

Abstract
Rheumatic heart disease (RHD), an autoinflammatory heart disease, was recently declared a global health priority by the World Health Organization. Here we report a genome-wide association study (GWAS) of RHD susceptibility in 1,163 South Asians (672 cases; 491 controls) recruited in India and Fiji. We analysed directly obtained and imputed genotypes, and followed-up associated loci in 1,459 Europeans (150 cases; 1,309 controls) from the UK Biobank study. We identify a novel susceptibility signal in the class III region of the human leukocyte antigen (HLA) complex in the South Asian dataset that clearly replicates in the Europeans (rs201026476; combined odds ratio 1.81, 95% confidence intervals 1.51–2.18, P = 3.48×10−10). Importantly, this signal remains despite conditioning on the lead class I and class II variants (P = 0.00033). These findings suggest the class III region is a key determinant of RHD susceptibility offering important new insight into pathogenesis while partly explaining the inconsistency of earlier reports.
Subject terms: Genome-wide association studies, Immunogenetics
Introduction
Rheumatic heart disease (RHD) is one of the leading causes of cardiovascular death and disability in children and young adults globally1,2. The disease is caused by an aberrant immunological response to Streptococcus pyogenes (also termed group A streptococcus), a process that causes scarring and thickening of the heart valves3. Beginning in childhood, RHD gradually causes the heart to fail, leading to complications including arrhythmias, stroke and early death3. A recent analysis by the Global Burden of Disease Consortium estimated 319,400 deaths and 10.5 million disability-adjusted life-years (DALYs) each year globally due to RHD2, a substantial disease burden, especially in comparison to other diseases with infectious aetiology4,5. In 2015, the highest age-standardised mortality due to RHD outside Oceania was observed in South Asia, with a total of 119,110 deaths in India alone2.
While much about the pathogenesis of RHD remains uncertain, the disease is generally considered to be autoimmune in nature with several factors relating to the pathogen itself and the environment of the host likely to impact risk6. In addition, host genetic variation is widely thought to play a role7, not least because of the higher concordance of acute rheumatic fever among monozygotic compared to dizygotic twins8. To date, two genome-wide association studies (GWAS) have been published: the first set in diverse populations in Oceania9, and the second in Aboriginal Australians10. Consistent with several studies predating the GWAS era, which linked the disease to the human leukocyte antigen (HLA) complex on chromosome 611, the Australian study found a signal that peaked in the class II region of HLA just below genome-wide significance, which was fine-mapped to a single nucleotide polymorphism (SNP) located within intron 1 of HLA-DQA110. The pre-GWAS results should be interpreted with considerable caution, given the variable genotyping approaches, small sample size, limited quality control and confounding due to genetic ancestry11. Across the pre-GWAS reports, there are no clear examples of the same HLA allele being associated with susceptibility in two or more studies11,12. Additionally, the specific classical alleles that best explained the Australian signal were not reported in any of the candidate gene studies11.
In contrast to the Australian study, however, our Oceanian study found negligible signal in the HLA complex9, a surprising finding given the putative role for HLA in the disease’s pathogenesis6. While we cannot be certain, it is possible this result represents a false negative, although it is notable the study was adequately powered to detect the large effect sizes that have been reported previously11. We speculate, therefore, that the negative result might be attributable to the substantial genetic heterogeneity within the study population, which could have diluted out a HLA signal, in which the underlying causal variants occurred on distinct background haplotypes in each of the ancestral groups. On balance, while we consider it highly likely that HLA variants contribute to RHD susceptibility, there is a clear need to clarify the causal variants of these association signals.
Here we report a GWAS of susceptibility to RHD limited to the South Asian population, motivated by the substantial burden of RHD within this region and the need to refine the HLA and other genetic signals previously associated with this disease. We identify in the South Asians a novel susceptibility signal in the class III region of the HLA complex that clearly replicates in a follow-up analysis of an independent European dataset derived from the UK Biobank, a resource selected for study on the basis of robust HLA reference data. Importantly, we show the class III signal remains apparent despite conditioning on the lead variants in class I and class II, suggesting that at least one underlying causal variant is situated in the class III region. This finding is significant, not only because of the numerous immunologic genes, including complement components, located in the class III region, but also since it likely goes some way to explaining the inconsistency of earlier reports.
Results
Genome-wide analysis
In total, 854 Northern Indians (510 cases and 344 controls) and 309 Fijian Indians (162 cases and 147 controls) passed QC and were included (see Supplementary Fig. S1; Supplementary Fig. S2). A single signal situated in the class III region of the HLA complex reached genome-wide significance (see Supplementary Fig. S3a; Supplementary Fig. S4a) with minimal evidence of residual confounding (λ = 0.9967; see Supplementary Fig. S3b). The top variant (rs201026476) in this region, with an imputation information metric score of 0.86 for the Fijian Indians and 0.87 for the Northern Indians, had a MAF of 0.15, and each copy of the minor allele was associated with a two-fold increased risk of disease (odds ratio, OR, 1.99, 95% confidence intervals, CI, 1.58–2.51, P = 7.45×10−9). The second and third strongest signals were found in the class I (HLA-B, rs3819306, P = 1.91×10−7) and class II (HLA-DQB1, rs28724238, P = 7.77×10−7) regions, respectively.
To further define this signal, we performed stepwise conditional analyses by adding the dose of each associated allele as a covariate to the model (see Supplementary Fig. S4). After conditioning on the class III signal, the strongest signal (rs3819306) was located in HLA-B (OR 1.39, 95% CI 1.20–1.61, P = 1.83×10−5; see Supplementary Fig. S4b). However, conditioning on the lead SNPs in HLA-B and HLA-DQB1, the lead SNP in class III remained associated with susceptibility (P = 0.00026) suggesting an independent effect (see Supplementary Fig. S4c). The previously reported rs927262210 was not associated with susceptibility (PLMM = 0.28).
To validate our findings, we examined the HLA locus in the European UK Biobank dataset (150 cases of mitral stenosis and 1309 controls; see Supplementary Table S1), combining the resulting association statistics from this analysis with those from the two South Asian populations (Fig. 1). The peak SNP in class III was associated with susceptibility in the UK Biobank data in the same direction (rs201026476, OR 1.54, 95% CI 1.14–2.10, PLMM = 0.0057), with a combined effect size that was consistent with the discovery analysis (OR 1.81, 95% CI 1.51–2.18, P = 3.48×10−10; Fig. 1a). The variant located in intron 4 of HLA-DQB1 (rs28724238, OR 1.75, 95% CI 1.42–2.15, P = 1.73×10−7) also replicated (PLMM = 0.017), as did the HLA-B signal, although in the combined analysis, the signal peaked at a SNP (rs9405084) located 1,286 base pairs upstream of HLA-B (OR 1.36, 95% CI 1.19–1.55, P = 3.39×10−6).
Figure 1.
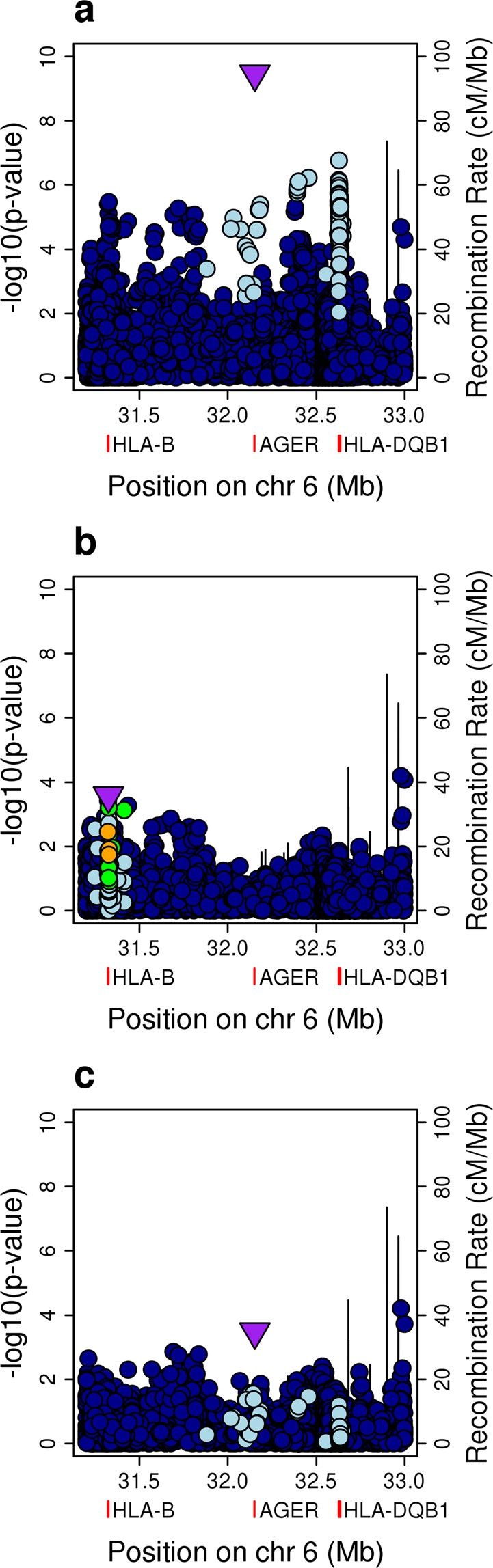
Meta-analysis of the South Asian and UK Biobank data following conditional analyses. (a) Unconditioned analysis. (b) Conditioned on the top SNP (rs201026476). (c) Conditioned on the top class I and class II SNPs (rs9405084 and rs28724238, respectively). For the HLA region, genomic position is plotted against the negative common logarithm of the P value from meta-analysis. The top class I (b) or class III SNP (a, c) following meta-analysis is shown by a purple triangle. Variants are coloured by linkage disequilibrium (LD), with the most associated variant averaged across the entire dataset (estimated r2: dark blue, 0–0.2; light blue, 0.2–0.4; green, 0.4–0.6; orange, 0.6–0.8; red, 0.8–1.0). The location of HLA-B, HLA-DQB1 and AGER are indicated by red rectangles below the x axis. The recombination rate is shown as a line plotted on the right-hand y-axis. These plots are based on those drawn by the widely used LocusZoom software.
The conditional analyses followed a similar pattern, although after conditioning on the top class III SNP, the strongest signal (rs432375, P = 6.40 × 10−5) was located 2,290 base pairs upstream of HLA-DOA, a HLA class II alpha chain paralogue, rather than at HLA-B (Fig. 1b). However, the class I signal remained apparent, with the lead SNP a coding variant within exon 1 of HLA-B (rs1050462, P = 0.00027). After conditioning on both rs9405084 (class I) and rs28724238 (class II), the class III signal was again maintained (rs201026476, P = 0.00033; Fig. 1c).
HLA imputation analysis
To further understand the potential functional variants across the HLA region, we imputed classical HLA alleles and amino acid polymorphisms at class I and class II loci. Using the T1DGC reference panel, a reasonably high proportion of variants were accurately imputed based on the R2 metric (proportion variants with R2 > 0.80: Fijian Indian, 91.74%; Northern Indian, 91.52%; European UK Biobank, 96.16%). For comparison, when using the Pan-Asian reference panel, imputation accuracy was significantly lower (proportion variants with R2 > 0.80: Fijian Indian, 73.10%; Northern Indian, 71.10%).
The strongest allelic signal in the class II region in the South Asian analysis mapped to the HLA-DQB1*03:03 allele (OR 1.90, 95% CI 1.41–2.55, P = 2.59 × 10−5; see Supplementary Fig. S4a; Supplementary Fig. S5a), an allele imputed with high accuracy (see Supplementary Table S2 online). While this signal was maintained in the combined European and South Asian analysis (OR 1.78, 95% CI 1.38–2.29, P = 1.00 × 10−5; Fig. 2a; see Supplementary Fig. S6a), it was weaker than that at the coding change at position 185 (Thr185Ile; rs1130399) of HLA-DQB1 (Fig. 3a), which was associated with a 1.5-fold increased risk of disease (OR 1.56, 95% CI 1.31–1.85, P = 3.95 × 10−7; Fig. 3b). There was also a signal at HLA-B*40:06 (P = 0.00048; Fig. 2a), although again the signal was slightly stronger at the coding change at position -16 (Val-16Leu; rs1050462) of HLA-B (P = 5.67 × 10−5; Fig. 3a; see Supplementary Fig. S6c online).
Figure 2.
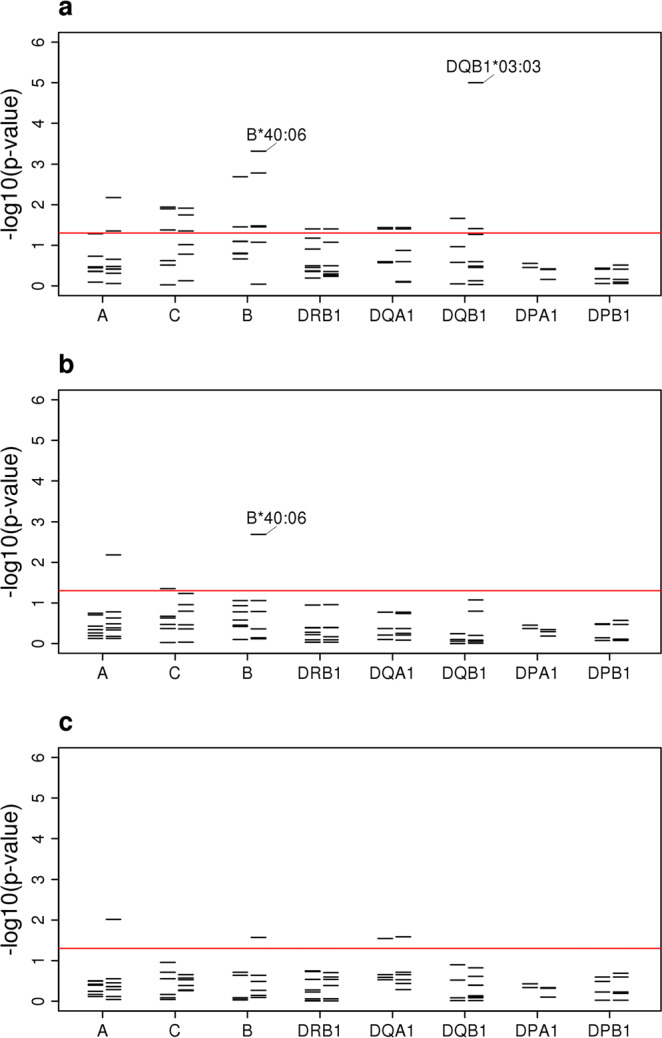
Classical HLA alleles associated with susceptibility to RHD within the South Asian and UK Biobank data following conditional analyses. (a) Unconditioned analysis. (b) Conditioned on the top SNP (rs201026476). (c) Conditioned on the top class I and class II SNPs (rs9405084 and rs28724238, respectively). For each locus, the negative common logarithm of the P value from LMM analysis is plotted with two-digit alleles to the left and four-digit alleles to the right defined by HLA imputation using SNP2HLA software with the T1DGC reference panel.
Figure 3.

Amino acid variants following HLA imputation. (a) For each locus, the negative common logarithm of the P value from LMM analysis is plotted for each amino acid polymorphism defined by HLA imputation. For HLA-DQB1 Thr185Ile and HLA-B Val-16Leu, the effect is shown in a single direction only. (b) Forest plot for the presence of isoleucine at position 185 in HLA-DQB1. For each population, the black squares centre on the odds ratio estimate from LMM on a logarithmic scale; the size of the square is proportional to the weight of the analysis. The horizontal line through each square corresponds to the confidence intervals. The black diamond centres on the combined effect estimate by fixed effects meta-analysis and stretches to the confidence intervals; the dashed line indicates no effect.
Overall, there was limited signal at the classical alleles and amino acids linked to susceptibility in the Australian study mentioned above, although we did observe an effect at the coding change at position 38 of HLA-DQB1 in the same direction (OR 0.87, 95% CI 0.76–0.99, P = 0.031; see Supplementary Table S3). Interestingly, however, HLA-DQB1*03:03 was the classical allele, with MAF > 0.5%, most associated with a self-reported history of rheumatic fever risk in a study by 23&Me (OR 1.28, 95% CI 1.05–1.55, P = 0.017)13, with an effect consistent in size and direction (combined OR 1.45, 95% CI 1.24–1.69, P = 4.05 × 10−6; see Supplementary Fig. S6b).
Finally, as in the SNP-based GWAS, the signal in class II was linked to the class III signal, while the class I signal was independent. After conditioning on the class III SNP, the strongest signal was HLA-B*40:06 (P = 0.0021; Fig. 2b). Conditioning on both the class I and class II SNPs, only a marginal signal remained at HLA-A*02:11 (P = 0.0096; Fig. 2c).
Discussion
In the first genome-wide association study of RHD to be reported outside of the Australia-Pacific region, we have resolved a complex HLA signal into its component parts. We have shown that a single HLA signal overlapping the class III region most likely comprises at least two independent coding or regulatory effects across the class I, II and III loci. While most studies to date have focused on the relationship between classical HLA alleles and susceptibility, our data suggest these signals are in fact more complex and cannot be attributed to the classical alleles alone. Indeed, based on annotations in Ensembl14, the effect of Thr185Ile in HLA-DQB1, as an example, is much more likely to be regulatory than coding, not least because it shows a strong negative association with expression of HLA-DQB1 itself14. Similarly, the independent lead class III variant (rs201026476), situated in the 3 prime UTR of the PBX2 (Pre-B-cell leukaemia transcription factor 2) gene has regulatory annotations and thus could impact expression of one or more of the numerous immunologic genes including complement components located in the class III region.
While the role of HLA polymorphism has long been suspected, there remains some doubt about the roles that individual alleles play in disease susceptibility across populations. Importantly, our analysis represents the first time HLA signals for RHD have been demonstrated with consistent direction and effect size in more than one ancestral group. Moreover, the signal at HLA-DQB1*03:03 in the 23&Me study, although based on self-reported rheumatic fever13, adds further weight to our findings. That our results differ from those reported in the Australian study10 is unsurprising, given there are likely to be substantial differences between the HLA loci of South Asians and Aboriginal Australians. Added to this, there were also a number of methodological differences, including the software employed for HLA imputation and linear mixed model analysis, which may exacerbate any disparity. Nonetheless, it is reassuring that both studies observed a signal at the coding change at position 38 of HLA-DQB1, raising the possibility that the two studies are tagging the same underlying causal variants. As noted above, we observed negligible HLA signal in our study set in Oceania including, beyond the Fijian Indian subgroup, the specific variants that associate with susceptibility in this South Asian analysis. Indeed, it may be difficult to fully unravel the contribution of HLA to RHD susceptibility in individuals of Oceanian ancestry until further HLA data are generated from these populations, enabling HLA imputation with a population-specific reference panel. Accordingly, we have begun efforts to develop such a panel by HLA typing a subset of our samples from individuals with Oceanian ancestry. Relating our findings to the HLA signals reported before the GWAS era is more difficult, not least because of the marked inconsistencies and the limitations of the studies themselves in addition to true geographical and ancestral differences. Interestingly, the presence of a signal in the class III region, which could have been differentially tagged in earlier studies, goes some way to explaining the inconsistencies of previously reported HLA associations.
This study has a few limitations. First, in comparison to some contemporary GWAS, our total sample size is relatively modest, and coupled with the small sizes of the individual subgroups (in particular the Fijian Indians), it is likely many variants with smaller effects will go undetected until larger collections are assembled. Nonetheless, our study was well powered to detect the vast majority of large effect variants reported in the candidate gene era11. Second, within Fiji, members of the general population were recruited as controls; these individuals did not undergo echocardiograms and therefore it is possible to have included a small number of undiagnosed cases of RHD. However, the prevalence of definite RHD among Fijians of Indian descent has been estimated at 3.6–4.4 cases per 1,00015,16 such that the impact of misclassification should be minimal. There may also be shortcomings associated with using the UK Biobank study, for there were no echocardiographic diagnoses available. However, specificity is likely to be regained by limiting the analysis to the mitral stenosis subgroup, an approach that is somewhat validated by the consistent replication of the South Asian signals.
Third, the genotyping array, containing ~230,000 variants following QC, was not very dense and contained, in comparison to other genotyping arrays, a limited number of variants within the HLA region. Despite this, overall HLA imputation accuracy was high when using the T1DGC reference panel. Imputation accuracy is highly dependent upon the reference panel used and as such, we have so far deliberately limited these analyses to the South Asians and Europeans for whom there are reasonable reference panels available. Fourth, this report is focused on the HLA locus because it was the only region of the genome that reached genome-wide significance in the South Asian analysis. Efforts are underway to combine these and other datasets in a genome-wide meta-analysis, facilitating follow-up of other regions, such as the immunoglobulin heavy chain locus9. Finally, at this stage, we cannot resolve the genetic determinants of sub-phenotypes, such as specific valve lesions, disease progression or complications, these are issues which larger-scale collaborative datasets should begin to tackle.
In summary, we report a major susceptibility locus for RHD in the HLA region, likely comprising at least two underlying causal variants, which strongly associates with susceptibility to RHD in South Asians and Europeans. These findings add substantially to the knowledge of the role of HLA polymorphism in susceptibility to this devastating and neglected disease. This not only has important ramifications for understanding the immunogenetic basis of the disease process, but also offers important new insight into pathogenesis.
Methods
Sample collections
For the South Asian analysis, genetic material was obtained with informed consent from cases and controls recruited to two distinct studies. Specifically, we expanded an existing collection in Northern India17–21, and we used samples from our existing collection of Pacific Islanders9, specifically the Fijians of Indian descent. Cases of RHD were defined on the basis of: a history of valve surgery for RHD, a definite RHD diagnosis by echocardiography, or borderline RHD diagnosis by echocardiography with prior acute rheumatic fever22.
In India, adults with incident or prevalent RHD were recruited as cases from a single large referral hospital, the Sanjay Gandhi Postgraduate Institute of Medical Sciences (SGPGIMS), Lucknow, Uttar Pradesh; recruitment was limited to patients with an echocardiographic diagnosis of RHD22. Controls were recruited based on normal echocardiograms and the absence of prior family history of rheumatic fever17–21. In total, DNA samples were obtained from 543 cases and 397 controls. Ethical approval for use of all samples obtained in India was granted by SGPGIMS, as well as the Oxford University Tropical Research Ethics Committee (OxTREC), and all experiments on these samples were performed in accordance with the relevant guidelines and regulations.
In Fiji, children and adults with incident or prevalent RHD were recruited as cases from either the Colonial War Memorial Hospital in Suva, or the Lautoka General Hospital in Lautoka, while members of the general population were recruited as controls, following the approach of the Wellcome Trust Case Control Consortium23. Accounting for approximately one third of the population, Fijians of Indian descent are a South Asian population who first came to Fiji from India in the 1870s under the British indentured labour scheme24. In total, DNA samples were obtained from 598 cases and 913 controls;9 of these, 170 cases and 158 controls were of Fijian Indian ancestry. Ethical approval for use of all samples obtained in Fiji was granted by the Fiji National Health Research Committee and the Fiji National Research Ethics Review Committee, as well as OxTREC, and all experiments on these samples were performed in accordance with the relevant guidelines and regulations.
Array genotyping and quality control
We obtained genetic material by sampling peripheral blood in both Fiji and India. Blood samples collected in India were stored in EDTA and frozen at −20 °C until transport to the laboratory facilities at Babasaheb Bhimrao Ambedkar University, Lucknow. Upon arrival, samples were stored at −80 °C until extraction using standard salting out procedures. Extracted DNA was prepared for analysis at the Wellcome Centre for Human Genetics (UK). The handling of blood samples collected in Fiji has previously been described, although it is noteworthy that a proportion of these samples underwent genome-wide amplification due to low DNA concentration9. From both collections, 1,268 DNA samples were genotyped at the Oxford Genomics Centre at ~300,000 variants using the HumanCore-24 BeadChip (Illumina Inc., USA). The resulting data were aligned to the forward strand of the Genome Reference Consortium Human Build 37.
After identifying and removing duplicated variants, the South Asian data was divided into two populations: Fijian Indian (n = 328) and Northern Indian (n = 940). We employed standard approaches to quality control (QC) the genotyping data25, with most steps performed using PLINK version 1.926 (see Supplementary Fig. S1; Supplementary Fig. S2).
Genome-wide imputation, association testing and meta-analysis
Imputation of genotypes not present on the array or missing was performed using the 1000 Genomes Project phase 3 reference panel27. We prephased the variants that had passed QC using SHAPEIT version 2 (r644)28 before performing genome-wide imputation using IMPUTE2 software29, excluding imputed SNPs with an information metric ≤0.4, and a minor allele frequency (MAF) ≤5%.
Genome-wide association analysis for the RHD phenotype was performed using a linear mixed model, as implemented in GCTA 1.24.4, which minimises confounding due to population structure, admixture and cryptic relatedness30. Additionally, genotypic sex was coded as a covariate for each population, as was sample type (non-amplified or whole genome amplified) for the Fijian Indians and genotyping batch for the Northern Indians. We assessed confounding using quantile-quantile plots and the test statistic inflation factor (λ), and used the accepted threshold for genome-wide significance (P < 5×10−8)31. Having estimated effect sizes by transformation32, we combined the resulting association statistics by genome-wide meta-analysis using inverse-variance-weighted fixed effects, as implemented in METASOFT33. Regional association plots, based on those drawn by the widely used LocusZoom software34, were generated for the data. We collated available data from published GWAS, including the Australian study10 and a study containing over 200,000 23&Me research participants of European ancestry, of which 1,115 were cases of self-reported rheumatic fever13.
HLA imputation analysis
HLA imputation was performed using SNP2HLA35, a software package that imputes classical HLA alleles and amino acid polymorphisms at class I (HLA-A, -B and -C) and class II (-DPA1, -DPB1, -DQA1, -DQB1 and -DRB1) loci from SNP data using the Type 1 Diabetes Genetics Consortium (T1DGC) reference panel. The T1DGC reference panel contains 5,868 SNPs and 4-digit classical HLA types for the eight loci listed above for 5,225 unrelated individuals of European ancestry. For comparison, HLA imputation was also performed with the Pan-Asian reference panel (n = 530)36; this comprises several underlying datasets with ancestry including: Singapore Chinese37; Chinese, Indian and Malaysian37; and Japanese and Han Chinese from the Phase II HapMap38. Association analyses mirrored those for the genotyping data using the imputed dosage data, rather than best-guess genotypes, but excluded alleles or amino acids with imputation accuracy R2 ≤ 0.3.
Conditional analysis
To identify secondary association signals, conditional association analyses in the SNP-based GWAS and the HLA region were performed with linear mixed models, as implemented in GCTA 1.24.4, using the same covariates as previously mentioned. Within the genome-wide dataset, we first identified the most strongly associated SNP following meta-analysis and performed stepwise iterative conditional regression, adding the dose of the associated SNP as a covariate to the model, to identify other independent signals. We also identified the most strongly associated HLA class I and class II SNPs within this same dataset and performed iterative conditional regression, adding the dose of each associated SNP as a covariate to the model, to identify additional independent signals. Conditional analyses in the HLA region were also performed by adding the dose of each of the previously mentioned SNPs as covariates to the model to see if there were additional signals attributable to HLA alleles or amino acids at each HLA locus.
Replication analysis
The replication analysis was based on the UK Biobank study, which contains genetic and phenotypic data collected on approximately 500,000 individuals from across the United Kingdom39. For the purpose of replication, we used mitral stenosis as a surrogate for RHD. Broadly, with the UK’s low prevalence of RHD, most diagnostic codes indicating RHD will represent other forms of valvular heart disease40. In contrast, codes indicating mitral stenosis, which is now a rare finding in the UK population41, are substantially more likely to indicate underlying RHD40, as the majority of mitral stenosis cases have underlying rheumatic aetiology22,42–45. Cases were therefore defined by self-report of mitral stenosis at enrolment or an International Statistical Classification of Diseases and Health Related Problems 10th Revision (ICD-10) code for rheumatic mitral stenosis (I05.0, rheumatic mitral stenosis; I05.2, rheumatic mitral stenosis with insufficiency) as a primary or other diagnosis in the hospital episode statistics or on a death certificate. Controls were selected from the remainder of the cohort, matched by age, ethnicity, deprivation index, birth outside the UK and recruitment centre, at a ratio of 1:10, beyond which the performance of linear mixed models deteriorates46. In total, we identified 196 cases and 1919 controls, of which 150 cases and 1309 controls were defined as Caucasian (i.e. European) by UK Biobank investigators (see Supplementary Table S1 online). These individuals had previously been genotyped at ~800,000 variants using the UK Biobank Axiom Array (Affymetrix, USA). These data were quality controlled by removal of individuals with missing rate > 2% and variants with missing rate > 1%, MAF < 5% or Hardy-Weinberg equilibrium (HWE) P < 1.0×10−9. The remaining preparation of the data, including genome-wide and HLA imputation, and the association analyses, mirrored the process used in the South Asian samples.
Supplementary information
Acknowledgements
We thank the investigators of the Pacific Islands Rheumatic Heart Disease Genetics Network which facilitated the sample collection in Fiji. We also thank the UK Biobank Resource (application number 11537), on which part of this research has been conducted. This work was supported by grants awarded to T.P. from the British Heart Foundation [PG/14/26/30509], the Medical Research Council [G1100449], the British Medical Association [Josephine Lansdell Grant 2018; Josephine Lansdell Grant 2012] and the National Institute for Health Research [ACF-2016-20-001], and to B.J.C. from the British Heart Foundation Centre of Research Excellence, Oxford [RE/13/1/30181]. A.J.M. was supported by a Wellcome Trust Fellowship with reference 106289/Z/14/Z and the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). A.V.S.H. is supported by a Wellcome Trust Senior Investigator Award [HCUZZ0] and by a European Research Council advanced grant [294557]. None of these funders had any role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We thank the High-Throughput Genomics Group at the Wellcome Centre for Human Genetics for generating the genotyping and sequencing data, subsidized by a core award from the Wellcome Trust [090532/Z/09/Z]. Computation used the Oxford Biomedical Research Computing (BMRC) facility, a joint development between the Wellcome Centre for Human Genetics and the Big Data Institute supported by Health Data Research UK and the NIHR Oxford Biomedical Research Centre. Financial support was provided by the Wellcome Trust Core Award Grant Number 203141/Z/16/Z. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author contributions
B.M., A.C.S., A.V.S.H. and T.P. organised and designed the study. B.M., A.C.S., A.V.S.H. and T.P. managed the study. B.M., N.G., S.K., J.K., M.L.P. and T.P. recruited the patients. K.A. and S.K. did the laboratory studies. K.A., A.J.M. and T.P. did the statistics and bioinformatics. K.A., B.M., B.J.C., A.J.M., A.V.S.H. and T.P. contributed to the interpretation of the results. K.A. wrote the first draft of the manuscript under the supervision of T.P. All authors contributed to revisions and approved the final version for publication.
Data availability
Genotype and phenotype data underlying the manuscript have been deposited in the European Genome-phenome Archive under accession numbers EGAS00001001881 (Fijian Indian data) and EGAS00001003565 (Northern Indian data). Some restrictions on access and usage apply and use is restricted to research focused on RHD.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-65855-8.
References
- 1.Lozano R, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. The Lancet. 2012;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watkins DA, et al. Global, regional, and national burden of rheumatic heart disease, 1990–2015. New England Journal of Medicine. 2017;377:713–722. doi: 10.1056/NEJMoa1603693. [DOI] [PubMed] [Google Scholar]
- 3.Marijon E, Mirabel M, Celermajer DS, Jouven X. Rheumatic heart disease. The Lancet. 2012;379:953–964. doi: 10.1016/S0140-6736(11)61171-9. [DOI] [PubMed] [Google Scholar]
- 4.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. The Lancet Infectious Diseases. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 5.Macleod CK, et al. Neglecting the neglected: the objective evidence of underfunding in rheumatic heart disease. Transactions of The Royal Society of Tropical Medicine and Hygiene. 2019;113:287–290. doi: 10.1093/trstmh/trz014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carapetis JR, et al. Acute rheumatic fever and rheumatic heart disease. Nature Reviews Disease Primers. 2016;2:15084. doi: 10.1038/nrdp.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muhamed B, Parks T, Sliwa K. Genetics of rheumatic fever and rheumatic heart disease. Nature Reviews Cardiology. 2020;17:145–154. doi: 10.1038/s41569-019-0258-2. [DOI] [PubMed] [Google Scholar]
- 8.Engel ME, Stander R, Vogel J, Adeyemo AA, Mayosi BM. Genetic susceptibility to acute rheumatic fever: a systematic review and meta-analysis of twin studies. PloS one. 2011;6:e25326–e25326. doi: 10.1371/journal.pone.0025326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parks, T. et al. Association between a common immunoglobulin heavy chain allele and rheumatic heart disease risk in Oceania. Nature Communications8, 14946, doi:10.1038/ncomms14946 https://www.nature.com/articles/ncomms14946#supplementary-information (2017). [DOI] [PMC free article] [PubMed]
- 10.Gray L-A, et al. Genome-Wide Analysis of Genetic Risk Factors for Rheumatic Heart Disease in Aboriginal Australians Provides Support for Pathogenic Molecular Mimicry. The Journal of Infectious Diseases. 2017;216:1460–1470. doi: 10.1093/infdis/jix497. [DOI] [PubMed] [Google Scholar]
- 11.Martin WJ, et al. Post-infectious group A streptococcal autoimmune syndromes and the heart. Autoimmunity Reviews. 2015;14:710–725. doi: 10.1016/j.autrev.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Bryant PA, Robins-Browne R, Carapetis JR, Curtis N. Some of the people, some of the time: susceptibility to acute rheumatic fever. Circulation. 2009;119:742–753. doi: 10.1161/CIRCULATIONAHA.108.792135. [DOI] [PubMed] [Google Scholar]
- 13.Tian C, et al. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nature Communications. 2017;8:599. doi: 10.1038/s41467-017-00257-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR. The Ensembl Regulatory Build. Genome Biology. 2015;16:56. doi: 10.1186/s13059-015-0621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Colquhoun SM, et al. Echocardiographic screening in a resource poor setting: Borderline rheumatic heart disease could be a normal variant. International Journal of Cardiology. 2014;173:284–289. doi: 10.1016/j.ijcard.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Steer AC, et al. High prevalence of rheumatic heart disease by clinical and echocardiographic screening among children in Fiji. The Journal of Heart Valve Disease. 2009;18:327–335. [PubMed] [Google Scholar]
- 17.Gupta U, et al. Association of angiotensin I-converting enzyme gene insertion/deletion polymorphism with rheumatic heart disease in Indian population and meta-analysis. Molecular and Cellular Biochemistry. 2013;382:75–82. doi: 10.1007/s11010-013-1719-2. [DOI] [PubMed] [Google Scholar]
- 18.Gupta U, et al. Signal transducers and activators of transcription (STATs) gene polymorphisms related with susceptibility to rheumatic heart disease in north Indian population. Immunology Letters. 2014;161:100–105. doi: 10.1016/j.imlet.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 19.Gupta U, et al. Influence of protein tyrosine phosphatase gene (PTPN22) polymorphisms on rheumatic heart disease susceptibility in North Indian population. Tissue Antigens. 2014;84:492–496. doi: 10.1111/tan.12440. [DOI] [PubMed] [Google Scholar]
- 20.Gupta U, et al. Association study of inflammatory genes with rheumatic heart disease in North Indian population: A multi-analytical approach. Immunology Letters. 2016;174:53–62. doi: 10.1016/j.imlet.2016.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Bhatt M, Kumar S, Garg N, Siddiqui MH, Mittal B. Influence of IL-1β, STAT3 & 5 and TLR-5 gene polymorphisms on rheumatic heart disease susceptibility in north Indian population. International Journal of Cardiology. 2019;291:89–95. doi: 10.1016/j.ijcard.2019.03.035. [DOI] [PubMed] [Google Scholar]
- 22.Reményi, B. et al. World Heart Federation criteria for echocardiographic diagnosis of rheumatic heart disease—an evidence-based guideline. Nature Reviews Cardiology9, 297–309, doi:10.1038/nrcardio.2012.7 https://www.nature.com/articles/nrcardio.2012.7#supplementary-information (2012). [DOI] [PMC free article] [PubMed]
- 23.The Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature447, 661–678, doi:10.1038/nature05911 https://www.nature.com/articles/nature05911#supplementary-information (2007). [DOI] [PMC free article] [PubMed]
- 24.Lal BV. Understanding the Indian indenture experience. South Asia: Journal of South Asian Studies. 1998;21:215–237. doi: 10.1080/00856409808723356. [DOI] [Google Scholar]
- 25.Lukacs Rita U, Goldstein Andrew S, Lawson Devon A, Cheng Donghui, Witte Owen N. Isolation, cultivation and characterization of adult murine prostate stem cells. Nature Protocols. 2010;5(4):702–713. doi: 10.1038/nprot.2010.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.The 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature526, 68–74, doi:10.1038/nature15393 https://www.nature.com/articles/nature15393#supplementary-information (2015). [DOI] [PMC free article] [PubMed]
- 28.Delaneau O, Howie B, Cox AJ, Zagury J-F, Marchini J. Haplotype estimation using sequencing reads. The American Journal of Human Genetics. 2013;93:687–696. doi: 10.1016/j.ajhg.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLOS Genetics. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Zaitlen NA, Goddard ME, Visscher PM, Price AL. Advantages and pitfalls in the application of mixed-model association methods. Nature Genetics. 2014;46:100–106. doi: 10.1038/ng.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sham PC, Purcell SM. Statistical power and significance testing in large-scale genetic studies. Nature Reviews Genetics. 2014;15:335–346. doi: 10.1038/nrg3706. [DOI] [PubMed] [Google Scholar]
- 32.Lloyd-Jones LR, Robinson MR, Yang J, Visscher PM. Transformation of summary statistics from linear mixed model association on all-or-none traits to odds ratio. Genetics. 2018;208:1397–1408. doi: 10.1534/genetics.117.300360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han B, Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. The American Journal of Human Genetics. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pruim RJ, et al. LocusZoom: regional visualization of genome-wide association scan results. Bioinformatics. 2010;26:2336–2337. doi: 10.1093/bioinformatics/btq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jia X, et al. Imputing Amino Acid Polymorphisms in Human Leukocyte Antigens. PLOS ONE. 2013;8:e64683. doi: 10.1371/journal.pone.0064683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okada Y, et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Human Molecular Genetics. 2014;23:6916–6926. doi: 10.1093/hmg/ddu387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pillai NE, et al. Predicting HLA alleles from high-resolution SNP data in three Southeast Asian populations. Human Molecular Genetics. 2014;23:4443–4451. doi: 10.1093/hmg/ddu149. [DOI] [PubMed] [Google Scholar]
- 38.de Bakker, P. I. et al. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nature Genetics38, 1166–1172, doi:10.1038/ng1885 https://www.nature.com/articles/ng1885#supplementary-information (2006). [DOI] [PMC free article] [PubMed]
- 39.Bycroft C, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–209. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katzenellenbogen JM, et al. Low positive predictive value of International Classification of Diseases, 10th Revision codes in relation to rheumatic heart disease: a challenge for global surveillance. Internal Medicine Journal. 2019;49:400–403. doi: 10.1111/imj.14221. [DOI] [PubMed] [Google Scholar]
- 41.d’Arcy JL, et al. Large-scale community echocardiographic screening reveals a major burden of undiagnosed valvular heart disease in older people: the OxVALVE Population Cohort Study. European Heart Journal. 2016;37:3515–3522. doi: 10.1093/eurheartj/ehw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olson LJ, Subramanian R, Ackermann DM, Orszulak TA, Edwards WD. Surgical pathology of the mitral valve: a study of 712 cases spanning 21 years. Mayo Clinic Proceedings. 1987;62:22–34. doi: 10.1016/S0025-6196(12)61522-5. [DOI] [PubMed] [Google Scholar]
- 43.Dare AJ, Harrity PJ, Tazelaar HD, Edwards WD, Mullany CJ. Evaluation of surgically excised mitral valves: Revised recommendations based on changing operative procedures in the 1990s. Human Pathology. 1993;24:1286–1293. doi: 10.1016/0046-8177(93)90261-E. [DOI] [PubMed] [Google Scholar]
- 44.Iung B, et al. A prospective survey of patients with valvular heart disease in Europe: The Euro Heart Survey on Valvular Heart Disease. European Heart Journal. 2003;24:1231–1243. doi: 10.1016/S0195-668X(03)00201-X. [DOI] [PubMed] [Google Scholar]
- 45.Iung B, et al. Valvular heart disease in the community: a European experience. Current Problems in Cardiology. 2007;32:609–661. doi: 10.1016/j.cpcardiol.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Zhou W, et al. Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nature Genetics. 2018;50:1335–1341. doi: 10.1038/s41588-018-0184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genotype and phenotype data underlying the manuscript have been deposited in the European Genome-phenome Archive under accession numbers EGAS00001001881 (Fijian Indian data) and EGAS00001003565 (Northern Indian data). Some restrictions on access and usage apply and use is restricted to research focused on RHD.