

Abstract
In this Concept article, recent advances are highlighted in the synthesis and applications of anomeric nucleophiles, a class of carbohydrates in which the C1 carbon bears a carbon–metal bond. First, the advantages of exploiting the carboanionic reactivity of carbohydrates and the methods for the synthesis of mono- and oligosaccharide stannanes are discussed. Second, recent developments in the glycosyl cross-coupling method resulting in the transfer of anomeric configuration from C1 stannanes to C-aryl glycosides are reviewed. These highly stereoretentive processes are ideally suited for the preparation of carbohydrate-based therapeutics and were demonstrated in the synthesis of antidiabetic drugs. Next, the application of the glycosyl cross-coupling method to the preparation of Se-glycosides and to glycodiversification of small molecules and peptides are highlighted. These reactions proceed with exclusive anomeric control for a broad range of substrates and tolerate carbohydrates with free hydroxyl groups. Taken together, anomeric nucleophiles have emerged as powerful tools for the synthesis of oligosaccharides and glycoconjugates and their future applications will open new possibilities to incorporate saccharides into small molecules and biologics.
Keywords: carbohydrates, copper, cross-coupling, palladium, synthetic methods
Graphical Abstract

Introduction
Carbohydrates occupy a central role in biology due to their important function as mediators of numerous cellular signaling and recognition events.[1] One of the most promising strategies to elucidate the roles of glycans in living organisms is the use of synthetic or designer (oligo)saccharides, often as truncated or modified probes, which perturb the biological and physico-chemical behavior of their natural cognates.[2] Access to these saccharides depends on synthetic techniques, at the heart of which lies chemical glycosylation.[3] The formation of a new glycosidic linkage is traditionally a two-step process involving anomeric derivatization of a saccharide donor followed by activation and coupling to an acceptor molecule.[4] Historically, anomeric derivatization involved enhancement of the nucleofugality of the C1-element to promote glycosylation with a nucleophilic acceptor. O-glycosylation is perhaps the most widely studied and utilized form of chemical glycosylation and successful syntheses of these natural glycosides often exploit electrophilic anomeric activation to promote new bond formation (Figure 1). Schmidt-type donors (trichloroacetimidates)[5] and thioethers are the most widely employed given their stability, ease of activation, and reliability under a variety of glycosylation conditions. Glycosyl halides followed by C1 alcohols are also frequently reported for their applications in halide ion-catalyzed and dehydrative glycosylation, respectively. The types of glycosylation reactions expand well-beyond natural C–O bond construction and many C–O surrogates, for example, C- and Se-glycosides, have also proved useful for probing the biological activities of natural carbohydrates.
Figure 1.

Number of O-glycosylation reactions reported in SciFinder with various pyranosyl glycosyl donors categorized by the nature of the leaving groups (query on April 5, 2018).
Despite the many distinct glycosylation methodologies described in the literature, the majority of them converge on the same mechanistic principle: nucleophilic substitution at the anomeric carbon.[6] Furthermore, subtle changes to the nature of the C1 leaving group, solvent, activating agent(s), saccharide identities, as well as temperature can determine whether or not the reaction proceeds through an SN1 or SN2 manifold.[6] Although there is a narrow mechanistic difference between these two analogous pathways, they can produce vastly different stereochemical outcomes. SN2-type pathways prompt stereochemical inversion dictated by the reactive intermediate allowing for predictable, stereocontrolled glycosylation; the SN1-type pathway, however, proceeds through a highly reactive oxocarbenium species 1a often resulting in complete loss of stereochemical integrity of the substrate (Scheme 1A). The stereochemical outcome of any given glycosylation reaction is, thus, highly sensitive toward minor systemic adjustments, making control of anomeric configuration a significant hurdle in preparative carbohydrate synthesis.[4]
Scheme 1.

(A) Classical displacement-based glycosylation methods. (B) Glycosyl cross-coupling method capitalizing on C1 nucleophiles.
Downstream applications of synthetic (oligo)saccharides and glycoconjugates seldom tolerate products of opposite anomeric selectivity. Taken together with the high demand for carbohydrate-based therapeutics and biological tools, it is not surprising that a large research endeavor has been initiated toward improving chemical glycosylation with a focus on developing predictable, stereocontrolled methodologies. Again, however, nearly all well-established strategies operate from the same paradigmatic approach: the anomeric stereoselectivities are governed by the inherent propensity of protecting groups installed around the saccharide core to direct the stereochemical outcome of the reaction. There are many instances in which this strategy is favorable and presents the opportunity to ensure highly stereoselective transformations. In terms of donor-controlled strategies, participation of C4-substituents is capitalized on to furnish α-galactosides in high selectivity and 1,2-trans-glycosylation is similarly facilitated by carbonyl-based groups (e.g., esters and carbamates) located at the C2 position. Other noteworthy examples include benzylidene protection of the hydroxyl groups at C4 and C6 in mannose donors to direct β-mannosylation.[7] In other cases, however, the strong influence of protecting groups serve to significantly impede the desired reaction outcome. This is especially true for many 1,2-cis-glycosylations[3a] as well as α-selective syntheses of sialic acids.[8]
The stereochemistry of chemical glycosylation can also be moderated by the acceptor’s nucleophilicity with weaker nucleophiles tending to react through a dissociative, SN1 pathway and stronger, more reactive nucleophiles preferring displacement reactions.[9] Similar to donor-controlled methodologies, the reactivity of the acceptor can be modulated to promote reactions in the pathway leading to the desired product stereochemistry through protecting-group manipulations. Although stereoselective glycosylation using this approach is possible, it, like donor-controlled techniques, still suffers in terms of practicality as well as poor synthetic efficiency-for instance, protecting group-based strategies typically require serial protection and deprotection steps which severely limits applicability in total synthesis. Overcoming the intrinsic bias of substrate reactivity using external control methods, for example, catalysts or additives, is well known and utilized but still falls short of enabling exclusive anomeric control.[5]
Taken together, it is evident that abating the fundamental challenges encountered in oligosaccharide synthesis still requires an intimate knowledge of the reactivity and selectivity principles of any given class of reagents. Thus, the practical implementation of the numerous glycosylation strategies developed over the last 50 years remains largely inaccessible to researchers outside of this esoteric subdivision of chemical synthesis.
Enhancing donor reactivity in the glycosylation reaction usually necessitates the introduction of a C1-group capable of acting as a nucleofuge (good leaving group), which inevitably augments the likelihood of an oxocarbenium-mediated glycosylation pathway, the end result of which can be a loss of donor stereochemical integrity. We envisaged an alternative resolution to the problem capitalizing on a process that occurs by retention of configuration as opposed to previous strategies aimed at stabilizing intermediates in the stereoinvertive pathway (Scheme 1B). In this manifold, the anomeric configuration of the product is directly correlated to the configuration of the substrate. The retentive process, in its ideal form, would also withstand the strong influence of saccharide protective groups and, therefore, allow the incorporation of saccharides containing free alcohol and amide moieties into the scope of viable substrates. Success of this methodology depends on three critical factors: (a) the reactions of interest must be stereospecific, (b) the substrates must be configurationally stable, and (c) both anomers of any desired carbohydrate must be accessible. During our initial investigations, we determined that anomeric stannanes-carbohydrates in which the C1 carbon contains a C–Sn linkage-met the abovementioned criteria as well as offered the ideal compromise between stability and stereospecific reactivity.
In this Concept article, we describe the development of glycosylation cross-coupling reactions involving anomeric nucleophiles. Specifically, we highlight our recent progress in the development of such reactions profiting from the privileged reactivity of anomeric stannanes which have resulted, thus far, in the preparation of C- and Se-glycosides and glycomimetics. Considering that this novel concept continues to provide viable solutions to the problems pervading carbohydrate synthesis, new applications will certainly emerge.
Synthesis of Anomeric Stannanes
Successful implementation of the envisioned glycosylation largely depended on the correct choice of metal to be installed at the C1 position. Prior work in the synthesis of anomeric nucleophiles has illuminated some of the critical problems associated with placing a metal at the anomeric carbon.[10] For instance, C1 organolithium reagents are widely used and are often accessible in high anomeric purity. However, their configurational stability is compromised when temperatures rise above −50°C leading to the formation of the thermodynamic product (e.g., the β anomer in the case of d-glucose and d-galactose).[11] Such instability renders these reagents impractical for use in the stereoretentive glycosyl cross-coupling reaction. Moreover, organolithiums tolerate a narrow range of functional groups. For example, in addition to their incompatibility with free hydroxyl and other biologically relevant groups, these nucleophiles are only compatible with 2-deoxy and 2-OH substrates-C2 benzyl ethers undergo rapid elimination resulting exclusively in glycal formation, as reported previously by Sinaÿ.[12] Despite these discouraging features, however, C1 organolithiums are easily generated from anomeric chlorides and can serve as precursors to more stable reagents, particularly or ganostannanes. Organoboranes, in the form of tetrahydropyran derivatives, have also been reported[13] but, given their sub strate-dependent reactivity as well as being able to act through both stereoretentive and stereoinvertive pathways, the extent of their future application warrants further evaluation.[14] Anomeric stannanes, previously reported by Falck,[15] Kessler,[16] and Vasella,[17] offer the ideal compromise between configurational stability, nucleophilicity, and ease of handling. Acyclic α-alkoxy substituted tin reagents have also been studied extensively and establish another viable platform for current investigations.[18]
We developed a synthetic scheme for the streamlined preparation of both α- and β-glycosyl stannanes of common monosaccharide building blocks from glycals 4 through a series of transformations capitalizing on stereoretentive and stereoinvertive reactions (Scheme 2). The preparation of 1,2-cis isomers 6 started with C1 alcohols that were converted into the corresponding thermodynamic α-chlorides 5 which were then advanced into stannanes 6 using nBuLi and lithium naphthalene followed by quenching with Bu3SnCl. In this sequence, deprotonation of the C2-alcohol is critical in order to prevent glycal formation. We performed all steps at −100°C although similar yields were obtained when this process was repeated at −78°C. The reason for maintaining lower temperatures of the cooling bath was to assure that the internal temperature did not exceed −50°C during the addition of Li/naphthalenide and nBuLi thus avoiding epimerization of the C1 anion. The synthesis of the 1,2-trans isomers 7 used 1,2-anhydro sugars derived from 4 that were opened by a nucleophilic tin source (Bu3SnMgMe) followed by hydrolysis of the resulting tin alkoxide to furnish 7. It is noteworthy to mention that this particular protocol can be carried out on a multigram-scale further highlighting the practical utility of these reagents. Similar transformations were applied to the synthesis of both anomers of 2-deoxy sugars. Scheme 2 lists selected examples of monosaccharides (out of 50 total) readily available using the sequence of reactions described above.
Scheme 2.

Synthesis of anomeric stannanes.
Although protective groups are often beneficial for directing regioselective manipulations at the desired positions on the carbohydrate, in the ideal synthesis, all biologically relevant transformations at C1 involve reagents with free OH and NH groups. We found that the benzyl groups could be easily removed from stannanes 12 under Birch conditions (Scheme 3). Furthermore, these reactions proceeded without erosion of anomeric configuration and, more importantly, with negligible levels of destannylation. The fully deprotected saccharides 13 can be easily purified by column chromatography on silica gel using ethyl acetate as an eluent and are obtained, in most cases, as solids. The crystalline form of free, unprotected glycosyl stannanes provides material with extended lifetime and ease of handling.
Scheme 3.
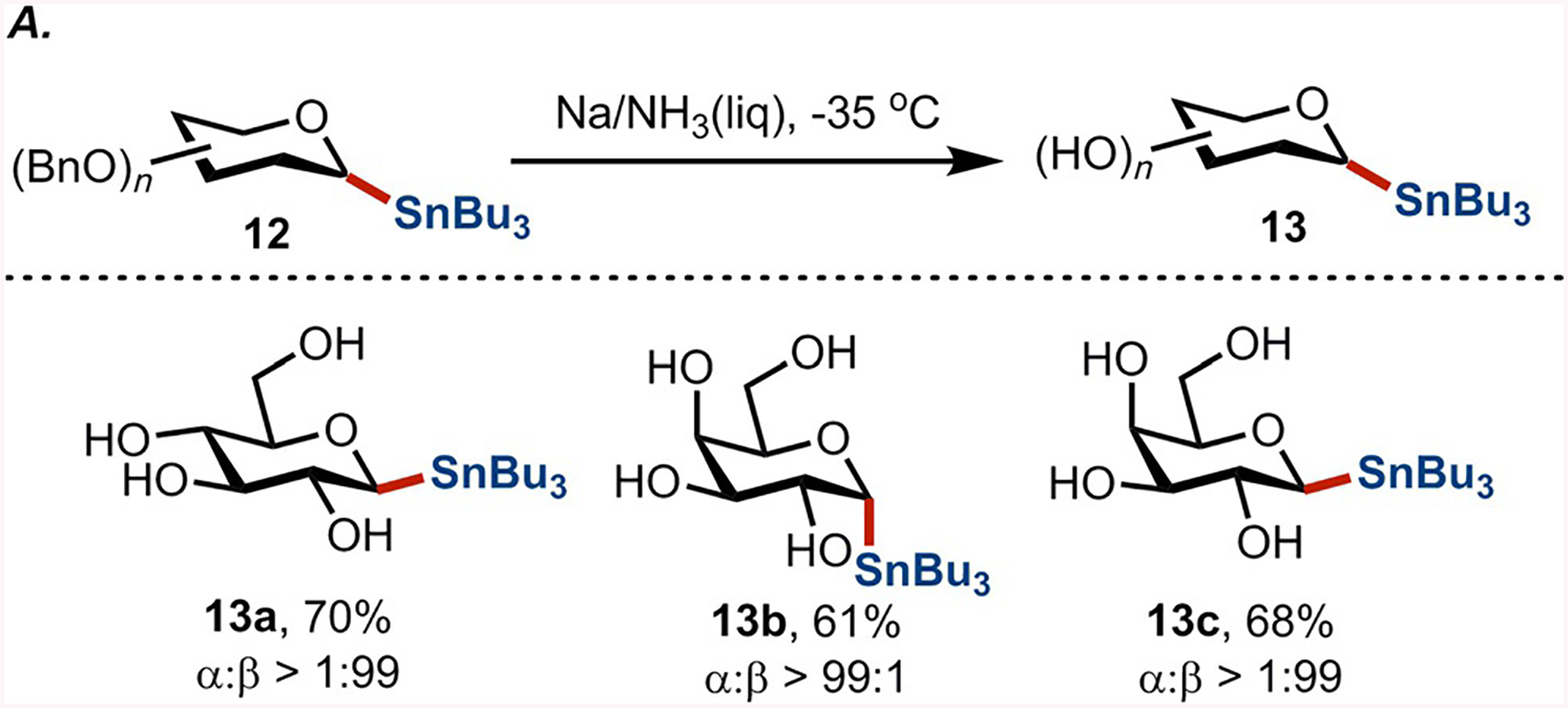
(A) Selected debenzylation products obtained after the Birch reduction. (B) Physical form of 11a at ambient conditions, >1 g quantity. (C) Fully deprotected d-glucose stannane 13a.
Anomeric stannanes are also compatible with classical glycosylation conditions: we demonstrated that Schmidt-type donors activated using TMSOTf or PdII catalysts can reliably extend the oligosaccharide chain using anomeric nucleophiles as glycosyl acceptors. Moreover, anomeric stannanes are tolerated under dehydrative glycosylation promoted by cyclic phosphonium anhydrides[19] and 1,2-anhydro-sugars can also be efficiently converted into the corresponding 2-OH glycosides using stannanes as the glycosyl acceptors.[20] With these conditions in hand, we were able to prepare a series of linear and branched saccharides.[20] Given the ease with which anomeric stannanes can be elaborated into oligosaccharides using established glycosylation techniques, while maintaining their configurational stability, the utility of this class of reagents is not restricted to the preparation of simple building blocks and is viable for the preparation of more complex starting materials.
One of the key points in analysis of anomeric stannanes is assignment of anomeric configuration. The most direct way to achieve this task is through analysis of 3J(HH) coupling constants between the C1-H and C2-H protons. Interestingly, the configuration at the anomeric position can also be ascertained by analysis of 1J(Sn-C) coupling constants between the C1 of the butyl group (not the C1 of the carbohydrate) and the 117Sn or 119Sn nuclei. The coupling constants observed for equatorial anomers fall in the range above 305 Hz, whereas axial anomers fall below 305 Hz for 117Sn nuclei. This observation is reminiscent of the experimentally observed 1J(C1-H1) for axial and equatorial anomers of pyranosides.
C-Glycosides
One of the most successful strategies to enhance in vivo stabilities of naturally-labile O-glycosides is replacement of the C–O bond with a more hydrolytically stable C–C surrogate resulting in the formation C-glycosides.[21] The preparation of this class of glycoconjugates has been recently reviewed with only a handful of methods enabling highly stereoselective C-glycosylation.[21d,22] Moreover, majority of these methods capitalize on anchimeric assistance of the C2 a group to obtain high selectivities and are consequently limited in scope. We wondered if anomeric stannanes would be suitable for the preparation C-glycosides through a stereoretentive process and thus focusing our initial work on establishing whether or not C1-stannanes could participate in the Stille reaction.[20,23] At the outset of these studies, we anticipated the competing β-elimination to plague the cross-coupling process. We found that removal of the C2 substituent and subsequent glycal formation could be prevented by a judicious selection of the appropriate phosphine ligand. An alternative solution to this problem would use substrates with free 2-OH, 2-NHR, and 2-deoxy sugars lacking suitable leaving groups; however, such an approach would inevitably narrow the range of compatible substrates and limit the reaction’s generality.
Catalyst optimization studies are presented in Table 1. Out of >50 different phosphine ligands, employment of Jackie-Phos L6, a ligand described by Buchwald,[24] most effectively minimized the formation of d-glucal while furnishing C-glyco-side 15 in excellent yield (94%) and exclusive selectivity. Other phosphines (e.g., tBu3P (L1), bidentate ligands L2 and L3)[25] resulted primarily in the formation of d-glucal. The conversion of 14 into glucal can occur at either the stage of organocopper or organopalladium intermediates as well as directly from C1-stannane 14. For example, amine bases (Et3N, Hünig’s base, pyridine) alone can promote destannylation and the formation of glycals in almost quantitative yield. Structurally similar ligands, such as L4 (XPhos) and L5 (tBuXPhos) turned out to be poorly selective and promoted only the elimination pathway.
Table 1.
Selected optimization of C-glycosyl cross-coupling.[a]
 | ||||
|---|---|---|---|---|
| Entry | Ligand | 15[b] [%] | d-Glucal[b] [%] | α:β[c] |
| 1 | L1 (10 mol%) | 9 | 42 | > 1:99 |
| 2 | L2 (5 mol%) | 6 | 19 | > 1:99 |
| 3 | L3 (5 mol%) | < 2 | 38 | ND |
| 4 | L4 (10 mol%) | < 2 | 24 | ND |
| 5 | L5 (10 mol%) | <2 | 10 | ND |
| 6 | L6 (10 mol%) | 94 | 25 | > 1:99 |
Optimization conditions: 14 (1.5 equiv), 3-iodotoluene (1.0 equiv), Pd2(dba)3 (2.5 mol%), CuCl (300 mol%), KF (200 mol%), solvent (0.03m).
NMR yield determined using internal standard (CHBr3).
Determined by analysis of the crude reaction mixture using 1H NMR spectroscopy.
In the cases where formation of C-glycoside 15 was observed, only the β anomer was formed. These results support the hypothesis that the phosphine ligands control the rate of elimination but have no effect on the stability of the putative organopalladium or organocopper intermediates at the anomeric carbon. Upon investigating this proposal computationally, we found that a bulky ligand not only prevents the ring flip leading to β-elimination of C2 substituent but also promotes the reductive elimination at Pd forming the product C–C bond. The special role of JackiePhos in this reaction was attributed to its ability to simultaneously suppress the competing degradative, β-elimination pathway leading to glucal while promoting a reductive elimination from Pd to give the product glycoside.
Scheme 4 depicts selected products of the glycosyl cross-coupling reaction with aryl iodides and bromides. This reaction proceeds with retention of anomeric configuration at C1 for a broad range of substrates and both anomers. For instance, both anomers of d-galactose can be prepared under the standardized conditions in excellent yields (16c and 16d). The reaction offers the opportunity to use glycosyl donors with free hydroxyl groups to introduce a saccharide at the desired positions in a predictable and fully controlled manner (Scheme 4B). Similarly, oligosaccharides performed well and the resulting glycosides were formed as a single anomer (Scheme 4B). The product configuration is controlled by the stannane substrate and no additional deprotection steps are necessary.
Scheme 4.

Scope of C-aryl glycoside synthesis by glycosyl cross-coupling.
The glycosyl cross-coupling reaction with anomeric stannanes presents a viable alternative to multi-step syntheses of bioactive C-glycosides. We were able to demonstrate that dapagliflizon 18b,[26] a SGLT2 inhibitor approved worldwide for the treatment of diabetes mellitus type 2,[27] could be prepared in one step with exclusive anomeric selectivity and 85% isolated yield (Scheme 5). Similarly, we were able to show that another gliflozin-type C-glycoside, empagliflozin,[28] could be prepared in a cross-coupling reaction with diaryliodonium triflate and β-d-glucose stannane in 79% yield and exclusive β selectivity.[29] Other applications of the glycosyl-cross coupling method involve the synthesis of TGE (tri-glucosylated enterobactin, Salmonella siderophore), on-resin glycosylation, and glycodiversification of peptides, small molecule drugs, and natural products.[20,30]
Scheme 5.

One-step synthesis of dapagliflozin. Reagents and conditions: (a) 14 (2 equiv), L4 (20 mol %), Pd2(dba)3 (5 mol%), CuCl (3 equiv), KF (2 equiv), 1,4-dioxane, 110°C, 72 h. (b) 13a (2 equiv), JackiePhos (20 mol%), Pd2(dba)3 (5 mol%), CuCl (3 equiv), KF (2 equiv), 1,4-dioxane, 110°C, 72 h.
Selenoglycosides
In continuation of our studies on the umpolung reactivity of anomeric stannanes, we wondered if this technology could be extended to carbon–heteroatom bond forming processes. Selenoglycosides are a unique class of carbohydrates in which the anomeric position is substituted with a selenium atom.[31] These compounds came into use in preparative carbohydrate chemistry in the 1990s.[32] Since then, selenoglycosides have also found use as anti-metastatic 19[33] and immunostimulatory agents 20[34] (Scheme 6A). The unique feature of these reagents is their propensity to undergo facile C-Se cleavage due the labile C–Se bond. Under the appropriate conditions, selenoglycosides can generate both anomeric radical and oxocarbenium intermediates. This property of selenoglycosides has been used in glycosylation reactions promoted by electrophilic reagents, such as AgOTf as well as in light-mediated strategies (Scheme 6B).[32b,35] The application of modern photoredox methods to the generation of anomeric radicals and cations poses an opportunity to revive the use of these multifaceted glycosyl donors in current preparative carbohydrate chemistry.[36]
Scheme 6.

Selected bioactive selenoglycosides and applications of Se-glycosides as glycosyl donors.
Classical methods for C–Se bond formation involve nucleophilic displacement from an anomeric bromide or through a series of manipulations of glycals and their corresponding 1,2-anhydro sugars. These protocols use protected glycosyl donors and generally provide the 1,2-trans isomers. The formation of 1,2-cis isomers is also reported but this transformation is only feasible with benzyl-protected donors. We proposed that the installation of a selenide at C1 could be easily accomplished using anomeric nucleophiles and the appropriate selenium donors.[37] To this end, we tested conditions using CuCl and KF as the standard reagents capable of transferring the glycoside group with high stereospecificity. Optimization studies (Table 2) primarily focused on identifying the relevant leaving group at the selenide donor. We determined the most convenient class of substrates to be the symmetrical diselenides (e.g., A) which furnished product 23 in 91% yield (entry 1). Under these conditions, transfer of only one group takes place and the second half of the substrate forms a selenol by-product. We then reasoned that, for the full utilization of selenium donors to take place, the addition of a mild oxidant (e.g., O2, or air) would be needed to regenerate the diselenide from the selenol by-product thus enabling the conversion of both selenium fragments into the product. When the reaction was conducted under air but with 0.5 equivalent of A, the isolated reaction yield was comparable with conditions employing a full equivalent of A (70%). Other substitutions on selenium were found to give variable yields depending on the nature of the leaving group. Sulfides B and C, for example, furnished selenoglycoside 23 in 79% and 88%, respectively, whereas a cyanide or phthalimide group on Se was less efficient (entry 9 and 10). From these studies, we converged on the use of symmetrical diselenides for simple substrates and seleno-sulfides for circumstances involving more complex substrates where diselenide regeneration under oxidative conditions might be prohibitively slow or result in undesired by-products.
Table 2.
Optimization of glycosyl cross-coupling.[a]
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Cu | Fluoride | Se | T [°C] | t [h] | Yield [%][b,c] |
| 1 | CuCI | KF | A | 110 | 24 | 91 |
| 2 | CuCI | None | A | 110 | 24 | 70 |
| 3 | None | KF | A | 110 | 24 | < 5 |
| 4d | CuCI | KF | A | 110 | 24 | 69 |
| 5 | CuCl | KF | A | 110 | 12 | 74 |
| 6 | CuCl | KF | A | 90 | 24 | 55 |
| 7 | CuCl | KF | B | 110 | 24 | 79 |
| 8 | CuCl | KF | C | 110 | 48 | 88 |
| 9 | CuCl | KF | D | 110 | 24 | 44 |
| 10 | CuCl | KF | E | 110 | 24 | 62 |
Reaction conditions: selenide A–E (0.100 mmol, 1.0 equiv), 14 (1.1 equiv), fluoride (2 equiv), CuX (1.5 equiv), and dry 1,4-dioxane (2 mL) under N2, 110°C;
isolated yield;
only β-anomer formed;
1 equiv of CuCl.
The synthesis of glycosyl selenides was accomplished with high degrees of stereocontrol from anomeric stannanes. Scheme 7 lists selected examples of the glycosyl cross-coupling with various saccharides. First, we demonstrated that common monosaccharides retain their anomeric configuration when coupled to (PhSe)2. Interestingly, the preparation of Se glycomimetics, such as Se-trehalose precursor 25n can be accomplished by taking advantage of the predetermined, transferable anomeric configuration of glycosyl stannane. Next, we demonstrated that Se-linked diglycosides can be formed under the standardized conditions for both anomers of the corresponding diselenide glycosides. The preparation of Se-trehalose precursor 25n is particularly interesting because the deprotected product is a non-hydrolysable analog to the natural non-reducing sugar, known to be an essential metabolite in pathogenic species, such as Mycobacterium tuberculosis.[38] Finally, we demonstrated that glycosyl groups can be incorporated stereospecifically into peptides (Scheme 8). These reactions were performed with either selenocysteine dimers 26 and resulted in exclusive anomeric control. This method is a practical strategy for accomplishing glycodiversification and bioconjugation with free saccharides (e.g., 27a) and a predetermined configuration of the resultant Se-glycosyl bond.
Scheme 7.

Scope of Se-glycoside synthesis by glycosyl cross-coupling.
Scheme 8.

Synthesis of Se-glycopeptides.
The proposed mechanism of the selenium coupling is represented in Scheme 9. The transfer of the pyranosyl group to CuI is mediated by the fluoride ion and proceeds with retention of anomeric configuration (28a→28b). The organocopper intermediate 28b can then undergo reactions by two pathways: direct displacement and cleavage of the Se–Se bond results in retention of configuration at the C1 positions through a putative transition state 31 (path A) or the Se–Se bond is cleaved first by oxidative addition and forms CuIII intermediate 33 followed by reductive elimination to 32 (path B). At this point, thermodynamic preferences of the intermediates and/or products can be excluded as stereochemical determinants given that both α and β anomers could be prepared independently and without erosion of anomeric configuration. Although more mechanistic work is needed to provide stronger support for either mechanism, the oxidative addition mechanism (path B) is less favored because formation of a CuIII intermediate, given its high oxidation state and presumed instability, is likely to undergo loss of stereochemical integrity and result in a mixture of anomers.
Scheme 9.

Proposed mechanism of stereoretentive Se-coupling.
Summary and Outlook
The anomeric stannanes present an intriguing class of molecules that have thus far demonstrated their utility in a number of challenging transformations in preparative carbohydrate chemistry. These reagents can now be accessed in any anomeric configuration of common monosaccharides, and they can be merged with classical glycosylation methods to provide access to complex oligosaccharide building blocks. Their propensity to undergo stereoretentive reactions can be exploited in the preparation of Se-linked glycans as well as C-glycosides. Through these privileged reactions of anomeric nucleophiles, our group has been able to reign unprecedented control over anomeric configuration and, given the proper selection of reaction conditions, catalyst, and ligand, can provide designer (oligo)saccharides in consistently high yield and exclusive selectivity. The range of potential applications of anomeric stannanes in target-oriented synthesis, late stage modifications of small molecule drugs and therapeutic-candidates, as well as seamless bioconjugation with peptides and proteins asserts these molecules as indispensable synthetic tools in modern carbohydrate synthesis.
Acknowledgements
This work was supported by the University of Colorado Boulder, the National Science Foundation (CAREER Award No. CHE-1753225), and the National Institutes of Health (U01GM125284).
Footnotes
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/chem.201803082.
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, Essentials of Glycobiology, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 2009; [PubMed] [Google Scholar]; b) Gabius H-J, The Sugar Code: Fundamentals of Glycosciences, Wiley-Blackwell, Hoboken, 2009. [Google Scholar]
- [2].Ernst B, Magnani JL, Nat. Rev. Drug Discovery 2009, 8, 661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Nigudkar SS, Demchenko AV, Chem. Sci 2015, 6, 2687–2704; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Bennett CS, Selective Glycosylations: Synthetic Methods and Catalysts: Synthetic Methods and Catalysts, Wiley, Hoboken, 2017; [Google Scholar]; c) Kulkarni SS, Wang C-C, Sabbavarapu NM, Podilapu AR, Liao P-H, Hung S-C, Chem. Rev 2018, 118, 8025–8104. [DOI] [PubMed] [Google Scholar]
- [4].Frihed TG, Bols M, Pedersen CM, Chem. Rev 2015, 115, 4963–5013. [DOI] [PubMed] [Google Scholar]
- [5].Zhu X, Schmidt RR, Angew. Chem. Int. Ed 2009, 48, 1900; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 1932. [Google Scholar]
- [6].Adero PO, Amarasekara H, Wen P, Bohé L, Crich D, Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Crich D, Sun S, J. Org. Chem 1996, 61, 4506–4507. [DOI] [PubMed] [Google Scholar]
- [8].Tanaka H, Nishiura Y, Takahashi T, J. Am. Chem. Soc 2006, 128, 7124– 7125. [DOI] [PubMed] [Google Scholar]
- [9].van der Vorm v) S., van Hengst JMA, Bakker M, Overkleeft HS, van der Marel GA, Codée JDC, Angew. Chem. Int. Ed 2018, 57, 8240–8244; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 8372–8376. [Google Scholar]
- [10].Somsák L, Chem. Rev 2001, 101, 81. [DOI] [PubMed] [Google Scholar]
- [11].Baryal KN, Zhu D, Li X, Zhu J, Angew. Chem. Int. Ed 2013, 52, 8012–8016; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2013, 125, 8170–8174. [Google Scholar]
- [12].Lancelin J-M, Morin-Allory L, Sinaÿ P, J. Chem. Soc. Chem. Commun 1984, 355–356. [Google Scholar]
- [13].Scheuermann ML, Johnson EJ, Chirik PJ, Org. Lett 2015, 17, 2716–2719. [DOI] [PubMed] [Google Scholar]
- [14].a) Molander GA, Wisniewski SR, J. Am. Chem. Soc 2012, 134, 16856–16868; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li L, Zhao S, Joshi-Pangu A, Diane M, Biscoe MR, J. Am. Chem. Soc 2014, 136, 14027–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Ye J, Bhatt RK, Falck JR, Tetrahedron Lett. 1993, 34, 8007–8010; [Google Scholar]; b) Belosludtsev YY, Bhatt RK, Falck JR, Tetrahedron Lett. 1995, 36, 5881–5882. [Google Scholar]
- [16].a) Wittmann V, Kessler H, Angew. Chem. Int. Ed. Engl 1993, 32, 1091–1093; [Google Scholar]; Angew. Chem 1993, 105, 1138–1140; [Google Scholar]; b) Burkhart F, Hoffmann M, Kessler H, Tetrahedron Lett. 1998, 39, 7699–7702. [Google Scholar]
- [17].Frey O, Hoffmann M, Wittmann V, Kessler H, Uhlmann P, Vasella A, Helv. Chim. Acta 1994, 77, 2060. [Google Scholar]
- [18].a) Ye J, Bhatt RK, Falck JR, J. Am. Chem. Soc 1994, 116, 1–5; [Google Scholar]; b) Falck JR, Bhatt RK, Ye J, J. Am. Chem. Soc 1995, 117, 5973–5982; [Google Scholar]; c) Mohapatra S, Bandyopadhyay A, Barma DK, Capdevila JH, Falck JR, Org. Lett 2003, 5, 4759–4762; [DOI] [PubMed] [Google Scholar]; d) Falck JR, Patel PK, Bandyopadhyay A, J. Am. Chem. Soc 2007, 129, 790–793; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Trépanier VÉ, Fillion E, Organometallics 2007, 26, 30–32; [Google Scholar]; f) He A, Falck JR, Angew. Chem. Int. Ed 2008, 47, 6586–6589; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 6688–6691; [Google Scholar]; g) Goli M, He A, Falck JR, Org. Lett 2011, 13, 344–346; [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Li H, He A, Falck JR, Liebeskind LS, Org. Lett 2011, 13, 3682–3685; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Mita T, Sugawara M, Hasegawa H, Sato Y, J. Org. Chem 2012, 77, 2159–2168; [DOI] [PubMed] [Google Scholar]; j) Wang R, Falck JR, Org. Biomol. Chem 2015, 13, 1624–1628; [DOI] [PubMed] [Google Scholar]; k) Dakarapu R, Falck JR, J. Org. Chem 2018, 83, 1241–1251. [DOI] [PubMed] [Google Scholar]
- [19].Dyapa R, Dockery LT, Walczak MA, Org. Biomol. Chem 2017, 15, 51–55. [DOI] [PubMed] [Google Scholar]
- [20].Zhu F, Rodriguez J, Yang T, Kevlishvili I, Miller E, Yi D, O’Neill S, Rourke MJ, Liu P, Walczak MA, J. Am. Chem. Soc 2017, 139, 17908–17922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Levy DE, Tang C, The Chemistry of C-Glycosides, Pergamon Press, Oxford, 1995; [Google Scholar]; b) Postema MHD, C-Glycoside Synthesis, CRC Press, Boca Raton, 1995; [Google Scholar]; c) Leclerc E, Pannecoucke X, Etheve-Quelquejeu M, Sollogoub M, Chem. Soc. Rev 2013, 42, 4270; [DOI] [PubMed] [Google Scholar]; d) Yang Y, Yu B, Chem. Rev 2017, 117, 12281–12356; [DOI] [PubMed] [Google Scholar]; e) Bokor É, Kun S, Goyard D, Tóth M, Praly J-P, Vidal S, Somsák L, Chem. Rev 2017, 117, 1687–1764. [DOI] [PubMed] [Google Scholar]
- [22].Kitamura K, Ando Y, Matsumoto T, Suzuki K, Chem. Rev 2018, 118, 1495–1598. [DOI] [PubMed] [Google Scholar]
- [23].Zhu F, Rourke MJ, Yang T, Rodriguez J, Walczak MA, J. Am. Chem. Soc 2016, 138, 12049–12052. [DOI] [PubMed] [Google Scholar]
- [24].Hicks JD, Hyde AM, Cuezva AM, Buchwald SL, J. Am. Chem. Soc 2009, 131, 16720–16734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Fors BP, Watson DA, Biscoe MR, Buchwald SL, J. Am. Chem. Soc 2008, 130, 13552–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN, J. Med. Chem 2008, 51, 1145–1149. [DOI] [PubMed] [Google Scholar]
- [27].Aguillón AR, Mascarello A, Segretti ND, de Azevedo HFZ, Guimaraes CRW, Miranda LSM, de Souza ROMA, Org. Process Res. Dev 2018, 22, 467–488. [Google Scholar]
- [28].a) Grempler R, Thomas L, Eckhardt M, Himmelsbach F, Sauer A, Sharp DE, Bakker RA, Mark M, Klein T, Eickelmann P, Diabetes Obes. Metab 2012, 14, 83–90; [DOI] [PubMed] [Google Scholar]; b) Wang XJ, Zhang L, Byrne D, Nummy L, Weber D, Krishnamurthy D, Yee N, Senanayake CH, Org. Lett 2014, 16, 4090. [DOI] [PubMed] [Google Scholar]
- [29].Yi D, Zhu F, Walczak MA, Org. Lett 2018, 20, 1936–1940. [DOI] [PubMed] [Google Scholar]
- [30].Yi D, Zhu F, Walczak MA, Org. Lett 2018, 20, 4627–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Witczak ZJ, Czernecki S in Advances in Carbohydrate Chemistry and Biochemistry, Vol. 53 (Ed.: Horton D), Academic Press, San Diego, 1998, pp. 143–199. [DOI] [PubMed] [Google Scholar]
- [32].a) Mehta S, Mario Pinto B, Tetrahedron Lett. 1991, 32, 4435–4438; [Google Scholar]; b) Mehta S, Pinto BM, J. Org. Chem 1993, 58, 3269–3276. [Google Scholar]
- [33].Bijian K, Zhang Z, Xu B, Jie S, Chen B, Wan S, Wu J, Jiang T, Alaoui-Jamali MA, Eur. J. Med. Chem 2012, 48, 143–152. [DOI] [PubMed] [Google Scholar]
- [34].McDonagh AW, Mahon MF, Murphy PV, Org. Lett 2016, 18, 552–555. [DOI] [PubMed] [Google Scholar]
- [35].Furuta T, Takeuchi K, Iwamura M, Chem. Commun 1996, 157–158. [Google Scholar]
- [36].Spell M, Wang X, Wahba AE, Conner E, Ragains J, Carbohydr. Res 2013, 369, 42–47. [DOI] [PubMed] [Google Scholar]
- [37].Zhu F, O’Neill S, Rodriguez J, Walczak MA, Angew. Chem. Int. Ed 2018, 57, 7091–7095; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 7209–7213. [Google Scholar]
- [38].O’Neill MK, Piligian BF, Olson CD, Woodruff PJ, Swarts BM, Pure Appl. Chem 2017, 89, 1223–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]