

Summary
Pseudomonas putida is recognized as a very promising strain for industrial application due to its high redox capacity and frequently observed tolerance towards organic solvents. In this research, we studied the metabolic and transcriptional response of P. putida KT2440 exposed to large‐scale heterogeneous mixing conditions in the form of repeated glucose shortage. Cellular responses were mimicked in an experimental setup comprising a stirred tank reactor and a connected plug flow reactor. We deciphered that a stringent response‐like transcriptional regulation programme is frequently induced, which seems to be linked to the intracellular pool of 3‐hydroxyalkanoates (3‐HA) that are known to serve as precursors for polyhydroxyalkanoates (PHA). To be precise, P. putida is endowed with a survival strategy likely to access cellular PHA, amino acids and glycogen in few seconds under glucose starvation to obtain ATP from respiration, thereby replenishing the reduced ATP levels and the adenylate energy charge. Notably, cells only need 0.4% of glucose uptake to build those 3‐HA‐based energy buffers. Concomitantly, genes that are related to amino acid catabolism and β‐oxidation are upregulated during the transient absence of glucose. Furthermore, we provide a detailed list of transcriptional short‐ and long‐term responses that increase the cellular maintenance by about 17% under the industrial‐like conditions tested.
We deciphered that a stringent response‐like transcriptional regulation program is frequently induced, which seems to be linked to the intracellular pool of 3‐hydroxyalkanoates (3‐HA) that are known to serve as precursors for polyhydroxyalkanoates (PHA). To be precise, P. putida is endowed with a survival strategy likely to access cellular PHA, amino acids, and glycogen in few seconds under glucose starvation to obtain ATP from respiration, thereby replenishing the reduced ATP levels and the adenylate energy charge.
![]()
Introduction
Pseudomonas putida is a very promising industrial host (Poblete‐Castro et al., 2012a; Nikel and Lorenzo, 2018). This strain is naturally endowed to adapt to different physicochemical and nutritional niches (Nikel et al., 2014) and offers strong metabolic redox power (Ebert et al., 2011), which enables it to endure high oxidative stress (Kim and Park, 2014; Nikel et al., 2016). Accordingly, the strain is immensely attractive not only for realizing biotransformation processes but also for the production of secondary or non‐native metabolites, lipids and even proteins in the industrial settings.
Often enough, the economic needs demand for industrial applications in large‐scale bioreactors (> 50 000 l) to exploit the economy‐of‐scale benefits, thereby reducing the manufacturing costs to a minimum (Takors, 2012). Accordingly, the strain constructs and process developments designed under well‐defined, appropriately controlled and homogenous laboratory‐scale conditions crucially need to perform equally well under poorly mixed industrial production scenarios (Takors, 2012; Lorenzo and Couto, 2019). To be precise, large‐scale substrate gradients of carbon or nitrogen sources generally reflect the physical limitations of mixing. Intentionally, aerobic industrial fed‐batch processes typically comprise a carbon‐ or nitrogen‐limited production period to keep the metabolic activity in the bioreactor within the technical limits of oxygen transfer and cooling.
Several studies have already addressed the scale‐up issue (Neubauer et al., 1995; Enfors et al., 2001; Neubauer and Junne, 2010), typically focusing on the phenotypic performance of microorganisms. In addition, present scale‐up studies are expected to exceed the conventional black box tests by understanding the subcellular responses in detail. Hence, the holistic cellular feedback on external heterogeneities should be predicted to prevent unwanted large‐scale performance losses a priori (Delvigne and Noorman, 2017). Accordingly, metabolic and transcriptional insights that specify the cellular feedback under large‐scale conditions are required to eventually derive detailed, large‐scale simulation models (Zieringer and Takors, 2018). Using an experimental set‐up comprising a stirred tank reactor (STR) and a plug flow reactor (PFR), such studies were already performed by Löffler et al. (2016) and Simen et al. (2017) investigating Escherichia coli. Notably, the pivotal role of ppGpp‐mediated stringent response was outlined orchestrating the metabolic and transcriptional regulation when cells were frequently exposed to glucose or ammonia starvation.
For P. putida, similar studies are missing. The same holds true for the fundamental question raised on the reaction of P. putida when repeatedly exposed to limiting glucose supply. Moreover, the putative existence of the stringent response and whether the alarmone ppGpp plays a similar role in P. putida as that in other bacteria (Hauryliuk et al., 2015) needs to be addressed.
Consequently, this study focuses on gaining a holistic picture of the metabolic and transcriptional response of the promising strain P. putida KT2440 when it is exposed to large‐scale stress conditions. For this purpose, cells were cultivated in the aforementioned STR‐PFR setup to decipher the transcriptional response and quantify the cellular maintenance demands. Furthermore, we conclude with guidelines for a putative genome reduction based on stress‐related energy expense. Finally, the interplay of starvation stress and medium‐chain‐length (mcl) polyhydroxyalkanoates (PHAs) will be unravelled, a particular trait of P. putida, which renders the strain as unique compared with the others.
Results
Cultivation in the STR‐PFR system
Cells of P. putida KT2440 were cultivated in biological triplicates in a scale‐down system combining a stirred tank reactor with a plug flow reactor (see Fig. 1). After an initial batch phase, each cultivation was operated in chemostat mode that was achieved by glucose limitation (dilution rate = growth rate of 0.19 ± 0.01 h−1). Steady states in the STR before connection of the PFR served as a reference. These steady states were characterized by the glucose levels in the medium below detection limit and were approved after constant conditions of dissolved oxygen tension (DOT), oxygen uptake rate (OUR) and carbon emission rate (CER) were monitored for minimum five residence times (refer to Fig. S1). Reference samples for the analysis of biomass, transcripts, intracellular nucleotides and supernatant were collected before connecting the PFR during the steady state in the STR. Through connection of the PFR via a pump, cells circulating through the PFR repeatedly experience glucose starvation after depleting the remaining low glucose carried over from the STR. The DOT levels in the PFR were measured online at ports P1, P2 and P5 and were maintained above 20% by aeration after port P1. On average, the cells circulating in the STR‐PFR compartment remained for about 6.2 min in the STR (Löffler et al., 2016) and 2.6 min in the PFR. The setup aimed to mimic the zones of different substrate availability in large‐scale bioreactors, thereby allowing to investigate cellular short‐ and long‐term responses at metabolic and transcriptional levels. The long‐term responses were observed by sampling the STR (port S) during 25 h. Accordingly, the adaptation of P. putida to the repeated PFR stimulus was monitored. Based on the converging online signals and biomass concentrations, a new steady state was attained after 25 h, which was in accordance with the observations of Löffler et al. (2016) for E. coli.
Fig. 1.
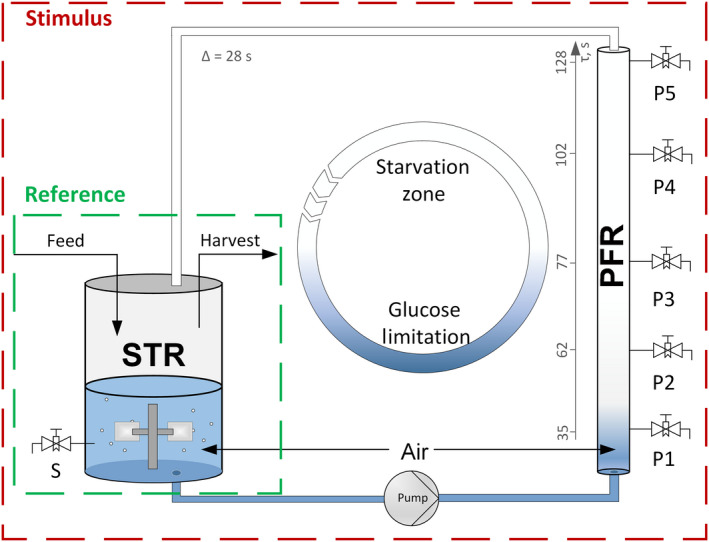
Setup of the scale‐down system comprising a stirred tank reactor (STR) that is operated in chemostat mode using glucose as the limiting factor (Reference condition). Via connection of the plug flow reactor (PFR), a stimulus in form of a substrate gradient is applied. The PFR was sampled at distinct time points at the ports P1, P2, P3, P4 and P5 simultaneously.
In addition, to record the short‐term response to a sudden glucose depletion, samples were withdrawn from the PFR ports P1–P5 after 15 min and 25 h. Neither glucose nor gluconate was found in STR and PFR; however, small amounts of 2‐ketogluconic acid (< 40 mg l−1) were detected in the supernatant, but they remained constant in the STR and PFR. During the course of the experiment, biomass (X) specific glucose (S) uptake rates persisted around an average value of 0.52 ± 0.03 . Considering the biomass concentration and the glucose consumption rate, the remaining glucose in the STR was presumably depleted at P1 in the PFR. However, direct experimental evidence is missing as the detection limit of the glucose enzyme kit is above 20 mg l−1.
Short‐term response to repeated glucose shortage
Metabolic response
The immediate metabolic dynamics on glucose shortage was monitored by measuring the intracellular nucleotides and metabolites of cells inside the PFR in the new steady state (25 h). Figure 2A illustrates the intracellular ATP, ADP and AMP kinetics in relation to the residence time in the PFR, revealing the quick decline of ATP and the concurrent rise of ADP and AMP levels immediately after glucose depletion (about 35 s). The nucleotide levels could have followed the expected trend of permanent decline as glucose shortage is directly linked to reduced ATP formation via glycolysis and the respiratory chain. However, the ATP levels recovered to 4.4 within 27 s, thereby reinstalling the pre‐shortage ATP‐to‐ADP ratio. Notably, the substrate limitation continued while cells remained in the PFR. The adenylate energy charge (AEC) is presented as a measure of the cellular energetic state. Figure 2B reveals that ATP and AEC courses are similar in the PFR. When the ATP pools were reduced (P1), AEC equally dropped from 0.77 (measured in the STR) to 0.7. Later, AEC recovered as fast as ATP to the pre‐shortage value.
Fig. 2.
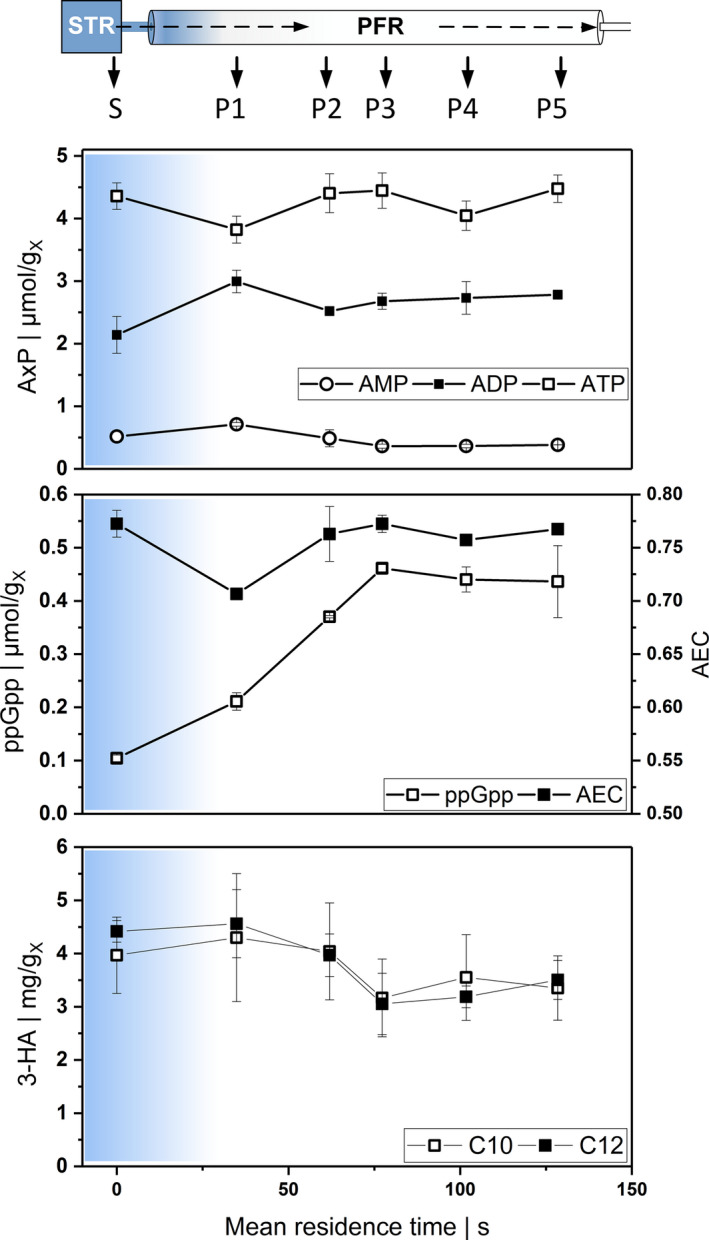
Metabolic response to transition from glucose limitation in the STR (S) to depletion (symbolized by blue gradient) and starvation in the PFR (P1‐P5). Samples are withdrawn after 25 h of repeated glucose starvation. X‐axis indicates the mean residence time (s) in the PFR compartment A: intracellular nucleotides AMP, ADP and ATP per gram CDW; (B): ppGpp per gram CDW and adenylate energy charge (AEC) calculated from nucleotides; (C): intracellular concentrations of 3‐hydroxydecanoic acid (C10) and 3‐hydroxydodecanoic acid (C12) per gram CDW.
Furthermore, we analysed intracellular ppGpp levels via HPLC. This second messenger is known to trigger the activation of the stringent response in E. coli (Ferenci, 2001; Löffler et al., 2016) and is anticipated to have the same function in P. putida (Liu et al., 2017; Mozejko‐Ciesielska et al., 2017). Figure 2B depicts the immediate increase of the intracellular ppGpp pool, from 0.1 to 0.4 within 77 s, after glucose depletion. Afterwards, the concentration remained constant until the end of the PFR.
In P. putida, the monomers of 3‐hydroxydecanoic acid (C10) and 3‐hydroxydodecanoic acid (C12) accumulate predominantly from different carbon sources in combination with nitrogen limitation (Huijberts et al. (1992), Sohn et al. (2010) and Mozejko‐Ciesielska et al. (2018)). In accordance, P. putida KT2440 accumulated the C10 and C12 monomers to 4.0 mgC10 gX ‐1 and 4.4 mgC12 gX ‐1 under glucose as sole limiting factor (STR, 25 h). Even more, the amounts of C10 and C12 decreased to 3.2 mgC10 gX ‐1 and to 3.1 mgC12 gX −1 within 77 s of glucose starvation in the PFR (see Fig. 2C). However, the apparent trend of intracellular 3‐HA reduction while cells suffer starvation in the PFR is statistically not significant (ANOVA P‐value > 0.05). Raw values with statistical analysis are provided in the Data S1.
Transcriptional response
To investigate whether the repeated glucose shortage affected the transcriptional regulation, a differential gene expression analysis was performed. Transcript profiles derived from the samples collected at the PFR (P1, P3, P5) were compared with those of the STR. Sampling was performed at 15 min and 25 h after the PFR was connected with the STR. The complete DEG analysis including log2‐fold changes and FDR‐adjusted P‐values can be found in the Data S1. In general, both the short‐term transcriptional response along the PFR and the long‐term culture adaptation were monitored. Figure 3A illustrates the amount of differentially expressed genes with a log2‐fold change ≥ 0.585 and a FDR < 0.01. By trend, the number of differentially expressed genes (DEGs) increased (i) with the duration of starvation (residence in the PFR) and (ii) with the frequency of the exposure to the glucose limitation. To be precise, first significant transcriptional changes occurred after 35 s of starvation in the PFR, both, in the ‘early’ (15 min) and in the ’late’ (25 h) phase of running the STR‐PFR experiment. After 77 s of starvation, 115 DEGs (up: 25; down: 90) were detected in the ‘early’ phase and 159 DEGs (up: 29; down: 130) in the ‘late’ phase. As aforementioned, the trend in relation to the time of starvation increased: 128 s of glucose depletion resulted in an ‘early’ response of 665 DEGs (up: 270; down: 395) whereas in the ‘late’ phase, 916 genes (up: 379; down: 537) were found as significantly differentially expressed.
Fig. 3.
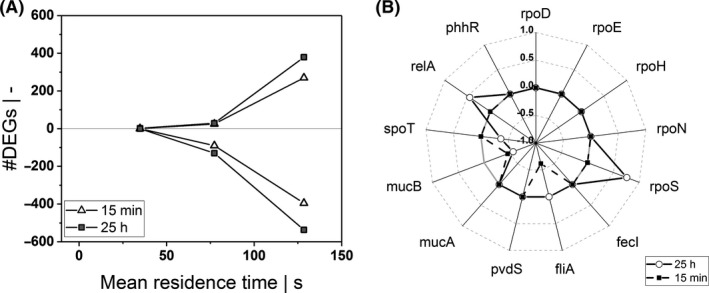
A: Amount of differentially expressed genes (DEGs) that are significantly (log2‐fold change > 0.585, FDR < 0.01) upregulated (positive) or downregulated (negative) as a function of mean residence time in the PFR after 15 min and 25 h relative to the STR analogs. B: log2‐fold changes (FDR < 0.01) of genes related to sigma factors at P5 after 15 min and 25 h.
Furthermore, log2‐fold changes (PFR‐P5, 25 h) of genes related to the sigma factors were identified and plotted in Fig. 3B (FDR < 0.01). The gene rpoS, coding for RNA polymerase sigma S factor and relA, known for its ppGpp synthase activity, were upregulated at port P5 in the ‘late’ phase. Besides relA, spoT plays an important role in the stringent response, too. This gene that encodes ppGpp hydrolase was downregulated in the ‘late’ stage.
Regulation of central carbon metabolism
To unravel the hidden regulatory patterns of transcriptional regulation, the DEGs were grouped according to their roles in central metabolism. Figure 4 shows heatmaps of these groups according to the log2‐fold change (FDR < 0.01) of the respective genes (refer to the Data S1 for log2‐fold changes and P‐values). A red or green colour represents down‐ or upregulation of each gene, respectively. Each heatmap is divided into long‐term (STR 15 min – STR 25 h) and short‐term changes (PFR P1 – P5, 25 h). In general, the most prominent dynamics is reflected by the short‐term transcriptional response along the PFR, whereas the long‐term transcriptional changes converged to the new steady state. The latter is elaborated in detail in the section Long‐term response to repeated glucose starvation.
Fig. 4.
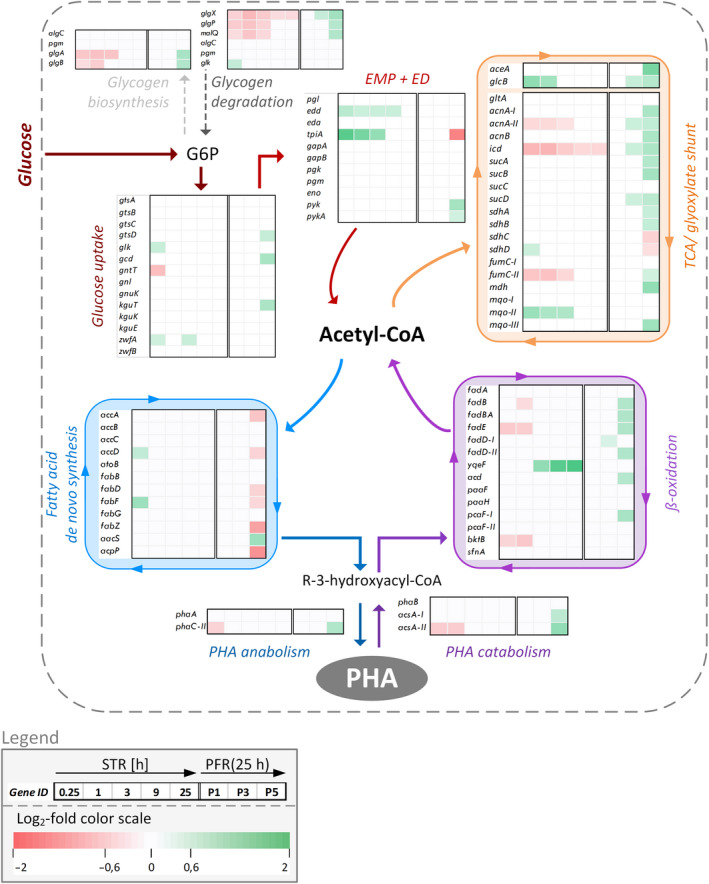
Pathway‐related heatmaps based on log2‐fold changes of differentially expressed genes (FDR < 0.01). Genes are grouped with respect to their metabolic function. The first five columns next to the gene ID in each heatmap indicate transcriptional changes after 15 min and 1, 3, 9 and 25 h with respect to the non‐perturbed reference in the STR. The last three columns depict transcript changes measured at PFR ports P1, P3 and P5 with respect to the STR analog after 25 h. The heatmap coding is presented in the legend. G6P = glucose 6‐phosphate; EMP = Embden–Meyerhof–Parnas pathway from glyceraldehyde‐3‐phosphate to pyruvate; ED = Entner–Doudoroff pathway; PHA = polyhydroxyalkanoate.
Within the ‘glucose uptake’ group, prominent short‐term transcriptional changes were found for glucose oxidation via gluconate and 2‐ketogluconate (2‐KG). Glucose dehydrogenase (gcd) and the 2‐KG related gene kguT are clearly upregulated at the exit of the PFR. In addition, upregulation was found for genes belonging to glycogen biosynthesis (glgA, glgB) and its degradation (glgX, glgP, malQ). Furthermore, glucose catabolism to pyruvate reveals only moderate transcriptional adaptation except for tpiA (log2‐fold = −1.7), coding for triosephosphate isomerase and pyk (log2‐fold = 0.7), coding for pyruvate kinase. In the tricarboxylic acid (TCA) cycle including the glyoxylate shunt, the transcriptional picture is more coherent. Almost all genes are repeatedly upregulated in the PFR; only succinate dehydrogenase enzymes (sdhC, sdhD) show the opposite trend.
Moreover, a clear transcriptional response of PHA metabolism can be observed. The short‐term dynamics in the PFR reveals upregulation of 6 out of 14 genes (fadB, fadBA, fadE, fadD‐II, acd, pcaF‐I) belonging to the β‐oxidation group; whereas, 6 downregulated (accA, accD, fabD, fabF, fabZ, acpP) and one upregulated gene (aacS) out of 12 genes as part of the fatty acid de novo synthesis are observed. Furthermore, phaC‐II, coding for a PHA polymerase subunit, as well as the acetyl‐coenzyme A synthetase genes (acsA‐I, acsA‐II) reveals upregulation.
To further identify transcriptional changes in the glucose‐starved metabolism, we applied two‐sample t‐statistics for sets (n > 3) using gage. Genes were grouped (n > 3) according to their metabolic pathway assignment and gene ontology (GO) (see Data S1). Resulting t‐values are illustrated in Fig. 5. In accordance with Fig. 4, the groups of β‐oxidation and TCA show statistically sound upregulation whereas fatty acid de novo synthesis reveals downregulation. Further upregulations were found for gene sets encoding amino acid catabolism (GO:0009063), PHA metabolism (anabolism + catabolism), glycogen catabolism and glucose uptake. However, amino acid biosynthesis (GO:0008652), proteolysis (GO:0006508) and EMP + ED are characterized by low t‐values which qualifies their transcriptional changes as low compared with the reference. Furthermore, the genes ppk, and ppkB, related to polyphosphate kinase, show significant upregulation at the outlet of the PFR (refer to Data S1).
Fig. 5.
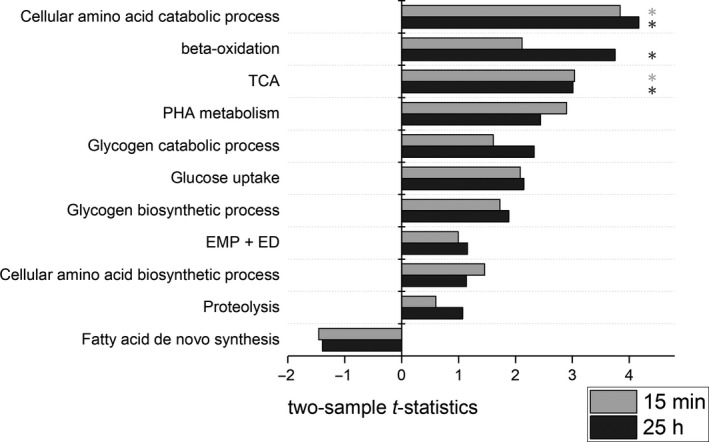
Enrichment analysis of specific gene sets using gage. Two‐sample t‐statistics for PFR‐P5 vs. STR after 15 min and 25 h of repeated glucose starvation are presented for each group. Functional groups with significant changes (FDR < 0.05) are indicated with an asterisk. The gene sets can be found in the Data S1.
Energetic burden of glucose starvation response
Besides transcriptional analysis, we estimated the energetic burden imposed by transcription and translation. Calculations are based on the computation of nucleotide and amino acid costs and on the estimation of ATP costs of gene expression (refer to section ‘experimental procedures’). Again, we compared the state in the STR to that of the PFR outlet (P5).
In total, we calculated 23.3 as additional ATP cost for differential gene expressions in the PFR, based on the counts of DEGs within 128 s. Thereof, 19.7 accounts for translation and 3.7 for transcription. With respect to the non‐growth‐associated maintenance (NGAM) demand of 140.8 for P. putida (van Duuren et al., 2013) within the time frame of 128 s, this equals an add‐on of 14.0% and 2.6% due to translation and transcription, respectively. The total add‐on increased NGAM to 164.1 .
As presented in Fig. 2C, the degradation of 3‐hydroxyalkanoates (3‐HA) was observed when the cells passed through the PFR. As a consequence, energy equivalents as well as C2‐carbons in the form of acetyl‐CoA were remobilized to serve as temporary energy and carbon sources. To balance the energy regain from degradation of 3‐HA, the following equation was used (adaptation from Reddy et al. (2014)):
| (1) |
Under carbon starvation, 3‐HAs are released from PHA granules by PHA depolymerase PhaZ (equals phaB (PP_5004)). Further activation via the ATP‐dependent ACS1 leads to hydroxyacyl‐CoA, which fuels the fatty acid β‐oxidation cycle (Ruth et al., 2008). Each cycle yields one acetyl‐CoA, reduces one FAD+ to FADH2 and one NAD+ to NADH, and eventually leads to Cn‐2 acyl‐CoA. Hence, oxidation of a C10 HA costs 2 ATP but generates 4 FADH2, 4 NADH and 5 acetyl‐CoA (Fulco, 1983). Moreover, the remobilized acetyl‐CoA is further fuelled into the TCA and oxidized to CO2, thereby generating additional 3 NAD(P)H, 1 FADH2 and 1 ATP (Nikel et al., 2015). In total, 568.9 are recuperated from the observed degradation of 3‐hydroxydecanoate (C10) and 3‐hydroxydodecanoate (C12) assuming the P/O ratio of 1.33.
To sum up the energetic burden of P. putida in the PFR, Fig. 6 illustrates the theoretical growth (GAM) and non‐growth‐associated maintenance (NGAM) demands (in total 715 ), additional ATP expenses for transcription and translation and ATP regeneration from PHA degradation in the starvation zone of the PFR. To be precise, the HA‐based ATP regain can cover 79.6% of the total ATP required for cellular maintenance in the starvation zone of the PFR. Noteworthy, the amount of acetyl‐CoA attained from degradation of C10 and C12 in the PFR (about 0.07 ) equals 32% of the glucose‐based acetyl‐CoA production in the STR (glucose uptake in 128 s = 0.11 ).
Fig. 6.
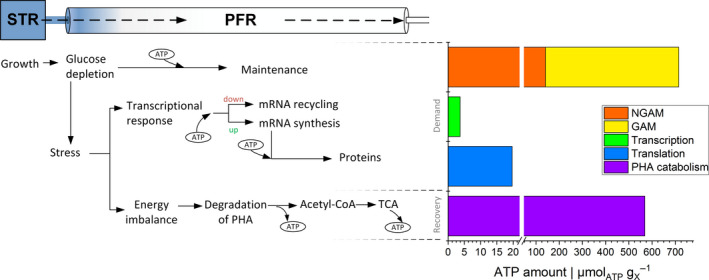
Scheme of short‐term response of P. putida to sudden glucose depletion at the transcriptional and metabolic levels. On the right side, theoretical ATP maintenance demand of cells growing at 0.19 h−1 and add‐on of ATP costs are presented in contrast to the ATP recovery from PHA degradation within a time frame of 128 s inside the PFR.
Long‐term response to repeated glucose starvation
Phenotypical adaptation
To qualify the long‐term adaptation, key parameters of the reference condition, that is, non‐perturbed P. putida growing with are listed in Table 1. Additionally, new steady state values are listed, which were attained after the STR‐PFR was operated for 25 h. The biomass‐to‐substrate yield YX/S remained unchanged (t‐test P‐value > 0.05), whereas the AEC decreased significantly (t‐test P‐value < 0.05) from 0.87 to 0.79 in the STR‐PFR system. Additionally, a trend of reduced PHA pools in the late phase compared with the early phase can be observed: the concentration of 3‐hydroxydecanoic acid (C10) significantly (t‐test P‐value < 0.05) decreased by 31% from 5.86 ± 1.04 to 4.04 ± 0.43 and 3‐hydroxydodecanoic acid (C12) from 5.0 ± 0.93 to 4.61 ± 0.29 (not significant). Further analysis revealed that 3‐HA yields on glucose decreased in a similar manner for YC 10 /S and YC 12 /S, respectively. Furthermore, we calculated the ATP demand for storing intracellular 3‐HA (3‐OH‐Cn) using Eq. 2:
| (2) |
Table 1.
Consequences of repeated glucose oscillation on key cultivation parameters. The ‘STR’ parameters (standard deviation in parenthesis) are derived in the reference steady state and the ‘STR + PFR’ parameters in the new steady state after 25 h.
| Condition | YX/S | AEC | Y C10/ X | Y C12/ X | Y C10/ S | Y C12/ S | ATP demand for 3‐HA synthesis | |
|---|---|---|---|---|---|---|---|---|
| g g‐1 | – | mg gX ‐1 | mg gX ‐1 | mg gS ‐1 | mg gS ‐1 S | mmolATP gX ‐1 h‐1 | ||
| C10 | C12 | |||||||
| STR | 0.38 (0.03) | 0.87 (0.01) | 5.86 (1.04) | 5.00 (0.93) | 2.57 (0.72) | 2.20 (0.64) | 0.18 | 0.18 |
| STR + PFR | 0.39 (0.03) | 0.79 (0.04) | 4.04 (0.43) | 4.61 (0.29) | 1.80 (0.34) | 2.04 (0.25) | 0.13 | 0.17 |
In theory, acetyl‐CoA is converted to malonyl‐CoA via an ATP‐dependent carboxyltransferase. Malonyl‐CoA is then transferred to an acyl‐carrier protein and incorporated with another acetyl‐CoA under split‐off of CO2 into fatty acid de novo synthesis that yields 3‐hydroxybutyrate (C4). For elongation purpose, further malonyl‐CoA molecules are added to form Cn+2 3‐hydroxyalkanoates. For energy costs of acetyl‐CoA, refer to Table S2. If we consider similar assumptions for the degradation of 3‐HA as aforementioned, the ATP demand for storing 5.86 at a growth rate of 0.19 h‐1is calculated to 0.18 Similarly, the continuous production of 5.0 will cost 0.18 . This minimal but constant production of 3‐hydroxyalkanoates accounts for 1.7% of the total ATP maintenance demand (NGAM + GAM). In the new large‐scale‐like steady state, the ATP costs account for only 1.4% of the maintenance.
Adaptation on transcriptional level
The transcriptional adaptation of the total culture is analysed by comparing the transcriptional patterns in the STR (sampling at port S) after 15 min and 25 h versus the reference condition without PFR link. Fig. 7 contours the relatively strong response after 15 min, indicated by 615 significant DEGs (up: 344 and down 271), which reduced to rather small changes (up: 38; down: 27) after 25 h. However, the majority of upregulated (95%) and 59% of downregulated genes of the late transcriptional responses are dominated by early transcriptional changes, which are consequently termed as ‘strategic’ adaptions. Among those, icd, coding for isocitrate dehydrogenase, glgX coding for the glycogen debranching enzyme, and yqeF, coding for acetyl‐CoA acetyltransferase, which is an important enzyme in the β‐oxidation cycle, were discovered. For completeness, a further differential expression analysis along with a gene set enrichment analysis can be found in the Data S1.
Fig. 7.
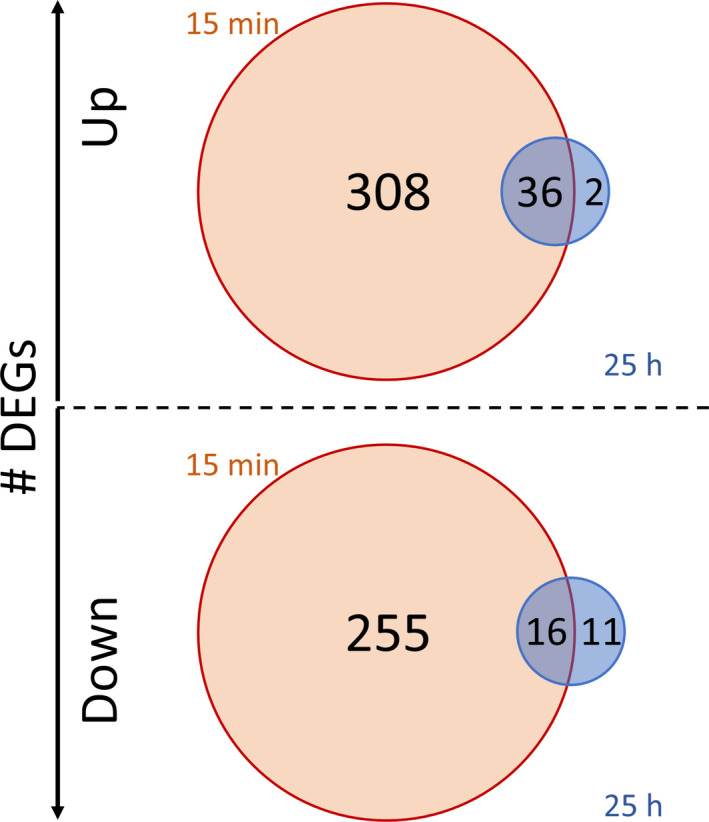
Amount of differentially expressed genes (DEGs) (FDR < 0.01) as part of the long‐term response to repeated glucose starvation indicated inside the circles. The gene expression profile in the STR at the late phase (25 h, blue) is compared with the analog profile measured in the early phase (after 15 min, red) in the STR. The intersection indicates the amount of common genes in both phases.
To further evaluate the transcriptional dynamics, ATP costs for mRNA synthesis and subsequent protein formation are estimated using the set of upregulated DEGs (FDR < 0.01) found after 25 h at port P5‐PFR. Table 2 lists the top 20 genes responsible for the highest ATP expense. ATP requirements are indicated relative to the NGAM and are separated in transcriptional and translational costs. The highest impact presents the RNA polymerase sigma factor encoded by rpoS with an addition of 1.22%. Moreover, one gene that is particularly important for flagellum‐dependent cell motility, fliC, accounts for over 0.85% of additional energy costs. Another ATP expensive reaction to sudden glucose shortage is the overexpression of oprF that is responsible for more than 0.92% of the additional energy costs. Together, these 20 genes account for 8.2% of the additional maintenance demand, which equals 55% of the total NGAM increase. The remaining 426 genes only represent 6.7% of maintenance increase, which renders Table 2, except for essential genes, as a valid recommendation list for genome reduction to minimize additional maintenance costs.
Table 2.
Top 20 ATP‐consuming upregulated genes at the PFR outlet (P5) after 25 h. ATP costs for transcription and translation as well as log2 fold changes are given for individual genes.
| Gene | Transcription | Translation | Σ | log2‐fold | Function |
|---|---|---|---|---|---|
| oprI | 0.67 | 1.48 | 2.15 | 0.30 | Major outer membrane lipoprotein |
| rpoS | 0.48 | 1.04 | 1.52 | 0.70 | RNA polymerase sigma S factor |
| rmf | 0.43 | 0.97 | 1.40 | 0.03 | Ribosome modulation factor |
| oprF | 0.38 | 0.84 | 1.23 | 0.41 | Porin F |
| fliC | 0.34 | 0.74 | 1.07 | 0.52 | Flagellin |
| oprB‐I | 0.31 | 0.67 | 0.99 | 0.53 | Carbohydrate‐selective porin |
| tufB | 0.26 | 0.57 | 0.82 | 0.68 | Elongation factor Tu‐B |
| fusA | 0.23 | 0.50 | 0.72 | 0.68 | Elongation factor G 1 |
| hpf | 0.21 | 0.46 | 0.67 | 0.42 | Ribosome hibernation promoting factor |
| rpsO | 0.19 | 0.42 | 0.61 | 0.46 | 30S ribosomal protein S15 |
| rplL | 0.18 | 0.39 | 0.57 | 0.23 | 50S ribosomal protein L7/L12 |
| yohC | 0.17 | 0.37 | 0.54 | 1.50 | Inner membrane protein |
| yeaG | 0.17 | 0.37 | 0.54 | 1.06 | Protein kinase |
| rplJ | 0.17 | 0.37 | 0.54 | 0.29 | 50S ribosomal protein L10 |
| rpsS | 0.17 | 0.37 | 0.54 | 0.32 | 30S ribosomal protein S19 |
| groL | 0.14 | 0.31 | 0.45 | 0.57 | Chaperonin GroEL |
| ygaU | 0.14 | 0.30 | 0.43 | 0.29 | Murein cross‐linking regulator |
| proC | 0.13 | 0.28 | 0.40 | 0.42 | Pyrroline‐5‐carboxylate reductase |
| rpoA | 0.12 | 0.26 | 0.37 | 0.51 | DNA‐directed RNA polymerase subunit alpha |
| tufA | 0.11 | 0.25 | 0.37 | 0.40 | Elongation factor Tu‐A |
| Σ | 4.98 | 10.96 | 15.94 |
Discussion
Dynamics of the adenylate energy charge
The well‐balanced AEC of P. putida in the starvation compartment of the PFR is noteworthy. The AEC decreased within the first 35 s but recovered to its initial state of > 0.75 even in absence of substrate. The measured AEC values are in accordance with recent studies: Vallon et al. (2015) reported an AEC of approximately 0.8 in mixed glucose/n‐butanol‐limited chemostat at growth rates between 0.1 and 0.3 h‐1 and Martínez‐García et al. (2014) reported an AEC > 0.75 for P. putida grown in shaking flasks. Chapman et al. (1971) concluded in their studies that growth in E. coli can occur only with AECs > 0.8. Accordingly, moderate AEC values between 0.5 and 0.8 ensure viability but no excessive growth. In our study, P. putida’s energy charge was close to or > 0.8 measured in the reference state and in the new steady state. As shown in Fig. 2, the cells were affected by the transient substrate depletion, but cellular countermeasures restored the AEC to physiological levels. For comparison, the ADP levels in E. coli were found to increase after exposing the cells to substrate starvation, eventually leading to decreased AECs < 0.7 (Löffler et al., 2016).
The stringent response in P . putida KT2440
We observed a strong correlation between glucose depletion and the intracellular amplification of ppGpp in P. putida. This alarmone is well known to initiate the stringent response (Traxler et al., 2011; Hauryliuk et al., 2015). Intracellular levels are typically regulated by RelA (ppGpp synthase) and SpoT (ppGpp hydrolase) homolog proteins (Potrykus and Cashel, 2008). As described by Gentry et al. (1993), the expression of sigma factor , encoded by rpoS, is also regulated by ppGpp, thus revealing its dominant role when E. coli enters the stationary phase after exponential growth.
In P. putida, the role of the stringent response is elucidated less compared with that in E. coli. Studies with P. putida and P. aeruginosa reported that ppGpp plays a crucial role in biofilm regulation (Liu et al., 2017) and quorum sensing (van Delden et al., 2001); however, its regulatory function during carbon starvation remains unclear. In our study, we observed the upregulation of rpoS at P5 in the PFR (see Fig. 3B) coinciding with increased levels of ppGpp. The trend of ppGpp dynamics was comparable to that of the E. coli studies (Löffler et al., 2016) although the maximum value of 0.46 in P. putida was lower than that in E. coli (> 1 ). The relA gene was always upregulated at P5, suggesting that the encoded ppGpp synthase was responsible for the increase of the alarmone levels. RelA is typically activated under nitrogen and amino acid starvation; whereas, the activity of SpoT is linked to phosphate, carbon or fatty acid starvation often offering both, ppGpp‐hydrolytic and synthetic activity (Hauryliuk et al., 2015). In accordance, the gene expression level of spoT is reduced in starving P. putida cells (P5, 25h), thus underlining its hydrolytic function. Furthermore, ppGpp is involved in the metabolism of fatty acids in starving E. coli (Traxler et al., 2008) and P. putida (Mozejko‐Ciesielska et al., 2017). As we observed downregulation of multiple fatty acid synthesis genes, along with upregulation of the ppGpp synthesis gene relA and several β‐oxidation‐related genes, both coinciding with rising ppGpp levels during starvation in the PFR, we conclude that ppGpp triggers regulatory programmes known from other bacteria in P. putida, as well. Additionally, we found strong upregulation of aceA and glcB indicating the amplification of the glyoxylate shunt activity, as it was recently revealed for E. coli, (Michalowski et al., 2017). From the aforementioned findings, we anticipate that ppGpp plays a similar role in P. putida than in other microbes such as in E. coli: ppGpp appears to be the alarmone of the stringent response. Maximum intracellular levels are slightly below the thresholds of other bacteria as reported by Lazzarini et al. (1971), Ryals et al. (1982) and Traxler et al. (2011). However, measurements of intracellular levels are rather error‐prone and there is no rationale that equal ppGpp levels need to be present in all bacteria. Though, a high threshold saves the bacteria from erroneously expressing stationary phase genes in transient nutritional starvation.
Consequences of repeated starvation
The repeated exposure of P. putida to glucose‐limiting conditions revealed short‐ and long‐term responses with partially opposite trends: After 25 h in STR, the total number of differentially expressed genes diminished to only 10% of the DEGs measured after 15 min. This reduction implies that cells have adapted in the long‐term to the repeated glucose shortage. In contrast, the short‐term response to glucose depletion continued to amplify in the PFR comprising 974 DEGs after 25 h versus 662 genes after 15 min. This phenomenon is in contrast to the findings of Löffler et al. (2016) who found reducing numbers of DEGs in E. coli after repeated PFR exposure. Indeed, the technical impacts such as 18 s extended exposure time in PFR or the application of different analytical tools (DESeq2 vs. edgeR) might cause some bias, but they may not explain the fundamental trend. However, one key feature of P. putida needs to be considered: the existence of the intracellular PHA buffer which, interestingly enough, was reduced by > 30% in case of the C10 3‐HA after repeated glucose starvation compared with the initial state. Hence, we anticipate that intracellular levels of the buffer additionally interact with the stringent response, which is apparently a unique link in P. putida.
The crucial role of 3‐HAs in Pseudomonas putida as a survival strategy
As aforementioned, 3‐hydroxyalkanoates can be polymerized to mcl‐PHA serving as intracellular carbon and energy depot to counteract sudden starvation conditions. This requires (i) the existence of said PHA pools and (ii) cellular ability to quickly access the storage.
Referring to (i) PHA formation is predominately described as a consequence of nitrogen limitation or use of fatty acids as sole carbon and energy source. Strictly speaking, we cannot show the existence of PHAs in P. putida KT2440 under glucose‐limited growth conditions but we give evidence to accumulated 3‐HAs with relatively low intracellular levels of 1.1% biomass weight (STR, 25 h). This content is slightly lower than the 2.7% found in steadily growing P. putida KT2440 on glycerol with a dilution rate of 0.12 h‐1 (Beckers et al., 2016). Noteworthy, the overall reduction of the 3‐HA content due to the repeated starvation could be part of the adaptation mechanism in P. putida. This would result in a reduction of 17% of ATP needs for 3‐HA de novo synthesis.
Considering (ii), the trend of a reduction of intracellular 3‐HAs as countermeasure to a decreased energy charge as well as significant expression of PHA‐related genes illustrates the high dynamics of PHA catabolism initiated under glucose starvation condition. The basic role of PHA synthesis as a storage of energetic building blocks has already been anticipated by Escapa et al. (2012) for P. putida KT2442 and by James et al. (1999) for Legionella pneumophila. Escapa et al. (2012) reported that wild‐type P. putida KT2442 formed more biomass than phaC1 (Poly (3‐hydroxyalkanoate) polymerase)‐deficient mutants when growing on octanoic acid. Thus, our findings are in accordance with those studies and further highlight the quick response that presumably starts with PHA depolymerization followed by immediate oxidation of the prevalent C10 and C12 monomers via β‐oxidation, and eventually, the oxidation of acetyl‐CoA to CO2 in the TCA, all in a matter of seconds. As depicted in Fig. 6, the theoretical 3‐HA‐based recovery of ATP can account for 79.6% of the cell’s ATP maintenance demand under glucose starvation assuming steady growth and applying the rather conservative P/O ratio of 1.33 (van Duuren et al., 2013; Hintermayer and Weuster‐Botz, 2017). However, if higher P/O ratios are considered, such as 1.75 (Nogales et al., 2008; Lieder et al., 2015; Beckers et al., 2016) or 1.85 (Hardy et al., 1993; Oberhardt et al., 2011), the total 3‐HA‐based recuperation of ATP would cover approximately 100% of ATP maintenance demand. Noteworthy, C10 and C12 pools are found to be low and almost equal under the carbon‐limiting conditions applied in this study. The observation contrasts usually measured high C10 to low C12 proportions of PHA (Beckers et al., 2016; Mozejko‐Ciesielska et al., 2018). However, the latter reflect the scenario of high external glucose levels which differs from the low external glucose conditions and the limited uptake rates of this study. For such a scenario, no reference compositions have been published so far. Considering equal levels of C10 and C12 pools under glucose‐limited conditions fragments of cellular phospholipids with similar chain length might have impaired the GC‐based detection of C12 3‐HAs additionally.
We further assume that NADPH, that is essential for growth, is partly regenerated by the oxidation of acetyl‐CoA derived from 3‐HAs under glucose starvation. Besides, a significant upregulation of transhydrogenase‐related genes (pntAB and pntB) in cells leaving the PFR (128 s) might result in an increased conversion from NADH to NADPH. Consequently, the catabolism of intracellular‐stored PHAs can be qualified as a very powerful and fast tactical measure to shift between ‘feast and famine’ lifestyles.
Transient carbon starvation intensifies catabolic reactions
The basic trend is as follows: key genes of de novo synthesis of fatty acids (Mozejko‐Ciesielska et al., 2018) are downregulated whereas β‐oxidation‐related genes are clearly amplified along the PFR. This observation underlines the shift from an anabolic to a catabolic fatty acid metabolism due to the sudden starvation stress. Noteworthy, phaC‐II (PP_5005) is exceptionally upregulated and encodes the integration of activated 3‐HA building blocks (R‐3‐hydroxyacyl‐CoA) into PHAs. These monomers are also the hydroxylated products of 3‐HA released from PHA granules via PHA depolymerase (phaB) and activated via acyl‐CoA synthetase (acsA‐I, acsA‐II) (Ruth et al., 2008), which was found to be amplified under the said starvation conditions. Accordingly, transient surplus of hydroxyacyl‐CoAs may have initiated the back integration which reflects the dual role of anabolism and catabolism in the PHA cycle (Doi et al., 1990; Uchino et al., 2007; Ren et al., 2009). Interestingly, the acetyl‐CoA fuelling into the TCA cycle is accompanied by the downregulation of succinate dehydrogenase and the upregulation of the glyoxylate shunt (aceA, aceK, glcB), which can be qualified as a measure to prevent further carbon loss via nonessential decarboxylation.
Besides oxidation of 3‐hydroxyalkanoates, further catabolic activity can be anticipated from transcriptomic data (see Fig. 5) such as the upregulation of the majority of TCA genes, which can only partly be explained by the short‐term‐boosted acetyl‐CoA flux caused by the breakdown of 3‐HAs. Most likely, the transcriptional pattern reflects the already initiated next step of starvation response, namely to access additional carbon sources such as intracellular amino acids. Indeed, there is a significant upregulation of genes coding for amino acid catabolism somehow reminding on the well‐known autophagy processes in eukaryotic cells (Hamasaki et al., 2003). In accordance, amino acid transport and metabolism were also found to be increased in transiently starving E. coli cells (Löffler et al., 2016). Additionally, glycogen might serve as carbon buffer in glucose grown P. putida cells as upregulation was found for both, glycogen degrading and synthesizing genes (refer to Fig. 4). Poblete‐Castro et al. (2012b) reported similar transcript changes in glycogen metabolism for decanoate‐grown P. putida KT2442 under nitrogen and carbon–nitrogen limitation. In summary, the coincidence of glucose starvation, ongoing maintenance demands, rapid restoration of adenylate energy levels, 3‐HA degradation and upregulation of gene sets with catabolic function build the basis for the hypothesis that P. putida actively degrades intracellular carbon buffers to deal with short‐term carbon (and ATP) shortage. ATP balancing further supports this hypothesis. Noteworthy, significant upregulation of polyphosphate kinase‐related genes indicates a further strategy to balance dropping ATP levels. Accordingly, we qualify the 3‐HA degradation programme as one of the short‐term responses only affecting cellular metabolism to a minor extent. As indicated by transcript analysis, 3‐HA degradation coincides with the degradation of intracellular amino acid pools and the access to glycogen pools as an apparent complementary measure to manage threatening starvation conditions. Therefore, the transcriptional regulation may be part of the stringent response programme in P. putida.
Engineering a streamlined P . putida strain for large‐scale application
Table 2 lists those genes, which were repeatedly upregulated, thereby utilizing ATP unnecessarily for transcription and translation. Noteworthy, the respective upregulated genes most likely mimic long‐term strategic cellular programmes that are redundant in large‐scale bioreactors when cells simply fluctuate between different zones. Transcriptional dynamics occur despite P. putida’s countermeasures of 3‐HA degradation, which only serve to prevent the precursor and ATP shortage.
In essence, Table 2 may serve as a guideline to genetically streamline P. putida for better performance in large‐scale as less ATP is wasted but possible deletion targets require careful consideration regarding metabolic consequences. For instance, the deletion of the flagellar system (fliC) may be an obvious target accounting for 0.85 % of additional energy costs. Recently, Martínez‐García et al. (2014) showed that the deletion of the major motility apparatus resulted in physiological advantages such as increased energy charge and improved tolerance to oxidative stress which was reflected by improved NAD(P)H supply. Moreover, Lieder et al. (2015) demonstrated improved recombinant protein production and energy status using the streamlined P. putida strains EM329, lacking flagellar genes and EM383, with additional elimination of the proviral DNA. Other promising deletions may comprise the knock‐out of non‐necessary carbon uptake and processing pathway which are numerously present in the degrading microbe P. putida and which are not necessary when the cell is growing in well‐defined synthetic media. Moreover, the knowledge of numerous non‐coding RNAs that were recently discovered in repeatedly starving P. putida KT2440 (Pobre et al., 2020) could complement the data obtained in this study regarding putative deletion targets to genetically optimize this strain for large‐scale application.
Conclusion
Despite being glucose‐limited, P. putida KT2440 accumulated PHA‐precursors 3‐hydroxyalkanoates to 1.1% of biomass, which already indicates the prominent role of the storage compound. We conclude that P. putida is able to access 3‐HAs, most presumably released from PHA granules, within seconds, and further oxidizes the intermediates to acetyl‐CoA, which is fuelled into TCA eventually to gain ATP via respiration. Noteworthy, P. putida may buffer the carbon shortage for approximately 5 min following this strategy. Consequently, the intracellular ATP pools and AECs are quickly restored, thereby resulting in a highly robust metabolism and energy status, a trait that is most welcome in large‐scale heterogeneously mixed bioreactors. To be precise, P. putida is already endowed with a gift other industrial hosts such as E. coli cannot offer. Under the conditions tested, P. putida only needs 0.4% of the consumed sugar to refill 3‐HA stores, which is considered as a minor expense given that P. putida continues to grow and to keep high energy status even when it is strictly glucose starved. Nevertheless, P. putida already initiates next steps of the carbon starvation programme as soon as 3‐HA levels start degrading. In particular, intracellular sources of amino acids, glycogen and polyphosphates seem to be addressed as part of the stringent response. Concomitantly, the transcriptional response even increases with lowering 3‐HA pools. These phenomena deserve further investigation to render P. putida as the optimum host for large‐scale application.
Experimental procedures
Strain and medium
The Pseudomonas putida strain KT2440 (Bagdasarian et al., 1981) (DSM‐6125, ATCC47054) was used in all experiments. Mineral salts medium (M12) (Vallon et al., 2013) was applied for the cultivations consisting of (per litre): 2.2 g (NH4)2SO4, 0.4 g MgSO4 × 7 H2O, 0.04 g CaCl2 × 2 H2O, 0.02 g NaCl, 2 g KH2PO4; and trace elements: 2 mg ZnSO4 × 7 H2O, 1 mg MnCl2 × 4 H2O, 15 mg Na3C6H5O7 × 2 H2O, 1 mg CuSO4 × 5 H2O, 0.02 mg NiCl2 × 6 H2O, 0.03 mg Na2MoO4 × 2 H2O, 0.3 mg H3BO3, 10 mg FeSO4 × 7 H2O (Merck, Darmstadt, Germany).
Cultivation
Prior to each bioreactor fermentation, a preculture was inoculated from the working cell bank (33% glycerol stock, stored at − 70 °C) and cultivated in a 500 ml baffled shake flask containing 50 ml M12 medium with 4 g l−1 glucose, 10 g l−1 3‐morpholino‐propanesulphonic acid (MOPS), and 0.5 g l−1 yeast extract (VWR International, Radnor, Pennsylvania). After 6–8 h of incubation (130 rpm, 30 °C, pH 7), the second preculture was performed in 1000 ml shaking flask containing 100 ml M12 medium with 4 g l−1 glucose and 10 g l−1 MOPS. For inoculation, the exponentially growing cells (about 0.9 gX l‐1) from the first preculture were applied. After incubation for 14 h, the total volume of the second preculture (about 1.0 gX l‐1) was used to start batch cultivation. The batch process was carried out in a 3 l stirred tank reactor (STR) (Bioengineering, Wald, Switzerland) containing 1.6 l M12 medium (inoculum included) with 15 g l−1 glucose, at 30 °C with total pressure of 1.5 bar and constant aeration of 1.5 l min−1. During fermentation, the pH was adjusted at 7 using 25% NH4OH (Carl Roth, Karlsruhe, Germany). Dissolved oxygen was maintained above 20% by controlling the stirrer speed. After depletion of initial glucose, the chemostat was started, thereby setting the constant feed (M12 medium with 18 g l−1 glucose) at 0.2 h−1 dilution rate. Antifoam (Struktol J 647, Schill + Seilacher, Hamburg, Germany) was added with 50 µl h−1. After attaining a steady state (t > five residence times), the plug flow reactor (PFR) was connected to the STR via a diaphragm metering pump (Sigma/1, ProMinent, Heidelberg, Germany), which continuously drained approx. 25% of the biosuspension through the PFR and back into the STR (see Fig. 1). The scale‐down setup was identical to that of Löffler et al. (2016) except for the different diaphragm metering pump (Sigma/1, ProMinent, Heidelberg, Germany) leading to slightly modified mean residence times in the PFR (see Table S5). Moreover, the aeration after port 1 of the PFR was 0.1 l min−1 leading to the total aeration of 1 vvm. PFR was heated to maintain a temperature of 30 °C.
Determination of biomass and organic acids
To quantify the cell dry weight (CDW) concentration, 4 × 1 ml of biosuspension was centrifuged with 14,000 rpm for 5 min at 4 °C, washed twice with demineralized water, transferred into pre‐weighed glass vials (1.5 ml, VWR International, Radnor, Pennsylvania) and eventually dried at 105 °C for 24 h. The weight was determined using a micro balance (XP26 Delta Range®, Mettler Toledo, Gießen, Germany). Enzyme kits (r‐biopharm AG, Darmstadt, Germany) were applied to quantify the organic acids, D‐glucose and D‐gluconic acid in the supernatant. 2‐ketogluconic acid was measured using isocratic HPLC equipped with the RI detector (1200Series, Agilent, Santa Clara, CA, USA) and a Rezex ROA‐Organic Acid H+ (300 × 7.8 mm) column (Phenomenex, Aschaffenburg, Germany) at 50 °C. 5 mM H3SO4 was used as a mobile phase at a rate of 0.4 ml min−1.
Nucleotide analysis
Two millilitre of biosuspension was quenched using precooled (−22 °C) perchloric acid (35% v/v) containing 80 µM EDTA and was incubated for 15 min at 6 °C while shaking (Cserjan‐Puschmann et al., 1999). Samples were processed and analysed with HPLC, as described by Löffler et al. (2016). Concentrations of AMP, ADP, ATP and ppGpp were normalized by dry biomass, and the adenylate energy charge (AEC) was calculated using the following approach (Atkinson, 1968):
Computation of nucleotide and amino acid energy costs
ATP costs of transcription and translation were estimated following the E. coli approach of Löffler et al. (2016). As P. putida’s central carbon metabolism differs from that of E. coli, several changes were applied. First, the phosphate/oxygen ratio (P/O) of 1.33 (van Duuren et al., 2013), as conversion factor of ATP gain from respiratory oxidation of NADH (and FADH), was used instead of 1.49. ATP costs for forming metabolic precursors are presented in Table S1. These energy costs were considered to calculate the ATP requirements for synthesizing nucleotide triphosphates (NTPs), listed in Table S2. In addition, Table S3 reveals ATP requirements for producing amino acids from the respective precursors. The stoichiometry of enzymatic reactions was derived from the Pseudomonas Genome Database (Winsor et al., 2016) collectively with the recently published metabolic flux balances of glucose grown P. putida KT2440 (Kohlstedt and Wittmann, 2019).
Analysis and quantification of 3‐HA
Intracellular 3‐hydroxyalkanoate monomers were methanolized prior to determination by GC‐FID. 8 ml of biosuspension was sampled into the precooled centrifuge tubes and immediately centrifuged with 7197 × g for 5 min at 4 °C. After one washing step with 9 ml demineralized H2O and a second centrifugation step, the cell pellet was deep‐frozen at − 70 °C. The frozen biomass was then subsequently lyophilized. For derivatization, 10–20 mg of lyophilized biomass was resuspended in 2 ml chloroform containing 0.05 g l−1 3‐methylbenzoic acid as the internal standard and 2 ml acid methanol (15% v/v sulphuric acid). After incubation at 97 °C (± 3 °C) for 5 h, the samples were cooled, 1 ml of demineralized water was added, and the samples were then centrifuged for phase separation. For quantification, standards of 3‐hydroxydecanoate and 3‐hydroxydodecanoate were derivatized in the same manner (see Fig. S2). The organic phase was analysed via GC‐FID. The GC system comprised an HP 5890 series II equipped with a Zebron ZB‐5MS column (30 m, 0.25 mm ID, 0.25 µm df). Therefore, 1 µl of the sample was injected with a split ratio of 20:1 at an inlet temperature of 280 °C. Initial oven temperature was maintained at 100 °C for 2 min, followed by a gradient of 8 °C/min until 280 °C and was maintained for 5 min. Sample preparation procedure was adopted from Eugenio et al. (2010). To compensate for the expected low intracellular 3‐HA pool sizes, the relatively large sampling volume of 8 ml biosuspension was used ensuring 3‐HA titres of 25 to 50 mg3‐HA l‐1 in the extracted samples. For quantitative analysis, the peak areas of analytes in the derived chromatograms were normalized by the respective peak area of the internal standard and correlated to the linear fit of the calibration. The resulting concentrations were normalized by the dry biomass weight used for extraction to obtain biomass‐specific intracellular concentrations.
RNA sequencing
Total RNA was extracted using the Trizol reagent (Ambion), following the protocol of the supplier, except for few modifications: For cell harvest, 2 ml of ice‐cold stop solution (5% water‐saturated phenol in ethanol) was added to 10 ml of bacterial culture, well mixed and centrifuged at 4000 × g, for 5 min at 4 °C. The supernatant was carefully removed, and the pellet was resuspended in 350 μl of lysis solution (0.2% lysozyme in Tris‐HCl, pH 7.5) and was then incubated for 5 min at 37 °C. Next, the cells were lysed in a FastPrep homogenizer using 0.5 ml of small glass beads and 0.5 ml of acid phenol, and were centrifuged at a maximum speed for 10 min at 4 °C. The aqueous phase was collected and treated with 10 U of Turbo DNase (Ambion), for 1 h at 37 °C. RNA extraction proceeded with Trizol treatment, and after precipitation in ethanol (with 300 mM sodium acetate), the RNA was resuspended in RNase‐free water. RNA integrity was evaluated by agarose gel electrophoresis, and its concentration was estimated spectrophotometrically (NanoDrop ND‐1000). Total RNA was sequenced (Illumina, paired‐end, 150 bp, 20M reads) by StabVida (Portugal). The rRNA was depleted from the samples with the RiboZero kit, and the sequencing libraries were constructed with the TruSeq kit (Illumina) for bacteria. The sequencing raw files are available at the Gene Expression Omnibus under the accession number http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE129947.
Data processing
Initially, we utilized fastqc v0.11.8 (Andrews, 2010) to perform quality control on the paired‐end raw sequencing reads before and after the data preprocessing. trimmomatic v0.39 (Bolger et al., 2014) was used to remove the technical sequences within the reads. Furthermore, we discarded the reads with an average Phred quality score below 20 units, and we expected a read length of 35 bases to reduce the mapping ambiguity. As a result, roughly 84.4% of the raw reads were left after the preprocessing procedure. Subsequently, the surviving paired‐end reads were aligned against the reference genome of P. putida KT2440 (NC_002947.4) obtained from the NCBI reference sequence database (O'Leary et al., 2016) using segemehl v0.3.4 (Hoffmann et al., 2009; Otto et al., 2014). Therefore, we require the reads to match to the reference sequence at least by 90% resulting in 83.9% of all the (preprocessed) sequencing reads that could be aligned against the reference genome (Fig. S3). Later, featurecounts v1.6.4 (Liao et al., 2014) was applied using the respective annotations for the selected reference sequence to enumerate the number of reads overlapping the transcripts of interest. We selected features characterized as ‘CDS’ and report the attribute ‘Name’ that corresponds to the RefSeq accession identifiers in order to ensure the further use of only protein‐coding transcripts in the downstream analysis. In total, 72.6% of the preprocessed reads could be assigned uniquely to the annotated features thus making up between 4.8 and 12 million counts per sample (see Supplementary Data, sheet ‘statistics’, Fig. S5).
Differential expression analysis
Initially, we assessed the data quality using the regularized log‐transformed count data, wherein we performed a principal component analysis (PCA) using a subset of 1000 genes with the highest standard deviation between samples (Fig. S5B–D). The deseq2 R‐package v3.8 (Love et al., 2014), available from Bioconductor (http://www.bioconductor.org), was used to perform the differential gene expression analyses. The resulting P‐values were adjusted for multiple testing according to Benjamini and Hochberg (1995) in order to control the false discovery rate (FDR). We identified genes as significantly differentially expressed by applying the FDR‐adjusted P‐values < 0.01 and of a fold change > 1.5 and < −1.5 (equals log2‐fold > 0.585 and < −0.585). Subsequently, we assigned gene information to the initial RefSeq accession identifiers by combing information from the NCBI RefSeq, UniProt (The UniProt Consortium, 2019) and the Pseudomonas genome database (Winsor et al., 2016). In addition, we used this database to categorize the genes into separate pathways and metabolic functions.
Gene set enrichment analysis
Similar to Löffler et al. (2016) and Simen et al. (2017), we performed functional enrichment tests using Bioconductor’s R package gage v3.5.2 (Luo et al., 2009) to identify significant up‐ or downregulated sets of genes. We used the Gene Ontology term annotations for P. putida obtained from the Pseudomonas genome database or grouped genes according to pathways/ metabolic function and applied the rlog‐transformed raw count data (see Data S1). We configured gage to conduct group‐on‐group comparisons (compare=”as.group”) between the reference and target condition equal to the differential expression analysis using two‐sample t‐test statistics (use.fold=F). The gene sets were considered to be significantly different with an FDR q‐value < 0.05 corrected after Benjamini and Hochberg (1995). The underlying data are provided in the Data S1 (see tab ‘GSEA’).
Conflict of interest
None declared.
Supporting information
Fig. S1. Physiological kinetics of P. putida KT2440 cultivated in a glucose‐limited chemostat before and after the connection of the PFR (time point 0 h). Error bars represent the standard deviation of three biological replicates.
Fig. S2. Calibration fit for GC standards. Derivatized 3‐hydroxydecanoate and 3‐hydroxydodecanoate were measured via GC FID. For normalization purpose, each area was normalized by the respective area of the internal standard (IS). This calibration range was suitable to determine the 3‐HA concentrations in the extracted cultivation samples.
Fig. S3. Read distribution of the samples withdrawn from the PFR (A) and the STR (B). The bars correspond to the read number before (raw) and after preprocessing (trimmed) as well as after the sequence alignment (aligned).
Fig. S4. Portion of assigned features to the total number of aligned reads for samples withdrawn from the PFR (A) and STR (B).
Fig. S5. (A) Scatterplot of the standard deviation across samples against the mean (calculated gene‐wise) using the regularized log transformation. The red line depicts the running median estimator (window‐width 10%). If there is no variance‐mean dependence, then the line should be approximately horizontal. (B) Individuals (samples) of 12 process time points plotted on principal component 1 (PC1) and 2 (PC2). Circles and squares correspond to the location of the sampling in the PFR and STR, respectively. Arrows follow the adaptation trajectories within the PFR. A potential steady state is indicated by the green ellipse. (C) Scree plot of the PCA illustrating the contribution of each principal component to the variance. (D) Barplot of the top 20 variables (genes) contributing to the variability of PC1 and PC2. The red reference line corresponds to the expected value if the contribution where uniform and is considered as important in contributing to the dimension.
Fig. S6. Boxplot of the Cook’s distances to see if one sample is consistently higher than the others (this is not the case).
Fig. S7. Results of the gene set enrichment analysis of both the short‐term and long‐term response. For that, samples (rows) withdrawn from the PFR (P1, P3, P5) were compared with the samples of the STR at the corresponding time points and samples withdrawn from the STR were compared to the reference condition in the STR. Gene sets were considered as significantly different with an FDR q‐value < 0.05, shown as cells with the value of the t‐statistic (indicated by the color scale) from GAGE indicating the magnitude of gene set level changes. Numbers at the bottom correspond to the size of the gene sets.
Table. S1. Energy balance for central metabolic precursors. ‘Energy produced’ is calculated from glucose until precursor. ‘ATP produced’ summarizes the energy produced based on the P/O ratio of 1.33. Negative values indicate consumption
Table. S2. Energy costs for de novo synthesis of nucleotide triphosphates (NTP). Calculation is based on the P/O ratio of 1.33.
Table. S3. Energy costs for amino acids (AA) based on their respective precursors. Total ATP costs summarize energy produced from glucose to precursor (see Table S1) and from precursor to AA. Amino acid composition for P. putida KT2440 is derived from Sohn et al. (2010).
Table. S4. Carbon share of biomass, PHA and CO2 (± standard deviation from three biological replicates) based on glucose uptake rate at µ = 0.194 0.01 h‐1. Carbon composition of biomass was taken from van Duuren, Jozef et al. (2013).
Table. S5. Parameters for characterizing the plug flow reactor.
Table. S6. Measured values of intracellular 3‐HA concentrations in experiment 1 (Exp. 1) and experiment 2 (Exp. 2), mean values and standard deviation (SD) in relation to the residence time in the PFR.
Data S1. The supplementary data file provides the underlying data of differential expression and gene set enrichment analysis.
Acknowledgements
We thank Andreas Freund and Alexander Dietrich for the technical support with the bioreactor setup and Mira Lenfers‐Lücker for assistance with the HPLC analysis. Furthermore, we want to thank Vikas Patil and Max Fischer (Institute of Technical Biology, University of Stuttgart) for their support with the GC analysis and Lorena Hägele for her excellent analytical and laboratory contribution. Moreover, we thank Victor de Lorenzo, Vitor Martins dos Santos, Sven Panke, Bernd Hauer, Ian Fotheringham and Andrea Herold for valuable comments and the other members of the project ‘EmPowerPutida’ for a great cooperation. The authors further gratefully acknowledge the funding of this work by the European Union’s Horizon 2020 research and innovation programme under grant agreement No 635536 and the Federal Ministry of Education and Research (BMBF) program 031A310 and 031L0164A to B.V.
Microbial Biotechnology (2020) 13(4), 1145–1161
Funding Information
European Union’s Horizon 2020 research and innovation programme under grant agreement No 635536 and the Federal Ministry of Education and Research (BMBF) program 031A310 and 031L0164A to B.V.
References
- Andrews, S. (2010) Babraham Bioinformatics – FastQC A Quality Control tool for High Throughput Sequence Data. URL https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. [Google Scholar]
- Atkinson, D.E. (1968) The energy charge of the adenylate pool as a regulatory parameter. Interaction with feedback modifiers. Biochemistry 7: 4030–4034. [DOI] [PubMed] [Google Scholar]
- Bagdasarian, M. , Lurz, R. , Rückert, B. , Franklin, F.C.H. , Bagdasarian, M.M. , Frey, J. , and Timmis, K.N. (1981) Specific‐purpose plasmid cloning vectors II. Broad host range, high copy number, RSF 1010‐derived vectors, and a host‐vector system for gene cloning in Pseudomonas. Gene 16: 237–247. [DOI] [PubMed] [Google Scholar]
- Beckers, V. , Poblete‐Castro, I. , Tomasch, J. , and Wittmann, C. (2016) Integrated analysis of gene expression and metabolic fluxes in PHA‐producing Pseudomonas putida grown on glycerol. Microb Cell Fact 15: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. , and Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc: Series B (Methodological) 57: 289–300. [Google Scholar]
- Bolger, A.M. , Lohse, M. , and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, A.G. , Fall, L. , and Atkinson, D.E. (1971) Adenylate energy charge in Escherichia coli during growth and starvation. J Bacteriol 108: 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cserjan‐Puschmann, M. , Kramer, W. , Duerrschmid, E. , Striedner, G. , and Bayer, K. (1999) Metabolic approaches for the optimisation of recombinant fermentation processes. Appl Microbiol Biotechnol 53: 43–50. [DOI] [PubMed] [Google Scholar]
- van Delden, C. , Comte, R. , and Bally, A.M. (2001) Stringent response activates quorum sensing and modulates cell density‐dependent gene expression in Pseudomonas aeruginosa . J Bacteriol 183: 5376–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Duuren, J.B.J.H. , Puchalka, J. , Mars, A.E. , Bucker, R. , Eggink, G. , Wittmann, C. , et al (2013) Reconciling in vivo and in silico key biological parameters of Pseudomonas putida KT2440 during growth on glucose under carbon‐limited condition. BMC Biotechnol 13: 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvigne, F. , and Noorman, H. (2017) Scale‐up/Scale‐down of microbial bioprocesses: a modern light on an old issue. Microb Biotechnol 10: 685–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi, Y. , Segawa, A. , Kawaguchi, Y. , and Kunioka, M. (1990) Cyclic nature of poly(3‐hydroxyalkanoate) metabolism in Alcaligenes eutrophus . FEMS Microbiol Lett 55: 165–169. [DOI] [PubMed] [Google Scholar]
- Ebert, B.E. , Kurth, F. , Grund, M. , Blank, L.M. , and Schmid, A. (2011) Response of Pseudomonas putida KT2440 to increased NADH and ATP demand. Appl Environ Microbiol 77: 6597–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enfors, S.‐O. , Jahic, M. , Rozkov, A. , Xu, B. , Hecker, M. , Jürgen, B. , et al (2001) Physiological responses to mixing in large scale bioreactors. J Biotechnol 85: 175–185. [DOI] [PubMed] [Google Scholar]
- Escapa, I.F. , García, J.L. , Bühler, B. , Blank, L.M. , and Prieto, M.A. (2012) The polyhydroxyalkanoate metabolism controls carbon and energy spillage in Pseudomonas putida . Environ Microbiol 14: 1049–1063. [DOI] [PubMed] [Google Scholar]
- de Lorenzo, V. , and Couto, J. (2019) The important versus the exciting: reining contradictions in contemporary biotechnology. Microb Biotechnol 12: 32–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Eugenio, L.I. , Escapa, I.F. , Morales, V. , Dinjaski, N. , Galán, B. , García, J.L. , and Prieto, M.A. (2010) The turnover of medium‐chain‐length polyhydroxyalkanoates in Pseudomonas putida KT2442 and the fundamental role of PhaZ depolymerase for the metabolic balance. Environ Microbiol 12: 207–221. [DOI] [PubMed] [Google Scholar]
- Ferenci, T. (2001) Hungry bacteria – definition and properties of a nutritional state. Environ Microbiol 3: 605–611. [DOI] [PubMed] [Google Scholar]
- Fulco, A.J. (1983) Fatty acid metabolism in bacteria. Prog Lipid Res 22: 133–160. [DOI] [PubMed] [Google Scholar]
- Gentry, D.R. , Hernandez, V.J. , Nguyen, L.H. , Jensen, D.B. , and Cashel, M. (1993) Synthesis of the stationary‐phase sigma factor sigma s is positively regulated by ppGpp. J Bacteriol 175: 7982–7989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamasaki, M. , Noda, T. , and Ohsumi, Y. (2003) The early secretory pathway contributes to autophagy in yeast. Cell Struct Funct 28: 49–54. [DOI] [PubMed] [Google Scholar]
- Hardy, G.P.M.A. , Joost Teixeira de Mattos, M. , and Neijssel, O.M. (1993) Energy conservation by pyrroloquinoline quinol‐linked xylose oxidation in Pseudomonas putida NCTC 10936 during carbon‐limited growth in chemostat culture. FEMS Microbiol Lett 107: 107–110. [DOI] [PubMed] [Google Scholar]
- Hauryliuk, V. , Atkinson, G.C. , Murakami, K.S. , Tenson, T. , and Gerdes, K. (2015) Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat Rev Microbiol 13: 298–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hintermayer, S.B. , and Weuster‐Botz, D. (2017) Experimental validation of in silico estimated biomass yields of Pseudomonas putida KT2440. Biotechnol J 12: 1600720. [DOI] [PubMed] [Google Scholar]
- Hoffmann, S. , Otto, C. , Kurtz, S. , Sharma, C.M. , Khaitovich, P. , Vogel, J. , et al (2009) Fast mapping of short sequences with mismatches, insertions and deletions using index structures. PLoS Comput Biol 5: e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijberts, G. N. , Eggink, G. , and Waard, P.d., Huisman, G.W., and Witholt, B., (1992) Pseudomonas putida KT2442 cultivated on glucose accumulates poly(3‐hydroxyalkanoates) consisting of saturated and unsaturated monomers. Appl Environ Microbiol 58: 536–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James, B.W. , Mauchline, W.S. , Dennis, P.J. , Keevil, C. W. , and Wait, R. (1999) Poly‐3‐hydroxybutyrate in Legionella pneumophila, an energy source for survival in low‐nutrient environments. Appl Environ Microbiol 65: 822–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , and Park, W. (2014) Oxidative stress response in Pseudomonas putida . Appl Microbiol Biotechnol 98: 6933–6946. [DOI] [PubMed] [Google Scholar]
- Kohlstedt, M. , and Wittmann, C. (2019) GC‐MS‐based 13C metabolic flux analysis resolves the parallel and cyclic glucose metabolism of Pseudomonas putida KT2440 and Pseudomonas aeruginosa PAO1. Metab Eng 54: 35–53. [DOI] [PubMed] [Google Scholar]
- Lazzarini, R.A. , Cashel, M. , and Gallant, J. (1971) On the regulation of guanosine tetraphosphate levels in stringent and relaxed strains of Escherichia coli . J Biol Chem 246: 4381–4385. [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G. K. , and Shi, W. (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930. [DOI] [PubMed] [Google Scholar]
- Lieder, S. , Nikel, P. I. , de Lorenzo, V. , and Takors, R. (2015) Genome reduction boosts heterologous gene expression in Pseudomonas putida . Microb Cell Fact 14: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , Xiao, Y. , Nie, H. , Huang, Q. , and Chen, W. (2017) Influence of (p)ppGpp on biofilm regulation in Pseudomonas putida KT2440. Microbiol Res 204: 1–8. [DOI] [PubMed] [Google Scholar]
- Löffler, M. , Simen, J.D. , Jager, G. , Schaferhoff, K. , Freund, A. , and Takors, R. (2016) Engineering E. coli for large‐scale production – Strategies considering ATP expenses and transcriptional responses. Metab Eng 38: 73–85. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. , and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, W. , Friedman, M.S. , Shedden, K. , Hankenson, K.D. , and Woolf, P.J. (2009) GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinform 10: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez‐García, E. , Nikel, P.I. , Aparicio, T. , and de Lorenzo, V. (2014) Pseudomonas 2.0: Genetic upgrading of P. putida KT2440 as an enhanced host for heterologous gene expression. Microb Cell Fact 13: 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalowski, A. , Siemann‐Herzberg, M. , and Takors, R. (2017) Escherichia coli HGT: Engineered for high glucose throughput even under slowly growing or resting conditions. Metab Eng 40: 93–103. [DOI] [PubMed] [Google Scholar]
- Mozejko‐Ciesielska, J. , Dabrowska, D. , Szalewska‐Palasz, A. , and Ciesielski, S. (2017) Medium‐chain‐length polyhydroxyalkanoates synthesis by Pseudomonas putida KT2440 relA/spoT mutant: Bioprocess characterization and transcriptome analysis. AMB Express 7(1): 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozejko‐Ciesielska, J. , Pokoj, T. , and Ciesielski, S. (2018) Transcriptome remodeling of Pseudomonas putida KT2440 during mcl‐PHAs synthesis: effect of different carbon sources and response to nitrogen stress. J Ind Microbiol Biotechnol 45: 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neubauer, P. , and Junne, S. (2010) Scale‐down simulators for metabolic analysis of large‐scale bioprocesses. Curr Opin Biotechnol 21: 114–121. [DOI] [PubMed] [Google Scholar]
- Neubauer, P. , Haggstrom, L. , and Enfors, S.O. (1995) Influence of substrate oscillations on acetate formation and growth yield in Escherichia coli glucose limited fed‐batch cultivations. Biotechnol Bioeng 47: 139–146. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , and de Lorenzo, V. (2018) Pseudomonas putida as a functional chassis for industrial biocatalysis: from native biochemistry to trans‐metabolism. Metab Eng 50: 142–155. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Martinez‐Garcia, E. , and de Lorenzo, V. (2014) Biotechnological domestication of pseudomonads using synthetic biology. Nat Rev Microbiol 12: 368–379. [DOI] [PubMed] [Google Scholar]
- Nikel, P.I. , Chavarria, M. , Fuhrer, T. , Sauer, U. , and de Lorenzo, V. (2015) Pseudomonas putida KT2440 strain metabolizes glucose through a cycle formed by enzymes of the entner‐doudoroff, Embden‐Meyerhof‐Parnas, and pentose phosphate pathways. J Biol Chem 290: 25920–25932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikel, P.I. , Chavarria, M. , Danchin, A. , and de Lorenzo, V. (2016) From dirt to industrial applications: Pseudomonas putida as a Synthetic Biology chassis for hosting harsh biochemical reactions. Curr Opin Chem Biol 34: 20–29. [DOI] [PubMed] [Google Scholar]
- Nogales, J. , Palsson, B.Ø. , and Thiele, I. (2008) A genome‐scale metabolic reconstruction of Pseudomonas putida KT2440: i JN746 as a cell factory. BMC Syst Biol 2: 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt, M.A. , Puchałka, J. , Martins dos Santos, V.A.P. , and Papin, J.A. (2011) Reconciliation of genome‐scale metabolic reconstructions for comparative systems analysis. PLoS Comput Biol 7: e1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary, N.A. , Wright, M.W. , Brister, J.R. , Ciufo, S. , Haddad, D. , McVeigh, R. , et al (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto, C. , Stadler, P.F. , and Hoffmann, S. (2014) Lacking alignments? The next‐generation sequencing mapper segemehl revisited. Bioinformatics 30: 1837–1843. [DOI] [PubMed] [Google Scholar]
- Poblete‐Castro, I. , Becker, J. , Dohnt, K. , dos Santos, V.M. , and Wittmann, C. (2012a) Industrial biotechnology of Pseudomonas putida and related species. Appl Microbiol Biotechnol 93: 2279–2290. [DOI] [PubMed] [Google Scholar]
- Poblete‐Castro, I. , Escapa, I.F. , Jäger, C. , Puchalka, J. , Lam, C.M.C. , Schomburg, D. , et al (2012b) The metabolic response of P. putida KT2442 producing high levels of polyhydroxyalkanoate under single‐ and multiple‐nutrient‐limited growth: highlights from a multi‐level omics approach. Microb Cell Fact 11: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pobre, V. , Graça‐Lopes, G. , Saramago, M. , Ankenbauer, A. , Takors, R. , Arraiano, C.M. , and Viegas, S.C. (2020) Prediction of novel non‐coding RNAs relevant for the growth of Pseudomonas putida in a bioreactor. Microbiology (Reading, England) 166: 149–156. [DOI] [PubMed] [Google Scholar]
- Potrykus, K. , and Cashel, M. (2008) (p)ppGpp: Still magical? Annu Rev Microbiol 62: 35–51. [DOI] [PubMed] [Google Scholar]
- Reddy, B.V. , Prasad, B.R. , Sinha, S.N. , and Ahmed, N. (2014) New mathematical derivations for calculation of ATP yield due to the complete oxidation of different types of fatty acids. Ind J Biochem Biophys 51: 52–57. [PubMed] [Google Scholar]
- Ren, Q. , de Roo, G. , Ruth, K. , Witholt, B. , Zinn, M. , and Thöny‐Meyer, L. (2009) Simultaneous accumulation and degradation of polyhydroxyalkanoates: futile cycle or clever regulation? Biomacromol 10: 916–922. [DOI] [PubMed] [Google Scholar]
- Ruth, K. , de Roo, G. , Egli, T. , and Ren, Q. (2008) Identification of two acyl‐CoA synthetases from Pseudomonas putida GPo1: one is located at the surface of polyhydroxyalkanoates granules. Biomacromol 9: 1652–1659. [DOI] [PubMed] [Google Scholar]
- Ryals, J. , Little, R. , and Bremer, H. (1982) Control of rRNA and tRNA syntheses in Escherichia coli by guanosine tetraphosphate. J Bacteriol 151: 1261–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simen, J.D. , Löffler, M. , Jager, G. , Schaferhoff, K. , Freund, A. , Matthes, J. , et al (2017) Transcriptional response of Escherichia coli to ammonia and glucose fluctuations. Microb Biotechnol 10: 858–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn, S.B. , Kim, T.Y. , Park, J.M. , and Lee, S.Y. (2010) In silico genome‐scale metabolic analysis of Pseudomonas putida KT2440 for polyhydroxyalkanoate synthesis, degradation of aromatics and anaerobic survival. Biotechnol J 5: 739–750. [DOI] [PubMed] [Google Scholar]
- Takors, R. (2012) Scale‐up of microbial processes: impacts, tools and open questions. J Biotechnol 160: 3–9. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium (2019) UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler, M.F. , Summers, S.M. , Nguyen, H.‐T. , Zacharia, V.M. , Hightower, G.A. , Smith, J.T. , and Conway, T. (2008) The global, ppGpp‐mediated stringent response to amino acid starvation in Escherichia coli . Mol Microbiol 68: 1128–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traxler, M.F. , Zacharia, V.M. , Marquardt, S. , Summers, S.M. , Nguyen, H.‐T. , Stark, S.E. , and Conway, T. (2011) Discretely calibrated regulatory loops controlled by ppGpp partition gene induction across the ‘feast to famine’ gradient in Escherichia coli . Mol Microbiol 79: 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino, K. , Saito, T. , Gebauer, B. , and Jendrossek, D. (2007) Isolated poly(3‐hydroxybutyrate) (PHB) granules are complex bacterial organelles catalyzing formation of PHB from acetyl coenzyme A (CoA) and degradation of PHB to acetyl‐CoA. J Bacteriol 189: 8250–8256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallon, T. , Glemser, M. , Malca, S.H. , Scheps, D. , Schmid, J. , Siemann‐Herzberg, M. , et al (2013) Production of 1‐Octanol from n ‐Octane by Pseudomonas putida KT2440. Chem Ing Tec 85: 841–848. [Google Scholar]
- Vallon, T. , Simon, O. , Rendgen‐Heugle, B. , Frana, S. , Mückschel, B. , Broicher, A. , et al (2015) Applying systems biology tools to study n‐butanol degradation in Pseudomonas putida KT2440. Eng Life Sci 15: 760–771. [Google Scholar]
- Winsor, G.L. , Griffiths, E.J. , Lo, R. , Dhillon, B.K. , Shay, J.A. , and Brinkman, F.S.L. (2016) Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44: D646–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zieringer, J. , and Takors, R. (2018) In silico prediction of large‐scale microbial production performance: constraints for getting proper data‐driven models. Comput Struct Biotechnol J 16: 246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Physiological kinetics of P. putida KT2440 cultivated in a glucose‐limited chemostat before and after the connection of the PFR (time point 0 h). Error bars represent the standard deviation of three biological replicates.
Fig. S2. Calibration fit for GC standards. Derivatized 3‐hydroxydecanoate and 3‐hydroxydodecanoate were measured via GC FID. For normalization purpose, each area was normalized by the respective area of the internal standard (IS). This calibration range was suitable to determine the 3‐HA concentrations in the extracted cultivation samples.
Fig. S3. Read distribution of the samples withdrawn from the PFR (A) and the STR (B). The bars correspond to the read number before (raw) and after preprocessing (trimmed) as well as after the sequence alignment (aligned).
Fig. S4. Portion of assigned features to the total number of aligned reads for samples withdrawn from the PFR (A) and STR (B).
Fig. S5. (A) Scatterplot of the standard deviation across samples against the mean (calculated gene‐wise) using the regularized log transformation. The red line depicts the running median estimator (window‐width 10%). If there is no variance‐mean dependence, then the line should be approximately horizontal. (B) Individuals (samples) of 12 process time points plotted on principal component 1 (PC1) and 2 (PC2). Circles and squares correspond to the location of the sampling in the PFR and STR, respectively. Arrows follow the adaptation trajectories within the PFR. A potential steady state is indicated by the green ellipse. (C) Scree plot of the PCA illustrating the contribution of each principal component to the variance. (D) Barplot of the top 20 variables (genes) contributing to the variability of PC1 and PC2. The red reference line corresponds to the expected value if the contribution where uniform and is considered as important in contributing to the dimension.
Fig. S6. Boxplot of the Cook’s distances to see if one sample is consistently higher than the others (this is not the case).
Fig. S7. Results of the gene set enrichment analysis of both the short‐term and long‐term response. For that, samples (rows) withdrawn from the PFR (P1, P3, P5) were compared with the samples of the STR at the corresponding time points and samples withdrawn from the STR were compared to the reference condition in the STR. Gene sets were considered as significantly different with an FDR q‐value < 0.05, shown as cells with the value of the t‐statistic (indicated by the color scale) from GAGE indicating the magnitude of gene set level changes. Numbers at the bottom correspond to the size of the gene sets.
Table. S1. Energy balance for central metabolic precursors. ‘Energy produced’ is calculated from glucose until precursor. ‘ATP produced’ summarizes the energy produced based on the P/O ratio of 1.33. Negative values indicate consumption
Table. S2. Energy costs for de novo synthesis of nucleotide triphosphates (NTP). Calculation is based on the P/O ratio of 1.33.
Table. S3. Energy costs for amino acids (AA) based on their respective precursors. Total ATP costs summarize energy produced from glucose to precursor (see Table S1) and from precursor to AA. Amino acid composition for P. putida KT2440 is derived from Sohn et al. (2010).
Table. S4. Carbon share of biomass, PHA and CO2 (± standard deviation from three biological replicates) based on glucose uptake rate at µ = 0.194 0.01 h‐1. Carbon composition of biomass was taken from van Duuren, Jozef et al. (2013).
Table. S5. Parameters for characterizing the plug flow reactor.
Table. S6. Measured values of intracellular 3‐HA concentrations in experiment 1 (Exp. 1) and experiment 2 (Exp. 2), mean values and standard deviation (SD) in relation to the residence time in the PFR.
Data S1. The supplementary data file provides the underlying data of differential expression and gene set enrichment analysis.