

Abstract
Exposure to high dose total body irradiation (TBI) can result in hematopoietic acute radiation syndrome (H-ARS), characterized by leukopenia, anemia, and coagulopathy. Death from H-ARS occurs from hematopoietic insufficiency and opportunistic infections. Following radiation exposure, red blood cells (RBC) undergo hemolysis from radiation-induced hemoglobin denaturation, causing the release of iron. Free iron can have multiple detrimental biological effects, including suppression of hematopoiesis. We investigated the impact of radiation-induced iron release on the bone marrow following TBI and the potential impact of the ACE inhibitor captopril, which improves survival from H-ARS. C57BL/6J mice were exposed to 7.9 Gy, 60Co irradiation, 0.6 Gy/min (LD70-90/30). RBC and reticulocytes were significantly reduced within 7 days of TBI, with the RBC nadir at 14-21 days. Iron accumulation in the bone marrow correlated with the time course of RBC hemolysis, with a ~10-fold increase in bone marrow iron at 14-21 days post-irradiation, primarily within the cytoplasm of macrophages. Iron accumulation in the bone marrow was associated with increased expression of genes for iron binding and transport proteins, including transferrin, transferrin receptor 1, ferroportin, and integrin alphaMbeta2. Expression of the gene encoding Nrf2, a transcription factor activated by oxidative stress, also increased at 21 days post-irradiation. Captopril did not alter iron accumulation in the bone marrow or expression of iron storage genes, but did suppress Nrf2 expression. Our study suggests that following TBI, iron is deposited in tissues not normally associated with iron storage, which may be a secondary mechanism of radiation-induced tissue injury.
Introduction
Exposure to total body irradiation (TBI) induces a number of adverse health effects, collectivly referred to as acute radiation syndrome (ARS). The hematopoietic system is especially sensitive to radiation-induced injury, and hematopoietic subsyndrome of ARS (H-ARS) can be observed in humans exposed to 4-6 Gy TBI [1]. Mortality from H-ARS is believed to be due to hematopoietic insufficiency (both leukopenia and anemia), opportunistic infection from immune suppression, and coagulation dysfunction [2-5].
Substantial research has been directed toward understanding hematopoietic cellular response to radiation. White blood cells and many hematopoietic progenitors undergo apoptosis in response to DNA damage from radiation exposure [5, 6]. However, reticulocytes and red blood cells (RBC) are resistant to radiation-induced apoptosis due to their lack of DNA and the absence of apoptotic machinery [7, 8]. Instead, reticulocytes and RBC undergo hemolysis as a result of oxidation and denaturation of hemoglobin (HGB) and morphological changes associated with altered mechanical properties of the cytoskeleton and membrane [5, 7, 8]. Although the loss of white blood cells has significant impact on immune function, the lysis of erythrocytes from radiation exposure results in deficits in oxygenation and, possibly even more significantly, the release of iron.
Iron is a vital nutrient, with critical functions at the cellular level in enzymatic reactions, mitochondrial respiration and oxygen transport in many organisms [9]. The most active use of iron is in the bone marrow for erythropoiesis, and 65-75% of the total iron is found in heme associated with hemoglobin in erythrocytes [10]. However, iron binding and transport in the body is tightly regulated because unbound iron can promote the generation of toxic free radicals through its interaction with oxygen, for example in Fenton and Haber-Weiss reactions [10-13]. Iron recycling is performed primarily by specialized macrophages, which normally ingest senescent RBC in the spleen [10, 14]. However, dietary studies of iron overload demonstrated that excessive iron can exceed the capacity of spleen and liver macrophages to maintain iron homeostasis. Excessive iron negatively impacts a number of biological functions, including the hematopoietic system, potentially through the induction of reactive oxygen species (ROS) [15]. Surplus iron also alters gene expression in macrophages, inhibiting normal antimicrobial activities and potentially contributing to pathogentic activities of these cells as in atherosclerosis [14, 16, 17]. Finally, the excessive iron can contribute to bone damage by inducing excessive osteoclast activity [18].
In a murine model of TBI, serum iron was demonstrated to be significantly elevated within one day following exposure to 7 Gy total body gamma radiation; iron levels in the serum increased through 20 days postirradiation [19, 20]. In a separate study of targeted femur irradiation in mice, iron levels were also increased in the serum and liver [18]. Therefore, both total body and localized exposure to radiation induced significant iron release from the lysis of erythrocytes.
Here we investigated the effect of TBI on iron levels in the bone marrow. Our data show increased iron depostion in the bone marrow of mice correlated with the loss of RBC and reticulocytes. Expression of iron binding and transport genes were increased in the bone marrow, correlating with the increased bone marrow iron. Previously we demonstrated in a murine model of H-ARS that captopril treatment improved survival following expsure to 7.5-7.9 Gy TBI (LD50/30) at least in part through improved recovery of several mature blood cell types, including RBC and reticulocytes [21-24]. We found that captopril did not affect radiation-induced iron deposition in the bone marrow or expression of iron-binding proteins. However, captopril treatment reduced indicators of oxidative stress in the bone marrow. These data provide insight into the effects of iron released from hemolysis following TBI, and suggest that released iron may be a target for radiation countermeasures.
Materials and Methods
Chemicals.
Reagents were obtained from Sigma-Aldrich (St. Louis, MO, USA) except where indicated.
Animals, irradiation, and captopril treatment.
All animal handling procedures were performed in compliance with guidelines from the National Research Council for the ethical handling of laboratory animals and were approved by the Armed Forces Radiobiology Research Institute (AFRRI) Animal Care and Use Committee. Female C57BL/6J mice were purchased from Jackson Laboratories (Bar Harbor, ME). Mice were kept in a barrier facility for animals accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. Mice were housed in groups of four. Animal rooms were maintained at 21 ± 2°C, 50% ± 10% humidity, and 12-h light/dark cycle with commercial freely available rodent ration (Harlan Teklad Rodent Diet 8604, Frederick, MD, USA). Animals were randomized to treatment groups for each experiment. Mice that were 12-14 weeks of age were placed in Lucite jigs and exposed to TBI in a bilateral gamma radiation field in the AFRRI high level 60Co facility as previously described [25]. The midline tissue dose to the mice was 7.79 Gy at a dose rate of 0.6 Gy/min. The alanine/electron spin resonance (ESR) dosimetry system [26] was used to measure dose rates (to water) in the cores of acrylic mouse phantoms. For all experiments, separate control groups were placed in Lucite jigs but without exposure to radiation (sham irradiation). Captopril (USP grade; Sigma-Aldrich) was dissolved at 0.055 g/L in acidified water [27] to deliver ~12 mg/kg/day [21, 24]. Captopril in the water was provided to animals from 2 days post-irradiation or sham irradiation for 14 consecutive days as previously described [21, 24]. As a control for captopril treatment, animals received acidified water (vehicle) without captopril. Survival studies utilized N = 16 animals per group, a minimum number required to obtain statistical significance [28].
Female GATA1low transgenic mice, that develop spontaneous myelofibrosis, were obtained from Jackson laboratories, and bred as previously described [29]. These mice were used at 13 months of age, when myelofibrosis had developed, to provide a model for a non-radiation bone marrow injury.
Blood cell analysis.
Complete blood counts (CBC) with differentials were obtained using an Advia 2120 Hematology Analyzer (Siemens, Tarrytown, NY, USA). Separate mice were used for each point (n = 1-5 per group), which we previously determined is a minimal number of animals required to provide statistical significance [24].
Histology and erythroblastic island scoring.
Dissected sternebrae surgically removed from euthanized animals were fixed in 10% neutral formalin overnight. Tissues were paraffin blocked and stained using standard methods for hematoxylin and eosin (H&E) by Histoserve (Germantown, MD, USA). Prussian blue staining, with nuclear fast red counterstain, was performed at the Walter Reed National Military Medical Center, Anatomic Pathology Laboratory (Bethesda, MD, USA). Erythroblastic island images were obtained with an Olympus DP73 camera, using Olympus cellSens imaging software (Olympus Life Science, Waltham, MA, USA). Prussian blue stained images bone marrow sections were digitally imaged using a Nikon Eclipse Ti microscope with a Nikon DSRi2 camera (Nikon Instruments, Inc, Melville, NY, USA). Elements software, version 4.51, (Nikon Instruments) was used for analysis of images. ImageJ software was used for histological image analysis (NIH, Bethesda, MD, USA; https://imagej.nih.gov/ij/download.html). Sections were evaluated for erythroblastic islands by a pathologist blinded to the treatment groups.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from bone marrow cells or liver tissue using TRIzol (ThermoFisher, Carlesbad, CA, USA) using phenol-chloroform extraction with silicone lubricant using a modified protocol [30], or using the Qiagen RNeasy kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s instructions. RNA (500 ng) was used with the iScript cDNA kit (Bio-Rad, Hercules, CA, USA) for cDNA synthesis. Quantitative PCR was carried out on a CFX96 real-time PCR detection system (Bio-Rad) and SYBR Green qPCR master mix (Bio-Rad). PCR reaction conditions were 3 min at 95.0°C, followed by cycles of 10 seconds at 95.0°C, 30 sec at 55.0°C for 39 total cycles (Bio-Rad CFX Manager 3.1 preloaded, CFX-2stepAmp protocol). The sequences for primers for target amplification are shown in Table 1. Relative gene expression to a reference gene was calculated using the ΔΔCq method [31, 32].
Table 1.
Primers for qPCR.
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Ccl2 | 5’-CCACTCACCTGCTGCTACTC-3’ | 5’-TGCTTGAGGTGGTTGTGGAA-3’ |
| Ccl3 | 5’-GAGCTGACACCCGACTG-3’ | 5’-TCAGGATTCAGTTCCAGGTC-3’ |
| Flvcr1 | 5’-GGCACAATATAAACACCGGGC-3’ | 5’-TCCGACTGTATAGACACCATGAC-3’ |
| Gapdh | 5’-ATGTGTCCGTTGTGGACTTG-3’ | 5’-GGTCCTCAGTGTAGCCCAAG -3’ |
| Hamp | 5’-CCTGAGCAGCACCACCTATC-3’ | 5’-GCAACAGATACCACACTGGGA-3’ |
| Hmox1 | 5’-GTTTGAGGAGCTGCAGGTGA-3’ | 5’-TGCCAACAGGAAGCTGAGAG-3’ |
| Itgam | 5’-AGAACACCAAGGACCGTCTG-3’ | 5’-AATCCAAAGACCTGGGTGCG-3’ |
| Lcn2 | 5’-GGACTACAACCAGTTCGCCA-3’ | 5’-CAAAGCGGGTGAAACGTTCC-3’ |
| Nfe2l2 | 5’-CCTCACCTCTGCTGCAAGTA-3’ | 5’-ACCTTGTACCGCCTCGTCTG-3’ |
| Tfrc1 | 5’-GCTCGTGGAGACTACTTCCG-3’ | 5’-AGAGAGGGCATTTGCGACTC-3’ |
| Trf1 | 5’-AAGTGCATCAGCTTCCGTGA-3’ | 5’-AGACCACACTGGCCTTGATG-3’ |
| Slc40a1 | 5’-TTCCTCCTCTACCTTGGCCA-3’ | 5’-CTGCCACCACCAGTCCATAG-3’ |
Murine sequences for Ccl2 (chemokine [C-C motif] ligand 2), Ccl3 (Chemokine [C-C motif] ligand 3), Flvcr1 (feline leukemia virus receptor; heme iron receptor), Gapdh (glyceraldehyde-3-phosphate dehydrogenase), Hamp (hepcidin-1), Hmox1 (heme oxygenase-1), Itgam (integrin subunit alpha M), Icam4 (intracellular adhesion molecule 4), Lcn2 (lipocalin-2; also known as neutrophil gelatinase associated lipocalin or siderocalin), Nefe2l2 (nuclear factor [erythroid-derived2]-like 2), Tfrc1 (transferrin receptor-1), Trf1 (transferrin-1), and Scl40a1 (ferroportin).
Statistical Analysis.
Kaplan–Meier plots were analyzed using Fisher Exact Tests to assess the differences in survival between the groups after irradiation (GraphPad Prism v7.1; LaJolla, CA, USA). P values less than 0.05 were considered to be statistically significant. Results are represented as means ± SEM. Two-way ANOVA with either Tukey’s or Sidak’s post-hoc tests were used for multiple comparisons.
Results
Delayed captopril administration improves 30-day survival but does not alter the nadirs of RBC, reticulocytes, or hemoglobin following TBI.
We previously reported that captopril (13-110 mg/kg/day) improved survival of C57BL/6J mice from H-ARS following TBI [21, 24]. Exposure of mice to 7.9 Gy (0.6 Gy/min) of 60Co TBI reduced survival to 12.5% at 30 days (Fig. 1A). Captopril treatment significantly improved survival (62.5%, p<0.05). The reduction in RBC was significant within 4 days post-irradiation (reduced by ~20% in radiation + vehicle and ~10% in radiation + captopril), and the nadir for RBCs occurred at ~21 days post-irradiation (Fig. 1B). In contrast, the nadir for reticulocytes occurred within ~4 days post-irradiation (<1% of basal levels for both irradiation groups) (Fig. 1C). Total HGB, which is a separate indicator of iron loss from RBC, decline is correlated with RBC decline (Fig. 1D). Recovery of RBC and HGB were observed in the captopril-treated group to near basal levels at 30 days post-irradiation, while reticulocytes were ~6-fold above basal levels at 30 days. Note that no animals survived past 25 days in the radiation + vehicle group, so no values were obtained after that time point. Together these data suggest that TBI induces erythrocyte hemolysis that is detectable within 4 days post-irradiation and is progressive through 21 days post-irradiation. Captopril did not affect hemolysis but improved recovery of RBC, reticulocytes and the hematocrit.
Figure 1. Time courses of mortality from 7.9 Gy TBI, hemoglobin depletion, and Fe release in the plasma.
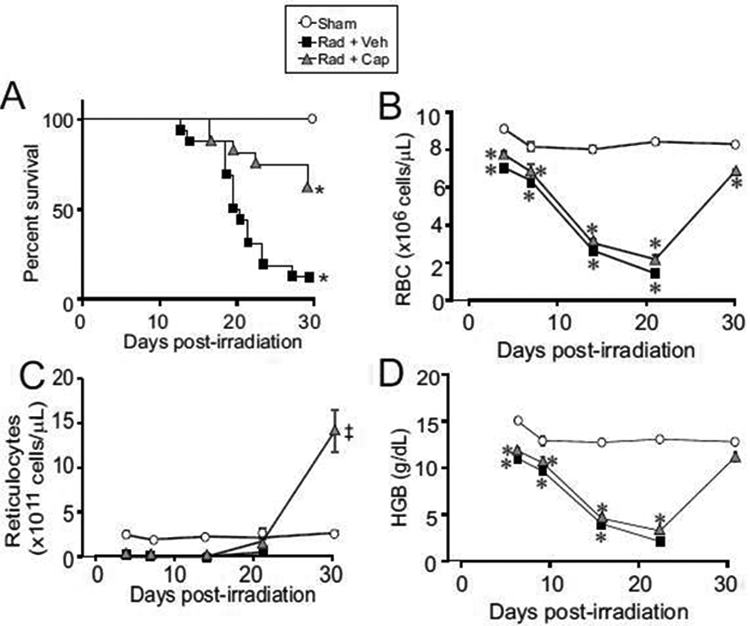
C57BL/6J mice, 12-14 weeks of age, were exposed to total body 60Co irradiation or sham irradiated (sham). Irradiated mice either received vehicle (Rad + Veh) or captopril (Rad + Cap). A. Kaplan-Meier Curves of the effects of reduced dosage and delayed administration of captopril on survival from TBI. For irradiated groups, n = 16- 20; for the sham irradiated group, n = 8. The percentage of surviving mice are shown. B-D. Blood was obtained at 3, 7, 14, 21 and 30 days post-irradiation for analysis and quantification of RBC (B), reticulocytes (C), and hemoglobin (HBG) (D). Data show means ± SEM, n = 3-5 per group, except for the 30 day time point for radiation + vehicle, which had only one animal (indicated by ‡). * indicates p < 0.05 compared with sham irradiated mice at the same time point. Figures A, B, and C were modified and are reproduced here with permission [24].
Iron accumulates in macrophages of the bone marrow following TBI.
Previous studies showed that dietary iron overload can lead to iron deposition in the bone marrow [15]. A recent study of TBI also showed that iron can be deposited in the bone marrow [33]. We examined bone marrow sections from mice exposed to TBI to determine whether radiation-induced hemolysis also increased in the bone marrow in response to TBI. Iron-containing lysosomes (siderosomes) are detectable by Prussian blue staining [34], and were visibly present in the bone marrow within 7 days following TBI (Fig. 2A-D). Numbers of stained cells, total area of staining, and the average size of Prussian blue-stained aggregates were determined and compared with control bone marrow to determine relative increases in bone marrow iron in siderosomes (Fig. 2B-D). We observed an ~8-10-fold increase in the number of cells with detectable Prussian blue staining granules at 7 days of radiation exposure compared with control, which were maximal at ~21 days post-irradiation (p<0.05, Fig. 2B). The total area of staining was reflective of the numbers of positively stained cells, also peaking at 21 days post-irradiation (p<0.05, Fig. 2C). Interestingly, the average size of stained aggregates in irradiated bone marrow also peaked at ~21 days post-irradiation at ~4.5-fold larger than in control bone (p<0.05, Fig. 2D). Captopril treatment had no significant effect on radiation-induced changes in Prussian blue staining.
Figure 2. Iron deposition in the bone marrow following TBI.

C57BL/6J mice, 12-14 weeks of age, were exposed to 7.9 Gy total body or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (13 mg/kg/day, Rad + Cap). Sternebrae were obtained euthanasia from sham and irradiated animals at 7, 14, 21, and 30 days post-irradiation. A. Representative images of Prussian blue-stained sternebrae sections from sham irradiated animals and 7.9 Gy total body irradiated animals at 14 days post-irradiation. 20× magnification is shown. B-D. Images were analyzed from four random fields from each slide. Number of Prussian blue positive staining cells per 20× field (B). Total Prussian bluelabeled area per 20× field (C). Average size of Prussian blue stained aggregate per 20× field. Data show means ± SEM, n=4-5 mice/group (D). Graphs show means ± SEM, n=4-5 mice/group. * indicates p<0.05 compared with sham irradiated animals. ‡ indicates only one animal in the irradiated vehicle group at 30 days post-irradiation; statistical analysis could not be performed with this time point with this treatment group. E. Representative images of Prussian blue-stained sternebrae sections from a GATA1low transgenic mouse with spontaneous myelofibrosis at 13 months of age, 20× magnification.
To determine whether iron deposition in the bone marrow was related to loss of normal cellularity, we examined the bone marrow from GATA1low mice. GATA1 deficiency in the hematopoietic system induces aberrant megakaryocytopoiesis, with hyperproliferating progenitors defective in terminal differentiation, leading to impaired erythropoiesis, anemia, myelofibrosis and bone marrow failure [29, 35, 36]. Prussian blue staining of bone marrow from 13-month-old GATA1low mice showed a loss of bone marrow cellularity, similar to mice exposed to TBI, but with no increase in iron deposition compared with control mice (Fig. 2E). These data suggest that the iron deposition in the bone marrow from mice exposed to TBI was not due solely to loss of cellularity in the bone marrow.
Normally, iron can be found within macrophages in the bone marrows, either within the cytoplasm in the form of free ferritin molecules or in the lysosome as aggregated ferritin and/or in complex with hemosiderin in siderosomes [37]. In pathological conditions of iron overloading, the endothelial cells of the sinusoids and plasma cells may also contain iron [37]. Histological analysis showed that Prussian blue staining was observed primarily within macrophages, with exclusion of stain from the nuclei, following TBI (Fig. 3). Smaller areas of staining were observed in some erythrocytes. This pattern of staining was observed at all time points post-irradiation (data not shown).
Figure 3. Increased Prussian blue staining of macrophages in the bone marrow following TBI.

C57BL/6J mice, 12-14 weeks of age, were exposed to 7.9 Gy total body 60Co irradiation or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (Rad + Cap). Tissues were obtained after euthanasia from sham and irradiated animals at 7, 14, 21, and 30 days post-irradiation. A. Representative images of Prussian blue-stained sternebrae sections from sham irradiated animals and 7.9 Gy total body irradiated animals at 14 days post-irradiation. Green arrows indicate Prussian blue-stained macrophages; black arrowheads indicate Prussian blue-stained erythrocytes. 90× magnification is shown. B,C. Expression of the macrophage chemokines Ccl2 (B) and Ccl3 (C) were determined by qRT-PCR of femoral bone marrow RNA at the indicated time points. Graphs show means ± SEM; n=3-5 independent animals/group. * indicates p<0.05 compared with sham irradiated animals. ‡ indicates only one animal in the irradiated vehicle group at 30 days post-irradiation; statistical analysis could not be performed with this time point with this treatment group.
Under conditions of excessive RBC damage, Kupffer cells (liver macrophages) provide substantial clearance of RBC and processing of iron [14]. Under situations of severe iron overload, such as during massive hemolysis, chemokine (C-C motif) ligand 2 (Ccl2) and chemokine (C-C motif) ligand 3 (Ccl3) are produced by the liver to attract additional macrophages [14, 38]. We therefore investigated the gene expression of these chemokines in response to TBI. Ccl2 and Ccl3 expression was significantly increased at 7 and 14 days post-irradiation (p<0.05; Fig. 3B-C). Both chemokines returned to near basal levels by 21 days post-irradiation. Captopril did not significantly alter radiation-induced expression of these chemokines.
TBI alters the regulation of genes for iron uptake and transfer.
Iron homeostasis requires the expression of specific genes for iron uptake and transport, and under conditions of hemolysis or iron overload, iron binding proteins are transcriptionally upregulated [14]. Monocytes, a cell type primarily responsible for the iron homeostasis, express transferrin, transferrin receptor, and ferroportin, among other genes, for iron uptake and binding [39]. We investigated the expression of genes for proteins involved in iron uptake, storage, and transport in the bone marrow in following TBI. Trf1, the gene encoding the transferrin protein that transports iron in the serum iron, was increased 2.5-fold at 7 days post-irradiation. Tfrc1 (transferrin receptor 1), which binds transferrin for internalization, was elevated ~3-4-fold at 7 and 14 days post-irradiation (p<0.05, Fig. 4A,B). Slc40a1 (ferroportin), for the export of iron, was also increased 4-fold at 7 and 14 days post-irradiation (p<0.05, Fig. 4C). The gene Itgam encodes integrin subunit alpha M (also known as Mac-1 or CD11b/CD18) that can bind to and import iron oxide nanoparticles [40, 41]. We found that Itgam was upregulated ~3-fold from 14-30 days post-irradiation (p<0.05, Fig. 4D). Lcn2 encodes lipocalin-2, a protein that can participate in transport and export of iron [42, 43]. In contrast with the increase of other genes, Lcn2 was down-regulated ~50% compared to basal levels in response to radiation at all time points (Fig. 4E). Finally, expression of the gene for feline leukemia virus receptor, Flvcr1, which encodes the macrophage receptor for uptake of heme iron [14], was not significantly altered by TBI (Fig. 4F). Interestingly, captopril treatment had little or no effect on radiation-induced regulation of most of these genes. Two exceptions were Itgam, which was reduced to near basal levels at 21 days post-irradiation, and Lcn2, which was increased to near basal levels at 21 days post-irradiation (Fig. 4D,E, respectively; p<0.05 compared with radiation + vehicle). Note that statistical comparisons with radiation + vehicle at the 30-day time point was not possible as only one animal survived at this time point in the vehicle-treated group.
Figure 4. Expression genes encoding iron binding and iron transport in the bone marrow following TBI.
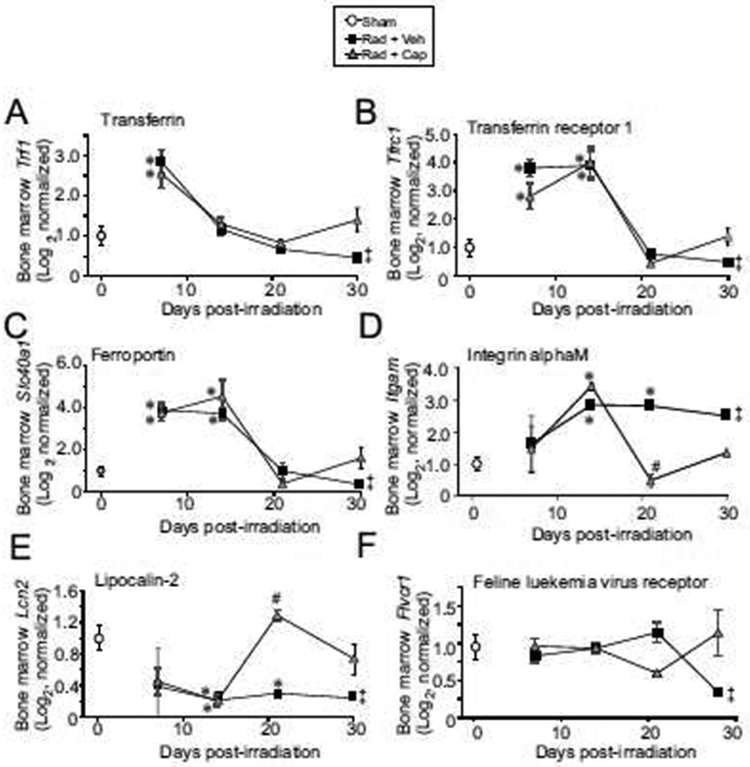
C57BL/6J mice, 12-14 weeks of age, were exposed to 7.9 Gy total body 60Co irradiation or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (Rad + Cap). RNA for qRT-PCR was purified from femoral bone marrow after euthanasia from sham and irradiated animals at 7, 14, 21, and 30 days post-irradiation. A. Tfr1; B. Tfrc1; C. Slc40a1; D. Itgam; E. Lcn2; F. Fiver1. Graphs show means ± SEM; n= 3-5 independent animals/group. * indicates p<0.05 compared with sham irradiated animals. # indicates p<0.05 from Rad + Veh group at the same time point. ‡ indicates only one animal in the irradiated vehicle group at 30 days post-irradiation; statistical analysis could not be performed with this time point with this treatment group.
Erythroblastic islands are suppressed following TBI.
Central macrophages, or “nurse macrophages” export stored iron to developing erythroid cells and provide a scaffold and environmental niche for erythroid development in the bone marrow [10, 39]. Sternebrae sections were analyzed for the formation of erythroblastic islands after TBI (Fig. 5A)[44]. Analysis showed that even when macrophages contained large iron complexes stained by Prussian blue, they were still able to form erythroid island complexes (Fig. 5A). Numbers of erythroblastic islands decreased to below detection at 7 days post-irradiation compared with control levels and remained more than 90% below basal levels for the duration of the experiment in radiation + vehicle treated animals (Fig. 5B). Captopril treatment resulted in increased levels of erythroid islands at 30 days post-irradiation to ~50% of basal levels. No statistical comparison could be made between captopril- and vehicle-treated groups at 30 days post-irradiation as only one animal survived in the vehicle group.
Figure 5. Erythroblastic island populations in the bone marrow following TBI.

C57BL/6J mice, 12-14 weeks of age, were exposed to 7.9 Gy total body 60Co irradiation or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (Rad + Cap). Sternebrae were obtained after euthanasia from sham and irradiated animals at 7, 14, 21, and 30 days post-irradiation. A. Representative images of H&E-stained sternebrae sections from an irradiated captopril-treated animal, 30 days post-irradiation. Green arrow indicates central macrophage without Prussian blue staining; black arrow indicates central macrophage with Prussian blue staining; blue arrows indicate erythroid cells. 40× magnification is shown. B. Erythroblastic islands were quantified at the indicated time points after radiation. Graphs show means ± SEM; n= 3-5 independent animals/group. ‡ indicates only one animal in the irradiated vehicle group at 30 days post-irradiation; statistical analysis could not be performed with this time point with this treatment group.
Captopril suppresses radiation-induced oxidative stress in the bone marrow.
Iron overload is associated with oxidative stress and activation of redox responses [15]. We hypothesized that iron deposition in the bone marrow could be associated with oxidative stress resulting from the production of ROS [15]. We investigated Nfe2l2 gene expression, which encodes Nrf2, in the bone marrow as a function of time post-irradiation as an indicator of oxidative stress response. Nfe2l2 mRNA levels remained at basal level until 21 days post-irradiation in vehicle treated animals, at which time Nfe2l2 expression increased to ~20-fold higher than basal levels (Fig. 6A; p<0.05). To confirm the regulation of Nfe2l2, we examined the expression of Hmox1, which is expressed in macrophages in response to the need for RBC processing and can also be regulated downstream of Nrf-2 activation [10, 14]. Hmox1 gene expression was significantly elevated from 7-21 days post-irradiation in vehicle-treated animals (Fig. 6B; p<0.05). Captopril treatment suppressed the increase in Nfe2l2 and a reduction of the Hmox1 expression at 21 days post-irradiation.
Figure 6. Expression genes for oxidative stress in the bone marrow following TBI.
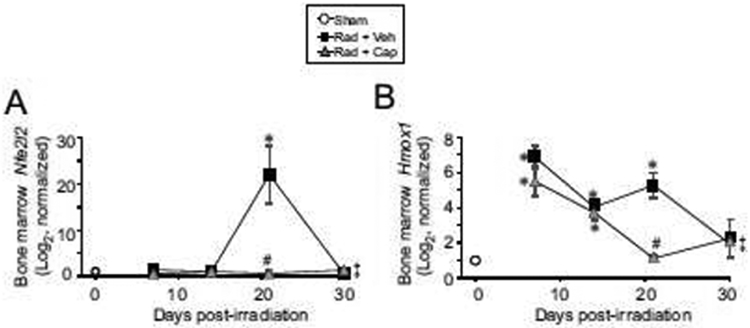
C57BL/6J mice, 12-14 weeks of age, were exposed to 7.9 Gy total body 60Co irradiation or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (Rad + Cap). RNA for qRT-PCR was purified from femoral bone marrow obtained from mice after euthanasia from sham and irradiated animals at 7, 14, 21, and 30 days post-irradiation. A. Nfe2l2; B. Hmox1. Graphs show means ± SEM; n= 3-5 independent animals/group. * indicates p<0.05 compared with sham irradiated animals. # indicates p<0.05 from Rad + Veh group at the same time point. ‡ indicates only one animal in the irradiated vehicle group at 30 days post-irradiation; statistical analysis could not be performed with this time point with this treatment group.
Expression of hepcidin is biphasically regulated following total body irradiation.
The hormone hepcidin is produced by the liver in response to serum iron concentrations. Hepcidin negatively regulates Fpn1 levels, resulting in a reduction of iron uptake from the diet in enterocytes and a reduction of transfer of iron out of macrophages to the plasma [14, 39]. Hepcidin was shown to be increased in mouse urine from 1-5 days following exposure to non-lethal levels of total body irradiation [45]. Increased levels of urinary hepcidin coincided with increased Hamp gene expression in the liver at 24-72 h post-irradiation [45]. We investigated iron deposition and Hamp expression in the livers of mice exposed to 6.85 Gy TBI (Fig. 7). Prussian blue staining of the livers could be seen within 11 days post-irradiation, with significant staining at 17-19 days post-irradiation (Fig. 7A). Prussian blue staining remained above baseline levels through 31 days post-irradiation. No significant differences were observed with captopril treatment (data not shown). Investigation of Hamp expression revealed biphasic regulation following TBI, with ~2.5-fold upregulation by 7 days post-irradiation, followed by a 70% decrease at 21 days post-irradiation (Fig. 7.B).
Figure 7. Prussian blue staining of the liver and Hamp expression following TBI.

C57BL/6J mice, 12-14 weeks of age, were exposed to 6.85 Gy total body 60Co irradiation or sham irradiated (sham). Irradiated mice received vehicle (Rad + Veh) or captopril (Rad + Cap). A. Representative images of Prussian blue-stained liver sections from sham irradiated animals and 7.7 Gy total body irradiated animals at the indicated times post-irradiation. 20× magnification is shown. B. RNA for qRT-PCR was purified from liver tissue obtained from mice after euthanasia from sham irradiated animals and from irradiated animals at 7, 14, 21, and 28 days post-irradiation. Graphs show means ± SEM; n= 4 independent animals/group. * indicates p<0.05 compared with sham irradiated animals.
Discussion
The hematopoietic system is sensitive to radiation damage, and mortality from 4-6 Gy TBI in humans is believed to occur from hematopoietic insufficiency. Ionizing radiation induces apoptosis in white blood cells, but RBC and reticulocytes undergo radiation-induced hemolysis, resulting in the release of iron in the blood [20]. However, the impact of the rapidly released iron on the bone marrow is not completely understood. Our data indicate there is an 8-10-fold increase in iron-containing hemosiderin in the bone marrow of mice following exposure to TBI as determined by Prussian blue staining. The time course of increased levels of iron within hemosiderin in the bone marrow correlated with radiation-induced loss of RBC and reticulocytes. Interestingly, a murine model of spontaneous bone marrow failure which also reduced levels of RBC, the GATA1low model of myelofibrosis, did not display increased iron deposition in the bone marrow, suggesting that the iron accumulation following TBI may be related to rapid hemolysis and not to the loss of normal bone marrow cellularity or anemia. Our data demonstrate increased expression of genes encoding proteins for iron binding, transport and storage, suggesting that an active process was induced for the sequestering levels iron in the bone marrow (Fig. 8). We previously demonstrated that captopril treatment improved survival and hematopoietic recovery in mice exposed to TBI [21-24]. In our current study, we found that captopril did not affect the levels or time course of iron deposition in the bone marrow. However, captopril was associated with improved recovery of erythroblastic islands and suppression of oxidative stress-related gene expression in the bone marrow following TBI.
Figure 8. Schematic of radiation-induced alterations in iron handling following total body irradiation.

Following exposure to lethal levels of total body irradiation, there is a depletion of ironcarrying RBC and reticulocytes due to radiation-induced hemolysis, resulting in increased levels of iron Fe2+ and Fe3+ in the serum. Our data indicate that Fe3+ levels increase in the bone marrow and liver, detectable by Prussian blue staining. The changes in iron in the serum result in biphasic regulation of hepcidin in the liver, with ~2.5 fold increase within 7 days post-irradiation, which can suppress iron transfer from enterocytes and macrophages. At the same time, there is increased expression of iron handling proteins in the bone marrow, including proteins for iron uptake, transport, and storage.
Macrophages are primarily responsible for iron recycling in the body [14]. Previous studies suggested that the macrophages are recruited to the liver from the bone marrow for chelation of excessive iron in dietary iron overload [38]. Our study found that following TBI and hemolysis, increased bone marrow iron was found primarily within macrophages. Interestingly, we found that genes for the expression of two macrophage chemoattractants, Ccl2 and Ccl3, increased in the bone marrow significantly after TBI [38]. The increased expression of monocyte recruitment genes in the bone marrow suggests that the bone marrow may produce signals to retain macrophages or recruit them from other tissues.
Our data indicate that there is a robust regulation of genes associated with iron binding, transport and storage in the bone marrow following TBI. We found increased expression of genes encoding Trf1, Tfrc1, and Slc40a1. We also identified the increase in Itgam, the gene encoding integrin alphaMbeta2, that binds iron oxide particles for internalization. In contrast with heme-binding Flvcr1, which was downregulated following radiation, Itgam expression was increased at 14-21 weeks post-irradiation, suggesting that uptake of iron oxide may be more important post-irradiation than uptake of heme iron. Additionally, gene expression of Lcn2, encoding lipocalin-2 which binds iron in siderophores, was suppressed ~50% following TBI. This suggests that iron released in response to radiation is not all contained within heme or ferritin complexes, potentially requiring the upregulation of different classes of iron-binding proteins. Our data indicate the simultaneous increase in enzymes for iron import, export and storage. This complex regulation of genes responsible for iron homeostasis has also been observed in the liver in rats following bile duct ligation to induce obstructive cholestasis [46]. Our current data does not allow the identification of individual cell types that express genes for iron uptake, export or storage, and it is possible that the genes encoding opposing enzymes are expressed differentially. Our data also show that Hamp, the gene encoding hepcidin, was biphasically regulated in the liver following TBI, with ~2.5 upregulation at 7 days post-irradiation followed by 70% suppression at 21 days post-irradiation. Hepcidin negatively regulates protein levels of Fpn1 by protein degradation to reduce export of iron by enterocytes and macrophages [14]. This suggests that although gene expression of Fpn1 is increased in the bone marrow at 7 days following radiation, the protein levels may be reduced. Further research is required to determine changes in iron binding protein levels following radiation exposure, and to identify specific cell types in which these protein levels are altered.
Our data suggest that captopril treatment did not impact expression of Hamp by the liver or many of the iron handling genes induced in the bone marrow following radiation exposure. Interestingly, captopril treatment suppressed Itgam expression and enhanced lipocalin-2 expression back to baseline levels, suggesting that captopril treatment may normalize some aspect of iron processing. Previous studies have demonstrated that Ang II can affect iron metabolism, increasing the levels of iron transporters and leading to increased iron tissue levels [47-49]. In a murine model of Ang II-induced hypertension, treatment with an Ang II receptor blocker reduced tissue levels of iron, which was attributed to reduced levels of Fpn1 and hepcidin [50]. Further studies are required to understand the mechanism by which captopril modulates Itgam and lipocalin-2, and to determine whether these genes impact survival following radiation exposure.
Iron dysregulation is associated with oxidative stress response that can activate downstream redox stress signaling [15, 43]. Nrf2, a primary redox responsive transcription factor, is activated by nuclear translocation in response to oxidative conditions, and gene expression of Nfe2l2 is also regulated by redox stress [51]. We observed increased Nfe2l2 expression at 21 days post-irradiation. That Nfe2l2 is not elevated during the entire time course of elevated iron in the bone marrow suggests that the increased iron itself does not induce Nfe2l2. Interestingly, treatment with captopril suppressed the expression of Nfe2l2, consistent with our previous findings showing that captopril decreased inflammatory responses induced by TBI [24]. In vivo and cell culture findings showed that Ang II signaling can induce oxidative stress and cellular damage [52-55]. In contrast, Ang II receptor blockers and ACE inhibitors have been shown to reduce oxidative stress in vivo in animal models [52, 56, 57]. The contribution of the Ang II pathway to oxidative stress following radiation exposure is not well understood, and further studies are needed to determine whether Ang II pathway activation may be involved in delayed tissue injuries after radiation exposure.
Previous reports demonstrated that experimentally induced iron overload in mice has deleterious effects on the hematopoietic system, with reduced proliferation of hematopoietic stem and progenitor cells in the bone marrow [15]. Studies of patients with iron overload, from myelodysplastic syndrome or ironoverload anemia, showed that excessive iron was associated with impaired erythroid progenitor proliferation and cytopenia [58, 59]. Excessive iron uptake also impacts the immune function of mature macrophages, suppressing normal antimicrobial activity [14, 60]. In both experimental animal studies and studies of patients with iron overload, iron chelation had beneficial effects on hematopoietic cell recovery [15, 59], and recent study demonstrated that iron chelation following TBI had beneficial effects for survival and hematopoietic recovery [33]. Additional studies are required to determine whether iron also accumulates in other tissues, and whether iron from radiation-hemolysis could be a general mechanism of tissue injury following TBI.
Highlights:
Radiation-induced hemolysis of RBC results in iron deposition in the bone marrow
Bone marrow iron deposition is associated with expression of iron binding proteins and upregulation of Nrf2, a redox stress response factor
Captopril reduces redox stress gene expression in the bone marrow after radiation
Acknowledgements
We would like to thank Drs. John Kalinich, Armed Forces Radiobiology Research Institute, and Dr. Michael R. Landauer for critically reading this manuscript. Thanks to Akira Isaac, Department of Pharmacology and Molecular Therapeutics, USUHS. This work was supported by Defense Medical Research and Development Program (DMRDP) DM178018, a Leukemia & Lymphoma Society grant to RMD, and National Institutes of Health (National Cancer Institute) grant R01 CA184168 (P.I. Albert J Fornace Jr, former P.I. Elliott Rosen). Some of the authors are employees of the U.S. Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C §101 defines a U.S. Government work as a work prepared by a military service member or employees of the U.S. Government as part of that person’s official duties. The views in this article are those of the authors and do not necessarily reflect the views, official policy, or position of the Uniformed Services University of the Health Sciences, the Armed Forces Radiobiology Research Institute, Department of the Navy, Department of Defense, or the U.S. Federal Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All authors confirm that there are no financial or non-financial conflicts of interest.
References
- [1].Macia IGM, Lucas Calduch A, Lopez EC. Radiobiology of the acute radiation syndrome. Rep Pract Oncol Radiother. 2011;16:123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kennedy AR, Maity A, Sanzari JK. A review of radiation-induced coagulopathy and new findings to support potential prevention strategies and treatments. Radiat Res. 2016;186:121–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Wagemaker G Heterogeneity of radiation sensitivity of hemopoietic stem cell subsets. Stem Cells. 1995; 13 Suppl 1:257–260. [DOI] [PubMed] [Google Scholar]
- [4].Singh VK, Romaine PL, Newman VL, Seed TM. Medical countermeasures for unwanted CBRN exposures: part II radiological and nuclear threats with review of recent countermeasure patents. Expert Opin Ther Pat. 2016;26:1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Peslak SA, Wenger J, Bemis JC, et al. Sublethal radiation injury uncovers a functional transition during erythroid maturation. Exp Hematol. 2011;39:434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cheki M, Shirazi A, Mahmoudzadeh A, Bazzaz JT, Hosseinimehr SJ. The radioprotective effect of metformin against cytotoxicity and genotoxicity induced by ionizing radiation in cultured human blood lymphocytes. Mutat Res. 2016;809:24–32. [DOI] [PubMed] [Google Scholar]
- [7].Puchala M, Szweda-Lewandowska Z, Kiefer J. The influence of radiation quality on radiation-induced hemolysis and hemoglobin oxidation of human erythrocytes. J Radiat Res. 2004;45:275–279. [DOI] [PubMed] [Google Scholar]
- [8].Zhang B, Liu B, Zhang H, Wang J. Erythrocyte stiffness during morphological remodeling induced by carbon ion radiation. PLoS One. 2014;9:e112624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Evstatiev R, Gasche C. Iron sensing and signalling. Gut. 2012;61:933–952. [DOI] [PubMed] [Google Scholar]
- [10].Andrews NC. Iron homeostasis: insights from genetics and animal models. Nat Rev Genet. 2000;1:208–217. [DOI] [PubMed] [Google Scholar]
- [11].Daher R, Karim Z. Iron metabolism: State of the art. Transfus Clin Biol. 2017;24:115–119. [DOI] [PubMed] [Google Scholar]
- [12].Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82-83:969–974. [DOI] [PubMed] [Google Scholar]
- [13].Valko M, Jomova K, Rhodes CJ, Kuca K, Musilek K. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch Toxicol. 2016;90:1–37. [DOI] [PubMed] [Google Scholar]
- [14].Nairz M, Theurl I, Swirski FK, Weiss G. "Pumping iron"-how macrophages handle iron at the systemic, microenvironmental, and cellular levels. Pflugers Arch. 2017;469:397–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chai X, Li D, Cao X, et al. ROS-mediated iron overload injures the hematopoiesis of bone marrow by damaging hematopoietic stem/progenitor cells in mice. Sci Rep. 2015;5:10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nairz M, Haschka D, Demetz E, Weiss G. Iron at the interface of immunity and infection. Front Pharmacol. 2014;5:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sullivan JL. Iron in arterial plaque: modifiable risk factor for atherosclerosis. Biochim Biophys Acta. 2009;1790:718–723. [DOI] [PubMed] [Google Scholar]
- [18].Zhang J, Zheng L, Wang Z, et al. Lowering iron levels protects against bone loss in focally irradiated and contralateral femurs through distinct mechanisms. Bone. 2019;120:50–60. [DOI] [PubMed] [Google Scholar]
- [19].Xie L-H, Zhang X-H, Hu X-D, Min X-Y, Zhou Q-F, Zhang H-Q. Mechanisms of an increased level of serum iron in gamma-irradiated mice. Radiat Environ Biophys. 2016;55:81–88. [DOI] [PubMed] [Google Scholar]
- [20].Zhang XH, Lou ZC, Wang AL, Hu XD, Zhang HQ. Development of serum iron as a biological dosimeter in mice. Radiat Res. 2013;179:684–689. [DOI] [PubMed] [Google Scholar]
- [21].Davis TA, Landauer MR, Mog SR, et al. Timing of captopril administration determines radiation protection or radiation sensitization in a murine model of total body irradiation. Exp Hematol. 2010;38:270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barshishat-Kupper M, Mungunsukh O, Tipton AJ, et al. Captopril modulates hypoxia-inducible factors and erythropoietin responses in a murine model of total body irradiation. Exp Hematol. 2011;39:293–304. [DOI] [PubMed] [Google Scholar]
- [23].Day RM, Davis TA, Barshishat-Kupper M, McCart EA, Tipton AJ, Landauer MR. Enhanced hematopoietic protection from radiation by the combination of genistein and captopril. Int Immunopharmacol. 2013;15:348–356. [DOI] [PubMed] [Google Scholar]
- [24].McCart EA, Lee YH, Jha J, et al. Delayed Captopril administration mitigates hematopoietic injury in a murine model of total body irradiation. Sci Rep. 2019;9:2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Davis TA, Mungunsukh O, Zins S, Day RM, Landauer MR. Genistein induces radioprotection by hematopoietic stem cell quiescence. Int J Radiat Biol. 2008;84:713–726. [DOI] [PubMed] [Google Scholar]
- [26].Xing L, Franz MG, Marcelo CL, Smith CA, Marshall VS, Robson MC. Amnion-derived multipotent progenitor cells increase gain of incisional breaking strength and decrease incidence and severity of acute wound failure. J Burns Wounds. 2007;7:e5. [PMC free article] [PubMed] [Google Scholar]
- [27].Escribano GMJ, Torrado Duran S, Torrado Duran JJ. [Stability of an aqueous formulation of captopril at 1 mg/ml]. 2005;29:30–36. [DOI] [PubMed] [Google Scholar]
- [28].Wooding WM. Planning Pharmaceutical Clinical Trials: Basic Statistical Principles. New York: John Wiley & Sons, Inc.; 1994. [Google Scholar]
- [29].Corey SJ, Jha J, McCart EA, et al. Captopril mitigates splenomegaly and myelofibrosis in the Gata1(low) murine model of myelofibrosis. J Cell Mol Med. 2018;22:4274–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mukhopadhyay T, Roth JA. Silicone lubricant enhances recovery of nucleic acids after phenol-chloroform extraction. Nucleic Acids Res. 1993;21:781–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. [DOI] [PubMed] [Google Scholar]
- [32].Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang X, Xing X, Liu H, et al. Ionizing radiation induces ferroptosis in granulocytemacrophage hematopoietic progenitor cells of murine bone marrow. Int J Radiat Biol. 2020:1–12. [DOI] [PubMed] [Google Scholar]
- [34].Dullmann J, Wulfhekel U, Nielsen P, Heinrich HC. Iron overload of the liver by trimethylhexanoylferrocene in rats. Acta Anatomica. 1992;143:96–108. [DOI] [PubMed] [Google Scholar]
- [35].Garcia P, Berlanga O, Vegiopoulos A, Vyas P, Frampton J. c-Myb and GATA-1 alternate dominant roles during megakaryocyte differentiation. J Thromb Haemost. 2011;9:1572–1581. [DOI] [PubMed] [Google Scholar]
- [36].Liew CW, Rand KD, Simpson RJ, et al. Molecular analysis of the interaction between the hematopoietic master transcription factors GATA-1 and PU.1. J Biol Chem. 2006;281:28296–28306. [DOI] [PubMed] [Google Scholar]
- [37].Wulfhekel U, Dullmann J. The diagnostic value of bone marrow iron. Folia Haematol Int Mag Klin Morphol Blutforsch. 1990;117:419–434. [PubMed] [Google Scholar]
- [38].Theurl I, Hilgendorf I, Nairz M, et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat Med. 2016;22:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sukhbaatar N, Weichhart T. Iron regulation: macrophages in control. Pharmaceuticals (Basel). 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].von Zur Muhlen C, von Elverfeldt D, Bassler N, et al. Superparamagnetic iron oxide binding and uptake as imaged by magnetic resonance is mediated by the integrin receptor Mac-1 (CD11b/CD18): implications on imaging of atherosclerotic plaques. Atherosclerosis. 2007;193:102–111. [DOI] [PubMed] [Google Scholar]
- [41].von zur Muhlen C, Fink-Petri A, Salaklang J, et al. Imaging monocytes with iron oxide nanoparticles targeted towards the monocyte integrin MAC-1 (CD11b/CD18) does not result in improved atherosclerotic plaque detection by in vivo MRI. Contrast Media Mol Imaging. 2010;5:268–275. [DOI] [PubMed] [Google Scholar]
- [42].Barasch J, Hollmen M, Deng R, et al. Disposal of iron by a mutant form of lipocalin 2. Nat Commun. 2016;7:12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Jung M, Mertens C, Tomat E, Brune B. Iron as a central player and promising target in cancer progression. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chasis JA. Erythroblastic islands: specialized microenvironmental niches for erythropoiesis. Curr Opin Hematol. 2006;13:137–141. [DOI] [PubMed] [Google Scholar]
- [45].Iizuka D, Yoshioka S, Kawai H, et al. Hepcidin-2 in mouse urine as a candidate radiationresponsive molecule. J Radiat Res. 2016;57:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kolouchova G, Brcakova E, Hirsova P, et al. Modification of hepatic iron metabolism induced by pravastatin during obstructive cholestasis in rats. Life Sci. 2011;89:717–724. [DOI] [PubMed] [Google Scholar]
- [47].Tajima S, Tsuchiya K, Horinouchi Y, et al. Effect of angiotensin II on iron-transporting protein expression and subsequent intracellular labile iron concentration in human glomerular endothelial cells. Hypertens Res. 2010;33:713–721. [DOI] [PubMed] [Google Scholar]
- [48].Ishizaka N, Saito K, Furuta K, et al. Angiotensin II-induced regulation of the expression and localization of iron metabolism-related genes in the rat kidney. Hypertens Res. 2007;30:195–202. [DOI] [PubMed] [Google Scholar]
- [49].Ishizaka N, Saito K, Noiri E, et al. Administration of ANG II induces iron deposition and upregulation of TGF-beta1 mRNA in the rat liver. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1063–1070. [DOI] [PubMed] [Google Scholar]
- [50].Tajima S, Ikeda Y, Enomoto H, et al. Angiotensin II alters the expression of duodenal iron transporters, hepatic hepcidin, and body iron distribution in mice. Eur J Nutr. 2015;54:709–719. [DOI] [PubMed] [Google Scholar]
- [51].Basak P, Sadhukhan P, Sarkar P, Sil PC. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol Rep. 2017;4:306–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Imanishi T, Tsujioka H, Akasaka T. Endothelial progenitor cells dysfunction and senescence: contribution to oxidative stress. Curr Cardiol Rev. 2008;4:275–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Luther JM, Brown NJ. The renin-angiotensin-aldosterone system and glucose homeostasis. Trends Pharmacol Sci. 2011;32:734–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Endtmann C, Ebrahimian T, Czech T, et al. Angiotensin II impairs endothelial progenitor cell number and function in vitro and in vivo: implications for vascular regeneration. Hypertension. 2011;58:394–403. [DOI] [PubMed] [Google Scholar]
- [55].Zhang M, Mao Y, Ramirez SH, Tuma RF, Chabrashvili T. Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience. 2010;171:852–858. [DOI] [PubMed] [Google Scholar]
- [56].Kangussu LM, Marzano LAS, Souza CF, Dantas CC, Miranda AS, Simoes ESAC. The renin-angiotensin system and the cerebrovascular diseases: experimental and clinical evidence. Protein Pept Lett. 2019. [DOI] [PubMed]
- [57].Sharma N, Anders HJ, Gaikwad AB. Fiend and friend in the renin angiotensin system: An insight on acute kidney injury. Biomed Pharmacother. 2019;110:764–774. [DOI] [PubMed] [Google Scholar]
- [58].Hartmann J, Braulke F, Sinzig U, et al. Iron overload impairs proliferation of erythroid progenitors cells (BFU-E) from patients with myelodysplastic syndromes. Leuk Res. 2013;37:327–332. [DOI] [PubMed] [Google Scholar]
- [59].Taoka K, Kumano K, Nakamura F, et al. The effect of iron overload and chelation on erythroid differentiation. Int J Hematol. 2012;95:149–159. [DOI] [PubMed] [Google Scholar]
- [60].Martins R, Maier J, Gorki AD, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. 2016;17:1361–1372. [DOI] [PubMed] [Google Scholar]