

Abstract
The relationship between commonly occurring genetic variants (G1 and G2) in the APOL1 gene in African Americans and different disease traits, such as kidney disease, cardiovascular disease, and pre-eclampsia, remains the subject of controversy. Here we took a genotype-first approach, a phenome-wide association study (PheWAS), to define the spectrum of phenotypes associated with APOL1 high-risk variants in 1,837 African American participants of Penn Medicine Biobank (PMBB) and 4,742 African American participants of Vanderbilt BioVU. In PMBB, outpatient creatinine measurement-based estimated glomerular filtration rate (eGFR) and multivariable regression models were used to evaluate the association between high-risk APOL1 status and renal outcomes. In meta-analysis of both cohorts, the strongest PheWAS associations were for the high-risk APOL1 variants and diagnoses codes for “renal dialysis” (OR=3.75, p=4.4×10−27) and “end stage renal disease” (OR=3.42, p=4.8×10−14). A number of phenotypes were associated with APOL1 high-risk genotypes in an analysis adjusted only for demographic variables; however, no associations were detected with non-renal phenotypes after controlling for CKD/ESRD status. Using calculated eGFR-based phenotype analysis in PMBB, APOL1 high-risk status was associated with prevalent CKD/ESRD/renal transplant (OR=2.27, 95% CI 1.67–3.08). In high-risk participants, the eGFR was 15.4 mL/min/1.73m2 lower (SE=2.3) than in low-risk participants (p=4.3×10−11). Although APOL1 high-risk variants are associated with a range of phenotypes, the risks for other associated phenotypes appear much lower and in our dataset are driven by a primary effect of renal disease.
Introduction
African Americans are at higher risk for developing chronic kidney disease (CKD) compared to European Americans, independent of socioeconomic and traditional clinical risk factors.1–3 This disparity has been attributed to the presence of high-risk alleles, termed G1 and G2, in the gene APOL1. APOL1 encodes for the protein apolipoprotein L1 (apoL1), a component of a subclass of HDL (high-density lipoprotein) particles.4 Circulating APOL1 is synthesized in the liver, but APOL1 is also expressed in the kidney, pancreas and brain5 and is widely expressed in various tissues, including the vascular endothelium, heart, lung, podocytes, proximal tubules, and arterial smooth muscle cells.6 ApoL1 has a role in innate immunity by protecting against infection by Trypanosoma brucei, a parasite transmitted by the tsetse fly. Trypanosomes endocytose apoL1 which forms pores on the lysosomal membranes of the trypanosomes and causes influx of chloride, swelling of the lysosome and lysis of the trypanosome.7 Two coding sequence variants in APOL1 have been shown to confer resistance against Trypanosoma brucei rhodesiense, which had previously developed resistance to wild type apoL1.8
The prevalence of the APOL1 variants in the African American population is substantial: approximately 35% carry one risk allele and 10–15% carry two risk alleles9–11. Homozygotes and compound heterozygotes with two copies of the high-risk APOL1 alleles have been shown to be at increased risk for prevalent11,12 and incident10 CKD, as well as earlier progression to ESRD.10,13–15 Specifically, the high-risk status is associated with a 10- to 17-fold higher odds for focal segmental glomerulosclerosis (FSGS),9,16 a 7-fold higher odds for hypertension-associated ESRD,9 and a 29- to 89-fold higher odds for HIV-associated nephropathy.16,17 Consistent with the human studies, animal model studies indicate that expression of the APOL1 high-risk variant in podocytes induces proteinuria, global glomerulosclerosis and renal failure in mice.8
The association of the APOL1 high-risk genotypes to other phenotypes has also been studied. A recent study with 5,204 African Americans from Mount Sinai’s BioMe biobank with replication in additional BioMe participants, Vanderbilt BioVU and Northwestern NUgene, showed an association between the APOL1 variant alleles and higher systolic blood pressure and earlier onset of hypertension.18 Although several studies have evaluated the association between APOL1 and increased risk for a broad spectrum of cardiovascular outcomes, such as coronary heart disease, stroke, heart failure, cardiovascular surgery, endovascular catheterization, coronary calcification, myocardial infarction, and preeclampsia, the published results are conflicting.12,19–21
Phenome-wide association analysis (PheWAS) is a new method in which associations between a single genetic variant with the spectrum of phenotypes are analyzed, using electronic healthy record (EHR)-based phenotyping. Since development of this method, it has been successfully used for multitudes of conditions to identify and confirm genotype-associated phenotypes. To explore the full spectrum of phenotypes associated with the APOL1 high-risk genotypes in an unbiased manner, we undertook this genotype-first analysis, using EHR data from the Penn Medicine Biobank (PMBB) and Vanderbilt BioVU cohorts to perform phenome-wide association testing. Then, to determine the individual-level risk for the associated phenotypes in African Americans in the PMBB population, we evaluated the association between APOL1 high-risk variant status and prevalent CKD and ESRD based on estimated glomerular filtration rate (eGFR) calculated from serum creatinine measurements. We also used longitudinal creatinine data to model changes in eGFR over time, in order to evaluate the association between APOL1 variant status and renal disease progression. In study participants with history of renal transplant, data on primary renal pathology were obtained to assess for the association between APOL1 high-risk variant status and etiology of renal disease.
Results
Amongst the 2,033 African Americans in the PMBB with completed whole-exome sequencing, 1,837 (47.5% male, mean age 58 years) had at least one outpatient serum creatinine measurement available for analyses. A total of 242 participants had at least 2 risk alleles (G1/G1, G1/G2, or G2/G2 APOL1 variants) and were considered high-risk and the remaining 1,595 participants with 0 or 1 APOL1 risk allele were considered low-risk (Table 1). Using the most recent laboratory data for each participant, the average eGFR in low-risk variant carriers was 79.3 mg/mL/1.73m2 compared to 67.5 mg/mL/1.73m2 in high-risk participants (p<0.001). Participants had a median of 14 serum creatinine measurements over a median of 5.5 years.
Table 1.
Baseline characteristics of genotyped African-American participants from Penn Medicine Biobank stratified by APOL1 risk variant status.
| Characteristic | 0 or 1 copies of APOL1 risk variant (n=1595) |
2 copies of APOL1 risk variants (n=242) |
p value* |
|---|---|---|---|
| Age, mean(SD), year | 58.6 (13.9) | 57.4 (12.2) | 0.10 |
| Male sex, n(%) | 747 (46.8) | 125 (51.7) | 0.16 |
| Medical History, n(%) | |||
| Tobacco use** | 837 (52.5) | 117 (48.4) | 0.44 |
| Hypertension | 692 (43.4) | 125 (51.7) | 0.02 |
| ESRD on dialysis | 115 (7.2) | 55 (22.7) | <0.001 |
| Renal transplant | 62 (3.9) | 39 (16.1) | <0.001 |
| HIV | 53 (3.3) | 9 (3.7) | 0.75 |
| Sickle cell | 36 (2.3) | 7 (2.9) | 0.54 |
| Clinical Variables, median (IQR) † | |||
| Systolic BP, mmHg | 130 (121–138) | 131 (122–138) | 0.37 |
| Diastolic BP, mmHg | 76 (72–81) | 77 (71–82) | 0.24 |
| BMI, kg/m2 | 32 (27–38) | 32 (27–37) | 0.90 |
| Hemoglobin, g/dL | 12.3 (10.8–13.4) | 12.2 (10.6–13.4) | 0.23 |
| HbA1c††, % | 6.0 (5.5–7.1) | 6.0 (5.6–7.1) | 0.43 |
| Serum creatinine, mg/dL | 1.04 (0.83–1.41) | 1.18 (0.90–2.06) | <0.001 |
| eGFR, mL/min/1.73 m2 | 79.3 (54.0–101.6) | 67.5 (35.8–95.9) | <0.001 |
Using chi-square test for categorical variables and non-parametric Wilcoxon Rank-Sum for continuous variables.
Using chi-square test for categorical variables and non-parametric Wilcoxon Rank-Sum for continuous variables.
Tobacco use defined as current or former smoker.
Tobacco use defined as current or former smoker.
Median calculated for group from mean values of outpatient measurements for each participant since entry into the biobank until last available visit
Median calculated for group from mean values of outpatient measurements for each participant since entry into the biobank until last available visit
Median HbA1c calculated from subjects with available measurements: n=1182 with 0 or 1 copies of risk variant and n=183 with 2 copies of risk variant
Median HbA1c calculated from subjects with available measurements: n=1182 with 0 or 1 copies of risk variant and n=183 with 2 copies of risk variant
Abbreviations: BMI = body mass index, BP = blood pressure, eGFR = estimated glomerular filtration rate, ESRD = end stage renal disease, HbA1c = hemoglobin A1c, HIV = human immunodeficiency virus, IQR = interquartile range, SD = standard deviation
Of the 4,742 African American study participants in BioVU (31.2% male, mean age 43 years), 616 participants were high-risk with 2 risk alleles, while 4,126 participants had 0 or 1 risk allele (Table 2). Besides in the renal parameters, there were no significant differences in other characteristics between low- and high-risk participants. A total of 4,667 BioVU participants had available creatinine measurements in the EHR that were used to calculate eGFR for adjusted analyses.
Table 2.
Baseline characteristics of genotyped African-American participants from Vanderbilt BioVU stratified by APOL1 risk variant status.
| Characteristic | 0 or 1 copies of APOL1 risk variant (n=4126) |
2 copies of APOL1 risk variants (n=616) |
P-value* |
|---|---|---|---|
| Age, mean(SD), year | 43.2 (21.6) | 45.2 (19.8) | 0.04 |
| Male sex, n(%) | 1286 (31.2) | 210 (33.9) | 0.16 |
| Medical History, n(%) | |||
| Hypertension | 1967 (52.6) | 344 (60.2) | <0.001 |
| ESRD on dialysis | 187 (6.4) | 98 (21.1) | <0.001 |
| Renal transplant | 144 (5.0) | 73 (16.6) | <0.001 |
| HIV | 152 (4.9) | 16 (3.3) | 0.14 |
| Sickle cell | 117 (5.5) | 15 (5.0) | 0.71 |
| Clinical Variables, median (IQR) | |||
| BMI†, kg/m2 | 28.8 (23.8–35.1) | 29.6 (24.6–35.9) | 0.026 |
Using chi-square test for categorical variables and non-parametric Wilcoxon Rank-Sum for continuous variables
Using chi-square test for categorical variables and non-parametric Wilcoxon Rank-Sum for continuous variables
Median calculated for group from mean values of inpatient and outpatient measurements for each participant since entry into the biobank until last available visit
Median calculated for group from mean values of inpatient and outpatient measurements for each participant since entry into the biobank until last available visit
Abbreviations: BMI = body mass index, ESRD = end stage renal disease, SD = standard deviation, IQR = interquartile range
Phenome-wide impact of APOL1 risk variants
Phenome-wide association testing22,23 was used to determine the association of the high- risk APOL1 genotypes to phenotypes as defined by presence of related diagnosis codes in the EHR in both the PMBB and BioVU cohorts. In PMBB, carrying two high-risk APOL1 alleles was associated with 15 of 232 phenotypes (using the pre-specified Bonferroni adjusted cut-off for significance p<2.1×10−4) (Figure 1). The strongest associations were with “kidney replaced by transplant” (OR=6.1, 95% CI 5.6–6.6, p=1.3×10−12) and “end stage renal disease” (OR=4.2, 95% CI 3.8–4.6, p=3.4×10−12). Several other kidney disease-associated phenotypes such as “anemia in chronic kidney disease”, “hypertensive heart and/or renal disease”, and “complications of cardiac/vascular device, implant and graft” passed the significance threshold.
Figure 1.
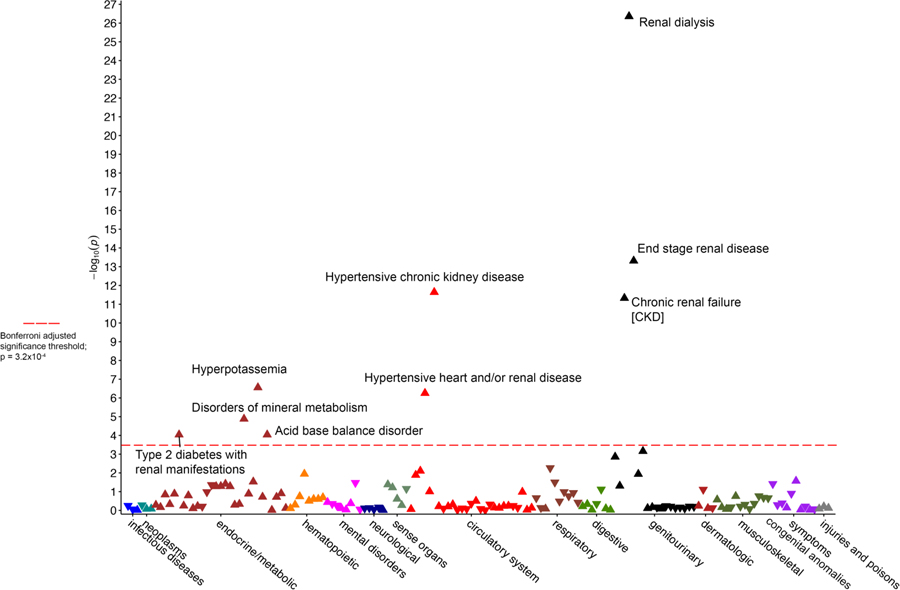
Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank. Red horizontal line designates minimum p value for statistical significance after correction for multiple testing (p=2.2×10−4)
In BioVU (Figure 2), carrying two high-risk APOL1 alleles also showed associations with phenotypes related to kidney disease. The strongest associations were with “renal dialysis” (OR= 3.6, 95% CI 2.7–4.8, p=6.7×10−19) and “end stage renal disease” (OR=3.0, 95% CI 2.3–3.9, p=1.1×10−15). As in PMBB, BioVU results also showed significant associations with a number of renal-associated phenotypes, such as “nephritis; nephrosis; renal sclerosis” and “chronic renal failure.”
Figure 2.
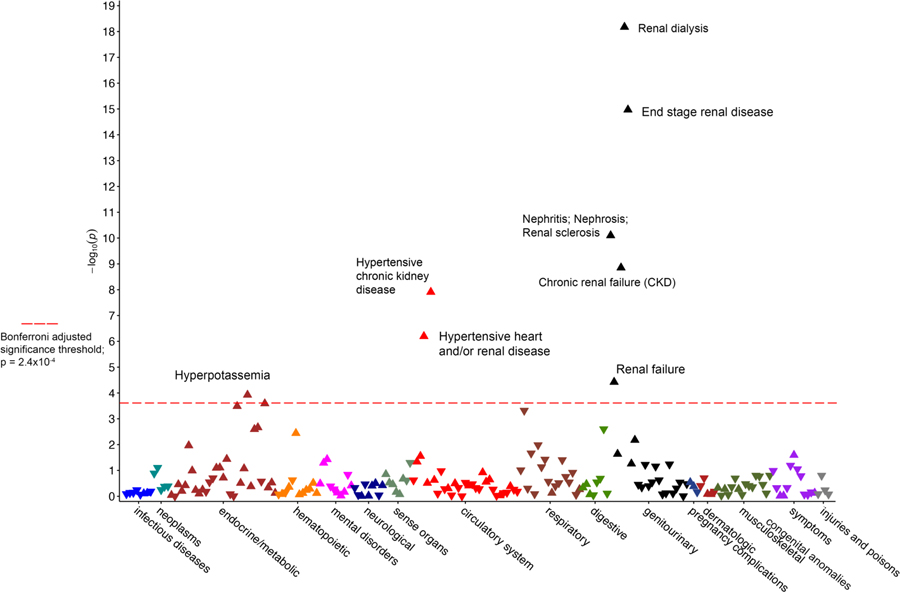
Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU. Red horizontal line designates minimum p value for statistical significance after correction for multiple testing (p=2.4×10−4).
Meta-analysis (Figure 3) of the PheWAS data from both cohorts showed strengthened associations with renal phenotypes, with an OR for “renal dialysis” of 3.8 (95% CI 2.9–4.8, p=4.4 × 10−27) and an OR for “end stage renal disease” of 3.4 (95% CI 2.5–4.7, p=4.8×10−14). We did not find any associations with non-renal phenotypes, such as those traditionally associated with CVD. Assuming a 5% CVD prevalence across the combined BioVU and PMBB cohorts (N=6,579) and the prevalence of the high-risk genotype of 13.0%, we had 80% power (with two-tailed error rate of 0.05) to detect an OR as low as 1.54.
Figure 3.
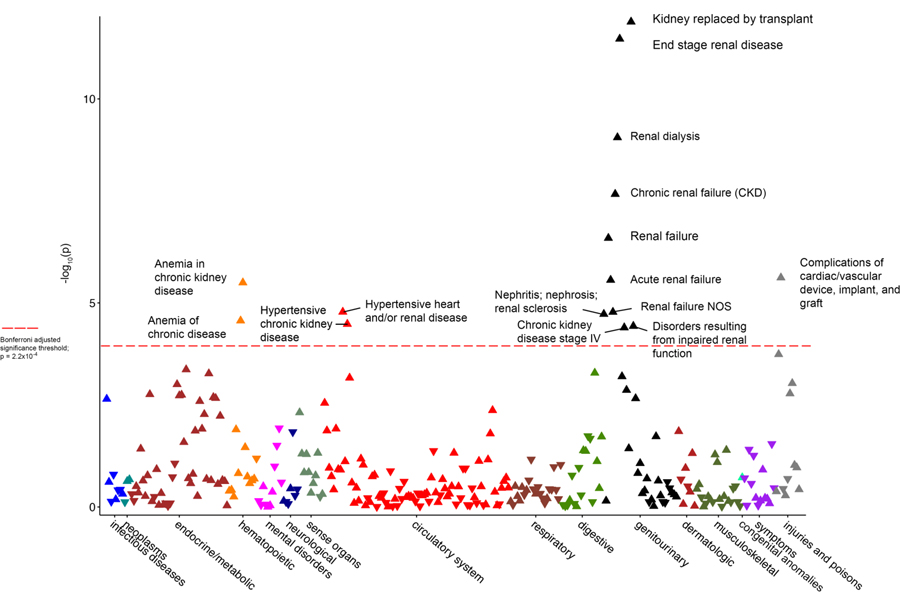
Manhattan plot of the meta-analysis of the combined Penn Medicine Biobank and Vanderbilt BioVU cohorts for phenome-wide association study data of the APOL1 high-risk genotypes. Red horizontal line designates minimum p value for statistical significance after correction for multiple testing (p=3.2×10−4)
We hypothesized that the associations between the APOL1 high-risk genotypes and the phenotypes that were statistically significant were mediated by renal disease. To formally demonstrate the dependence of the phenome-wide significant associations on kidney disease, we conducted sensitivity analyses using CKD/ESRD status as a binary covariate (defined as an eGFR<60 mL/min/1.73m2 using the most recent creatinine to calculate eGFR; eGFR was set to zero for participants with at least two diagnosis codes for dialysis or renal transplant) in the PheWAS. After controlling for CKD/ESRD, we observed a significant association with the high-risk genotype and “end stage renal disease” in PMBB (Figure S1) but no association with any phenotypes met statistical significance in BioVU (Figure S2).
Separately, the PheWAS model was additionally adjusted for each participant’s most recent eGFR in the PMBB and BioVU. When the eGFR was set to zero for participants with dialysis or renal transplant, no phenotypic associations reached significance in PMBB (Figure S3) while “gastrointestinal hemorrhage” just surpassed the significance threshold in BioVU with an OR of 0.5 and p=2.1×10−4 (Figure S4). In contrast, when the most recent eGFR was used without adjusting for dialysis or transplant status, the associations with the phenotypes “kidney replaced by transplant” (OR=6.0, 95% CI 5.3–6.7, p=5.8×10−7), “end-stage renal disease” (OR=6.5, 95% CI 5.2–7.9, p=4.1×10−7), and “renal dialysis” (OR=5.9, 95% CI 5.0–6.7, p=5.9×10−5) remained significant in the PMBB (Figure S5) after correction for multiple testing, likely signifying that individuals with these outcomes have essentially had their eGFR “normalized” by dialysis or renal transplant. Similarly, in BioVU, the associations with “renal dialysis” (OR=3.6, 95% CI 2.1–6.0, p=1.2×10−6) and “end stage renal disease” (OR=2.8, 95% CI 1.7–4.4, p=2.0×10−5) remained significant (Figure S6).
To explore the risk per copy of variant APOL1 allele, the PheWAS analyses were repeated using an additive model in PMBB (Figure S7) and BioVU (Figure S8). Results generally showed weaker associations with the same phenotypes that met significance in the recessive model. Further, there was no significant association with any phenotype after adjusting separately for CKD/ESRD status (Figures S9 and S10). When adjusting for most recent eGFR (Figures S11 and S12), only the association with “renal dialysis” in BioVU met statistical significance (OR=1.9, 95% CI 1.4–2.6, p=4.1×10−5).
Supplementary information is available at Kidney International’s website.
Association of APOL1 with clinical measurements
In addition to the diagnosis codes extracted from the EHR, we examined the association of the APOL1 high-risk genotypes with clinical measurements (BMI, systolic blood pressure, diastolic blood pressure) and lipid measurements extracted from the EHR. While there was no association with these measurements in the PMBB (Table S1), the high-risk genotype was associated with marginally higher total cholesterol (p<0.001), higher LDL cholesterol (p<0.001), lower HDL cholesterol (p=0.03), and higher triglyceride level (p<0.001) compared to participants with the low-risk genotype in BioVU (Table S2).
APOL1 and risk for renal disease
We further examined the presence of renal disease in the PMBB population using laboratory measurements along with diagnosis codes (Table 3). When using serum creatinine-based eGFR data for each participant, those with the APOL1 high-risk genotypes had increased odds of CKD or ESRD/transplant (OR=2.3, 95% CI 1.7–3.1, p=1.5×10−7). This was driven by the association with ESRD/renal transplant (OR=4.0, 95% CI 2.8–5.7, p=6.3×10−14), as the high-risk genotypes were not associated with CKD (OR=1.3, 95% CI 0.9–1.9, p=0.20), when individuals with ESRD or transplant were excluded.
Table 3.
Odds ratios (with 95% confidence intervals) for renal outcomes with presence of APOL1 high-risk variants in Penn Medicine Biobank using an unadjusted and adjusted logistic regression model.
| Model 1* | Model 2** | |||
|---|---|---|---|---|
| OR (95% CI) | p value | OR (95% CI) | p value | |
| CKD† or ESRD/renal transplant†† | 2.29 (1.71–3.07) | 2.58 × 10−8 | 2.27 (1.67–3.08) | 1.53 × 10−7 |
| CKD† | 1.36 (0.94–1.96) | 0.11 | 1.29 (0.87–1.90) | 0.20 |
| ESRD/renal transplant†† | 3.86 (2.76–5.40) | 2.78 × 10−15 | 3.97 (2.77–5.69) | 6.31 × 10−14 |
Model 1 is adjusted for age, age2, gender, and principal components 1–10 of genetic ancestry
Model 1 is adjusted for age, age2, gender, and principal components 1–10 of genetic ancestry
Model 2 additionally adjusts for mean body mass index, mean blood glucose, mean systolic blood pressure, and smoking status (current/former versus never)
Model 2 additionally adjusts for mean body mass index, mean blood glucose, mean systolic blood pressure, and smoking status (current/former versus never)
CKD defined as eGFR<60 mL/min/1.73 m2 and excluding those with the presence of a diagnosis code for dialysis or renal transplant
CKD defined as eGFR<60 mL/min/1.73 m2 and excluding those with the presence of a diagnosis code for dialysis or renal transplant
ESRD/transplant defined as eGFR < 15 mL/min/1.73 m2 or presence of at least two diagnosis codes for dialysis or renal transplant
ESRD/transplant defined as eGFR < 15 mL/min/1.73 m2 or presence of at least two diagnosis codes for dialysis or renal transplant
Abbreviations: CI = confidence interval, CKD = chronic kidney disease, eGFR = estimated glomerular filtration rate, ESRD = end stage renal disease, OR = odds ratio
Aside from binary CKD and ESRD/renal transplant status, we performed linear regression modeling to examine the impact of the APOL1 high-risk genotypes on quantitative renal function. After setting the eGFR for participants with ESRD or transplant to zero to reflect renal-replacement, high-risk genotype status was associated with eGFR that was on average 15.4 mL/min/1.73m2 lower (SE=2.3, p=4.0×10−15) than in in low-risk participants (Table 4). Due to the uncertainties in estimating eGFR≥60 mL/min/1.73m2 calculated from serum creatinine, we repeated the analyses excluding participants with normal renal function (n=1,219), and demonstrated that among the 618 participants with CKD, the high-risk APOL1 variant was associated with 13.2 mL/min/1.73 m2 lower eGFR (p=8.0×10−13). We did not observe differences in eGFR decline by APOL1 genotype.
Table 4.
Association between APOL1 high-risk variant status and eGFR in Penn Medicine Biobank using multivariable rank-based regression and eGFR slope using multivariable linear regression.
| Model 1* | Model 2** | ||||
|---|---|---|---|---|---|
| Effect Estimate (SE) | p value | Effect Estimate (SE) | p value | ||
| eGFR | |||||
| Overall | n=1837 | −16.00 (2.21) | 6.28 × 10−13 | −15.36 (2.32) | 4.30 × 10−11 |
| CKD/ESRD† | n=618 | −11.95 (2.22) | 1.05 × 10−7 | −13.16 (2.24) | 7.74 × 10−9 |
| eGFR slope | |||||
| Overall | n=1362 | 3.46 × 10−4 (5.70 × 10−4) | 0.54 | 4.57 × 10−4 (5.74 × 10−4) | 0.43 |
| CKD†† | n=354 | 2.94 × 10−4 (1.04 × 10−3) | 0.78 | 1.88 × 10−4 (1.04 × 10−3) | 0.86 |
Model 1 is adjusted for age, age-squared, gender, and principal components 1–10 of genetic ancestry
Model 1 is adjusted for age, age-squared, gender, and principal components 1–10 of genetic ancestry
Model 2 additionally adjusts for mean body mass index, mean blood glucose, mean systolic blood pressure, and smoking status (current/former versus never)
Model 2 additionally adjusts for mean body mass index, mean blood glucose, mean systolic blood pressure, and smoking status (current/former versus never)
CKD/ESRD defined as eGFR<60 mL/min/1.73 m2 or presence of at least two diagnosis codes for dialysis or renal transplant
CKD/ESRD defined as eGFR<60 mL/min/1.73 m2 or presence of at least two diagnosis codes for dialysis or renal transplant
CKD defined as eGFR≥15 mL/min/1.73 m2 and <60 mL/min/1.73 m2
CKD defined as eGFR≥15 mL/min/1.73 m2 and <60 mL/min/1.73 m2
Abbreviations: ß = unstandardized coefficient, CI = confidence interval, eGFR = estimated glomerular filtration rate, SE = standard error
APOL1 and renal pathology
To understand the renal histological phenotypes associated with the APOL1 high-risk genotypes, pathology records for the 191 participants in the PMBB with renal transplants were reviewed (Table 5). The proportion of subjects with focal segmental glomerulosclerosis (FSGS) and hypertensive renal disease was significantly higher in the APOL1 high-risk group (p=0.01). On the other hand, fewer subjects carried the diagnosis of diabetic nephropathy in the high-risk group.
Table 5.
Disease etiology of native kidney in renal transplant patients obtained from pathology reports stratified by APOL1 risk variant carrier status.
| Renal pathology, No. (%) |
APOL1 low-risk n=62 |
APOL1 high-risk n=39 |
|---|---|---|
| Focal Segmental Glomerulosclerosis* | 10 (16.1) | 15 (38.5) |
| Diabetic Nephropathy | 19 (30.1) | 6 (15.4) |
| Hypertension-associated Nephropathy | 6 (9.7) | 8 (20.5) |
| HIV-Associated Nephropathy | 1 (1.6) | 1 (2.6) |
| Lupus nephritis | 8 (12.9) | 1 (2.6) |
| Membranous nephropathy | 3 (4.8) | 0 (0) |
| Unknown/other | 15 (24.2) | 8 (20.5) |
Abbreviations: HIV = human immunodeficiency virus
Difference in proportions significant with p=0.01
Difference in proportions significant with p=0.01
Discussion
In this study, we used a genotype-first approach to understand the phenotypic associations of carrying two high-risk APOL1 alleles. The PMBB and BioVU are unique sources of EHR data including laboratory results and diagnosis codes, and genomic data for several thousands of participants from academic health systems. This rich source of data allowed us to evaluate the impact of the APOL1 high-risk variant in over 6,000 community-dwelling African Americans. With only the use of diagnosis codes, the PheWAS analyses highlighted robust associations between APOL1 with primarily kidney and kidney-associated pathologies.
Prior studies have shown an association between the APOL1 high-risk status with CKD. In 2,571 African Americans in SPRINT (Systolic Blood Pressure Intervention Trial), the high-risk genotype was associated with a higher odds for CKD at baseline with an OR=1.37 (95% CI 1.08–1.73).12 Arguably, SPRINT trial participants were at higher risk for CKD since hypertension at baseline was a requirement for participation. In the Dallas Heart Study (1,825 African Americans), the proportion of participants with CKD was 1.5% for European Americans, 1.7% for African Americans with zero or one APOL1 risk allele, and 6.7% for African Americans with two risk alleles.11 Results from our study confirm this association in a population-setting, showing that African Americans with two risk alleles have an over 200% increase in odds of having prevalent ESRD or renal transplant and an eGFR that is on average 15.4 mL/min/1.73m2 lower than the eGFR in low-risk African Americans. In the 3,067 African Americans in the Atherosclerosis Risk in Communities (ARIC) Study,10 participants with two APOL1 high-risk alleles had a 51% higher risk for incident CKD (hazard ratio=1.51, 95% CI 1.01–2.24) compared to those with zero or one risk allele.
Analyses in this study also revealed an association between APOL1 high-risk status and 3.2 times higher odds with prevalent ESRD or renal transplant as defined by an eGFR<15 mL/min/1.73m2 or by presence of diagnosis codes. However, we did not find an association with APOL1 and rate of eGFR decline. This is in contrast with previous studies such as SPRINT12, in which the high-risk APOL1 variant was negatively associated with eGFR slope (beta coefficient=−3.57, p=0.0029). In 3,030 adults (mean age 35 years) of the Coronary Artery Risk Development in Young Adults (CARDIA) study, African Americans with two APOL1 risk alleles experienced a 0.45% and 0.38% faster eGFR decline compared to Whites and African Americans with the low-risk APOL1 alleles, respectively.24 Of the 3,067 African Americans in the ARIC study, all with normal renal function at baseline10, those with two risk alleles had a 1.92-fold higher risk for developing incident ESRD (95% CI 1.19–3.10) compared to those with zero or one risk allele. Parsa et al.,13 investigated APOL1 high-risk status in both the African American Study of Kidney Disease and Hypertension (AASK) cohort and the Chronic Renal Insufficiency Cohort (CRIC). The authors found that African Americans in AASK with two APOL1 risk alleles were twice as likely to progress to ESRD or have a doubling of serum creatinine (HR=1.88, p<0.001) during the follow-up period. Additionally, in CRIC, African Americans with two APOL1 risk alleles had a faster eGFR decline compared to Whites (−1.32 mL/min/1.73 m2, p<0.001 in subjects with diabetes, and −1.05 mL/min/1.73 m2, p=0.005 in subjects without diabetes). As with SPRINT, the AASK cohort is also limited in generalizability as participants had to meet specific enrollment criteria. The CRIC cohort recruited participants with CKD, and baseline eGFR was much lower (41.3±14.8 mL/min/1.73m2 in diabetic participants and 46.6±17.4 in non-diabetics) than in the populations presented here.
Interestingly, meta-analysis of our data revealed a significant association of the APOL1 high-risk genotype with “type 2 diabetes with renal manifestations” (OR=1.7, 95% CI 1.3–2.2, p=9.1×10−5). Prior reports showed conflicting results between APOL1 high-risk genotype and diabetic kidney disease. As patients with diabetes rarely undergo kidney biopsy to determine the cause of their CKD, future histological analysis-based studies are needed to understand this relationship.
Results from our study suggest an association between the APOL1 high-risk genotype and hypertension. In our PheWAS, the high-risk variant was associated with the phenotypes “hypertensive heart and/or renal disease” and “hypertensive chronic kidney disease.” Although more granular phenotyping would allow us to discern whether the variant was associated to phenotypes with a primary effect on hypertension versus a primary effect on renal disease and secondary effect of hypertension, given that the association disappeared after adjusting for kidney disease indicates that it is likely mediated by a renal pathway. The association with disease phenotypes related to hypertension is consistent with findings from a recent study using the Mount Sinai BioMe biobank (n=5,204), with replication in additional members of BioMe, Vanderbilt BioVU and Northwestern NUgene, in which the APOL1 risk alleles are associated with higher systolic blood pressure and earlier diagnosis of hypertension in an additive pattern.18
Compared to prior reports in which renal histological analyses was unavailable, we had the opportunity to analyze pathological changes in transplant patients. While the sample size is limited, for the 191 participants with renal transplant, there was a significantly higher number of FSGS cases in the APOL1 high-risk group. Our results are in line with prior animal model25 and histopathological studies26 that reported focal and global sclerosis as the most specific lesions associated with APOL1 variant carriers.9,16
Strengths of this study include the large EHR-based cohorts (PMBB and BioVU) used to define the range of phenotypes associated with the APOL1 high-risk genotype. With over 6,000 participants across both cohorts, results from this study are broadly generalizable to the wider population. Further, participants in PMBB cohort had a lengthy follow-up (median 5.5 years) with high number of creatinine measurements (median 14) per participant that allowed for robust longitudinal modeling of eGFR. Some limitations of this study must be acknowledged. APOL1 has been shown to be associated with increased proteinuria.27 Measurement of proteinuria is variable in EHR data but would be of interest for future studies to explore. Secondly, the PheWAS approach does not test the association of the APOL1 high-risk variant with any given disease, as defined in the traditional sense. To determine the association with a specific disease entity, for example CVD, using a composite of phenotypes, each defined by a set of diagnosis codes, and more specific EHR data, such as laboratory data, would be a more precise method to evaluate that association. We used a Bonferroni correction for multiple testing which sets a more conservative threshold for statistical significance to protect from Type I error by dividing the original p-value (0.05), by the number of analyses on the dependent variable (number of phenotypes with more than 5% prevalence). This method may overcorrect for these associations because of the lack of independence across phenotypes when each is defined by a phecode. Prior reports28–30 show mixed findings on the association between the APOL1 high-risk genotype and CVD. Differences in study design, definition of CVD outcomes, and assessment of confounders, particularly the confounding effect APOL1-associated CKD on development of CVD, make it difficult to conclude whether an association may exist. A recent study from the Million Veteran Program was able to detect only minimal associations between APOL1 genotype status and CVD in over 50,000 African Americans. The effect estimates were extremely small, to the point of being clinically insignificant, and the association was driven by the underlying CKD. 31 Our results are in keeping with these findings and suggests little to no independent association between APOL1 and CVD.
The APOL1 high-risk variant in our two population-based cohorts is associated with a highly significant 3–6-fold increase in incidence of renal disease. We did not detect an association with non-renal traits suggesting that APOL1 likely works in renal-specific pathways. Future studies of the pathways affected by APOL1 may offer new targets for therapy in the at-risk population.
Methods
Study Populations
Participants in the Penn Medicine Biobank were recruited from clinical practice sites throughout the University of Pennsylvania Health System. Participants were consented for bio-specimen storage and access to EHR data. Whole exome sequences were generated from DNA extracted from stored buffy coats by the Regeneron Genetics Center and mapped to Genome Reference Consortium Build 38 (GRCh38) as described previously.32 Samples with low coverage (<75% of targeted bases achieving 20x coverage; n=46), discordance between reported and genetically determined sex (n=112), high heterozygosity (n=97), high missingness (>5% of targeted bases; n=14), genetic evidence of sample duplication (n=89), or cryptic relatedness (closer than third degree relatives; n=149) were removed.
BioVU is Vanderbilt University Medical Center’s DNA repository that is linked to de-identified electronic medical records.33 Imputation of genome-wide genotype platforms were performed using the Haplotype Reference Consortium (HRC) reference on the Michigan Imputation Server with minimac3 as described elsewhere.34,35 Standard genotype data quality control, as described previously,25 was performed, including examination of marker and sample genotyping efficiency, allele frequency calculations, and tests of Hardy-Weinberg equilibrium. SNPs with a significant deviation from Hardy Weinberg equilibrium (p<1×10−6) were removed. Samples with a call rate by individual of <95% and/or low-quality SNPs/markers with a call rate of <95% were removed.36 A strand flipping protocol was added for the MEGA platform.37
The study was approved by each institution’s Institutional Review Board.
APOL1 Genotyping
Three single nucleotide polymorphisms within the last exon of APOL1 constitute the G1 (rs73885319, rs60919145; both missense mutations) and G2 (rs71785313; a 6-base pair deletion) haplotypes. Because of the difficulty in sequencing the G2 allele, rs12106505 (r2~0.82), was used as a proxy in the PMBB and BioVU cohorts.38 The APOL1 variant allele homozygotes or compound heterozygotes, labeled “high-risk,” were defined as carriers of 2 copies of rs73885319 (G1), 2 copies of rs12106505 (G2), or 1 copy of each. The carriers of 1 or zero APOL1 risk alleles were considered “low-risk.”
Data Collection
For the PMBB cohort, the International Classification of Diseases, Ninth Revision (ICD-9) and Tenth Revision (ICD-10) disease diagnosis codes and procedural codes were extracted. Additionally, all creatinine values measured in the outpatient setting were extracted for participants from the time of enrollment in the Biobank until July 1, 2017; values greater than 20 mg/dL were removed, as these were presumed to be lab or data entry errors. Outpatient values for body mass index (BMI), systolic and diastolic blood pressure, random glucose, and lipid panel measurements were also extracted from the EHR.
For each available creatinine value, the eGFR was calculated using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI)39 equation: GFR = 141× min(SCr/κ, 1)α × max(SCr/κ, 1)−1.209 × 0.993Age × 1.018 [if female] × 1.159, where SCr is the serum creatinine in mg/dL, κ is 0.7 for females and 0.9 for males, and α is −0.329 for females and −0.411 for males.
Pathology records for participants in the PMBB with renal transplants were reviewed to determine the disease etiology underlying their renal failure.
For the BioVU cohort, ICD-9 diagnosis codes and procedural codes were extracted from the EHR. Inpatient and outpatient values for serum creatinine, BMI, systolic and diastolic blood pressure, and lipid measurements were extracted from the EHR for each participant. The eGFR was calculated using the CKD-EPI equation using serum creatinine measurements as above.
Phenome-wide Association Study (PheWAS)
A phenome-wide association study (PheWAS) approach was used to determine the phenotypes associated with the APOL1 high-risk variants.22 In PMBB, ICD-10 encounter diagnoses were mapped to ICD-9 using a combination of the Center for Medicare and Medicaid Services 2017 General Equivalency Mappings (https://www.cms.gov/Medicare/Coding/ICD10/2017-ICD-10-CM-and-GEMs.html) and manual curation and then mapped to distinct phecodes using the R package PheWAS.23 A phecode for a phenotype required the presence of two or more relevant ICD-9 codes; individuals with only one ICD-9 code were excluded from analyses for that phenotype. Each phenotype was tested for an association with the high-risk genotype using a logistic regression model adjusted for age, age2, sex, and principal components 1–10 of genetic ancestry using the R package PheWAS.23 We considered only phenotypes with more than 5% prevalence in the cohort, which corresponded to 233 phenotypes, and used a Bonferroni correction to adjust for multiple testing (p=2.2×10−4). In conditional analyses, the logistic regression model was repeated first adjusting for CKD/ESRD status (eGFR<60 mg/mL/1.73m2; the eGFR was set to zero for participants with at least two diagnosis codes for dialysis or renal transplant) and, second, for eGFR as a continuous variable using the most recent eGFR for each participant. The PheWAS analyses was also repeated using an additive model for the APOL1 risk variants.
In BioVU, ICD-9 codes were similarly mapped to distinct disease phenotypes using phecodes. Each phenotype was tested for its association with the high-risk genotypes using logistic regression, adjusting for age, age2, sex, and the first three principal components of genetic ancestry. The PheWAS analysis used 206 phenotypes after excluding phenotypes with less than 5% prevalence, to examine disease associations using an algorithm that translates ICD-9 billing codes found in the EHR to phecodes as described previously.40 A p-value for significance of 2.4×10−4 was used. In conditional analyses, the logistic regression model was repeated as in PMBB, first adjusting for CKD/ESRD status and, second, for the most recent available eGFR value for each participant. The PheWAS analyses was then repeated using an additive model for the APOL1 risk variants.
A meta-analysis using both the PMBB and BioVU datasets was completed using fixed effects meta-analysis as implemented in the PheWAS package. There were a combined 153 phenotypic traits tested (Table S3), and a p-value for significance of 3.2×10−4 was used.
To further explore the association of APOL1 with measurements associated with renal disease and CVD, a linear regression model was used with continuous values for median BMI, median systolic blood pressure, median diastolic blood pressure, highest total cholesterol, highest LDL-cholesterol, lowest HDL-cholesterol, and median triglyceride levels.
Statistical Analysis
Continuous variables were summarized as means ± standard deviations or medians with interquartile ranges and differences between groups assessed with the Wilcoxon rank-sum test. Categorical variables were summarized with counts and percentages, and differences between groups assessed with the chi-square test.
Association testing for eGFR as a continuous trait was performed using a recessive model, comparing the eGFR for high-risk APOL1 participants with 2 risk alleles to low-risk carriers of 0 or 1 risk alleles. The eGFR was set to 0 mL/min/1.73m2 for all participants with at least two diagnoses codes indicating end-stage renal disease, hemodialysis, peritoneal dialysis or renal transplant. A rank-based regression model was used for the last available eGFR per participant, adjusted for age, age2, sex, and principal components 1–10 of genetic ancestry. Then, the model was additionally adjusted for mean BMI, mean glucose, mean systolic blood pressure, and smoking status (current/former smoker versus never smoker).
A linear mixed effects model with BLUP (Best Linear Unbiased Predictor) estimates of intercepts and slopes41 was used to model eGFR slope for each PMBB participant using eGFRs calculated from all available outpatient creatinine. A multivariable linear regression model, adjusted for covariates as described above, was then used with eGFR slope as the outcome.
In the PMBB cohort, a logistic regression model was used to determine the odds ratio for those with any renal impairment or history of renal transplant (eGFR<60 mg/mL/1.73m2 or presence of at least two diagnosis codes for dialysis or renal transplant), CKD (eGFR≥15 and <60 mg/mL/1.73m2 excluding those with a diagnosis code for dialysis or renal transplant), and ESRD or renal transplant (eGFR<15 mg/mL/1.73m2 or presence of at least two corresponding diagnosis codes), for those with the APOL1 high-risk genotypes compared to low-risk participants. This model was adjusted first for age, age2, sex, and the first ten principal components of genetic ancestry, then additionally adjusted for mean BMI, mean glucose, mean systolic blood pressure, and smoking status.
All analyses were completed using R version 3.2.2 (Vienna, Austria) or STATA version 14.2 (StataCorp, College Station, Texas).
Supplementary Material
Table S1. Association between APOL1 high-risk genotypes using a recessive model with BMI, blood pressure, and lipid levels in the Penn Medicine Biobank.
Table S2. Association between APOL1 high-risk genotypes using a recessive model with BMI, blood pressure, and lipid levels in Vanderbilt BioVU.
Table S3. List of phenotypes (n=153) as defined by ICD codes included in meta-analysis phenome-wide association study of Penn Medicine Biobank and BioVU cohorts.
Figure S1. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for CKD status.
Figure S2. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for CKD status.
Figure S3. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for most recent eGFR with the eGFR set to zero for participants with at least two diagnosis codes for dialysis or renal transplant.
Figure S4. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for most recent eGFR with the eGFR set to zero for participants with at least two diagnosis codes for dialysis or renal transplant.
Figure S5. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for most recent eGFR.
Figure S6. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for most recent eGFR.
Figure S7. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model.
Figure S8. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model.
Figure S9. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model and adjusting for CKD/ESRD status.
Figure S10. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model and adjusting for CKD/ESRD status.
Figure S11. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model and adjusting for most recent eGFR.
Figure S12. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model and adjusting for most recent eGFR.
Acknowledgements
Work in the lab of KS is supported by NIH R01 DK105821, DK076077, DK087635, DP3 DK108220. SMD is supported by the US Department of Veterans Affairs (IK2-CX001780). This publication does not represent the views of the Department of Veterans Affairs or the United States Government. Work in the lab of CM Stein and QF is supported by the following grants: NIH GM109145, GM131770, R01 GM120523, T32 GM007569. BioVU work was supported by R01 LM010685 and P50-GM115305. BioVU received and continues to receive support through the National Center for Research Resources (UL1-RR024975), which is now the National Center for Advancing Translational Sciences (UL1-TR000445).
Footnotes
Statement of Competing Financial Interests
The Susztak lab receives support from Boehringer Ingelheim, GlaxoSmithKline, Regeneron, Gilead, Merck and ONO Pharmaceutical for work not related to this project.
References
- 1.McClellan W, Tuttle E. Racial differences in the incidence of hypertensive end-stage renal disease (ESRD) are not entirely explained by differences in the prevalence of hypertension. American Journal of Kidney Disease 1988; 12: 285–290. [DOI] [PubMed] [Google Scholar]
- 2.Cowie CC, Port FK, Wolfe RA et al. Disparities in incidence of diabetic end-stage renal disease according to race and type of diabetes. N. Engl. J. Med 1989; 321: 1074–1079. [DOI] [PubMed] [Google Scholar]
- 3.Lipworth L, Mumma MT, Cavanaugh KL et al. Incidence and predictors of end stage renal disease among low-income blacks and whites. PLoS ONE 2012; 7: e48407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruggeman LA, O’Toole JF, Sedor JR. Identifying the Intracellular Function of APOL1. Journal of the American Society of Nephrology 2017; 28: 1008–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ejaz S. Transcription and translation of APOL1 variants. Biosci. Rep 2017; 37: BSR20170647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Madhavan SM, O’Toole JF, Konieczkowski M et al. APOL1 localization in normal kidney and nondiabetic kidney disease. Journal of the American Society of Nephrology 2011; 22: 2119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pérez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005; 309: 469–472. [DOI] [PubMed] [Google Scholar]
- 8.Beckerman P, Susztak K. APOL1: The Balance Imposed by Infection, Selection, and Kidney Disease. Trends Mol Med 2018; 24: 682–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Genovese G, Friedman DJ, Ross MD et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010; 329: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foster MC, Coresh J, Fornage M et al. APOL1 variants associate with increased risk of CKD among African Americans. Journal of the American Society of Nephrology 2013; 24: 1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman DJ, Kozlitina J, Genovese G et al. Population-based risk assessment of APOL1 on renal disease. Journal of the American Society of Nephrology 2011; 22: 2098–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Langefeld CD, Divers J, Pajewski NM et al. Apolipoprotein L1 gene variants associate with prevalent kidney but not prevalent cardiovascular disease in the Systolic Blood Pressure Intervention Trial. Kidney International 2015; 87: 169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parsa A, Kao WHL, Xie D et al. APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med 2013; 369: 2183–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grams ME, Rebholz CM, Chen Y et al. Race, APOL1 Risk, and eGFR Decline in the General Population. Journal of the American Society of Nephrology 2016; 27: 2842–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanji Z, Powe CE, Wenger JB et al. Genetic variation in APOL1 associates with younger age at hemodialysis initiation. Journal of the American Society of Nephrology 2011; 22: 2091–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopp JB, Nelson GW, Sampath K et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. Journal of the American Society of Nephrology 2011; 22: 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasembeli AN, Duarte R, Ramsay M et al. APOL1 Risk Variants Are Strongly Associated with HIV-Associated Nephropathy in Black South Africans. Journal of the American Society of Nephrology 2015; 26: 2882–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nadkarni GN, Galarneau G, Ellis SB et al. Apolipoprotein L1 Variants and Blood Pressure Traits in African Americans. J Am Coll Cardiol 2017; 69: 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito K, Bick AG, Flannick J et al. Increased burden of cardiovascular disease in carriers of APOL1 genetic variants. Circ. Res 2014; 114: 845–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mukamal KJ, Tremaglio J, Friedman DJ et al. APOL1 Genotype, Kidney and Cardiovascular Disease, and Death in Older Adults. Arteriosclerosis, thrombosis, and vascular biology 2016; 36: 398–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen TK, Appel LJ, Grams ME et al. APOL1 Risk Variants and Cardiovascular Disease: Results From the AASK (African American Study of Kidney Disease and Hypertension). Arteriosclerosis, thrombosis, and vascular biology 2017; 37: 1765–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denny JC, Bastarache L, Ritchie MD et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol 2013; 31: 1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics 2014; 30: 2375–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peralta CA, Bibbins-Domingo K, Vittinghoff E et al. APOL1 Genotype and Race Differences in Incident Albuminuria and Renal Function Decline. Journal of the American Society of Nephrology 2016; 27: 887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beckerman P, Bi-Karchin J, Park ASD et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med 2017; 23: 429–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Larsen CP, Beggs ML, Saeed M et al. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Mod. Pathol 2015; 28: 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen TK, Tin A, Peralta CA et al. APOL1 Risk Variants, Incident Proteinuria, and Subsequent eGFR Decline in Blacks with Hypertension-Attributed CKD. Clin J Am Soc Nephrol 2017; 12: 1771–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson TW, Freedman BI. The Apolipoprotein L1 Gene and Cardiovascular Disease. Methodist Debakey Cardiovasc J 2016; 12: 2–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Estrella MM, Parekh RS. The Expanding Role of APOL1 Risk in Chronic Kidney Disease and Cardiovascular Disease. Semin. Nephrol 2017; 37: 520–529. [DOI] [PubMed] [Google Scholar]
- 30.Grams ME, Surapaneni A, Ballew SH et al. APOL1 Kidney Risk Variants and Cardiovascular Disease: An Individual Participant Data Meta-Analysis. Journal of the American Society of Nephrology 2019; 30: 2027–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bick AG, Akwo E, Robinson-Cohen C et al. Association of APOL1 Risk Alleles With Cardiovascular Disease in Blacks in the Million Veteran Program. Circulation 2019; 140: 1031–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dewey FE, Gusarova V, Dunbar RL et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N. Engl. J. Med 2017; 377: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roden DM, Pulley JM, Basford MA et al. Development of a Large-Scale De-Identified DNA Biobank to Enable Personalized Medicine. Clinical Pharmacology & Therapeutics 2008; 84: 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Das S, Forer L, Schönherr S et al. Next-generation genotype imputation service and methods. Nat. Genet 2016; 48: 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fuchsberger C, Abecasis GR, Hinds DA. minimac2: faster genotype imputation. Bioinformatics 2015; 31: 782–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turner S, Armstrong LL, Bradford Y et al. Quality control procedures for genome-wide association studies. Haines JL, Korf BR, Morton CC, et al., eds. Curr Protoc Hum Genet 2011; Chapter 1: Unit1.19–1.19.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J, Samuels DC, Shyr Y et al. StrandScript: evaluation of Illumina genotyping array design and strand correction. Hancock J, ed. Bioinformatics 2017; 33: 2399–2401. [DOI] [PubMed] [Google Scholar]
- 38.Genovese G, Friedman DJ, Pollak MR. APOL1 variants and kidney disease in people of recent African ancestry. Nat Rev Nephrol 2013; 9: 240–244. [DOI] [PubMed] [Google Scholar]
- 39.Levey AS, Stevens LA, Schmid CH et al. A New Equation to Estimate Glomerular Filtration Rate. Ann. Intern. Med 2009; 150: 604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denny JC, Ritchie MD, Basford MA et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics 2010; 26: 1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson GK. That BLUP is a Good Thing: The Estimation of Random Effects. Statistical Science 1991; 6: 15–32. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Association between APOL1 high-risk genotypes using a recessive model with BMI, blood pressure, and lipid levels in the Penn Medicine Biobank.
Table S2. Association between APOL1 high-risk genotypes using a recessive model with BMI, blood pressure, and lipid levels in Vanderbilt BioVU.
Table S3. List of phenotypes (n=153) as defined by ICD codes included in meta-analysis phenome-wide association study of Penn Medicine Biobank and BioVU cohorts.
Figure S1. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for CKD status.
Figure S2. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for CKD status.
Figure S3. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for most recent eGFR with the eGFR set to zero for participants with at least two diagnosis codes for dialysis or renal transplant.
Figure S4. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for most recent eGFR with the eGFR set to zero for participants with at least two diagnosis codes for dialysis or renal transplant.
Figure S5. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Penn Medicine Biobank using a recessive model and adjusting for most recent eGFR.
Figure S6. Manhattan plot for phenome-wide association study data of the APOL1 high-risk genotypes in the Vanderbilt BioVU using a recessive model and adjusting for most recent eGFR.
Figure S7. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model.
Figure S8. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model.
Figure S9. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model and adjusting for CKD/ESRD status.
Figure S10. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model and adjusting for CKD/ESRD status.
Figure S11. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Penn Medicine Biobank using an additive model and adjusting for most recent eGFR.
Figure S12. Manhattan plot for phenome-wide association study data of the APOL1 risk allele in the Vanderbilt BioVU using an additive model and adjusting for most recent eGFR.