

Abstract
Current methods for chromosome painting via fluorescence in situ hybridization (FISH) are costly, time consuming, and are limited in complexity. In contrast to conventional sources of probe, Oligopaints are computationally designed, synthesized on microarrays, and amplified by PCR. This approach allows for precise control over the sequences they target, which can range from a few kilobases to entire chromosomes with the same basic protocol. We have utilized the flexibility and scalability of Oligopaints to generate low-cost and renewable chromosome paints for Drosophila, mouse, and human chromosomes. These Oligopaint libraries can be customized to label any genomic feature(s) in a chromosome-wide manner. Additionally, this method is compatible with sequential FISH to label entire genomes with a single denaturation step. Here, we outline a protocol and considerations to scale the Oligopaint technology for fluorescent labeling of whole chromosomes.
Keywords: FISH, Oligopaint, chromosome painting, oligonucleotide
1. Introduction
The discovery of nonrandom chromosome folding and positioning in the nucleus [1, 2] has precipitated ongoing efforts to determine the contribution of this spatial organization to gene function. Fluorescence in situ hybridization (FISH) has been instrumental in relating gene positioning to transcriptional outputs, as seen at the chromosomal level with X-inactivation [3–7] and with the positioning of genes relative to their chromosome and to the nuclear periphery [8–13]. With advances in computing power and next-generation sequencing, however, bulk genomic assays such as Hi-C [14–17] have outpaced the implementation of traditional FISH methodologies in elucidating the structure and spatial characteristics of chromosomes. FISH, on the other hand, can more readily capture single-cell variability that is lost with ensemble genomic assays and inherently adds positioning information [18–20]. The Oligopaints pipeline for FISH utilizes in silico screening of candidate probe sequences for favorable criteria, including uniqueness and melting temperature, that can be easily synthesized as short and renewable oligo probes [21, 22]. Oligopaints avoid issues with traditional FISH probes that use genomic DNA or degenerate primers for random amplification by avoiding non-unique and repetitive sequences [23–27]. This greatly diminishes batch effects common with FISH, ultimately leading to more reproducible results.
Oligopaints has already been successfully employed by a number of groups to visualize relatively small genomic regions (5–300Kb) [14, 19, 28–33]. Recently, we have successfully scaled the Oligopaint technology for the visualization of large genomic regions (several Mbs) up to entire chromosomes for several species without added cost or loss in resolution (Fig. 1). Compared to conventional chromosome painting strategies, we find that Oligopaints greatly reduce background signal permitting accurate measurements of chromosome size, shape, and position in the nucleus [34]. Indeed, this strategy has been used in cells and tissues in both flies and worms to identify factors necessary for large-scale chromosome folding [32, 34]. Finally, multi-Mb Oligopaints can be further functionalized by assigning multiple primer pairs to each probe, allowing a single genomic sequence to be used for multiple probe sets (Fig. 2). For example, oligos assigned to a chromosome can be selectively amplified to interrogate different scales of chromosome organization, from whole chromosomes to sub-chromosomal domains and individual genes. The control over probe production is also crucial when generating allele-specific probes, which can address biological questions regarding allele-specific alterations in structure and/or expression [28]. Here, we outline a protocol and considerations to scale the Oligopaint technology for fluorescent labeling of whole chromosomes.
Fig. 1.
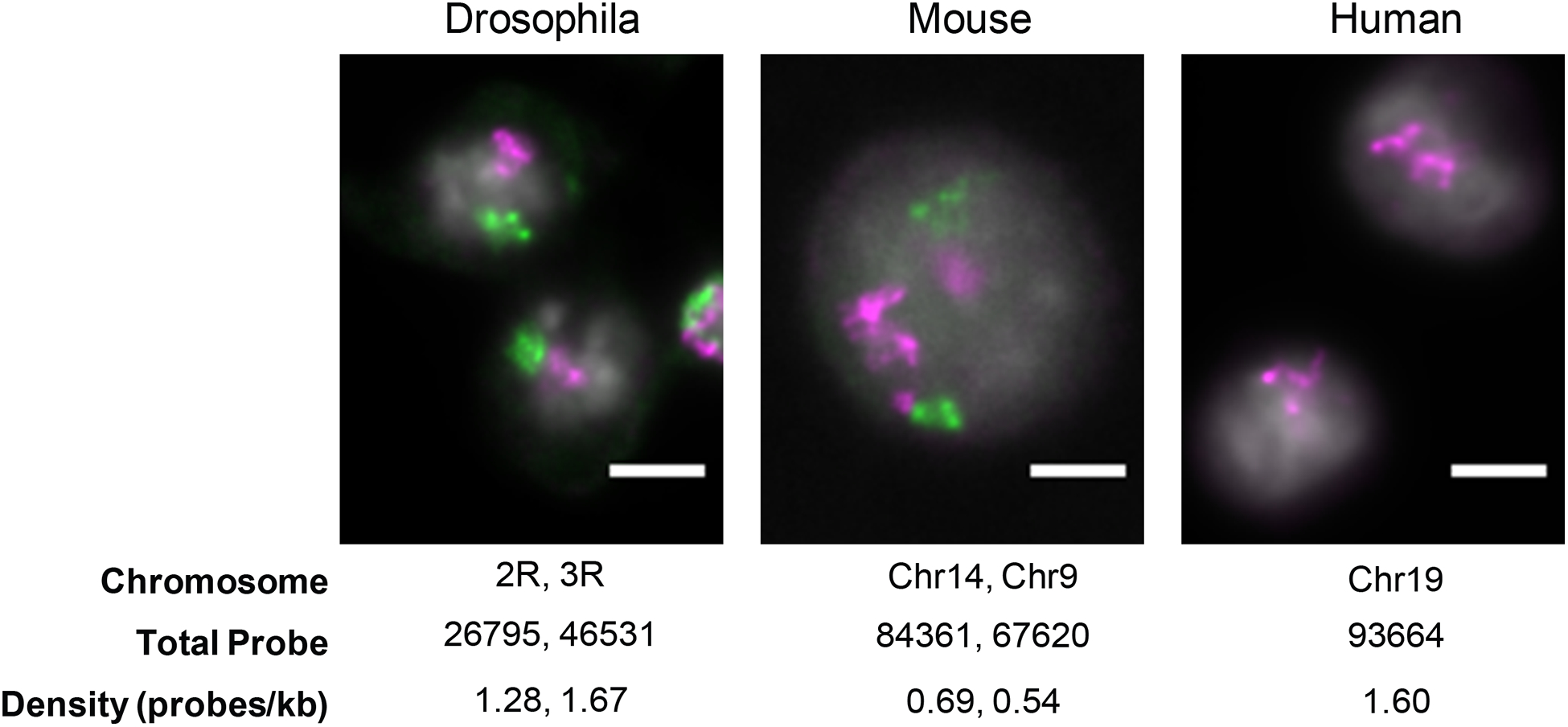
Representative images from a single z-slice of chromosome territories in Drosophila (Kc167 cells), mouse (3T3 cells), and human (HCT-116 cells) interphase nuclei. Chromosome target, total probe count, and average density of probe labeling across each chromosome are given. Scale bar, 5 μm.
Fig. 2.
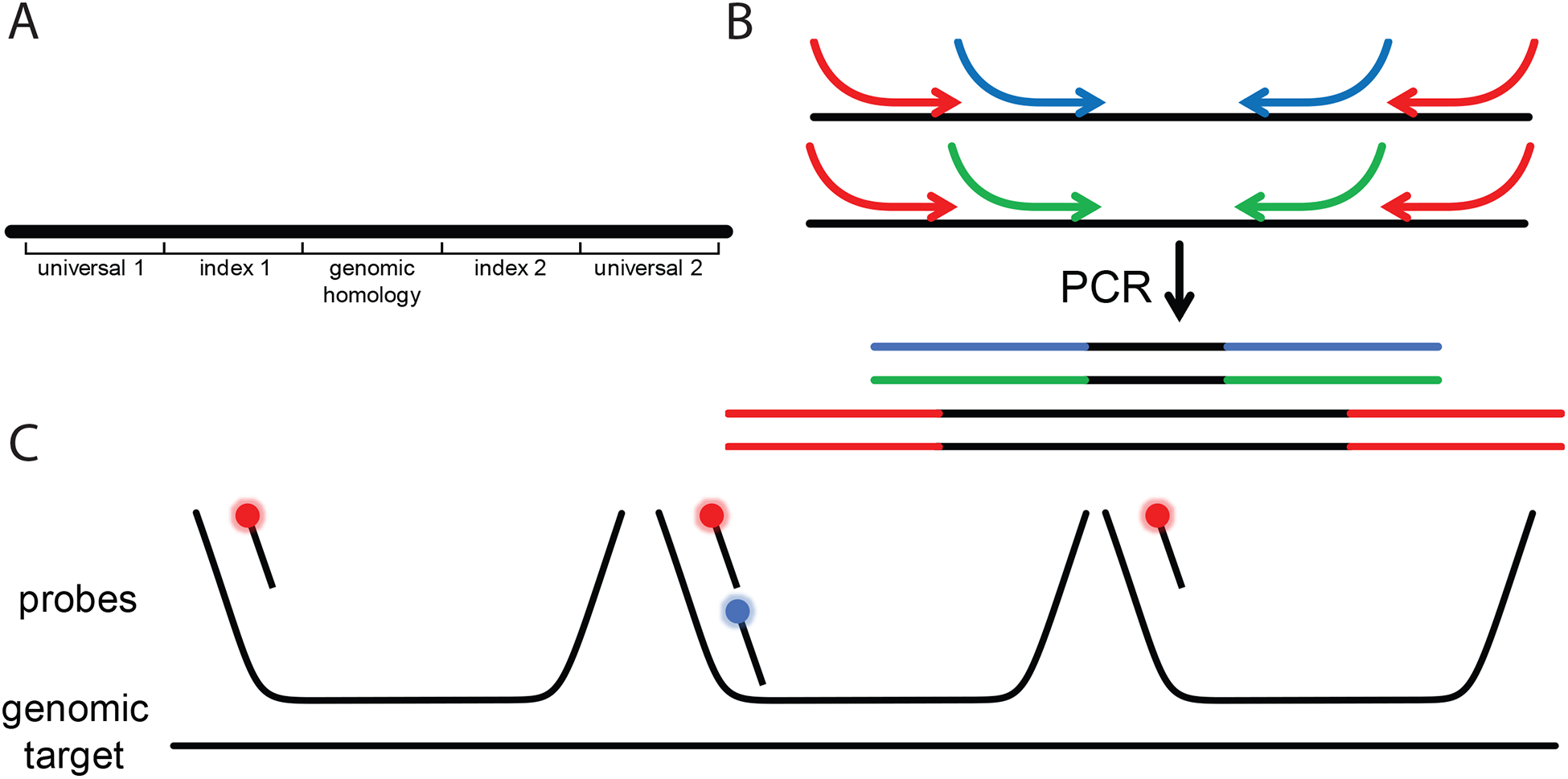
Design, production, and detection of Oligopaints. A Probe sequences with homology to specific genomic regions are flanked with a pair of indices. For chromosome paints, an additional pair of “universal” indices are used across all probe sequences. B The unique indices allow the same oligo pool to be used for the creation of multiple Oligopaints. Primers to the universal indices (red) amplify the entire pool, and the nested primers (blue and green) amplify subsets of the pool, leading to the generation of chromosome- and region-specific Oligopaints, respectively. The primers contain extra “overhang” sequences, usually the secondary binding site and T7 primer sequence, which are incorporated into the probe PCR product. C Unlabeled primary probes hybridized to their genomic target can be detected using labeled secondary probes. With the proper probe design, simultaneous detection of the chromosome- and region-specific probes can be accomplished using two different secondary probes (red and blue, respectively).
2. Materials
2.1. Complex oligo pools synthesized on microarray from commercial vendor
2.2. Initial PCR Amplification
Complex oligo pool
Phusion High-Fidelity PCR Master Mix
Forward primer
Reverse primer
PCR purification kit
2.3. Secondary PCR Amplification
Forward primer with secondary binding sequence on 5’ end (see Note 1).
Reverse primer with T7 promoter sequence on 5’ end: TAATACGACTCACTATAGGG.
PCR clean up kit.
Taq polymerase and appropriate reaction buffer.
10 mM dNTP mix.
2.4. T7 Reaction
HiScribe T7 Quick High Yield RNA Synthesis Kit
RNaseOUT recombinant ribonuclease inhibitor (see Note 2)
2.5. Reverse Transcription
Maxima H Minus RT Transcriptase
100 mM dNTP Mix: add 100 mM dNTPs in equal ratio to each other.
2.6. Probe Purification
DNA Clean and Concentrator-100 kit (see Note 3)
1 M NaOH
0.5 M EDTA
Oligo binding buffer
100% ethanol
2.7. Sample Fixation
Poly-L-lysine coated slides (see Note 4)
10x PBS solution:
1x PBS solution: dilute 10x PBS solution with ddH2O.
Fixation solution: Add 10 ml of 16% (vol/vol) paraformaldehyde, 26 ml of ddH2O, and 4 ml of 10x PBS.
Coplin staining jar for slides
Kim wipes
2.8. Sample Permeabilization (see Note 5)
Permeabilization solution: Add 200 μl of Triton X-100 to 40 ml of ddH2O. Prepared fresh.
70% ethanol: Add 12 ml of 100% ethanol to 28 ml of ddH2O
90% ethanol: Add 4 ml of 100% ethanol to 36 ml of ddH2O.
100% ethanol: Set aside 50 ml.
20x SSC: 3 M NaCl, 300 mM sodium citrate
4x SSCT: For 1 L, add 200 ml of 20x SSC, 2 ml of Tween-20, and 798 ml of ddH2O.
2x SSCT + 50% formamide: Combine 20 ml of 4x SSCT with 20 ml of formamide (see Note 6).
2.9. Sample Pretreatment
Water bath set at 92°C (or lower temperatures, see Note 7)
Water bath set at 60°C
Heat block, mostly immersed in 92°C water bath. Top of the heat block should be above the water to rest the slide.
2x SSCT + 50% formamide. Fill a coplin jar with this solution and place into each water bath.
2.10. Hybridization of Primary Probe
2.11. Post-Hybridization Wash
2x SSCT: For 1 L, add 100 ml of 20x SSC, 1 ml of Tween-20, and 899 ml of ddH2O.
0.2x SSC: For 1 L, add 10 ml of 20x SSC and 990 ml of ddH2O.
2.12. Hybridization of Secondary Probe
Secondary probe: a fluorophore-labeled oligo with reverse complementarity to the secondary binding site.
Dextran sulfate mix (see above).
Formamide
2.13. Post-Secondary Wash and Mounting
2x SSCT (see above).
0.2x SSC (see above).
Hoechst 33342, 10 mg/ml.
3. Methods
3.1. Oligopaint probe design considerations:
Generation of probe sequences: The original Oligopaints were designed using the BLAST algorithm [21], but newer Oligominer pipeline takes advantage of next-generation sequence alignment, making Oligopaint design more feasible without the need for a high performance computing cluster [22]. Premined probe sequences to popular model organisms are publicly available [35], but any genomic sequence in fasta format can be mined for probes. Importantly, the quality and quantity of probe is highly dependent on the quality and completeness of the genome build. Oligopaint probes, in general, can tolerate some mismatches along the hybridization sequence, but while this allows Oligopaint probes to be used across different genomes containing polymorphisms relative to the reference genome, improper screening of repetitive sequences can lead to the inadvertent design and non-specific probes.
Probe density: Density of probes targeting a genomic region can be relaxed when targeting multi megabases to entire chromosomes compared to sub-megabase genomic targets. While the original Oligopaint and Oligominer bioinformatics pipeline will automatically find all candidate probes across a region of interest, with a typical density of 5–20 probes per kilobase (probes/kb), we have found that ~0.5 to ~2 probes/kb is sufficient to label entire chromosomes [34] (see Fig. 1).
Multiplexing probe design and production: Because Oligopaints are made by primer-based amplification, nested primers can be used to assign a probe sequence for multiple purposes (see Fig. 2a,b). For instance, a single oligo pool can be used to differentially label different chromosomes or different regions of an entire chromosome. This has recently been used to segment the chromosome into megabase- and kilobase-sized chunks [32].
Multiplex probe labeling: Since the primer sequences are incorporated into the probe themselves, they can be used to dock “secondary” fluorophore-labeled probes for indirect labeling, akin to the use of primary and secondary antibody pairs for immunolabeling applications [21, 28]. As an example, whole chromosomes in Drosophila cells can be hybridized with unlabeled primary probe, and then secondary probes can simultaneously label the whole chromosome as well as sub-Mb regions within it [34] (see Fig. 2c). This approach to labeling also simplifies probe production, as one would only need to synthesize one chromosome probe to enable ad hoc labeling of features of interest of that chromosome. Finally, these secondary probes can either be bleached [19] or stripped off with competing unlabeled oligos called “toeholds” [31, 34] to allow for these fluorescent dyes to be reassigned to other secondaries for the detection of many features in the same cell. This strategy has worked well to achieve genome-wide 5-color chromosome painting in Drosophila nuclei [34].
3.2. Probe Production
PCR amplify the oligo pool by assembling a 50 μl reaction with the following: 1.25 μl of 20 μM forward primer, 1.25 μl of 20 μM reverse primer, 1 μl of 100 pg/μl oligo pool, 21.5 μl of ddH2O, and 25 μl of 2x Phusion Master Mix.
Run PCR with the following cycle: initial denaturation of 98°C for 3 min, then 30 cycles of 98°C for 5 s and 72°C for 15 s, then 72°C for 2 min.
Purify the reaction using a PCR purification kit and measure the DNA concentration. Dilute to 20 ng/μl.
Perform second PCR to attach a T7 sequence and secondary binding sequence, using the oligo pool PCR product as a template (see Note 12). Assemble a 200 μl reaction with the following: 0.4 μl of 200 μM forward primer containing secondary binding sequence, 0.4 μl 200 of μM reverse primer containing T7 sequence, 4 μl of 10 mM dNTP mix, 1 μl of 20 ng/μl oligo pool PCR product, PCR buffer, Taq polymerase, and ddH2O. The 200 μl reaction can be split across multiple tubes.
Run a standard PCR reaction based on the manufacturer’s suggestion, such as the following: initial denaturation of 94°C for 5 min, then 35 cycles of 94°C for 45 s, 56°C for 30 s, and 72°C for 30 s, then 72°C for 5 min.
Combine the PCR reactions, purify, and elute in 50 μl. A typical yield is at least 30 ng/μl.
Synthesize RNA in a 20 μl reaction with the following components from the HiScribe T7 kit: 7 μl of the PCR product, 2 μl of each ribonucleotide, 2 μl of 10x T7 buffer, and 2 μl of T7 polymerase. Add 1 μl of RNAseOUT to help prevent RNA degradation. Incubate at 37°C overnight.
The following day, bring the T7 reaction to room temperature and assemble a 150 μl reverse transcription (RT) reaction directly with the T7 reaction mix. Include the following: 20 μl of T7 sample, 7.5 μl of 200 μM forward primer containing the secondary sequence, 9.6 μl of a 100 mM dNTP mix, 30 μl 1x RT buffer, 2 μl of Maxima H Minus enzyme, 1.5 μl RNaseOUT, and 79.4 μl of ddH2O (see Note 13).
Incubate the RT reaction at 50°C for 2 hours.
Degrade the RNA using the following alkaline hydrolysis reaction: add 75 μl of EDTA and 75 μl of 1 M NaOH directly to the RT reaction. Heat at 95°C for 10 min.
Purify the probe by adding the following: 600 μl of oligo binding buffer and 1.2 ml of 100% ethanol to the RT-EDTA-NaOH mix (see Note 14). Vortex briefly, then purify with the DNA Clean and Concentrator-100 kit using manufacturer protocol. Elute the probe in 150 μl of ddH2O.
Measure the concentration of the probe on a spectrophotometer. Convert the concentration to pmol/μl by using the formula: [concentration of probe in pmol/μl] = [concentration of probe in ng/μl] * 3.03 * (1/number of nucleotides).
3.3. Sample Fixation
Harvest cells in normal culture conditions and create a cell suspension (see Note 15).
Place slides in a container that can be closed.
Seed slides with a ~200 μl aliquot of cells from the suspension (see Note 4).
Carefully move slides to incubator and allow them to settle for 30 min to 4 hours. Remember to keep them at culture conditions (temperature and humidity).
Make fixative fresh, add to coplin jar.
Quick wash of slides in 1x PBS coplin jar, switch out after every batch (10) of slides
Transfer slides to fixative and incubate for 10 minutes
Transfer slides to PBS for another quick rinse then either store at 4 °C or proceed to next steps.
3.4. Sample Permeabilization (see Note 5)
Create fresh permeabilization and ethanol mixes and add to coplin jars.
Transfer slide to coplin jar with permeabilization mix and incubate for 15 min.
Quickly rinse slide with 1x PBS, then transfer slide to coplin jar with 70% ethanol and incubate for 2 min.
Transfer slide to coplin jar with 90% ethanol and incubate for 2 min.
Transfer slide to coplin jar with 100% ethanol and incubate for 2 min.
Transfer slide to coplin jar with 2x SSCT and proceed to next steps (see Note 16)
3.5. Sample Pretreatment
Preheat water baths, and allow the coplin jars to reach set temperature.
Transfer slide to 2x SSCT + 50% formamide at room temperature and allow to incubate for 5 min.
Transfer slide to submerged 92°C coplin jar containing 2x SSCT + formamide and incubate for 2.5 min.
Transfer slide to submerged 60 °C coplin jar containing 2x SSCT + formamide and incubate for 20 min.
Prepare the hybridization mix containing probe, formamide, dNTPs, and dextran sulfate.
After the 20 min incubation, transfer slide to empty coplin jar to dry and allow slides to come to room temperature for 5 min. Keep slides in this coplin jar until ready to mount (see Note 17).
3.6. Hybridization of Probe
Dry excess buffer from borders of slide with a kimwipe, while avoiding the sample.
Add the hybridization mix to a coverslip, then invert slide onto coverslip. The coverslip will adhere to the slide, and then the slide can be reinverted such that the coverslip is facing up.
Apply rubber cement to the borders of the coverslip to create a seal between the coverslip and the slide.
Allow rubber cement to dry completely (~10 min); otherwise, it will bubble up and hybridization mix will leak out
Place the slide facing up onto 92°C heat block and incubate for 2.5 min (see Note 7)
Remove the slide and place in a humidified chamber to prevent drying of slides. We use an old pipette box wherein the slides are placed on top of the rack and water is placed on the bottom of the box.
Store the humidified chamber in a 37°C incubator overnight for at least 16 hours.
3.7. Post-Primary Hybridization Wash
Prepare coplin jars with 2xSSCT in water baths set at 60°C.
Remove slides from chamber and start removing the rubber cement and coverslips. A razor blade can be used to slip under the coverslip, and gently lift the coverslip away from the slide.
After removal of the coverslip, transfer the slide to 2xSSCT solution at room temperature for a quick rinse.
Transfer slide to 2x SSCT solution at 60 °C and incubate for 15 min.
Transfer slide to coplin jar containing 2x SSCT at room temperature and incubate for 10 min. (see Note 18)
Transfer slide to coplin jar containing 0.2x SSCT at room temperature and incubate for 10 min.
Transfer slide to coplin jar containing 2x SSCT and prepare for secondary hybridization.
3.8. Hybridization of Secondary Probe
Prepare hybridization mix for secondary probe. (see Note 19)
Remove slide from coplin jar and wipe borders of slide dry, avoiding the sample area.
Add hybridization mix to coverslip, then invert slide onto coverslip. The coverslip will adhere to the slide. Reinvert and apply rubber cement to seal. Allow to dry completely.
Transfer slide to humidified chamber and incubate at room temperature away from light, for 30 min to 2 hours.
3.9. Post-Secondary Hybridization Wash
Prepare coplin jars with 2xSSCT in water baths set at 60°C.
Take slides out of the chamber and start removing the rubber cement and coverslips. A razor blade can be used to slip under the coverslip, and gently lift the coverslip away from the slide.
After removal of the coverslip, place slide in 2xSSCT solution at room temperature for a quick rinse.
Transfer slide to 2x SSCT solution at 60°C and incubate for 15 min.
Transfer slide to coplin jar containing 2x SSCT at room temperature and incubate for 10 min (see Note 18)
Transfer slide to coplin jar containing 0.2x SSCT at room temperature and incubate for 10 min.
Transfer slide to coplin jar containing 2x SSCT and prepare for mounting.
3.10. Mount for imaging
Remove slide from coplin jar and wipe borders of slide dry, avoiding the sample.
Add 15 μl of mounting medium to a new coverslip, then invert slide onto the coverslip. The coverslip will adhere to the slide. Remove excess medium and seal coverslip to the slide using nail polish.
4. Notes
We use secondary binding sequences that have been previously published [28].
If using a different RNase inhibitor, make sure it can withstand the 50°C reverse transcriptase step, as some RNAse inhibitors are heat labile.
If the reaction is scaled up or down, the columns can be changed to improve the workflow. The main consideration when choosing columns is the binding capacity.
Some samples have difficulty adhering to slides during the settling period. One alternative is to grow cells directly on the coverslip by placing the coverslips in a 6-well plate, then culturing cells in the well. Fixation and permeabilization can be done in the well itself. It is preferable to seed the slides or coverslips with cells prior to fixation since this allows greater control over cell density and clumpiness, which may impact downstream microscopy analysis. Alternatively, especially in the case of cells grown in suspension, cytospinning of cells can be used to attach cells onto slides.
The goal with the permeabilization is to help the probes reach their target by partially solubilizing the cell and nuclear membranes. As such, different sample types require different permeabilization protocols [28, 32, 36], or is sometimes the case with cultured cells, may not require any permeabilization whatsoever. If there are no permeabilization methods published in the literature, one can leave out the permeabilization step as a first run, then add different permeabilization conditions to see if probe detection improves.
Formamide should be stored at 4°C. Formamide quality is very important for efficient hybridization, and keeping the formamide at 4°C will help prevent its degradation for several months.
This water bath is set for both the pre-denaturation and denaturation steps. Samples can be denatured using lower temperatures and longer times; we have also successfully tested 80°C at 5 min. Lower temperatures may help with specific sample types and/or with the preservation of epitopes for detection via immunofluorescence. Likewise, increased formamide concentrations can be increased to help with the denaturation of the genome at lower temperatures.
The amount of probe that is needed can vary depending on the sample type and the probe density. We find that it is good to test a range of amounts, from 50 pmol to 200 pmol. The amount of dNTPs can be varied with the amount of probe paint to find an acceptable signal-to-noise. Probes to multiple chromosomes can be added simultaneously as long as probes contain different secondary binding sites. However, we have found that too much total probe can cause degradation of overall signal across all probes. The amount of probe that can be added simultaneously will have to be determined empirically, but as a rule of thumb, we find that adding more than ~600 pmol of probe can lead to loss of signal.
PVSA is macromolecular crowder, similar to dextran sulfate, that assists in hybridization [37]. PVSA is not required for hybridization if dextran sulfate is used, but it can improve the signal-to-noise detection of FISH signals.
We find that dNTPs also improves hybridization, similar to reported uses of salmon sperm DNA and other carriers. We prefer dNTPs over other carriers to avoid signal when using DAPI to stain DNA.
In order to reduce background and improve signal to noise, hybridization temperature above 37°C can be used. Please keep in mind the Tm of the probe sequences to determine the maximum temperature that can be used to avoid loss of probe binding.
We routinely use a second PCR step to produce more material for RNA synthesis, but this is not necessary. The initial PCR step can be used for T7 reaction as long as the forward and reverse primers containing the secondary binding site and T7 site, respectively, are used.
Fluorophore-labeled primers can also be used to create probes with conjugated dyes. We have observed increased background signal using these types of probes, and instead opt for an unlabeled primary/labeled secondary probe strategy. Also, if labeled primers are used, a different RNA degradation step may be required since alkaline hydrolysis can lead to loss of the fluorophore.
Manufacturer protocol suggests using double the ethanol, which does lead to higher DNA concentration readings, but may be due to unincorporated primers and dNTPs. The amount of ethanol outlined in the protocol is more stringent in purifying oligos 80 nucleotides and longer.
Cell concentration will have to be determined empirically due to variations in cell sizes. The main objective is to minimize overlapping cells while maximizing cell density in a microscopic field of cells.
Air-drying the slide after the last ethanol step and prior to submerging in 2x SSCT is optional but may improve the signal; however, this step may require optimization as some samples or some environments may lead to overdrying. When this step is included, do not dry slides beyond 2 minutes.
Slides at this step do not tend to overdry, possibly due to the residual 2x SSCT and formamide. This step tends to improve signal, but if slides do seem to dry out, slides can be placed back into the coplin jar containing room temperature 2x SSCT + formamide.
Care should be taken not to leave samples at 60°C in SSCT or at room temperature in 0.2x SSC beyond the recommended time, as high temperatures and low salt conditions can lead to loss of probe. Washes at room temperature with 2xSSCT can be extended up to a couple of hours in order to reduce background.
Secondary hybridization requires less probe and less formamide for primary probe detection, due to the fact that the secondary binding site is already single stranded. The original hybridization mix, however, can be used as well. We have also tested shorter incubation times of 30 minutes and also hybridization temperatures of 37°C and have seen good signal in most cases.
References
- 1.Cremer T, Cremer C, Baumann H et al. (1982) Rabl’s model of the interphase chromosome arrangement tested in Chinese hamster cells by premature chromosome condensation and laser-UV-microbeam experiments. Hum Genet 60(1):46–56. [DOI] [PubMed] [Google Scholar]
- 2.Cremer T, Cremer M (2010) Chromosome territories. Cold Spring Harb Perspect Biol 2(3):a003889. doi: 10.1101/cshperspect.a003889 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eils R, Dietzel S, Bertin E et al. (1996) Three-dimensional reconstruction of painted human interphase chromosomes: active and inactive X chromosome territories have similar volumes but differ in shape and surface structure. J Cell Biol 135(6 Pt 1):1427–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Clemson CM, Hall LL, Byron M et al. (2006) The X chromosome is organized into a gene-rich outer rim and an internal core containing silenced nongenic sequences. Proc Natl Acad Sci U S A 103(20):7688–7693. doi: 10.1073/pnas.0601069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Splinter E, de Wit E, Nora EP et al. (2011) The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev 25(13):1371–1383. doi: 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen C, Blanco M, Jackson C et al. (2016) Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science 354(6311):468–472. doi: 10.1126/science.aae0047. [DOI] [PubMed] [Google Scholar]
- 7.Chaumeil J, Le Baccon P, Wutz A et al. (2006) A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev 20(16):2223–2237. doi: 10.1101/gad.380906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mahy NL, Perry PE, Gilchrist S et al. (2002) Spatial organization of active and inactive genes and noncoding DNA within chromosome territories. J Cell Biol 157(4):579–589. doi: 10.1083/jcb.200111071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahy NL, Perry PE, Bickmore WA (2002) Gene density and transcription influence the localization of chromatin outside of chromosome territories detectable by FISH. J Cell Biol 159(5):753–763. doi: 10.1083/jcb.200207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Branco MR, Pombo A (2006) Intermingling of chromosome territories in interphase suggests role in translocations and transcription-dependent associations. PLoS Biol 4(5):e138. doi:05-PLBI-RA-1043R2 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finlan LE, Sproul D, Thomson I et al. (2008) Recruitment to the nuclear periphery can alter expression of genes in human cells. PLoS Genet 4(3):e1000039. doi: 10.1371/journal.pgen.1000039 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Steensel B, Belmont AS (2017) Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169(5):780–791. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shah S, Takei Y, Zhou W et al. (2018) Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell 174(2):376.e16. doi: 10.1016/j.cell.2018.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao SS, Huntley MH, Durand NC et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159(7):1665–1680. doi: 10.1016/j.cell.2014.11.021 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lieberman-Aiden E, van Berkum NL, Williams L et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326(5950):289–293. doi: 10.1126/science.1181369 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dekker J, Mirny L (2016) The 3D Genome as Moderator of Chromosomal Communication. Cell 164(6):1110–1121. doi: 10.1016/j.cell.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fraser J, Williamson I, Bickmore WA et al. (2015) An Overview of Genome Organization and How We Got There: from FISH to Hi-C. Microbiol Mol Biol Rev 79(3):347–372. doi: 10.1128/MMBR.00006-15 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Flyamer IM, Gassler J, Imakaev M et al. (2017) Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544(7648):110–114. doi: 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang S, Su J, Beliveau BJ et al. (2016) Spatial organization of chromatin domains and compartments in single chromosomes. Science 353(6299):598–602. doi: 10.1126/science.aaf8084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bintu B, Mateo LJ, Su J et al. (2018) Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362(6413). doi: 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beliveau BJ, Joyce EF, Apostolopoulos N et al. (2012) Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc Natl Acad Sci U S A 109(52):21301–21306. doi: 10.1073/pnas.1213818110 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beliveau BJ, Kishi JY, Nir G et al. (2018) OligoMiner provides a rapid, flexible environment for the design of genome scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115(10):E2192. doi: 10.1073/pnas.1714530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scalenghe F, Turco E, Edström JE et al. (1981) Microdissection and cloning of DNA from a specific region of Drosophila melanogaster polytene chromosomes. Chromosoma 82(2):205–216. [DOI] [PubMed] [Google Scholar]
- 24.Carter NP, Ferguson-Smith MA, Perryman MT et al. (1992) Reverse chromosome painting: a method for the rapid analysis of aberrant chromosomes in clinical cytogenetics. Journal of medical genetics 29(5):299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meltzer PS, Guan XY, Burgess A et al. (1992) Rapid generation of region specific probes by chromosome microdissection and their application. Nat Genet 1(1):24–28. doi: 10.1038/ng0492-24. [DOI] [PubMed] [Google Scholar]
- 26.Rabbitts P, Impey H, Heppell-Parton A et al. (1995) Chromosome specific paints from a high resolution flow karyotype of the mouse. Nat Genet 9(4):369–375. doi: 10.1038/ng0495-369. [DOI] [PubMed] [Google Scholar]
- 27.Liehr T, Heller A, Starke H et al. (2002) Microdissection based high resolution multicolor banding for all 24 human chromosomes. Int J Mol Med 9(4):335–339. [PubMed] [Google Scholar]
- 28.Beliveau BJ, Boettiger AN, Avendano MS et al. (2015) Single-molecule super-resolution imaging of chromosomes and in situ haplotype visualization using Oligopaint FISH probes. Nat Commun 6:7147. doi: 10.1038/ncomms8147 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanborn AL, Rao SSP, Huang S et al. (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci U S A 112(47):6456. doi: 10.1073/pnas.1518552112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boettiger AN, Bintu B, Moffitt JR et al. (2016) Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529(7586):418–422. doi: 10.1038/nature16496 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bintu B, Mateo LJ, Su J et al. (2018) Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362(6413). doi: 10.1126/science.aau1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fields BD, Nguyen SC, Nir G et al. (2018) A Multiplexed DNA FISH strategy for Assessing Genome Architecture in C. elegans. bioRxiv:397471. doi: 10.1101/397471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nir G, Farabella I, Pérez Estrada C et al. (2018) Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. bioRxiv:374058. doi: 10.1101/374058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosin LF, Nguyen SC, Joyce EF (2018) Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet 14(7):e1007393. doi: 10.1371/journal.pgen.1007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lab Wu (2017) Oligopaints - Harvard Medical School. https://oligopaints.hms.harvard.edu/.
- 36.Lee JH, Daugharthy ER, Scheiman J et al. (2015) Fluorescent in situ sequencing (FISSEQ) of RNA for gene expression profiling in intact cells and tissues. Nat Protoc 10(3):442–458. doi: 10.1038/nprot.2014.191 [doi]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Frei AP, Bava F, Zunder ER et al. (2016) Highly multiplexed simultaneous detection of RNAs and proteins in single cells. Nat Methods 13(3):269–275. doi: 10.1038/nmeth.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]