Pf is critical for several stages of the P. aeruginosa biofilm life cycle (Rice et al., 2009; Secor et al., 2015) and is a key contributor to the formation of small colony variants and virulence in vivo (Ilyina, 2015; Sweere et al., 2019). Three putative TA loci have been predicted in the genome of the model strain P. aeruginosa PAO1 by bioinformatic analysis (Williams et al., 2011), and HigB/HigA on the chromosome was shown to be a type II TA system that controls biofilm formation and virulence (Li et al., 2016, Wood and Wood, 2016; Zhang et al., 2018, Guo et al., 2019). In the present study, we characterized the type II TA system PfiT/PfiA in the Pf4 prophage of PAO1 and found that it controls the production of the Pf4 phage. PfiT greatly inhibits cell growth, and PfiA neutralizes the toxicity of PfiT through direct protein–protein interactions. The pfiA and pfiT genes are cotranscribed and the PfiAT complex, but not antitoxin PfiA, autoregulates the TA operon by binding to the palindrome 5′‐AATTCN5 GTTAA‐3′, overlapping the −35 region of the TA operon. The deletion of the toxin pfiT gene induced the production of Pf4 phage by increasing the expression of the replication initiation factor gene, and the phages released from the toxin pfiT‐deleted strain can overcome the immunity supplied by the phage repressor Pf4r. To the best of our knowledge, this is the first experimental evidence that a TA system in a filamentous phage controls phage production.
Results
PfiT and PfiA in the Pf4 prophage form a TA pair
We recently reannotated the Pf4 genome during the identification of the phage excisionase gene xisF4 (Li et al., 2019). Two neighbouring genes that are only 9 bp apart, PA0729 and Rorf0727, are located at the right end of the Pf4 prophage. Rorf0727 encodes a protein of 83 aa that belongs to the Phd antitoxin family (here, we renamed it PfiA), and PA0729 encodes a protein of 115 aa that belongs to the ParE toxin family (here, we renamed it PfiT; Fig. 1A). To determine whether they constitute a bona fide TA pair, open reading frames of the two genes were cloned into plasmid pMQ70 to obtain pMQ70‐pfiA and pMQ70‐pfiT, respectively, using the primers listed in Table S1. Expression of pfiT or pfiA was induced in PAO1 with 10 mM l‐arabinose. Cell growth (turbidity) and cell viability (CFU ml−1) were measured over time. Overexpression of pfiT in PAO1 led to not only growth inhibition but also cell death (Fig. 1B,C). In contrast, overexpression of pfiA did not affect cell growth or cell death. To further assess whether PfiA can block the toxicity of PfiT, we cloned the coding region of pfiA and pfiT to construct pMQ70‐pfiAT, which was used to coexpress pfiA and pfiT in PAO1. Coexpression of pfiT with pfiA showed similar growth and cell viability compared with the empty vector pMQ70 (Fig. 1B,C), indicating that PfiA neutralized PfiT toxicity. Thus, PfiA functions as an antitoxin to prevent the growth‐inhibitory effect of toxin PfiT. Since most TA systems are cotranscribed, we then conducted a primer extension assay using the oligonucleotide FAM‐pfiT‐r (Table S1), which is complementary to pfiT, to search for the transcription start of the TA operon. As shown in Fig. 1D, the major extension product is 700 nt in size, indicating that pfiA and pfiT are cotranscribed and that the transcriptional start site of the pfiAT operon is 127 bp upstream of pfiA (Fig. 1A,D). Collectively, these results demonstrated that the antitoxin PfiA and toxin PfiT form a type II TA pair.
Fig. 1.
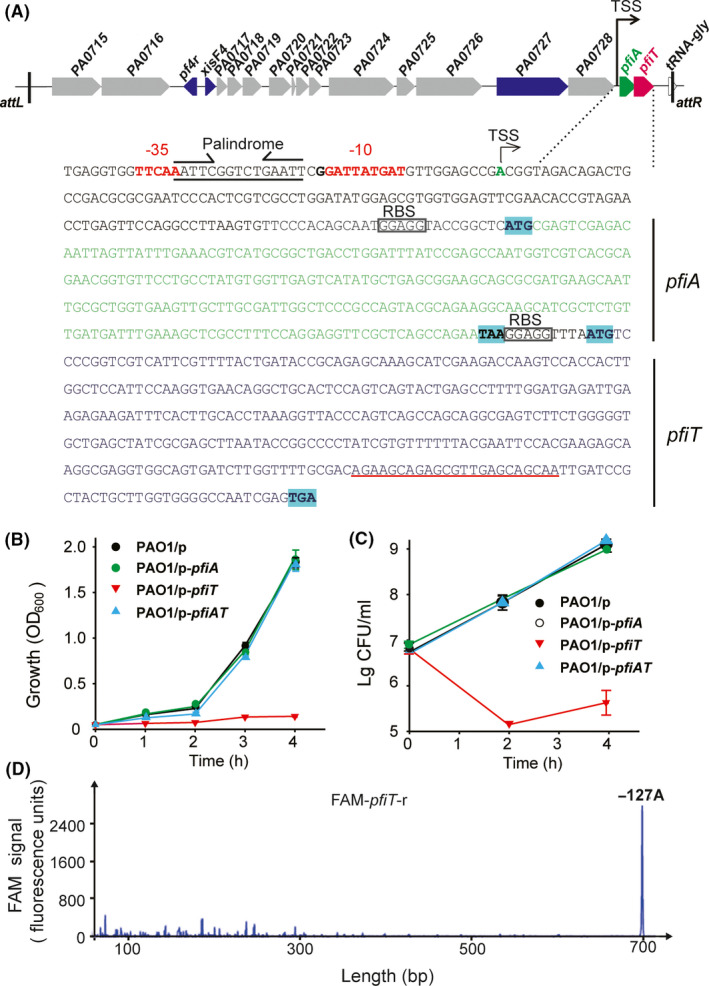
PfiA and PfiT form a type II TA pair. (A) Location and sequence of pfiAT within prophage Pf4. The ‘attL’ and ‘attR’ indicate the left and right attachment sites respectively. The antitoxin pfiA is shown by the green arrow and letters, while the toxin pfiT is shown by the red arrow and letters. Below, the sequence analysis of the pfiAT operon is indicated. The −10 and −35 regions are shown in red letters. The transcriptional start site (TSS) and RBS are also shown. (B) Growth (turbidity) and (C) viability (CFU ml−1) of PAO1 wild‐type carrying the pMQ70‐based plasmids were determined with 10 mM l‐arabinose added at a turbidity of 0.1 at 600 nm. ‘p’ indicates plasmid pMQ170. Three independent cultures of each strain were tested, and error bars indicate the standard error of the mean (n = 3). (D) The TSS of pfiAT was determined with a 5′‐end FAM‐labelled primer, which is underlined in red in A. The x‐axis indicates the length of the cDNA with FAM, and the y‐axis indicates the fluorescence intensity of the FAM signal.
PfiA interacts with PfiT in vivo
For most type II TA systems, the toxin interacts with the antitoxin directly to form a protein complex in vivo. To test whether PfiA binds to PfiT, a pull‐down assay was performed with pET28b‐pfiAT‐His to coexpress a C‐terminal hexahistidine‐tagged (His‐tagged) PfiT with untagged antitoxin PfiA. As expected, affinity purification revealed that another protein was pulled down along with His‐tagged PfiT (expected size ~ 13.81 kDa) using Ni‐NTA agarose beads and subsequent tricine‐SDS‐PAGE (Fig. 2A), and the size of this protein was consistent with the size of the PfiA antitoxin (~ 9.44 kDa). To further determine the interaction between PfiA and PfiT, a bacterial two‐hybrid (BATCH) assay based on the physical interaction of the T18 and T25 catalytic domains was conducted. An in‐frame translational fusion between the T18 catalytic domain and pfiA was performed to generate pUT18C‐pfiA, and a similar fusion between the T25 catalytic domain and pfiT was also generated (pKT25‐pfiT). For the positive control, a fragment encoding a 35 aa leucine zipper was translationally fused to the T25 and T18 catalytic domains to generate pKT25‐zip and pUT18C‐zip respectively. For the negative control, the empty vector pKT25 without an insert and pUT18C‐zip were used. Consistent with the above pull‐down assay, pKT25‐pfiT and pUT18C‐pfiA showed clear β‐galactosidase activity, indicating that the interaction between PfiA and PfiT occurred (Fig. 2B). Taken together, PfiA and PfiT form a complex in vivo, and the inhibitory effect of PfiA to PfiT is likely due to the direct interaction between them.
Fig. 2.
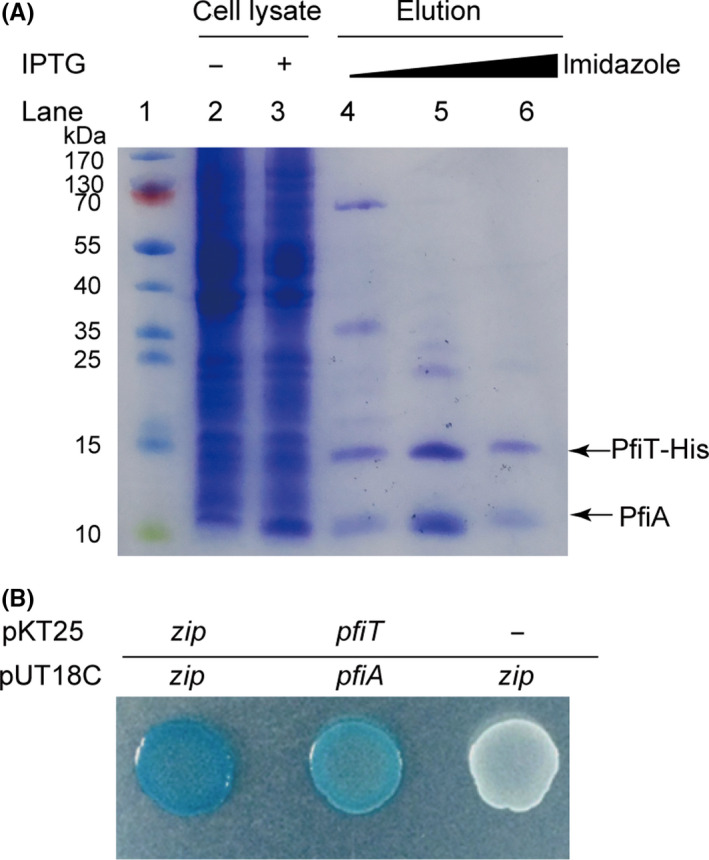
PfiA interacts with PfiT in vivo.
A. Tricine‐SDS‐PAGE showed that the antitoxin PfiA was copurified with His‐tagged PfiT‐His from pET28b‐pfiAT‐His in E. coli BL21 (DE30).
B. Bacterial two‐hybrid assay to assess the interactions between PfiA and PfiT. Cells harbouring the pKT25‐zip and pUT18C‐zip plasmids were used as positive controls, and cells harbouring the pKT25 (without insert) and pUT18C‐zip plasmids were used as negative controls.
The PfiAT complex controls pfiAT transcription
Type II antitoxins alone or in complex with toxins can bind to their promoters and negatively regulate the transcription of the TA operon. To test whether PfiA affects TA promoter activity, we transcriptionally fused a 254 bp promoter region to lacZ and integrated it into the chromosome of PAO1 via a mini‐CTX plasmid according to a previously reported method (Hoang et al., 2000). We also constructed TA deletion mutants in PAO1. Two mutants, ∆pfiT and ∆pfiAT, were constructed and confirmed by PCR and DNA sequencing (Fig. 3A). We tried to knock out pfiA in this experiment, but no correct strain was obtained after extensive effort, indicating that this antitoxin may not be able to be removed due to the strong toxicity of the toxin. Then, the promoter activity was determined in the PAO1 wild‐type strain and the two deletion mutant strains. The β‐galactosidase activity in PAO1 wild‐type cells was 185.51 ± 15.54 Miller units (MU), and it increased to 662.82 ± 15.93 MU in the ∆pfiAT cells (Fig. 3B). These results showed that PfiT/PfiA negatively regulates its own promoter activity. However, there was no significant change in the promoter activity between the ∆pfiT and ∆pfiAT cells (664.73 ± 46.18 MU versus 662.82 ± 15.93), suggesting that antitoxin PfiA alone may not be sufficient for the autoregulation of the TA operon. To further investigate this, PfiA and the PfiAT TA complex were produced via pHERD20T‐pfiA and pHERD20T‐pfiAT in the two deletion mutant reporter strains, and the promoter activity was determined. Consistent with the above results, only a slight decrease in β‐galactosidase activity was observed when PfiA was overexpressed compared with the empty vector in both reporter strains. However, a significant decrease in β‐galactosidase activity was observed when the PfiAT complex was coexpressed compared with the empty vector (Fig. 3C). In addition, the autoregulation of the PfiT/PfiA TA pair was determined with EMSA. A PCR product of 254 bp, which included the promoter region of the TA operon, was used to bind with PfiA or the PfiAT complex. The PfiAT complex specifically bound to the pfiAT promoter region (Fig. 3D, lanes 1–4). However, no binding to the promoter region was observed for PfiA in the absence of the toxin (Fig. 3D, lanes 8–10), and the binding appeared when the PfiAT TA complex was added (Fig. 3D, lanes 5–7). Thus, the PfiAT complex represses the transcription of the pfiAT operon by binding to the TA promoter region, and PfiT functions as a corepressor of PfiA.
Fig. 3.
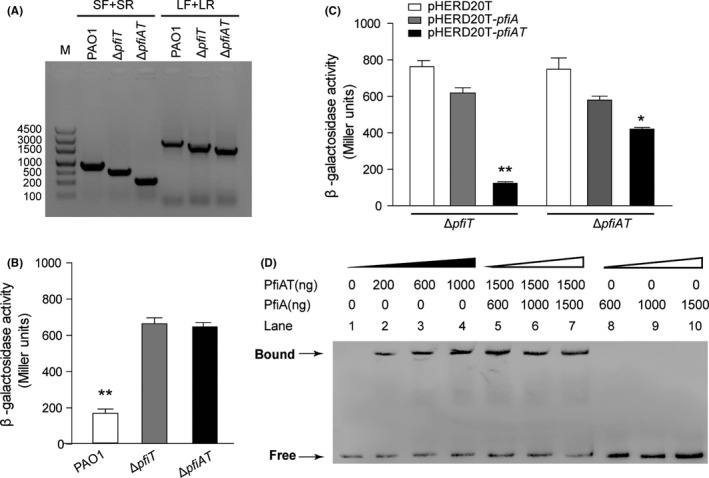
The PfiAT complex represses the pfiAT operon.
A. Deletions of toxin gene pfiT and TA operon pifAT were confirmed with PCR method using the primer pairs LF/LR and SF/SR. M indicates DNA marker.
B. The β‐galactosidase activity of the PpfiAT–lacZ reporter was determined in strains PAO1, ΔpfiT and ΔpfiAT.
C. The β‐galactosidase activity of PpfiAT–lacZ was determined in strains ΔpfiA and ΔpfiAT carrying pHERD20T, pHERD20T‐pfiA and pHERD20T‐pfiAT. Arabinose (10 mM) was added to induce the expression of genes for 3 h at OD600 ~ 0.1. Three independent cultures of each strain were tested, and error bars indicate the standard error of the mean (n = 3).
D. EMSA showed that antitoxin PfiA alone could not bind to the promoter of the pfiAT operon. The PfiAT complex bound to the promoter region of the pfiAT operon in a concentration‐dependent manner. *P < 0.05, **P < 0.01.
The PfiAT complex binds to 5′‐AATTCN5GAATT‐3′ in the pfiAT promoter
Bioinformatic analysis of the pfiAT operon identified a palindromic sequence, 5′‐AATTC GGTCT GAATT‐3′, overlapping the predicted −35 region of pfiAT (Fig. 1A). To determine the exact binding site of the PfiAT complex, a DNase I footprinting assay was employed using the 300 bp promoter region of pfiAT and the purified PfiAT complex. The results showed that the region containing the palindrome was specifically protected from DNase I digestion by the PfiAT complex (Fig. 4A). To further confirm the DNA‐binding ability of the PfiAT complex to the palindrome in vivo, we constructed a series of lacZ reporter plasmids with different mutations in the palindromic sequence. Plasmids pLP170‐M1‐pfiAT and pLP170‐M2‐pfiAT contain one mutation each in the left arm and right arm, respectively, and pLP170‐M3‐pfiAT contains mutations in both arms (Fig. 4B). Then, the β‐galactosidase activities were determined in both PAO1 wild‐type and ∆pfiAT strains. All mutations in these constructs increased the β‐galactosidase activities significantly in PAO1 cells, which contain the pfiAT operon, indicating that the palindromic sequence is critical for the promoter activity of the pfiAT operon. In addition, mutation of the palindromic sequence had no effect on β‐galactosidase activity in ∆pfiAT cells, showing that the PfiAT complex binds to the palindromic sequence. Taken together, the PfiAT complex represses its own expression by binding to 5′‐AATTCN5 GAATT‐3′ in the pfiAT promoter.
Fig. 4.
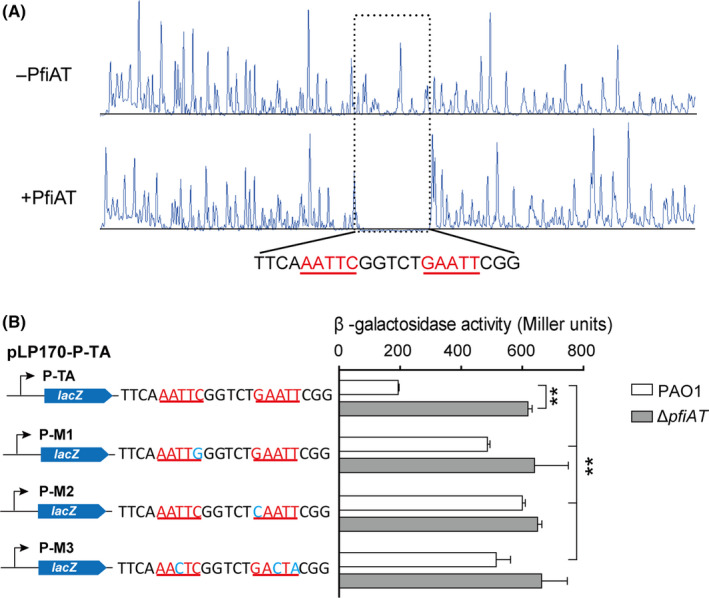
PfiAT binds to inverted repeats in the promoter of the pfiAT operon.
A. DNase I footprinting assay demonstrated that the PfiAT complex bound to the DNA motif containing 5′‐AATTCN5 GAATT‐3′.
B. The promoter activities of the mutated promoters were determined in strains PAO1 and ΔpfiAT. Three independent cultures of each strain were used, and error bars indicate standard deviation. **P < 0.01.
PfiT inhibits Pf4 replication by inducing PA0727
To probe the physiological function of the PfiAT TA pair, we investigated Pf4 production by the deletion mutant ∆pfiT. Specifically, wild‐type PAO1 and ∆pfiT cells were cultured statically in LB medium to form pellicle biofilms, and the supernatant was collected at different time points to determine the plaque‐forming units (PFU) in the Pf4 deletion strain (∆Pf4). As shown in Fig. 5A, deletion of pfiT greatly increased Pf4 phage production over time (left) and increased Pf4 phage production by approximately 100,000‐fold compared with the wild‐type at 6 h (right). To explore how PfiA regulates Pf4 phage production, qRT‐PCR was used to quantify the expression of Pf4 genes in the wild‐type and ∆pfiT strains. The amplification efficiencies of the primer sets used in qRT‐PCR lie between 89.3 and 106.7% (Fig. S1). Since the autorepression of the TA pair was disrupted in the ∆pfiT strain, as expected, we found that the expression of antitoxin pfiA was induced 19.68 ± 4.15‐fold when pfiT was deleted (Fig. 5B). In addition, the phage excisionase coding gene xisF4 and replication initiation protein‐coding gene PA0727 were induced 71.00 ± 0.52‐fold and 16.23 ± 1.67‐fold, respectively, when pfiT was deleted, but not the phage repressor coding gene pf4r (Fig. 5B). However, the excision of the Pf4 prophage was not induced in the ∆pfiT cells (data not shown). Therefore, disruption of the cooperativity of PfiA and PfiT induced the replication of the Pf4 phage by inducing PA0727 expression, thus increasing phage production.
Fig. 5.
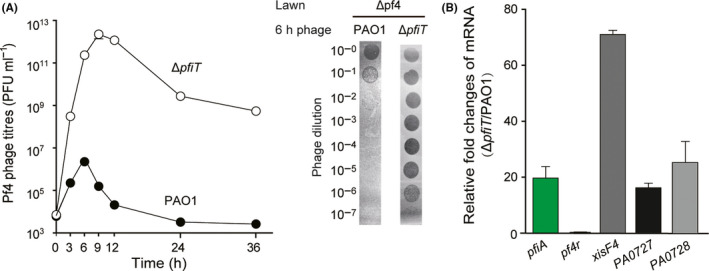
Deletion of pfiT activates Pf4 production.
A. Pf4 phage titres were determined on ΔPf4 lawns using pellicle supernatants from strains PAO1 and ΔpfiT. Strains were cultured in 6‐well plates under static conditions. Three independent cultures of each strain were used, and error bars indicate standard deviation. Plaque formation by the phage lysates at 6 h is also shown. Phage lysates were serially diluted, and 10 μl samples were spotted on ΔPf4 lawns.
B. Relative fold changes of mRNA of pfiA, pf4r, xisF4 and PA0727 in strain ΔpfiT versus wild‐type strain PAO1. RNA was extracted from the static pellicle culture at 6 h.
PfiT coordinates Pf4r in conferring immunity to Pf4
We have found that the phage repressor Pf4r confers immunity to Pf4 (Li et al., 2019). To test whether infection of the phages from the wild‐type PAO1 and ∆pfiT cells is both inhibited by Pf4r, the production of phage Pf4 was induced by overexpressing XisF4 via pHERD20T‐xisF4 in wild‐type PAO1 and ∆pfiT hosts. Similar phage titres of phages were obtained from supernatant of the two strains after induction with 10 mM arabinose for 4 h (Fig. 6 left panel). Then, the Pf4 phages were used to infect the ∆Pf4 strain with overexpressing pf4r. Consistent with our earlier work, overexpression of pf4r in the ∆Pf4 host strain provided higher immunity (~10,000‐fold higher) than the empty vector for the Pf4 phage released from wild‐type PAO1. In contrast, the immunity against phage infection was greatly reduced for the phage released from ∆pfiT cells, approximately 10‐fold higher than the empty vector. This result suggested that PfiT is involved in both phage production and phage immunity.
Fig. 6.
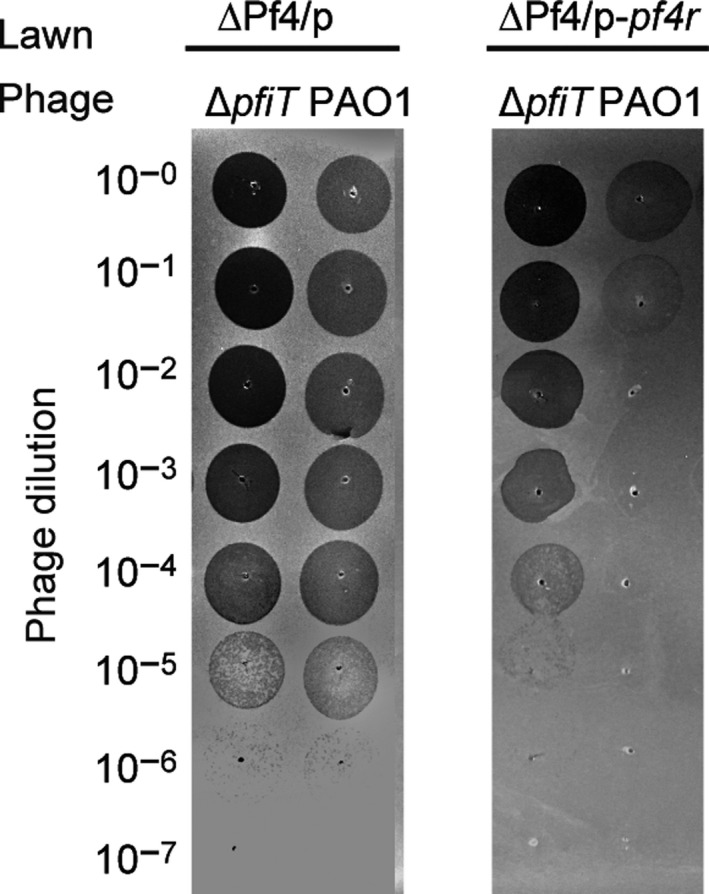
PfiT coordinates Pf4r in conferring immunity to Pf4. Plaque formation by the phage lysates from strains PAO1 and ΔpfiT carrying pHERD20T‐xisF4. Phages were collected 4 h after 10 mM arabinose was added to the planktonic cultures of each strain at the beginning. Phage lysates were serially diluted, and 10 μl samples were spotted on lawns of strain ΔPf4 carrying pHERD20T or pHERD20T‐pf4r.
Discussion
In this study, we provided evidences that the Pf4 prophage encoded type II TA pair controls the production of filamentous phages in PAO1. The results were as follows: (i) PfiT is a toxin, and its toxicity can be neutralized by its cognate antitoxin PfiA; (ii) PfiA and PfiT interact with each other directly, and the PfiAT complex binds to the 5′‐AATTCN5 GAATT‐3′ palindrome in the pfiAT operon; (iii) mutation of pfiT increases production of Pf4 phage by inducing the expression of the replication initiation protein; and (iv) PfiT coordinates Pf4r in conferring immunity to Pf4. Therefore, we proved that the toxin harboured in the prophage inhibits filamentous phage production in P. aeruginosa, and it also contributes to cell immunity to Pf4 phage infection, extending the physiological roles of the type II TA system.
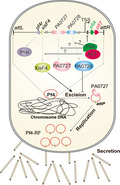
A schematic of our understanding of how the PfiT/PfiA TA system controls the production of the Pf4 phage and further affects virulence and biofilm formation in PAO1 is shown in Fig. 7. In certain typical type II TA systems, the antitoxins act as transcriptional repressors and adopt N‐terminal DNA‐binding domains such as helix–turn–helix, ribbon–helix–ribbon and AbrB‐type domains (Chan et al., 2016). However, PfiA has a truncated N‐terminus without these domains, which is similar to other Phd family antitoxins. The binding of the Phd family antitoxins to target sites requires the help of a toxin (Guerout et al., 2013), and a fully folded conformation where all secondary structure elements are formed after binding to the toxin (Cherny and Gazit, 2004; Garcia‐Pino et al., 2010). Here, we found that the binding of PfiA to the TA promoter region also requires PfiT. The binding of PfiT to the PfiA antitoxin may stabilize the N‐terminal domain, and change the allosteric and intrinsic disorder and thus control transcription regulation. A similar mechanism was observed in different TA systems, including Doc/Phd, CcdB/CcdA, RelE/RelB and VapC/VapB (Magnuson and Yarmolinsky, 1998; Afif et al., 2001; Overgaard et al., 2008; Winther and Gerdes, 2012). In some other TA systems, such as HigB/HiA and HicB/HicA, toxins are repressors of antitoxins and function in the transcriptional repression of the TA operon (Turnbull and Gerdes, 2017; Guo et al., 2019). ParE family toxins function as gyrase inhibitors and inhibit cell division by targeting GyrB (Jiang et al., 2002). We did not observe aberrant cell division when pfiT was overexpressed in PAO1, which was likely due to the low sequence similarity between PfiT and well‐characterized ParE family toxins. Homologs of PfiT are also found in other Pseudomonas strains, and the cellular target of PfiT in Pseudomonas will be investigated in future studies.
Fig. 7.
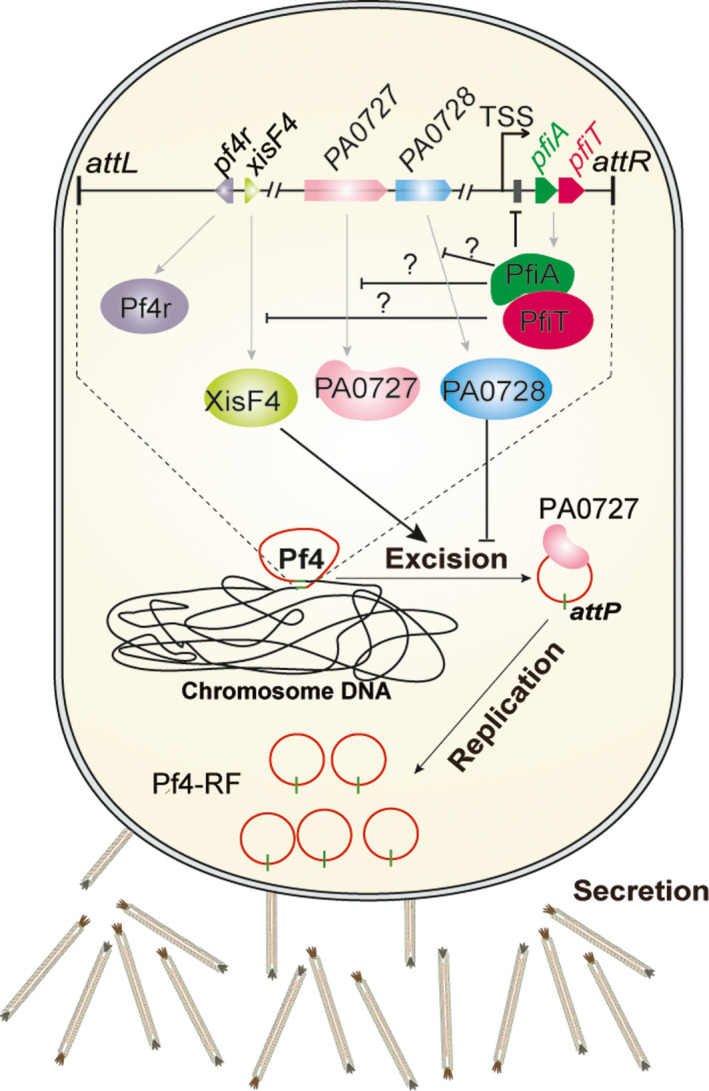
A proposed model of the PfiT/PfiA TA system controlling Pf4 production. The expression of the PfiAT complex autorepresses the expression of the pfiAT operon. Disruption of pfiT induced the expression of pfiA, xisF4 and PA0727 (replication initiation factor), but had no effect on the expression of pf4r. The induced XisF4 and PA0727 thus promote the excision and replication of Pf4, after which these phages are secreted from the cell. The secreted Pf4 phage will work as a biofilm contributor and host immunity stimulator.
Filamentous phages are considered some of the simplest life forms on earth, and they have relatively smaller genomes (7 ~ 12 kb) compared with dsDNA tailed phage. Filamentous phages such as Pf4 and Pf5 are integrated as prophages in the genomes of PAO1 and PA14 respectively. In PAO1, Pf4 is integrated between PA0714 and tRNAGly, while Pf5 is integrated inside the coding region of the PA14_49040 gene. The genome regions that encode phage replication, structure and assembly genes in the prophages Pf4 and Pf5 share much higher sequence identity than accessory gene regions (Li et al., 2019). The PfiT/PfiA TA system is located at the end of the Pf4 prophage and is not found in the Pf5 prophage. A unique feature of the Pf4 phage in PAO1 is the ability to cause superinfection (Rice et al., 2009), and no superinfection was reported for the Pf5 phage in PA14. In this study, we found that the Pf4 phage released from the toxin pfiT‐deleted strain can still efficiently infect PAO1 cells overexpressing the phage repressor gene pf4r. Although no similar palindromic sequence was identified in other regions of the Pf4 genome, the expression levels of the excisionase gene xisF4 and replication initiation factor PA0727 were induced significantly in the pfiT mutant strain, while no change in the expression level of the repressor gene pf4r was observed.
TA systems have a broad and important impact on bacterial physiology and bacterial pathogenicity by influencing developmental cascades such as the switch from planktonic to biofilm cells and/or the activation of the expression of virulence genes (Guo et al., 2019). Pf4 phage production was mainly found during PAO1 biofilm formation. Here, we found that the type II TA system PfiT/PfiA encoded by Pf4 controls Pf4 production, as the deletion of the toxin induces the production of Pf4. Indeed, the ratio between the toxin and the antitoxin was greatly changed in the PAO1 WT and in the pfiT deletion mutant strains, as the deletion of pfiT also induced the expression of pfiA. Under conditions when more PfiA is present, PfiA is likely to induce Pf4 replication. On the other hand, most type II antitoxins are usually unstable. Under specific conditions, PfiA can be degraded by certain proteases to free PfiT. Accordingly, free PfiT is able to inhibit the replication of the phage. Thus, the ratio between the toxin and the antitoxin seems important in the regulation of Pf4 production. This could either enable the bacterial host cells to control Pf4 production or equip the phage to trigger its own replication when needed. However, there are still unsolved questions that need to be addressed to obtain a better understanding of the role of the TA system in controlling phage production during biofilm formation.
Experimental procedures
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids are listed in Table 1, and primers are listed in Table S1. E. coli and P. aeruginosa PAO1 strains were grown in Luria–Bertani (LB) medium at 37°C. Cells harbouring plasmids with the indicated resistance genes were cultured in medium supplemented with the following antibiotics at the indicated concentrations: kanamycin (50 µg ml−1), tetracycline (50 µg ml−1), gentamycin (30 µg ml−1) and carbenicillin (100 µg ml−1).
Table 1.
Bacterial strains and plasmids used in this study.
| Strains/plasmids | Description | Source |
|---|---|---|
| DH5α | F‐φ80lacZ∆M15 ∆(lacZYA‐argF)U169 recA1 endA1 hsdR17(rk − , mk+)phoA supE44 thi‐1 gyrA96 relA1 tonA | Novagen |
| BTH101 | F‐, cya‐99, araD139, galE15, galK16, rpsL1 (Strr), hsdR2, mcrA1, mcrB1 | Euromedex Kit |
| PAO1 | Wild‐type | Stover et al. (2000) |
| ΔPf4 | Whole Pf4 prophage removed from PAO1 host chromosome | Li et al. (2019) |
| ΔpfiT | pfiT deletion mutant derived from PAO1 chromosome | This study |
| ΔpfiAT | pfiAT deletion mutant derived from PAO1 chromosome | This study |
| PAO1:: PpfiAT‐lacZ | LacZ reporter strain | This study |
| ΔpfiT:: P pfiAT‐lacZ | LacZ reporter strain | This study |
| ΔpfiAT::P pfiAT‐lacZ | LacZ reporter strain | This study |
| Plasmids | ||
| pET28b | KmR, expression vector | Novagen |
| pET28b‐pfiA | KmR, pfiA in pET28b NcoI/Hind III | This study |
| pET28b‐pfiAT | KmR, pfiAT in pET28b NcoI/Hind III | This study |
| pMQ70 | ApR, CarR, expression vector with araC‐PBAD promoter | Shanks et al. (2006) |
| pMQ70‐pfiA | ApR, CarR, pfiA in pMQ70 SacI/HindIII | This study |
| pMQ70‐pfiT | ApR, CarR, pfiT in pMQ70 SacI/KpnI | This study |
| pMQ70‐pfiAT | ApR, CarR, pfiAT in pMQ70 SacI/KpnI | This study |
| pHERD20T | ApR, CarR, expression vector with araC‐PBAD promoter | Qiu et al. (2008) |
| pHERD20T‐pfiA | ApR, CarR, pfiA in pHERD20T NcoI/salI | This study |
| pHERD20T‐pfiAT | ApR, CarR, pfiAT in pHERD20T EcoRI/HindIII | This study |
| pHERD20T‐pf4r | ApR, CarR, xisF4 in pHERD20T NcoI/HindIII | Li et al. (2019) |
| pKT25‐zip | KmR; derived from pKT25. Sequence coding for the leucine zipper region of the GCN4 yeast protein. Positive control | Karimova et al. (1998) |
| pKT25‐pfiT | KmR; expression vector for pilT. | This study |
| pUT18C | ApR; derived from pUC19. Plac–MCS(HindIII–SphI–PstI–SalI–XbaI–BamHI–SmaI–KpnI–SacI–EcoRI)–T18 | Karimova et al. (1998) |
| pUT18C‐zip | ApR; derived from pUC19. Sequence coding for the leucine zipper region of the GCN4 yeast protein. Positive control. | Karimova et al. (1998) |
| pUT18C‐pfiA | ApR; expression vector for pfiA. | This study |
| pEX18AP | ApR, oriT +, sacB +, gene replacement vector | Hoang et al. (1998) |
| pFLP2 | ApR, Flp recombinase‐expressing plasmid | Hoang et al. (1998) |
| pPS856 | ApR, GmR; for amplifying gentamycin resistance cassette | Hoang et al. (1998) |
| pEX18AP‐pfiT | GmR, CarR, for deleting pfiT | This study |
| pEX18AP‐pfiAT | GmR, CarR, for deleting pfiAT | This study |
| mini‐CTX‐LacZ | TetR, integration vector for single‐copy, chromosomal lacZ fusions; Ω‐FRT‐attP‐MCS, ori, int, and oriT | Becher and Schweizer (2000) |
| pCTX‐PpfiAT‐lacZ | TetR, −313 bp relative to translational start site of pfiAT cloned into mini‐CTX‐lacZ | This study |
| pLP170 | CarR, promoterless‐lac Z | Pesci et al. (1997) |
| pLP170‐pfiAT | Wild‐type promoter of pfiAT fused into the lacZ of pLP170 | This study |
| pLP170‐M1‐pfiAT | FP1 mutant promoter of pfiAT fused into the lacZ of pLP170 | This study |
| pLP170‐M2‐pfiAT | FP3 mutant promoter of pfiAT fused into the lacZ of pLP170 | This study |
| pLP170‐M3‐pfiAT | FP4 mutant promoter of pfiAT fused into the lacZ of pLP170 | This study |
Construction of deletion mutants in PAO1
The gene deletion strain was constructed as described previously in P. aeruginosa (Hoang et al., 1998). Briefly, the upstream and downstream regions of pfiT and pfiAT were amplified through PCR from PAO1 genomic DNA. The gentamycin resistance gene cassette was amplified through PCR from the plasmid pPS856. These three amplicons were then ligated into the suicide plasmid pEX18Ap using the ClonExpress II One Step Cloning Kit (Vazyme, Nanjing, China). In‐frame deletion mutants were obtained via homologous recombination using the sucrose resistance selection method. The gentamycin resistance cassette was removed from the chromosome as described previously (Hoang et al., 1998). Finally, the correct mutants were confirmed by PCR and DNA sequencing.
Construction of reporter strains
The full coding regions of pfiA, pfiT and pfiAT were PCR‐amplified from PAO1 genomic DNA, and the PCR products were purified and ligated into the vectors pMQ70, pHERD20T and pET28b using the Vazyme ClonExpress II One Step Cloning Kit. For construction of promoter–reporter strains, the 254 bp upstream of pfiAT was amplified by PCR and ligated into the plasmid mini‐CTX‐lacZ. The correct plasmids were transformed into the PAO1, ΔpfiT and ΔpfiAT hosts and integrated into chromosomes at the attB site near the tRNASer sequence using a previously described method (Becher and Schweizer, 2000). Then, the tetracycline selection marker was removed as described (Hoang et al., 1998).
β‐Galactosidase activity assay
Specific β‐galactosidase activities of strains PAO1, ΔpfiT and ΔpfiAT harbouring the pfiAT promoter were determined by monitoring the absorbance at 420 nm using a Pro200 Multi‐Detection Microplate Reader (Tecan, Männedorf, Switzerland) using the Miller assay method (Miller, 1972). Overnight cultures were diluted 100‐fold in LB with or without carbenicillin (50 μg ml−1) and grown at 37 °C to an OD600 of 1.0, and then, β‐galactosidase activity was determined. To determine the promoter activity of pfiAT in ΔpfiT and ΔpfiAT carrying pHERD20T‐derived plasmids, overnight strains were diluted to OD600 ~ 0.1 and grown in LB supplemented with carbenicillin and 10 mM arabinose. After induction for 3 h, cells were collected to determine β‐galactosidase activity.
Bacterial two‐hybrid (BACTH) assay
The BACTH assay was conducted as described (Battesti and Bouveret, 2012) to investigate the interaction between PfiA and PfiT in vivo. The coding regions of pfiA and pfiT were cloned into pUT18C and pKT25 respectively. The recombinant plasmids were cotransformed into E. coli BTH101 (cya‐99) competent cells with selection for kanamycin and ampicillin resistance. Then, 10 µl of overnight culture was spotted on LB plates supplemented with kanamycin, ampicillin, IPTG (1 mM) and X‐gal (40 µg ml−1). The colonies grew for 20 h. The negative and positive controls were included as we described previously (Yao et al., 2018).
Primer extension
The 5′‐end FAM (6‐carboxyfluorescein)‐labelled primer FAM‐pfiT‐r was ordered from Invitrogen (Carlsbad, CA, USA). Total RNA was isolated from PAO1 wild‐type cells. The extension reactions were carried out with 10 μg of total RNA, 2 × 10−4 pmol of FAM‐pfiT‐r and 37.5 U of AMV reverse transcriptase (Promega, Madison, USA). The reaction mixture was incubated at 42°C for 90 min, and the products were concentrated with centrifugal filter units (Millipore, Bedford, MA, USA) before being loaded into an ABI3730 DNA Analyzer (Applied Biosystems, Foster City, CA, USA).
Protein purification
Proteins PfiA and PfiAT were purified from the E. coli BL21 (DE3) strain containing plasmid pET28b‐pfiA or pET28b‐pfiAT respectively. One litre of LB supplemented with kanamycin was inoculated with 10 ml of overnight culture, and the bacteria were grown with shaking at 37°C. IPTG 0.5 mM was added at OD600 0.5, and all the cells were collected by centrifugation after induction for 6 h. The subsequent steps of protein extraction from the collected pellet were performed as previously described (Liu et al., 2015).
Electrophoretic mobility shift assay (EMSA)
The DNA probe of the promoter region of pfiAT was amplified from the genomic DNA of the PAO1 strain using the primer pair pfiAT‐promoter‐f/r (Table S1). The purified DNA fragments were labelled with biotin by using the Biotin 3′‐End DNA Labeling Kit (Thermo Scientific, Rockford, USA). Then, the biotin‐labelled DNA fragments (0.25 pmol) were mixed with the purified proteins and incubated at 25°C for 2 h to perform binding reactions. The binding reaction components were added following the protocol as described in the LightShiſt Chemiluminescent EMSA Kit (Thermo Scientific, Rockford, USA). The binding reaction samples were run on a 6% polyacrylamide gel in 0.5 × Tris‐borate EDTA (TBE) and were then transferred to nylon membranes. The membranes were visualized using the Chemiluminescence Nucleic Acid Detection Module Kit (Thermo Scientific).
DNase I footprinting assay
The FAM‐labelled probe was generated by amplifying the promoter region of pfiTA using the 5′‐end FAM‐labelled forward primer (pfiTA‐FAM‐f) and the reverse primer (pfiAT‐promoter‐r; Table S1). For each reaction, 200 ng of FAM‐labelled probes was mixed with a series of amounts of PfiAT protein complex, and the mixtures were incubated for 30 min at 25°C. Then, a series of concentrations of DNase I (NEB, M0303S) was added to cleave the DNA probes. A series of different incubation time points was employed to achieve the best cutting efficiency. The reaction was stopped by adding 200 mM EDTA. Finally, the DNA was cleaned using the QIAEX II Gel Extraction Kit (Qiagen, Hilden, Germany). The data were obtained and analysed as described before (Wang et al., 2012).
Phage production and plaque assay
Strains were grown overnight and adjusted to an OD600 of 0.05 in 4 ml of LB in a 6‐well plate. Pf4 phages were collected over time. In brief, two‐millilitre culture from planktonic or pellicle PAO1 and ΔpfiT strains was centrifuged at 12 000 rpm for 2 min. Then, the supernatants were filtered with 0.22 μm filters (Millipore Corporation, Billerica, MA, USA) to obtain pure Pf4 phage solutions. The top‐layer agar method was used to obtain bacterial lawns as previously described (Eisenstark, 1967).
Quantitative reverse transcription real‐time PCR (qRT‐PCR)
Strains grown for phage production were collected by centrifugation (12 000 rpm for 1 min) after keeping static for 6 h. The collected cell pellets were used for RNA extraction using an RNA extraction kit (Tiangen, Beijing, China). cDNA synthesis was conducted using reverse transcription kit with supplied random primers (Promega, Madison, WI, USA). The reverse transcription reaction mixes were incubated with procedures: room temperature incubation for 10 min, 42°C for 15 min, 95°C for 5 min and on ice for 5 min. Total cDNA (50 ng) was used for qRT‐PCR using the Step One Real‐Time PCR System. The level of the 16S rRNA gene transcript was used to normalize the gene expression data. The amplification efficiency of each primer set used was tested (Fig. S1), and they were comparable. Fold changes in the concentrations of the targets were calculated as follows: 2−( Ct target Δ pfiT−Ct 16S rRNA Δ pfiT )/2−( Ct target PAO1−Ct 16s rRNA PAO1).
Author contributions
YL, YG and XW conceived and designed the study; YL, XL, KT, WW and YG acquired, analysed and interpreted the data; and YL, YG and XW wrote the manuscript.
Conflicts of interest
The authors declare no conflict of interest.
Supporting information
Table S1. Primers used in this study.
Fig. S1 . Real‐time PCR standard curves and amplification efficiencies of primers in Fig. 3B. The genomic DNA of PAO1 was 10‐fold serial diluted and RT‐PCR was performed for gene amplification. The threshold cycle (CT) of each concentration was used as Y‐axis and the log of input DNA was used as X‐axis, and the real‐time PCR standard curves were calculated, and the amplify efficiencies were calculated based on the following formula: E = (10−1/slope–1) × 100.
Acknowledgements
This work was supported by the National Science Foundation of China (31625001, 91951203 and 31970037), by the National Key R&D Program of China (2018YFC1406500), by National Science Foundation of Guangdong Province (2017A030313125), and by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020301). X.W. is a recipient of the 1000‐Youth Elite Program (the Recruitment Program of Global Experts in China).
Microbial Biotechnology (2020) 13(4), 1132–1144
Funding information
This work was supported by the National Science Foundation of China (31625001, 91951203 and 31970037), by the National Key R&D Program of China (2018YFC1406500), by National Science Foundation of Guangdong Province (2017A030313125), and by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020301).
Contributor Information
Yunxue Guo, Email: yunxueguo@scsio.ac.cn.
Xiaoxue Wang, Email: xxwang@scsio.ac.cn.
References
- Afif, H. , Allali, N. , Couturier, M. , and Van Melderen, L. (2001) The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison‐antidote system. Mol Microbiol 41: 73–82. [DOI] [PubMed] [Google Scholar]
- Asadulghani, M. , Ogura, Y. , Ooka, T. , Itoh, T. , Sawaguchi, A. , Iguchi, A. , et al (2009) The defective prophage pool of Escherichia coli O157: Prophage‐prophage interactions potentiate horizontal transfer of virulence determinants. Plos Pathog 5: e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battesti, A. , and Bouveret, E. (2012) The bacterial two‐hybrid system based on adenylate cyclase reconstitution in Escherichia coli . Methods 58: 325–334. [DOI] [PubMed] [Google Scholar]
- Becher, A. , and Schweizer, H.P. (2000) Integration‐proficient Pseudomonas aeruginosa vectors for isolation of single copy chromosomal lacZ and lux gene fusions. Biotechniques 29: 948–950, 952. [DOI] [PubMed] [Google Scholar]
- Chan, W.T. , Espinosa, M. , and Yeo, C.C. (2016) Keeping the wolves at bay: antitoxins of prokaryotic type II toxin‐antitoxin systems. Front Mol Biosci 3: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny, I. , and Gazit, E. (2004) The YefM antitoxin defines a family of natively unfolded proteins ‐implications as a novel antibacterial target. J Biol Chem 279: 8252–8261. [DOI] [PubMed] [Google Scholar]
- Christensen‐Dalsgaard, M. , and Gerdes, K. (2006) Two higBA loci in the Vibrio cholerae superintegron encode mRNA cleaving enzymes and can stabilize plasmids. Mol Microbiol 62: 397–411. [DOI] [PubMed] [Google Scholar]
- Eisenstark, A. (1967) Bacteriophage techniques In Methods in Virology. Maramorsch K., and Koprowski H. (eds). New York: Academic Press, pp. 449–524. [Google Scholar]
- Fineran, P.C. , Blower, T.R. , Foulds, I.J. , Humphreys, D.P. , Lilley, K.S. , and Salmond, G.P.C. (2009) The phage abortive infection system, ToxIN, functions as a protein‐RNA toxin‐antitoxin pair. Proc Natl Acad Sci USA 106: 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Pino, A. , Balasubramanian, S. , Wyns, L. , Gazit, E. , De Greve, H. , Magnuson, R.D. , et al (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142: 101–111. [DOI] [PubMed] [Google Scholar]
- Guerout, A.M. , Iqbal, N. , Mine, N. , Ducos‐Galand, M. , Van Melderen, L. , and Mazel, D. (2013) Characterization of the phd‐doc and ccd toxin‐antitoxin cassettes from Vibrio superintegrons . J Bacteriol 195: 2270–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y. , Quiroga, C. , Chen, Q. , McAnulty, M.J. , Benedik, M.J. , Wood, T.K. , and Wang, X. (2014) RalR (a DNase) and RalA (a small RNA) form a type I toxin‐antitoxin system in Escherichia coli . Nucleic Acids Res 42: 6448–6462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, Y.X. , Sun, C.L. , Li, Y.M. , Tang, K.H. , Ni, S.W. , and Wang, X.X. (2019) Antitoxin HigA inhibits virulence gene mvfR expression in Pseudomonas aeruginosa . Environ Microbiol 21: 2707–2723. [DOI] [PubMed] [Google Scholar]
- Harms, A. , Brodersen, D.E. , Mitarai, N. , and Gerdes, K. (2018) Toxins, targets, and triggers: an overview of toxin‐antitoxin biology. Mol Cell 70: 768–784. [DOI] [PubMed] [Google Scholar]
- Hayes, F. (2003) Toxins‐antitoxins: plasmid maintenance, programmed cell death, and cell cycle arrest. Science 301: 1496–1499. [DOI] [PubMed] [Google Scholar]
- Hazan, R. , and Engelberg‐Kulka, H. (2004) Escherichia coli mazEF‐mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol Genet Genomics 272: 227–234. [DOI] [PubMed] [Google Scholar]
- Hoang, T.T. , Karkhoff‐Schweizer, R.R. , Kutchma, A.J. , and Schweizer, H.P. (1998) A broad‐host‐range Flp‐FRT recombination system for site‐specific excision of chromosomally‐located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212: 77–86. [DOI] [PubMed] [Google Scholar]
- Hoang, T.T. , Kutchma, A.J. , Becher, A. , and Schweizer, H.P. (2000) Integration‐proficient plasmids for Pseudomonas aeruginosa: Site‐specific integration and use for engineering of reporter and expression strains. Plasmid 43: 59–72. [DOI] [PubMed] [Google Scholar]
- Ilyina, T.S. (2015) Filamentous bacteriophages and their role in the virulence and evolution of pathogenic bacteria. Mol Genet Microbiol Virol 30: 1–9. [Google Scholar]
- Jiang, Y. , Pogliano, J. , Helinski, D.R. , and Konieczny, I. (2002) ParE toxin encoded by the broad‐host‐range plasmid RK2 is an inhibitor of Escherichia coli gyrase. Mol Microbiol 44: 971–979. [DOI] [PubMed] [Google Scholar]
- Karimova, G. , Pidoux, J. , Ullmann, A. , and Ladant, D. (1998) A bacterial two‐hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci USA 95: 5752–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski, M.A. , Dwyer, D.J. , Hayete, B. , Lawrence, C.A. , and Collins, J.J. (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130: 797–810. [DOI] [PubMed] [Google Scholar]
- Kroll, J. , Klinter, S. , Schneider, C. , Voss, I. , and Steinbuchel, A. (2010) Plasmid addiction systems: perspectives and applications in biotechnology. Microb Biotechnol 3: 634–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnherr, H. , Maguin, E. , Jafri, S. , and Yarmolinsky, M.B. (1993) Plasmid addiction genes of bacteriophage‐P1‐Doc, which causes cell‐death on curing of prophage, and Phd, which prevents host death when prophage is retained. J Mol Biol 233: 414–428. [DOI] [PubMed] [Google Scholar]
- Li, M. , Long, Y. , Liu, Y. , Liu, Y. , Chen, R. , Shi, J. , et al (2016) HigB of Pseudomonas aeruginosa enhances killing of phagocytes by up‐regulating the type III secretion system in ciprofloxacin induced persister cells. Front Cell Infect Microbiol 6: 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Liu, X. , Tang, K. , Wang, P. , Zeng, Z. , Guo, Y. , and Wang, X. (2019) Excisionase in Pf filamentous prophage controls lysis‐lysogeny decision‐making in Pseudomonas aeruginosa . Mol Microbiol 111: 495–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X. , Li, Y. , Guo, Y. , Zeng, Z. , Li, B. , Wood, T.K. , et al (2015) Physiological function of Rac prophage during biofilm formation and regulation of Rac excision in Escherichia coli K‐12. Sci Rep 5: 16074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyczak, J.B. , Cannon, C.L. , and Pier, G.B. (2002) Lung infections associated with cystic fibrosis. Clin Microbiol Rev 15: 194–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnuson, R. , and Yarmolinsky, M.B. (1998) Corepression of the P1 addiction operon by Phd and Doc. J Bacteriol 180: 6342–6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda, H. , Tan, Q. , Awano, N. , Wu, K. , and Inouye, M. (2012) YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli . Mol Microbiol 84: 979–989. [DOI] [PubMed] [Google Scholar]
- Miller, J.H. (1972) Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Mruk, I. , and Kobayashi, I. (2014) To be or not to be: regulation of restriction‐modification systems and other toxin–antitoxin systems. Nucleic Acids Res 42: 70–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura, T. , and Hiraga, S. (1983) Mini‐F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci USA 80: 4784–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overgaard, M. , Borch, J. , Jorgensen, M.G. , and Gerdes, K. (2008) Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol Microbiol 69: 841–857. [DOI] [PubMed] [Google Scholar]
- Palleroni, N.J. (1984) Pseudomonadaceae In Bergey's Manual of Systematic Bacteriology. Kreig N.R., and Holt J.G. (eds). Baltimore, MD: Williams & Wilkins, pp. 141–199. [Google Scholar]
- Pecota, D.C. , and Wood, T.K. (1996) Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J Bacteriol 178: 2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci, E.C. , Pearson, J.P. , Seed, P.C. , and Iglewski, B.H. (1997) Regulation of las and rhl quorum sensing in Pseudomonas aeruginosa . J Bacteriol 179: 3127–3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu, D. , Damron, F.H. , Mima, T. , Schweizer, H.P. , and Yu, H.D. (2008) PBAD‐based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl Environ Microbiol 74: 7422–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice, S.A. , Tan, C.H. , Mikkelsen, P.J. , Kung, V. , Woo, J. , Tay, M. , et al (2009) The biofilm life cycle and virulence of Pseudomonas aeruginosa are dependent on a filamentous prophage. ISME J 3: 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts, R.C. , Strom, A.R. , and Helinski, D.R. (1994) The parDE operon of the broad‐host‐range plasmid RK2 specifies growth‐inhibition associated with plasmid loss. J Mol Biol 237: 35–51. [DOI] [PubMed] [Google Scholar]
- Secor, P.R. , Sweere, J.M. , Michaels, L.A. , Malkovskiy, A.V. , Lazzareschi, D. , Katznelson, E. , et al (2015) Filamentous bacteriophage promote biofilm assembly and function. Cell Host Microbe 18: 549–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanks, R.M.Q. , Caiazza, N.C. , Hinsa, S.M. , Toutain, C.M. , and O'Toole, G.A. (2006) Saccharomyces cerevisiae‐based molecular tool kit for manipulation of genes from gram‐negative bacteria. Appl Environ Microbiol 72: 5027–5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover, C.K. , Pham, X.Q. , Erwin, A.L. , Mizoguchi, S.D. , Warrener, P. , Hickey, M.J. , et al (2000) Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406: 959–964. [DOI] [PubMed] [Google Scholar]
- Sweere, J.M. , Van Belleghem, J.D. , Ishak, H. , Bach, M.S. , Popescu, M. , Sunkari, V. , et al (2019) Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 363: eaat9691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull, K.J. , and Gerdes, K. (2017) HicA toxin of Escherichia coli derepresses hicAB transcription to selectively produce HicB antitoxin. Mol Microbiol 104: 781–792. [DOI] [PubMed] [Google Scholar]
- Wang, X. , and Wood, T.K. (2011) Toxin‐antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl Environ Microbiol 77: 5577–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X.X. , and Wood, T.K. (2016) Cryptic prophages as targets for drug development. Drug Resist Update 27: 30–38. [DOI] [PubMed] [Google Scholar]
- Wang, X.X. , Kim, Y. , Ma, Q. , Hong, S.H. , Pokusaeva, K. , Sturino, J.M. , and Wood, T.K. (2010) Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1: 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Cen, X.F. , Zhao, G.P. , and Wang, J. (2012) Characterization of a new GlnR binding box in the promoter of amtB in Streptomyces coelicolor inferred a PhoP/GlnR competitive binding mechanism for transcriptional regulation of amtB . J Bacteriol 194: 5237–5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams, J.J. , Halvorsen, E.M. , Dwyer, E.M. , DiFazio, R.M. , and Hergenrother, P.J. (2011) Toxin‐antitoxin (TA) systems are prevalent and transcribed in clinical isolates of Pseudomonas aeruginosa and methicillin‐resistant Staphylococcus aureus . FEMS Microbiol Lett 322: 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winther, K.S. , and Gerdes, K. (2012) Regulation of enteric vapBC transcription: induction by VapC toxin dimer‐breaking. Nucleic Acids Res 40: 4347–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood, T.L. , and Wood, T.K. (2016) The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiologyopen 5: 499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, J.Y. , Guo, Y.X. , Wang, P.X. , Zeng, Z.S. , Li, B.Y. , Tang, K.H. , et al (2018) Type II toxin/antitoxin system ParEso/CopAso stabilizes prophage CP4So in Shewanella oneidensis . Environ Microbiol 20: 1224–1239. [DOI] [PubMed] [Google Scholar]
- Zeng, Z. , Liu, X. , Yao, J. , Guo, Y. , Li, B. , Li, Y. , et al (2016) Cold adaptation regulated by cryptic prophage excision in Shewanella oneidensis . ISME J 10: 2787–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.Y. , Xia, B. , Li, M. , Shi, J. , Long, Y.Q. , Jin, Y.X. , et al (2018) HigB reciprocally controls biofilm formation and the expression of type iii secretion system genes through influencing the intracellular c‐di‐gmp level in Pseudomonas aeruginosa . Toxins 10(11): 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primers used in this study.
Fig. S1 . Real‐time PCR standard curves and amplification efficiencies of primers in Fig. 3B. The genomic DNA of PAO1 was 10‐fold serial diluted and RT‐PCR was performed for gene amplification. The threshold cycle (CT) of each concentration was used as Y‐axis and the log of input DNA was used as X‐axis, and the real‐time PCR standard curves were calculated, and the amplify efficiencies were calculated based on the following formula: E = (10−1/slope–1) × 100.