

Abstract
Introduction:
Plasma kallikrein is a mediator of vascular leakage and inflammation. Activation of plasma kallikrein can induce features of diabetic macular edema (DME) in preclinical models. Human vitreous shows elevated plasma kallikrein levels in patients with DME. Because of the incomplete response of some patients to anti-VEGF agents, and the treatment burden associated with frequent dosing, there is still considerable need for VEGF-independent targeted pathways.
Areas covered:
This review covers the role of plasma kallikrein in the pathogenesis of DME and the therapeutic potential of plasma kallikrein inhibitors. It discusses early clinical studies of plasma kallikrein pathway modulation for DME, which have been associated with some improvement in visual acuity but with limited improvement in macular edema. This review also highlights KVD001, which is furthest along the development pathway, THR-149, which has recently completed a phase 1 study, and oral agents under development.
Expert opinion:
Plasma kallikrein inhibitors have a potential role in the treatment of DME, with mixed functional/anatomic results in early clinical trials. Given the large unmet need in DME treatment, further studies are warranted.
Keywords: Diabetic retinopathy, diabetic macular edema, vascular permeability, plasma kallikrein, vascular endothelial growth factor
1. Introduction
There is an alarming increase in diabetes mellitus worldwide. The World Health Organization (WHO) estimated approximately 442 million adults with diabetes worldwide in 2014 [1]. Along with rising prevalence, complications of diabetes have precipitously increased. Uncontrolled diabetes leads to a range of microvascular complications such as vision loss, kidney failure, and lower-limb amputations. Overall, these complications significantly increase morbidity and mortality.
Diabetic retinopathy (DR) is one of the most common complications of diabetes. A meta-analysis of 35 studies involving 28,896 people with diabetes predicts approximately 93 million people with any DR, 17 million with proliferative DR and 21 million people with diabetic macular edema [2]. Diabetic macular edema (DME) leads causes of blindness among working-age adults of most developed countries [3]. The Early Treatment Diabetic Retinopathy Study (ETDRS) classification, a gold standard, defined clinically significant macular edema (CSME) as (i) the presence of retinal thickening at or within 500 μm of the center of the macula or (ii) hard exudates at or within 500 μm of the center of the macula if associated with adjacent retinal thickening or (iii) a zone of retinal thickening of 1 disc area in size, or any part of which is within 1 disc diameter of the center of the macula. A follow-up Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR) demonstrated a 25-year incidence of ME and CSME of 29% and 17%, respectively. The strongest predictors of increased ME and CSME risk were hyperglycemia, followed by blood pressure [4]. In a Diabetes Control and Complications Trial (DCCT)-Epidemiology of Diabetes Interventions and Complications (EDIC) study, the 30-year incidence of CSME was reported to be 14.5% [5] in type 1 diabetes. Similar to WESDR, this study demonstrated an increase in HbA1c as the strongest risk factors of DME, followed by longer duration of diabetes, greater age and diastolic blood pressure [5]. The incidence of DME in people with type II diabetes is about 13.9% [6].
Prior to the development of currently available intravitreal (IVT) anti-VEGF medications, nearly half of patients with DME lost two or more lines of visual acuity within two years [7]. Even with current treatment, however, a large unmet need persists, particularly to address limited visual outcomes and treatment burden of frequent IVT injection. This review discusses the status of current treatment, and the development of new therapies targeting plasma kallikrein, which may play a role in the pathogenesis of DME.
2. Pathogenesis of DME
Chronic hyperglycemia structurally affects the three major components of the retinal microvasculature, the pericytes, endothelial cells and capillary basement membrane. Initially, damage to the pericytes, responsible for regulating capillary perfusion, leads to vascular dysregulation and microaneurysm formation [8]. Damage to endothelial cells, responsible for maintaining the blood-retinal barrier, leads to macular extracellular fluid. The thickening of the capillary basement membrane and increased deposition of extracellular matrix components ensue [3,8,9]. Ultimately, persistent retinal micro-vasculature damage leads to capillary nonperfusion and retinal ischemia, resulting in the upregulation of angiogenic factors, such as vascular endothelial growth factor (VEGF) [10].
DR is mediated in part by inflammation, as leukostasis, pros-taglandin upregulation and macrophage accumulation occur [11]. People with diabetes exhibit leukostasis, i.e. adherence of large and rigid leukocytes to vascular endothelium, as well as activation of adhesion molecules on retinal vasculature. This may result in ischemia, vascular occlusion and an increase in toxic superoxides, proteolytic enzymes, produced by these leukocytes [12]. These intertwined processes contribute to endothelial dys-function, vascular permeability, retinal vascular nonperfusion and, angiogenesis, with resulting vision loss. Inflammatory mediators such as VEGF, tumor necrosis factor-alpha, interleukin-6, interleukin-8, cyclo-oxygenase-2, and monocyte chemotactic protein-1 may also be upregulated in DME [11].
VEGF-A mediates angiogenesis by promoting endothelial cell migration, proliferation, and survival, as well as inflammation through an increase in microvascular permeability and leukocyte adhesion. Intravitreal anti-VEGF-A therapy is the first-line treatment for DME. Ranibizumab (Lucentis®, Genentech, Inc.), off-label bevacizumab (Avastin®, Genentech), and aflibercept (Eylea®, Regeneron Pharmaceuticals, Inc., which also inhibits VEGF-B and PlGF) are currently available commonly used anti-VEGF-A medications for DME. These agents bind and neutralize VEGF-A, decreasing angiogenic drive and vascular permeability, respectively reducing neovascularization and DME [3,10].
While anti-VEGFs are first-line treatment for DME, IVT corticosteroids also play a role in DME management. Corticosteroids reduce inflammatory cytokines and act on several pathogenic pathways in DME [13]. In pseudophakic eyes, DRCR.net Protocol-I demonstrated that off-label IVT triamcinolone acetonide (TA) was as effective at improving best-corrected visual acuity (BCVA) as ranibizumab [14]. The approved slow-release dexamethasone implant and extended-release nonbioerodible fluocinolone acetonide have demonstrated similar effects in pseudophakic eyes with DME [15]. A metanalysis of 521 DME eyes also demonstrated that dexamethasone implant improved visual acuity similar to anti-VEGFs with a significant improvement in anatomical outcomes [16]. Nevertheless, the dexamethasone implant is often used as a second line therapy in anti-VEGF nonresponding eyes or pseudophakic eyes, given the risk of corticosteroid-associated cataract and ocular hypertension [17,18].
In addition to VEGF-A, angiopoietin 2 (Ang2) is upregulated in patients with DME [19]. While Ang-1 activates tyrosine kinase with immunoglobulin-like domains (Tie-2) to maintain vascular stability, Ang-2 acts as a competitive antagonist to Ang-1, leading to a subsequent breakdown of blood-retinal barrier and inflammation [20]. Phase 2 studies involving a bispecific antibody, which targets both Ang2 and VEGF-A, demonstrated superior visual acuity gain compared to ranibizumab monotherapy [21], suggesting Ang2 inhibition as another potential treatment option for the DME.
Although the introduction of IVT anti-VEGF-A agents has led to meaningfully improved outcomes in DME, a large unmet need persists, particularly from the treatment burden of frequent IVT injection and incomplete response in some patients [22]. For example, incomplete resolution of macular edema was noted in approximately 1/3 of participants receiving anti-VEGF therapy at 1 and 2 years [23,24]. Furthermore, ‘real world’ studies based on electronic medical records (EMR) and claims data have demonstrated that DME patients may be undertreated, receiving as few as 2–6 IVT anti-VEGF injections in the first year [25–29], with mean gains of only 5 letters on average [27,30]. These limitations have fostered an interest in alternate pathways, including plasma kallikrein. Plasma kallikrein is known to be highly upregulated in vitreous of patients with DME [31], and thus can serve as a target for future drug development. Interestingly the plasma kallikrein pathway may be independent of VEGF, suggesting a potential role in anti-VEGF non-responders. Moreover, inhibition of plasma kallikrein can target a multitude of pathogenic mechanisms involved in DME and DR pathogenesis, such as vascular leakage, neovascularization and inflammation.
3. Plasma kallikrein
Plasma kallikrein is a serine protease synthesized mainly in the liver as a proenzyme prekallikrein (PK). The mRNA for plasma kallikrein or PK is expressed in a variety of tissues such as the brain, heart, lung, kidneys, adrenal glands, pancreas, spleen, prostate glands, and ovaries. PK is also known as the Fletcher factor due to its involvement in the fletcher trait, a condition with markedly prolonged activated partial thromboplastin time (aPTT). PK is encoded by a single gene localized on a q34-q35 region of the long arm of chromosome 4 [32]. PK is a single chain gamma-globulin zymogen with a molecular weight of 85–88 kDa, and a plasma concentration of ~490 nM. The N terminal region of PK consists of four contagious repeats composed of 4 groups of 90–91 amino acids arranged in ‘apple domains’ (A1-A4), Figure 1. The N-terminal of the PK lacks intrinsic activity and mainly involved in the recruitment of certain proteins [33]. The majority of PK circulates in plasma as a complex with alpha globulin, high molecular weight kininogen (HK). The apple domains, A1 and A4, serve as binding sites for HK. The proteolytic processing of PK leads to activated plasma kallikrein via activated factor XII (αFXIIa) on the negatively charged surface, factor XII fragment (βFXIIa) in the fluid phase and prolycarboxypeptidase on endothelial cells. Plasma kallikrein then cleaves HK to liberate bradykinin. While bradykinin directly activates the B2 receptor, the cleavage of bradykinin by carboxypeptidase generates des-arg9 bradykinin (DABK) leads to activation of B1 receptors (Figure 2).
Figure 1.
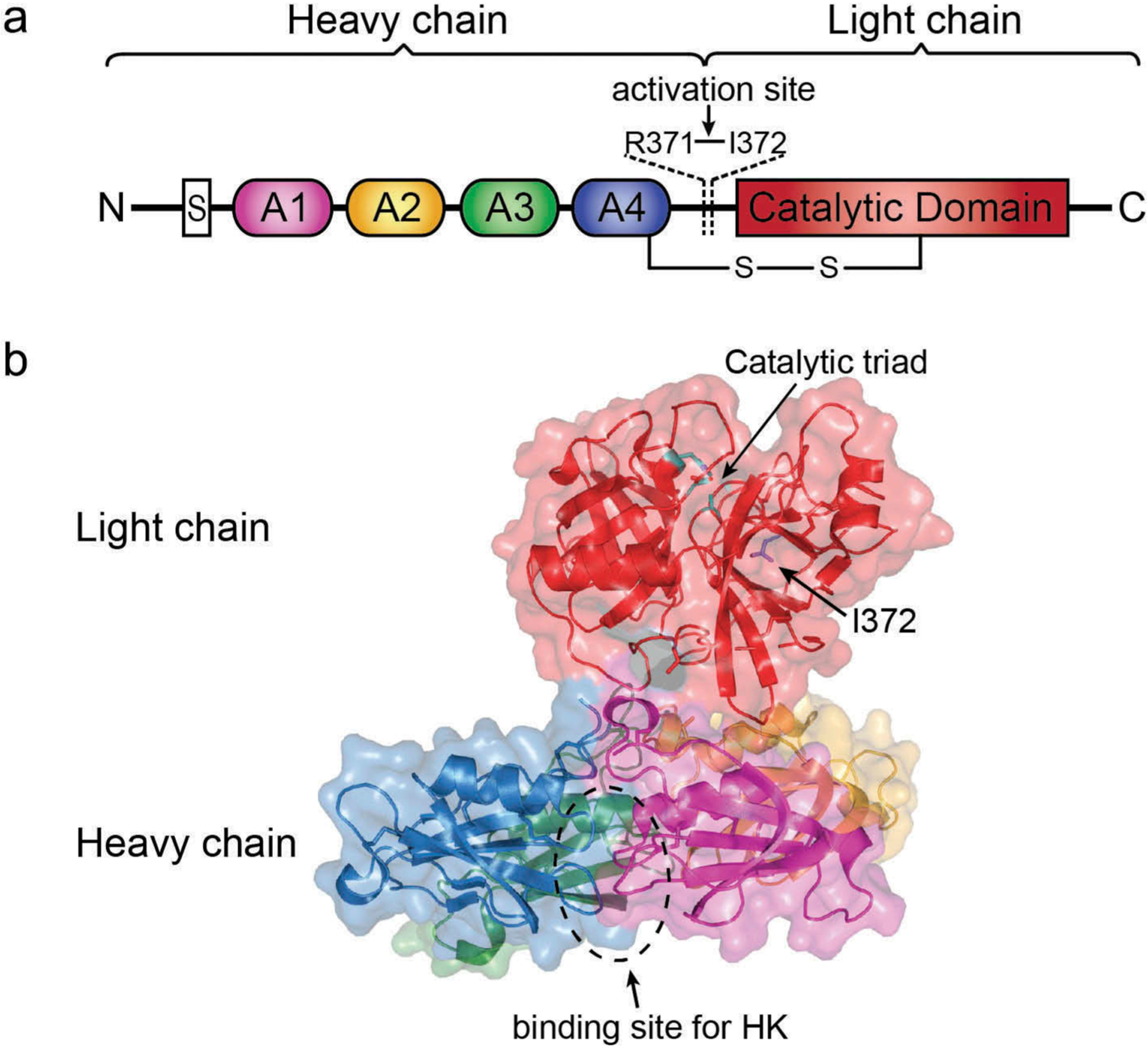
Structure of human full-length plasma kallikrein as determined by X-ray crystallography. (a) Domain architecture of PK. The activation site through cleavage between Arg371 and Ile372 is indicated. (b) A cartoon diagram of activated plasma kallikrein (PDB entry 6I44) with transparent surface rendering. The heavy chain consists of four apple domains colored as magenta (A1 domain), yellow (A2 domain), green (A3 domain), blue (A4 domain). The binding site for HK is located in the A1-A4 interface circled by a dotted line. The light chain consists of the trypsin-like serine protease catalytic domain colored as red with cyan marking the catalytic triad residues. This figure was generated using PyMOL [34].
Figure 2.

Schematic showing the activation of plasma kallikrein and downstream pathways. The FXII and plasma prekallikrein (PK) are proteolytically activated to FXIIa and plasma kallikrein, respectively. Plasma kallikrein then cleaves high molecular weight kininogen (HK) to release the bradykinin which may be further cleaved to des-Arg9-bradykinin (DABK). The bradykinin binds to bradykinin B2 receptors (B2R) and DABK binds to bradykinin (B1R) receptors to further activate the molecular cascade which subsequently leads to the increase in permeability and vasodilation. FXII-factor XII, FXIIa-factor XIIa, eNOS-endothelial nitric oxide synthase, iNOS-inducible nitric oxide synthase, PLA2-phospholipase A2, PGI2-prostacyclin, NO-nitric oxide, MPAK-mitogen-activated protein kinase; modified from Liu J, Feener EP. Plasma kallikrein-kinin system and diabetic retinopathy. Biological chemistry. 2013;394(3):319–328 [49].
The proteolytic processing of PK plays a vital role in both health and disease. For example, in hereditary angioedema (HAE), there is a deficiency of C1, an endogenous inhibitor of plasma kallikrein, and consequently, uncontrolled bradykinin-mediated edema ensues. The kallikrein-kinin system is activated in response to vascular injury and leads to a local increase in bradykinin, which further results in pro-inflammatory effects such as vascular permeability, vasodilation, and immune cell activation [35].
4. The role of plasma kallikrein in DR and DME
Human vitreous shows elevated levels of plasma kallikrein in patients with DME [31]. Specifically, PK and plasma kallikrein were increased two-fold and 11.0-fold respectively, in vitreous from DME patients compared to nondiabetic patients [31]. Also, vitreous fluids of patients with proliferative diabetic retinopathy (PDR) demonstrate an increase in plasma kallikrein, FXII and HK [36,37]. In preclinical models, activation of plasma kallikrein can induce features of DME, while inhibition of plasma kallikrein can ameliorate these findings [38]. Similarly, in plasma prekallikrein knockout mice, there is decreased diabetes-induced retinal vascular permeability [39].
The mechanism for intraocular plasma kallikrein elevation is unclear. Some propose that these proteins cross the blood-retinal barrier through DR-associated increased vascular permeability. Rodent models provide some supporting evidence. For example, in a rat model, injection of plasma kallikrein into the vitreous cavity results in both retinal vascular permeability and retinal edema, which is more pronounced in diabetic rats [40]. Moreover, the injection of plasma kallikrein accelerates vascular permeability in streptozotocin-induced diabetic mice. Treatment of diabetic mice with a small molecule inhibitor ASP-440 or C1 inhibitor show improved diabetes-induced retinal vascular permeability [40]. The increase in vascular permeability is mainly attributed to an increase in bradykinin and its receptors B1 and B2 [41,42]. There is also a potential involvement of endothelial nitric oxide synthase (eNOS) because it’s inhibition by N(G)-nitro-L-arginine methyl ester (L-NAME) results in a decrease in vascular permeability.
The reduced vascular perfusion can lead to retinal ischemia, and subsequently trigger the neovascularization response, PDR. This severe neovascular form of DR is associated with vitreous hemorrhage, retinal detachment and severe vision loss. It is not well understood how plasma kallikrein system is involved in retinal neovascularization; however, plasma kallikrein is reported to be angiogenic in other tissues, including the cornea [43]. The neovascular response of plasma kallikrein may be mediated via bradykinin and B1 receptor agonism because the knockout of the B1 receptor impaired the angiogenic response [44]. Some evidence also indicates the potential role of carbonic anhydrase-1 (CA-1) in elevated plasma kallikrein in PDR. There is an increase in CA-1 in vitreous of PDR patients and intravitreal injection of CA-1 increases permeability of diabetic rats; treatment with plasma kallikrein inhibitors block CA-1 induced retinal vascular permeability [37].
Plasma kallikrein mediated retinal edema may represent a VEGF-independent pathway, as blockade of VEGF receptor-2 in mice does not interfere with bradykinin-induced retinal edema [45,46]. Furthermore, systemic inhibition of plasma kallikrein decreased VEGF-induced retinal edema by 57% and 53% in mice and rats, respectively [47]. Similarly, VEGF-induced retinal vascular permeability and retinal edema were reduced by 68% and 47% respectively in plasma prekallikrein knock-out mice compared to wild type mice [47]. Although increases in plasma kallikrein were noted alongside increases in VEGF in vitreous samples of DME patients, a meaningful proportion of patients had elevated vitreous levels of plasma kallikrein in the absence of VEGF elevation [31]. Interestingly, when these high plasma kallikrein/low VEGF vitreous samples were intravitreally injected into diabetic rat eyes, there was increased retinal vascular permeability which could be blocked by bradykinin inhibition, but not by VEGF-A inhibition [31]. This suggests that the plasma kallikrein pathway may have an independent role from VEGF in DME pathophysiology.
Some studies suggest that there is a direct correlation between PK, plasma kallikrein and HbA1c, serum fructosamine. In contrast, an inverse relationship is reported between plasma kallikrein system and aPTT shortening in diabetes, suggesting an increase in plasma kallikrein in diabetes is mediated via activation of the intrinsic pathway [48]. Activation of plasma kallikrein in DME and PDR patients affects important functions such as innate inflammation, blood flow and coagulation [49]. The inflammatory response is mediated via the activation of B1 receptors [50], upregulation of nuclear factor-κB, and with potential involvement of leukostasis [42].
5. Plasma kallikrein inhibition
Several small molecules and bicyclic peptides targeting the plasma kallikrein/kinin system are currently under investigation for the treatment of DME via intravitreal, oral and topical administrations.
5.1. KVD001
KVD001 (KalVista Pharmaceuticals) is a highly potent and selective plasma kallikrein inhibitor, currently being developed as an IVT therapy. The pharmacodynamic effects of plasma kallikrein inhibition were assessed in a VEGF induced retinal edema in a mouse model. Systemic and oral administration of small molecule plasma kallikrein inhibitors (VA999272, KV998052, and KV998054) led to inhibition of retinal vascular permeability in mice [51]. Pharmacokinetics of KVD001 was evaluated in rabbits and monkeys following repeat intravitreal dosing. Sustained and high levels of KVD001 were observed in the retina with a calculated half-life of approximately 7 days [52]. Retinal levels of KVD001 were maintained above the half-maximal inhibitory concentration (IC50) for inhibition of plasma kallikrein for 28 days. The co-administration of an anti-VEGF agent did not affect the pharmacokinetic profile of KVD001. These data provided evidence that clinical exposure to KVD001 may be maintained at or above therapeutic levels with monthly or less frequent intervals.
Safety and preliminary efficacy of intravitreally injected KVD001 (1, 3 and 10 μg/eye) have been assessed in an open-label, single ascending dose clinical study [53]. Diabetic patients with visual acuity (VA) of 20/40 – 20/400 Snellen equivalent, and central subfield thickness (CST) of ≥305 μm (females) or ≥320 μm (males) were enrolled. All patients had received prior IVT anti-VEGF; however, these individuals were sub-optimal responders to current anti-VEGF treatment. In 12 patients who completed the 12-week study, a single administration of KVD001 resulted in mean VA improvement of 0.7, 1.0, 1.9, 2.8 and 4.1 letters compared to baseline at day 7, 14, 28, 56 and 84, respectively. Although the mean change in CST was marginal, none of the patients showed increased CST over 10% above baseline values. KVD001-related adverse events were noted at a low dose (3 patients) and a high dose (8 patients). The mid-dose (3 patients) showed mild ocular inflammation and a severe increase in the intraocular pressure immediately following administration. Authors acknowledge that the absence of a control group, a small number of patients per group, and only a single dose study may limit the interpretation of the observed signs of efficacy.
A Phase 2 clinical trial enrolled approximately 123 anti-VEGF treatment-experienced patients with persistent macular edema and reduced BCVA (NCT03466099). This sham-controlled, double-masked clinical trial evaluated two doses of KVD001, 6 μg and 3 μg, injected at baseline and then monthly over 3 months with 3 additional months of follow-up. Efficacy endpoints included BCVA, CST, and the diabetic retinopathy severity scale (DRSS). While the 6 μg dose of KVD001 showed +2.6 letters improvement over the sham treatment, the data did not reach the statistical significance (p = 0.223). The lower dose of KVD001, 3 μg showed a difference of +1.5 letters when compared to sham treatment (p = 0.465) [54]. No significant differences were noted in the secondary endpoints of CST or DRSS. However, the study population demonstrated a protective effect against the vision loss; 32.5% of patients treated with 6 μg of KVD001 (p = 0.042) showed vision loss when compared to 54.5% patients receiving a sham treatment [54]. Interestingly, after excluding patients with most severe vision loss (<55 letters) at baseline, the remaining 70% of the total patient population showed a difference in BCVA of 4.9 letters (p = 0.056) at the 6 μg dose. Further study is under consideration.
Currently, Kalvista is developing two oral candidates for HAE, and lists one oral candidate for DME in its early stage programs. While the phase 1 study for KVD900 suggests a favorable pharmacokinetic profile with maximum plasma concentration one hour following an oral administration of the capsule [55], it is not known whether oral agents cross the blood-retinal barrier sufficiently to yield appropriate levels. Further studies will help define therapeutic role of oral inhibitors in DME. While IVT therapy may be used in combination with anti-VEGF in broad DME population, an oral product may provide opportunities to significantly reduces the burden to patients and health care providers.
5.2. THR-149
THR-149 (Oxurion NV) is a potent human plasma kallikrein bicyclic peptide inhibitor developed using a phage display-based selections of constrained peptide libraries combined with rationally designed synthetic modifications. The pharmacodynamic effect of a bicyclic peptide was assessed in a streptozotocin (STZ) induced retinal permeability in a rodent model [56]. A significant reduction in retinal vascular leakage was observed at 4 weeks, compared to the vehicle-treated group, following an intravitreal administration of a bicyclic peptide inhibitor (100 μg/eye). Interestingly, this effect was comparable to the one obtained with a soluble VEGF-trap positive control. Following IVT administration to rabbits, a lead bicyclic peptide showed a long residence time in the eye, with a half-life of 39 ± 2 h. Importantly, peptide concentrations achieved in the eye were predicted to reach virtually complete inhibition of human plasma kallikrein. Preclinical safety of bicyclic peptide was assessed upon systemic administration in rats and cynomolgus non-human primates. No overt toxicological effect observed at the highest tested systemic dose of 125 μg (0.43 − 0.60 mg/kg) in rats and 1.25 mg (0.42 − 0.49 mg/kg) in cynomolgus non-human primates[56].
Most recently, THR-149 has undergone phase 1 open-label, a multicenter, non-randomized trial at 3 ascending dose levels (5, 22, 130 μg/eye) in 12 DME subjects with a history of response to prior anti-VEGF or corticosteroid treatment (NCT03511898). There were no dose-limiting toxicities or treatment-emergent serious adverse events. After a single IVT injection, there was rapid improvement in mean BCVA (3.9 letters on day 1, 7.5 letters on day 14), which was maintained until the end of the study (6.4 letters on day 90). Minimal change in CST was observed and reported to be within the variability of measurement [57]. On optical coherence tomography, macular volume correlated with improvement in BCVA (Oxurion Press Release 9 September 2019); an additional clinical study is currently planned.
5.3. RZ402
RZ402 (Rezolute Bio) is a potential once-daily orally administered small molecule plasma kallikrein inhibitor in preclinical development for DR and DME. RZ402 is shown to be a potent and highly selective protease inhibitor using in vitro assays, and in rat, monkey, and human plasma. It suppresses retinal vascular leakage in multiple clinically-relevant animal models of macular edema as effectively as an anti-VEGF agent [58]. It maintains efficacious concentrations for the intended 24-hour dosing interval following oral administration and reported to be safe and well-tolerated, with broad therapeutic margins [59,60]. Overall, these results support further assessment of the potential of RZ402 as an oral therapy for patients with DME.
5.4. VE-3539
Verseon Corp. has developed a series of plasma kallikrein inhibitors as a potential treatment of DME and other related eye disorders. Verseon’s plasma kallikrein inhibitors are potent in functional in vitro assays due to their single-digit nanomolar potency and hundred-fold selectivity over other serine proteases. Verseon’s candidates have been reported to have favorable pharmacokinetics and bioavailability for oral dosing as prodrugs, and reported to be efficacious in multiple preclinical in vivo models, including human plasma kallikrein and VEGF induced models [61]. In preclinical studies, the compound VE-3539 inhibited retinal thickening and retinal vascular leakage, which are key phenotypes observed in DME patients [62]. Verseon is expecting to bring the first candidate to the clinic in 2020.
6. Potential side effects of plasma kallikrein therapy
The pharmacological action of plasma kallikrein is mainly mediated by (1) plasma kallikrein driving regional blood flow via bradykinin-induced B2 receptor activation, and (2) intravascular thrombus formation via factor XI activation after an injury. Therefore, anti-kallikrein treatment could have adverse effects on hemodynamic changes induced by vasoconstrictor agents [63]. Animal models and ex-vivo human plasma samples from genetic knock-out of components of kallikrein system have exhibited changes in cardiovascular processes such as increased partial thromboplastin time [64], arotic aneurysm [65], increased blood pressure [66], decreased blood coagulation [67]. Considering paradoxical nature and complexity of kallikrein kinin system, careful considerations should be given to better understanding of involvement of kallikrein system in disease pathology, stage of the disease, and duration of inhibition of kallikrein system required for effectiveness. While genetic models of kallikrein deficiency in the relevant preclinical disease model can be beneficial in assessing potential side-effects, a clinical monitoring strategy for any cardiovascular events seem to be an important component of developing anti-kallikrein therapy. Locally administered therapies may mitigate some systemic risk, if extraocular levels remain low.
7. Conclusion
In summary, the pathogenesis and management of DR and DME are complex, involving multiple pathways. While anti-VEGF agents have revolutionized treatment, there is still an unmet need for alternative therapies to address treatment burden and limited efficacy outcomes. With the growing incidence of diabetes and DME, the search for therapeutic advancements takes on greater urgency. Modulation of the plasma kallikrein pathway has resulted in mixed functional/anatomic results in early clinical trials. Further study is warranted.
8. Expert opinion
Although anti-VEGF therapy has revolutionized the treatment of DME, there remains a large unmet need to address limited visual outcomes and treatment burden. Plasma kallikrein is a mediator of vascular leakage and inflammation, and there is evidence that plasma kallikrein is involved in DME pathogenesis in a VEGF independent fashion, as well as a VEGF interdependent fashion. Activation of plasma kallikrein can induce features of DME in preclinical models, and human vitreous shows elevated plasma kallikrein levels in patients with DME.
Consequently, plasma kallikrein inhibitors are expected to show potential as both monotherapy and combination therapy in primary and refractory cases of DME, respectively. In this way, plasma kallikrein inhibitors could reduce treatment burden and improve visual outcomes in DME, with the potential to treat cases refractory to current treatment modalities.
In two phase 1 studies and one phase 2 study, IVT plasma kallikrein inhibitors have shown early signs of safety, but mixed functional/anatomic efficacy. Specifically, these studies have shown modest improvement in BCVA. Furthermore, the phase 2 KVD001 study suggested a protective effect against vision loss, as well as greater improvement in those patients with less severe vision loss at baseline. Missing the DRSS endpoint is not surprising, given the relatively short nature of this six-month study. However, the lack of convincing improvement in macular edema, as measured by CST, is concerning, especially for a therapy thought to affect vascular permeability.
The early clinical trial results do not correlate with preclinical studies, although animal models of DME have limitations. It is unclear if additional dosage, alternative delivery methods or other plasma kallikrein inhibitors could result in more robust anatomic outcomes. As noted above, oral plasma kallikrein inhibitors are being developed for assessment in DME. Given the large unmet need in DME treatment, further study is warranted.
Article Highlights.
Despite impressive advancements in treating diabetic retinopathy over the past two decades with anti-vascular endothelial growth factor (anti-VEGF-A) therapy, there is a great need to reduce the treatment burden from frequent injections and to improve visual outcomes.
Plasma kallikrein is a mediator of vascular leakage and inflammation. Activation of plasma kallikrein can induce features of DME in preclinical models, and human vitreous shows elevated plasma kallikrein levels in patients with DME.
There is preclinical and clinical evidence that plasma kallikrein is involved in DME pathogenesis in a VEGF independent fashion, as well as a VEGF interdependent fashion. Consequently, plasma kallikrein inhibitors have potential as both monotherapy and combination therapy in primary and refractory cases of DME, respectively.
In early clinical studies of DME, plasma kallikrein pathway modulation is associated with some improvement in visual acuity but limited improvement in macular edema. It is unclear if additional dosage, alternative delivery methods or other plasma kallikrein inhibitors could result in more robust anatomic and visual outcomes. Given the large unmet need in DME treatment, further study is warranted.
Acknowledgments
The authors would like to thank Dr. Qianyi Luo for help with figures.
Funding
Research in Bhatwadekar lab is supported by funding by an NIH-NEI grant R01EY027779 and a pilot and feasibility grant from the Center for Diabetes and Metabolic Diseases (CDMD), Indiana University.
Footnotes
Declaration of interest
T Ciulla has an employment relationship with, and equity ownership in, Clearside Biomedical. This work was undertaken in his role as Volunteer Clinical Professor at Indiana University School of Medicine, and does not reflect any views or opinions of this corporation or management. V Kansara has an employment relationship with, and equity ownership in, Clearside Biomedical. This work does not reflect any views or opinions of thiscorporation or management. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
One reviewer has received honorarium from Allergan, Bayer, Boehringer Ingelheim, Genetech, Novartis, Oculis and Roche for consultancy and lecture fees.
Peer reviewers on this manuscript have no other relevant financial or other relationships to disclose
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Organization WH. WHO Global report on diabetes. World Health Organization; 2016. [Google Scholar]
- 2.Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35 (3):556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This is a metanalysis study that provides excellent information about the global prevalence of diabetic retinopathy and risk factors.
- 3.Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: pathophysiology, screening, and novel therapies. Diabetes Care. 2003. September;26(9):2653–2664. [DOI] [PubMed] [Google Scholar]
- 4.Klein R, Knudtson MD, Lee KE, et al. The Wisconsin epidemiologic study of diabetic retinopathy XXIII: the twenty-five-year incidence of macular edema in persons with type 1 diabetes. Ophthalmology. 2009;116(3):497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hainsworth DP, Bebu I, Aiello LP, et al. Risk factors for retinopathy in type 1 diabetes: the DCCT/EDIC Study. Diabetes Care. 2019;42 (5):875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein R, Klein BE, Moss SE, et al. The Wisconsin epidemiologic study of diabetic retinopathy. XV. The long-term incidence of macular edema. Ophthalmology. 1995. January;102(1):7–16. [DOI] [PubMed] [Google Scholar]
- 7.Ferris FL 3rd, Patz A. Macular edema. A complication of diabetic retinopathy. Surv Ophthalmol. 1984. May;28:Suppl:452–61. [DOI] [PubMed] [Google Scholar]
- 8.Ciulla TA, Harris A, Latkany P, et al. Ocular perfusion abnormalities in diabetes. Acta Ophthalmol Scand. 2002. October;80(5):468–477. [DOI] [PubMed] [Google Scholar]
- 9.Ehrlich R, Harris A, Ciulla TA, et al. Diabetic macular oedema: physical, physiological and molecular factors contribute to this pathological process. Acta Ophthalmol. 2010. May;88(3):279–291. [DOI] [PubMed] [Google Scholar]
- 10.Morello CM. Etiology and natural history of diabetic retinopathy: an overview. Am J Health Syst Pharm. 2007. September 1;64(17 Suppl 12):S3–7. [DOI] [PubMed] [Google Scholar]
- 11.Hussain RM, Ciulla TA. Treatment strategies for refractory diabetic macular edema: switching anti-VEGF treatments, adopting corticosteroid-based treatments, and combination therapy. Expert Opin Biol Ther. 2016;16(3):365–374. [DOI] [PubMed] [Google Scholar]
- 12.Miyamoto K, Ogura Y. Pathogenetic potential of leukocytes in diabetic retinopathy. Semin Ophthalmol. 1999. December;14(4):233–239. [DOI] [PubMed] [Google Scholar]
- 13.Campochiaro PA, Hafiz G, Mir TA, et al. Pro-permeability factors in diabetic macular edema; the diabetic macular edema treated with Ozurdex trial. Am J Ophthalmol. 2016;168:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elman MJ, Aiello LP, Beck RW, et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology. 2010;117(6):1064–1077. e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iluvien | home 2020. [cited Jan 21 2020]. Available from: https://iluvien.com/
- 16.He Y, Ren X-J, Hu B-J, et al. A meta-analysis of the effect of a dexamethasone intravitreal implant versus intravitreal anti-vascular endothelial growth factor treatment for diabetic macular edema. BMC Ophthalmol. 2018;18(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zur D, Iglicki M, Loewenstein A. The role of steroids in the management of diabetic macular edema. Ophthalmic Res. 2019;62(4):231–236. doi: 10.1159/000499540. [DOI] [PubMed] [Google Scholar]
- 18.Kim EJ, Lin WV, Rodriguez SM, et al. Treatment of diabetic macular edema. Curr Diab Rep. 2019;19(9):68. [DOI] [PubMed] [Google Scholar]
- 19.Patel J, Hykin P, Gregor Z, et al. Angiopoietin concentrations in diabetic retinopathy. Br J Ophthalmol. 2005;89(4):480–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Campochiaro PA, Peters KG. Targeting Tie2 for treatment of diabetic retinopathy and diabetic macular edema. Curr Diab Rep. 2016;16(12):126. [DOI] [PubMed] [Google Scholar]
- 21.Sahni J, Patel SS, Dugel PU, et al. Simultaneous inhibition of angiopoietin-2 and vascular endothelial growth factor-a with faricimab in diabetic macular edema: BOULEVARD phase 2 randomized trial. Ophthalmology. 2019. August;126(8):1155–1170. doi: 10.1016/j.ophtha.2019.03.023. [DOI] [PubMed] [Google Scholar]
- 22.Hussain RM, Ciulla TA. Emerging vascular endothelial growth factor antagonists to treat neovascular age-related macular degeneration. Expert Opin Emerg Drugs. 2017. September;22(3):235–246. [DOI] [PubMed] [Google Scholar]
- 23.Wells JA, Glassman AR, Ayala AR, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema: two-year results from a comparative effectiveness randomized clinical trial. Ophthalmology. 2016. June;123(6):1351–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diabetic Retinopathy Clinical Research N, Wells JA, Glassman AR. et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med. 2015. March 26;372(13):1193–1203. Available from: https://www.ncbi.nlm.nih.gov/pubmed/25692915 [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This is a multicenter clinical study that compared three different anti-VEGF treatments for DME. Overall the treatment effect was similar when the initial visual acuity loss was mild. However, aflibercept was more effective when the initial visual acuity was at worse.
- 25.Blinder KJ, Dugel PU, Chen S, et al. Anti-VEGF treatment of diabetic macular edema in clinical practice: effectiveness and patterns of use (ECHO study report 1). Clin Ophthalmol. 2017;11:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dugel PU, Layton A, Varma RB. Diabetic macular edema diagnosis and treatment in the real world: an analysis of medicare claims data (2008 to 2010). Ophthalmic Surg Lasers Imaging Retina. 2016. March;47(3):258–267. [DOI] [PubMed] [Google Scholar]
- 27.Holekamp NM, Campbell J, Almony A, et al. Vision outcomes following anti-vascular endothelial growth factor treatment of diabetic macular edema in clinical practice. Am J Ophthalmol. 2018;191:83–91. [DOI] [PubMed] [Google Scholar]
- 28.Kiss S, Liu Y, Brown J, et al. Clinical utilization of anti-vascular endothelial growth-factor agents and patient monitoring in retinal vein occlusion and diabetic macular edema. Clin Ophthalmol. 2014;8:1611–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.VanderBeek BL, Shah N, Parikh PC, et al. Trends in the care of diabetic macular edema: analysis of a national cohort. PloS One. 2016;11(2):e0149450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ciulla TA, Bracha P, Pollack J, et al. Real-world outcomes of anti-vascular endothelial growth factor therapy in diabetic macular edema in the United States. Ophthalmol Retina. 2018. December;2(12):1179–1187. [DOI] [PubMed] [Google Scholar]
- 31.Kita T, Clermont AC, Murugesan N, et al. Plasma kallikrein-kinin system as a VEGF-independent mediator of diabetic macular edema. Diabetes. 2015. October;64(10):3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study demonstrates that actions of plasma kallikrein are independent of VEGF.
- 32.Beaubien G, Rosinski-Chupin I, Mattei M, et al. Gene structure and chromosomal localization of plasma kallikrein. Biochemistry. 1991;30(6):1628–1635. [DOI] [PubMed] [Google Scholar]
- 33.Kolte D, Shariat-Madar Z. Plasma kallikrein inhibitors in cardiovascular disease. Cardiol Rev. 2016;24(3):99–109. [DOI] [PubMed] [Google Scholar]
- 34.Version 2.0. Schrödinger, LLC; The PyMOL molecular graphics system.
- 35.Couture R, Blaes N, Girolami JP. Kinin receptors in vascular biology and pathology. Curr Vasc Pharmacol. 2014. March;12(2):223–248. [DOI] [PubMed] [Google Scholar]
- 36.Gao -B-B, Chen X, Timothy N, et al. Characterization of the vitreous proteome in diabetes without diabetic retinopathy and diabetes with proliferative diabetic retinopathy. J Proteome Res. 2008;7 (6):2516–2525. [DOI] [PubMed] [Google Scholar]
- 37.Gao -B-B, Clermont A, Rook S, et al. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007;13(2):181. [DOI] [PubMed] [Google Scholar]; •• This research article demonstrates the role of carbonic anhydrase 1 in diabetic retinopathy and the potential involvement of plasma kallikrein in retinal edema.
- 38.Murugesan N, Fickweiler W, Clermont AC, et al. Retinal proteome associated with bradykinin-induced edema. Exp Eye Res. 2019;186:107744. [DOI] [PubMed] [Google Scholar]
- 39.Clermont A, Murugesan N, Zhou Q, et al. Plasma kallikrein mediates vascular endothelial growth factor–induced retinal dysfunction and thickening. Invest Ophthalmol Vis Sci. 2016;57(6):2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clermont A, Chilcote TJ, Kita T, et al. Plasma kallikrein mediates retinal vascular dysfunction and induces retinal thickening in diabetic rats. Diabetes. 2011. May;60(5):1590–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This preclinical study shows that elevation of plasma kallikrein leads to retinal thickening and vascular dysfunction. The treatment with plasma kallikrein inhibitor decreased retinal vascular permeability.
- 41.Lawson SR, Gabra BH, Guérin B, et al. Enhanced dermal and retinal vascular permeability in streptozotocin-induced type 1 diabetes in Wistar rats: blockade with a selective bradykinin B1 receptor antagonist. Regul Pept. 2005;124(1–3):221–224. [DOI] [PubMed] [Google Scholar]
- 42.Phipps J, Feener E. The kallikrein–kinin system in diabetic retinopathy: lessons for the kidney. Kidney Int. 2008;73(10):1114–1119. [DOI] [PubMed] [Google Scholar]; • This review article summarizes how the plasma kallikrein system is involved in diabetic retinopathy and also compares its involvement in diabetic nephropathy.
- 43.Parenti A, Morbidelli L, Ledda F, et al. The bradykinin/B1 receptor promotes angiogenesis by up-regulation of endogenous FGF-2 in endothelium via the nitric oxide synthase pathway. Faseb J. 2001;15(8):1487–1489. [PubMed] [Google Scholar]
- 44.Emanueli C, Bonaria Salis M, Stacca T, et al. Targeting kinin B1 receptor for therapeutic neovascularization. Circulation. 2002;105(3):360–366. [DOI] [PubMed] [Google Scholar]
- 45.Kita T, Clermont AC, Fujisawa K, et al. Identification of a VEGF-independent and plasma kallikrein-kinin-dependent pathway of retinal vascular permeability in diabetic macular edema. Invest Ophthalmol Vis Sci. 2012;53(14):2431.22427588 [Google Scholar]
- 46.Kita T, Clermont AC, Murugesan N, et al. Plasma kallikrein-kinin system as a VEGF-independent mediator of diabetic macular edema. Diabetes. 2015;64(10):3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clermont A, Murugesan N, Zhou Q, et al. Plasma kallikrein mediates vascular endothelial growth factor-induced retinal dysfunction and thickening. Invest Ophthalmol Vis Sci. 2016. May 1;57(6):2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This research study demonstrates an increase in plasma kallikrein in individuals with DME. Moreover, this study also reports that the effect of plasma kallikrein in DME pathogenesis is independent of VEGF.
- 48.Lippi G, Franchini M, Targher G, et al. Epidemiological association between fasting plasma glucose and shortened APTT. Clin Biochem. 2009;42(1–2):118–120. [DOI] [PubMed] [Google Scholar]
- 49.Liu J, Feener EP. Plasma kallikrein-kinin system and diabetic retinopathy. Biol Chem. 2013;394(3):319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review article gives detailed information on the plasma kallikrein system and diabetic retinopathy. The article discussed the kallikrein system, the potential role of bradykinin and the role of plasma kallikrein inhibitors.
- 50.Abdouh M, Khanjari A, Abdelazziz N, et al. Early upregulation of kinin B1 receptors in retinal microvessels of the streptozotocin-diabetic rat. Br J Pharmacol. 2003;140(1):33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murugesan N, Clermont AC, Rushbrooke LJ, et al. A novel oral plasma kallikrein (PKal) inhibitor KV123833 blocks VEGF-mediated retinal vascular hyperpermeability in a murine model of retinal edema (# 3464) 2018.
- 52.[cited 2020 Jan 21]. Available from: http://ir.kalvista.com/node/7656/html
- 53.Sun JK, Maturi RK, Boyer DS, et al. One-time intravitreal injection of KVD001, a plasma kallikrein inhibitor, in patients with central involved diabetic macular edema and reduced vision: an open-label phase 1B study. Ophthalmol Retina. 2019;3(12):1107–1109. [DOI] [PubMed] [Google Scholar]; • This is an open-label study where the safety, pharmacokinetics and potential efficacy of plasma kallikrein inhibitor was evaluated.
- 54.KalVista pharmaceuticals reports phase 2 clinical trial results in patients with diabetic macular edema | business wire. 2019. December 09.
- 55.KVD900 for HAE | kalVista Pharmaceuticals, Inc. 2020.
- 56.Teufel DP, Bennett G, Harrison H, et al. Stable and long-lasting, novel bicyclic peptide plasma kallikrein inhibitors for the treatment of diabetic macular edema. J Med Chem. 2018. April 12;61 (7):2823–2836. [DOI] [PubMed] [Google Scholar]
- 57.Retina Society. Results of a Phase 1, Open-Label, Dose-Escalation Study of THR-149 for the Treatment of DME. Dr. Pravin Dugel, London: ·15 September 2019 [Google Scholar]
- 58.Phipps JA, Clermont AC, Sinha S, et al. Plasma kallikrein mediates angiotensin II Type 1 receptor–stimulated retinal vascular permeability. Hypertension. 2009;53(2):175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.[cited Oct 20 2019]. Available from: https://www.rezolutebio.com/pipeline/pki-portfolio-rz402
- 60.[cited Jan 21 2010]. Available from: https://www.rezolutebio.com/pipeline/pki-portfolio-rz402
- 61.[cited Oct 21 2019]. Available from: https://www.verseon.com/programs/amd
- 62.Calton MA, Ma JA, Chang E, et al. An orally dosed plasma kallikrein inhibitor decreases retinal vascular permeability in a rat model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2018;59(9):3576. [Google Scholar]
- 63.Bryant J, Shariat-Madar Z. Human plasma kallikrein-kinin system: physiological and biochemical parameters. Cardiovasc Hematol Agents Med Chem. 2009;7(3):234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Damas J The brown Norway rats and the kinin system. Peptides. 1996;17(5):859–872. [DOI] [PubMed] [Google Scholar]
- 65.Kaschina E, Stoll M, Sommerfeld M, et al. Genetic kininogen deficiency contributes to aortic aneurysm formation but not to atherosclerosis. Physiol Genomics. 2004;19(1):41–49. [DOI] [PubMed] [Google Scholar]
- 66.Oh-Ishi S Biological regulation by the kallikrein-kinin system: a study with a kininogen-deficient rat strain. Nihon Yakurigaku Zasshi. 1993;101(4):209–218. [DOI] [PubMed] [Google Scholar]
- 67.Lombardi AM, Sartori MT, Cabrio L, et al. Severe prekallikrein (Fletcher factor) deficiency due to a compound heterozygosis (383Trp stop codon and Cys529Tyr). Thromb Haemost. 2003;90(12):1040–1045. [DOI] [PubMed] [Google Scholar]