

Abstract:
Vascular remodeling is one of the most critical complications caused by hypertension. Previous studies have demonstrated that rosuvastatin has anti-inflammatory, antioxidant, and antiplatelet effects and therefore can be used to treat cardiovascular disease. In this study, we explored the beneficial effects of rosuvastatin in reversing aortic remodeling in spontaneously hypertensive rats. After treating with different doses of rosuvastatin, its antilipid, antiapoptosis, and anti-inflammatory effects were determined. We also examined whether rosuvastatin can improve the structure and function of the aorta. We found that rosuvastatin treatment of spontaneously hypertensive rats for 2 months at 2 different doses can effectively reduce the media thickness of the aorta compared with the control group. Similarly, rosuvastatin improved the vascular relaxation function of the aortic rings at a high level of acetylcholine in vitro. Mechanistically, it was found that rosuvastatin increased the expression of endothelial nitric oxide synthase and plasma nitrite/nitrate levels. Besides, rosuvastatin suppressed the apoptosis and inflammation and upregulated the expression of gap-junction complex connexin 43 both in media and endothelium. Finally, rosuvastatin inhibited the AT1R/PKCα/HSP70 signaling transduction pathway. In summary, these findings demonstrated that rosuvastatin could improve the vascular structure and function mainly by increasing endothelial nitric oxide synthase expression and preventing apoptosis and inflammation. This study provided evidence that rosuvastatin has beneficial effects in reversing the remodeling of the aorta due to hypertension.
Key Words: rosuvastatin, hypertension, vascular remodeling, apoptosis, connexin 43
INTRODUCTION
Hypertension has been considered as one of the leading global diseases worldwide, responsible for 7.6 million deaths each year.1 It is a complex disease and characterized by chronic and excessive pressure on arteries. Raised blood pressure often causes hemodynamic disorders, neurohumoral changes, and local endocrine environment changes. These changes may form a crucial pathological basis that can aggravate hypertension and induce the damage of target organs, including heart, brain, kidney, and arterial blood vessels.2–4 Therefore, patients with hypertension often have higher risks of cardiocerebrovascular events, such as coronary heart disease, cerebral hemorrhage, and cerebral infarction.5,6
As an adaptive response to hypertension, arterial remodeling is one of the most important pathological changes during the progress of hypertension. Normally, such adaptively structural changes are accompanied by a narrowing of the vascular lumen, increased media thickness (MT), and stiffness of the aorta. Mechanistically, apoptosis, inflammation, and fibrosis are proved to be critical in the process of arterial remodeling induced by hypertension.7 Therefore, treatments that can reverse these pathological changes could be beneficial for patients with hypertension in attenuating arterial remodeling and target organ damage.
Although rosuvastatin is clinically used for decreasing low-density lipoprotein cholesterol (LDL-C), it also has many benefits in treating cardiovascular diseases. Some basic researches have been well performed to verify such effects and explore the potential mechanisms. For instance, rosuvastatin was found to attenuate the pressure overloaded cardiac hypertrophy through TGF-β/Smads pathways and Akt-ERK1/2-GATA4 pathways.8,9 Our previous study also demonstrated that rosuvastatin could prevent the myocardial injury in spontaneously hypertensive rats (SHRs) through PKCα/β2 pathway.10 In addition to this, rosuvastatin was also beneficial in vascular function. A study by Colucci et al11 reported that rosuvastatin could prevent Ang II-induced changes in the resistance arteries, including their structure and function. However, its effect on the hypertension-induced remodeling of large blood vessels has not been fully investigated. So, we designed this study to observe the effect of rosuvastatin on the structure and function of the aorta in SHR. We hypothesized that rosuvastatin could protect the aorta against vascular remodeling by increased endothelial nitric oxide synthase (eNOS) expression, inhibiting apoptosis and inflammation, and promoting Cx43 expression in SHR.
METHODS
Experimental Animals
One-year-old male SHRs and age-matched normotensive Wistar-Kyoto (WKY) rats were purchased from the Laboratory Animal Center of Zhejiang Chinese Medical University. The animal procedures were conducted in compliance with the institutional guidelines of Zhejiang Chinese Medical University and approved by the Animal Experimentation Ethics Committee of Zhejiang Chinese Medical University. According to the previous study,10 a total of 36 SHRs were randomly assigned to 3 groups namely: the SHR group, SHR with a low dose of rosuvastatin (10 mg/kg/d) (SHR + LD group), and SHR with a high dose of rosuvastatin (40 mg/kg/d) (SHR + HD group). Each group consisted of 12 SHRs. Rosuvastatin was dissolved in saline and given through gavage administration for 8 weeks. The SHR group was given the same dose of the vehicle. Age-matched WKYs were treated with the same dose of the vehicle as to the normal-tensive group (n = 12). Efforts were taken to ensure minimal suffering of rats in all the experiments.
Measurement Systolic Blood Pressure
Systolic blood pressure (SBP) was measured at the beginning and every 4 weeks with the tail-cuff method in all experimental rats.12 Briefly, the rats were allowed to adapt to the restraint cages, which was preheated to 37°C for 15 minutes before the measurement. Next, the tail was fixed and kept in close contact with a noninvasive blood pressure device (softronBP98A; Chengdu; China) under the same temperature. Each measurement was repeated 3 times for accuracy.
Measurement of Lipid Profiles
At the end of the experiment, the blood samples from all rats of 4 groups were collected after overnight fasting. Then, the blood samples were centrifugated at 3000g for 10 minutes at 4°C. The plasma samples were obtained and stored at −80°C. The concentrations of total cholesterol (TC), LDL-C, high-density lipoprotein cholesterol (HDL-C), and triglycerides (TG) were measured using commercial kits (Cat No.A110-2-1, Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Measurement of Plasma Nitrite/Nitrate Levels
To determine the NO production of plasma, the nitrite/nitrate levels, a stable metabolite of NO, were measured. The plasma was first obtained in the same way mentioned above. Plasma nitrite/nitrate concentrations were detected using the colorimetric assay kit (Cat No.S0024, Beyotime, Haimen, China) following the manufacturer's protocol. The nitrite/nitrate levels were determined by measuring the absorbance at 540 nm and calculated referring to the standard curve.
Measurement of Vascular Relaxation Function
For measuring the endothelium-dependent relaxation of the aorta, concentration–response curves were generated with a cumulative stimulation of acetylcholine (Ach) following previous studies.13,14 Briefly, the thoracic aortas were excised and quickly put in the cold modified Krebs–Henseleit solution (KHS). The composition of the solution in mM was as follows: NaCl: 118.3, KCl: 4.7, MgSO4: 1.2, NaHCO3: 25, CaCl2·2H2O: 2.5, KH2PO4: 1.2, d-Glucose: 5.5, and EDTA: 0.026. The aortic samples were cut into arterial ring segments of 4 mm width and immersed in a bath solution aerated with 95% O2 and 5% CO2 at 37°C. Each artery was stretched by 2 hooks, one fixed and other attached to a force–displacement transducer (Grass Instrument Co, Quincy, MA). The signals of the transducer were recorded using the MacLab data acquisition system (Version 3.5, ADI Instruments, Australia). The arterial rings were equilibrated with the tension of 1 g for at least 45 minutes. The bath solution was replaced every 15 minutes during the equilibrium procedure. Subsequently, the integrity of the artery was tested by stimulating with KCl (80 mM) for 5 minutes at intervals of 10 minutes. In the event of 2 consecutive equal contractions, the aortas were considered stable. After washing the surplus K+ solution, the aortic rings were stimulated with a cumulative concentration of Ach (10−10 − 10−5M). The ratios of endothelium-dependent relaxation were recorded. The response curve was acquired using GraphPad Prism 7.0 (GraphPad Software, CA). Each curve was plotted using the average result of 3 individual experiments for each group and expressed as mean ± SD.
Histopathological Analyses
The isolated thoracic aorta tissue was fixed with 4% paraformaldehyde for 24 hours. Then, it was dehydrated and embedded in paraffin. The aortas were cut into slices of 4 μm and prepared for hematoxylin and eosin (H&E) staining to measure the MT and luminal diameter (LD) of the aorta. Photomicrographs were determined by computer-aided image analysis systems of Image-Pro Plus version 6.0 software (Media Cybernetics, Silver Spring, MD). For each section, 3 random fields of aortic rings were selected and measured for MT and LD. The ratio of MT/LD was then calculated.
Immunofluorescence and TUNEL Staining
To localize connexin 43 (Cx43) and apoptosis in the thoracic aorta, immunofluorescence staining was performed. Paraffinized sections of aortas were then stained with anti-Cx43 antibody (1:200, Abcam, ab11370) overnight at 4°C. After this, staining with secondary antibody conjugated with fluorescein isothiocyanate (1:1000, MultiSciences, 70-GAR4881) for 1 hour at room temperature was performed. Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) staining was performed using in situ cell death detection kit (Roche Applied Science, IN) on paraffin-embedded sections following the manufacturer's protocol. Distribution of Cx43 and apoptosis on sections of the thoracic aorta was detected and analyzed under ×200 magnification using a fluorescence photomicroscope (Leica, Wetzlar, Germany). Five random fields were chosen for the calculation of the relative expression of Cx43 and TUNEL-positive cells for each slide. The calculation of Cx43 was expressed as the relative expression between 4 groups. The percentage of apoptosis cells was expressed as the ratio of TUNEL-positive (green) and the total number of cells stained using 4,6-diamino-2-phenylindole hydrochloride (DAPI) (blue). α-SMA (red) was also stained with primary antibody (1:200, CST, 19245) to reflect the position of the vascular smooth muscle cell (SMC).
Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA from the tissues was extracted using TRIzol (Invitrogen, CA) and converted into cDNA with PrimeScriptTM RT Master Mix (Takara, Tokyo, Japan). qPCR was subsequently performed to measure the relative mRNA expression of genes using UltraSYBR Mixture (CWBIO, Beijing, China) on the Viia 7 system (Applied Biosystems, CA). The cycling conditions were 95°C predenaturation for 10 minutes, followed by 40 cycles; each cycle included 95°C denaturation for 15 seconds and 60°C extension for 1 minute. The relative transcription levels of genes were calculated using the ΔCt method and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences of target genes were synthesized by TSINGKE (Hangzhou, Zhejiang, China) and are listed in Table 1.
TABLE 1.
List of Primer Sequence of Target Gene Used in RT-qPCR

Western Blotting
The protein expression was determined by Western blotting. First, the tissues of the thoracic aorta were macerated with radioimmunoprecipitation assay (RIPA) buffer and phenylmethanesulfonyl fluoride (PMSF) (100:1). The extraction of protein was performed with a tissue grinder, and supernatant was collected for the quantification using a bicinchoninic acid assay kit (Beyotime). The same amount of proteins (20 μg) was loaded on a polyacrylamide gel for electrophoresis followed by transfer to a 0.22 μm thick polyvinylidene fluoride membrane (Bio-Rad, CA). The membrane was blocked using 5% nonfat milk for 1 hour at room temperature and incubated with corresponding primary antibodies against eNOS (1:1000, CST, 9586), p-eNOS (1:1000, CST, 9571s), Akt (1:1000, CST, 4691), p-Akt (1:1000, CST, 4060), cleaved caspase-3 (1:1000, CST, 9662), Bcl-2 (1:200, Santa Cruz, SC-492), Bax (1:200, Santa Cruz, 20067), angiotensin II type 1 receptor (AT1R) (1:1000, Abcam, 124505), p-PKCα (1:200, Santa Cruz, SC-12356), PKCα (1:200, Santa Cruz, SC-8393), heat shock protein 70 (HSP70) (1:1000, CST, 4873), and GAPDH (1:5000, MultiSciences, 70-ab011-100) overnight at 4°C. In the end, the membrane was incubated with secondary anti-rabbit (1:5000, MultiSciences, 70-GAM0072) or anti-mouse (1:5000, MultiSciences, 70-GAM007) antibodies at room temperature for 1 hour. The bands were detected by electrochemiluminescence reagents (Beyotimes) using the Amersham Imager 600 system (GE Healthcare, Buckinghamshire, England). GAPDH was used as an internal loading control in our study.
Statistical Analysis
All the experimental results were expressed as mean ± SD. Differences between the 4 groups were compared by one-way ANOVA with Tukey's post hoc test. P-value < 0.05 was considered as a statistical difference.
RESULTS
Rosuvastatin Did Not Lower the SBP in SHRs
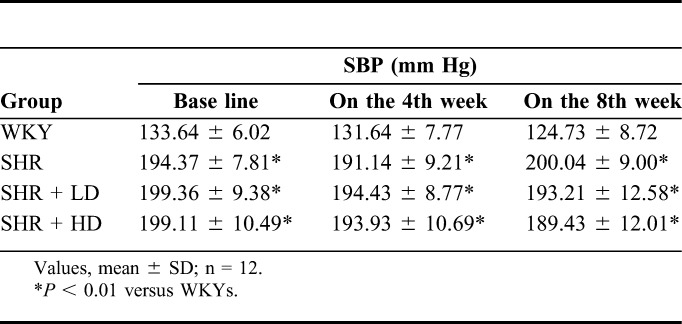
In the first steps, we examined whether rosuvastatin treatment can influence the blood pressure of SHRs. As shown in Table 2, the SBP of SHRs at baseline was much higher than that in age-matched WKY (P-value < 0.01). Continuous treatment of SHRs with either a lower or higher dose of rosuvastatin did not reduce the SBP significantly even after 2 months compared with the baseline, indicating that rosuvastatin did not have any antihypertensive effect in this study.
TABLE 2.
Changes of SBP
Rosuvastatin Improved the Lipid Profiles in SHRs
After 2 months of rosuvastatin treatment, the lipid profiles of all rats were verified. As shown in Table 3, SHRs have lower TC and LDL-C levels than WKYs (P-value < 0.01). Rosuvastatin treatment significantly lowered the levels of TC and LDL-C in SHRs (P-value < 0.01). Meanwhile, a high dose of rosuvastatin treatment upregulated the HDL-C levels in SHRs (P-value < 0.05). But, for TG levels, there were no differences between the 4 groups.
TABLE 3.
The Effect of Rosuvastatin on the Lipid Profiles

Rosuvastatin Upregulated the Expression of eNOS and Plasma Nitrite/Nitrate Levels in SHRs
We further examined the total and phosphorylated forms of eNOS, as well as its mRNA expression. Our results suggested that the mRNA level of eNOS was lower in SHRs as compared to WKYs (P-value < 0.01). Treatment with a low or high dose of rosuvastatin significantly upregulated eNOS mRNA level in SHRs (Fig. 1A). In addition, we also found that a high dose of rosuvastatin significantly increased the total and phosphorylated forms of eNOS (P-value < 0.01), as shown in Figures 1B–D. The ratio of p-eNOS/eNOS in the SHRs was also upregulated by rosuvastatin treatment, as shown in Figure 1E. However, plasma nitrite/nitrate concentrations detected by the total nitric oxide assay showed that the nitrite/nitrate levels in SHRs were lower than WKYs (Fig. 1F). Two different doses of rosuvastatin treatment both significantly increased the nitrite/nitrate levels compared with the SHR group. But, no significant difference was observed between 2 rosuvastatin groups.
FIGURE 1.
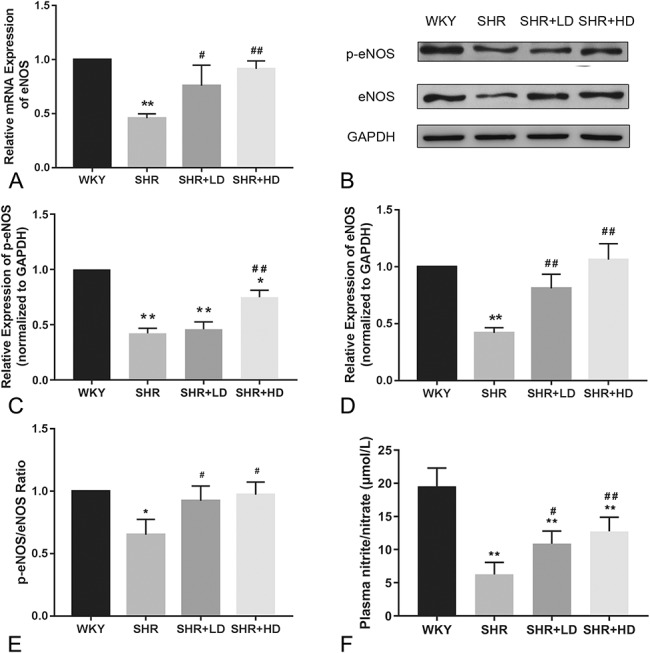
Effect of rosuvastatin on the expression and activation of eNOS in SHRs. A, The mRNA level of eNOS measured by RT-qPCR. B, Representative Western blotting image of p-eNOS and eNOS expression in the 4 groups. C and D, Quantification results of phosphorylated and total forms of eNOS by Western blotting. E, The ratio of p-eNOS/eNOS by Western blotting. F, The results of plasma nitrite/nitrate concentration in the 4 groups by total nitric oxide assay (n = 4 for each group). *P < 0.05 versus WKY group, **P < 0.01 versus WKY group, #P < 0.05 versus SHR group, ##P < 0.01 versus SHR group.
Rosuvastatin Improved the Endothelium-Dependent Vascular Relaxation in SHRs
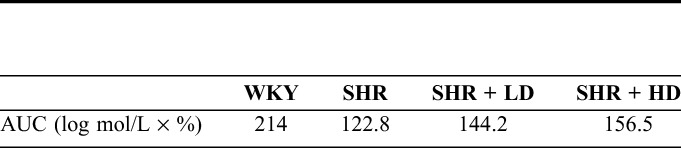
To further explore the effect of rosuvastatin on vascular relaxation function, 3 of the thoracic aortas from each group were dissected and subjected to endothelium-dependent relaxation tests. As shown in Figure 2, the WKYs had better endothelium-dependent relaxation function responding to the cumulative concentration of Ach than the SHRs. The most significant difference between the 2 groups was observed when the cumulative concentration of Ach reached 10−5 M. However, the relaxation function was restored in both rosuvastatin groups. The relaxation ratios of the aorta in the SHR + LD group and SHR + HD group were significantly higher than those in the SHR group at the cumulative concentration of 10−5 M. We also calculated the area under curve (AUC) values for 4 curves, as shown in Table 4. The AUC value of the SHR group was much smaller than the WKY group, whereas the AUC values of the SHR + LD group and SHR + HD group were both larger than that of the SHR group.
FIGURE 2.
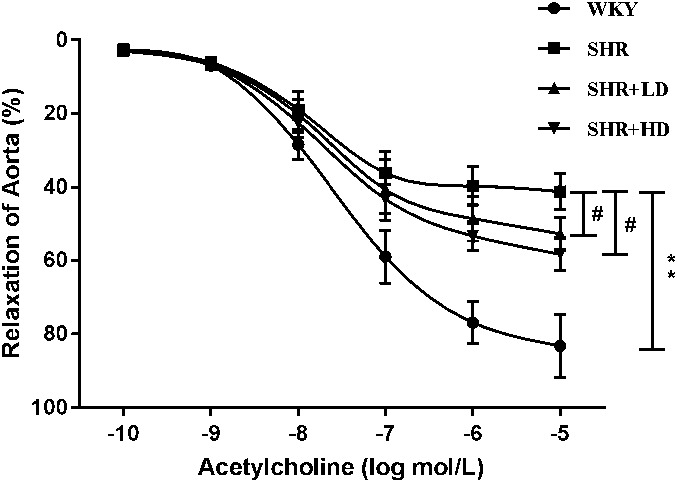
Effect of rosuvastatin on the endothelium-dependent relaxation of the aorta in response to acetylcholine in the 4 groups. Data are expressed as the mean ± SD (n = 3 each group). **P < 0.01 versus WKY group, #P < 0.05 versus SHR group.
TABLE 4.
The AUC Value of Each Endothelium-Dependent Relaxation Curve for Four Groups
Rosuvastatin Reversed the Remodeling of Aortic Structure in SHRs
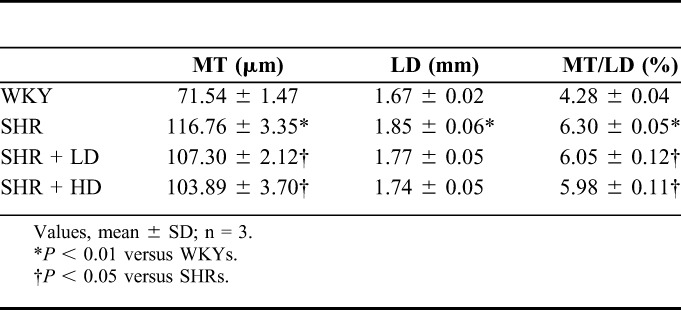
The changes in MT and LD of the thoracic aorta were shown in Table 5. SHRs displayed a higher MT and LD as compared to WKYs. The ratio of MT/LD in SHRs was higher as compared to WKYs. Treatment with a low or high dose of rosuvastatin significantly reduced MT compared with the SHR group (both P-value < 0.05). However, the level of LD after rosuvastatin treatment was not significantly decreased compared with the SHR group. Finally, in comparison to the ratio of MT/LD between the SHR group and 2 rosuvastatin groups, a significant reduction was observed (both P-value < 0.05).
TABLE 5.
Measurement of MT and LD
Rosuvastatin Suppressed Apoptosis in the Aorta of SHRs
As compared to WKYs, the expression of cleaved caspase-3 in SHRs was increased, whereas the ratios of p-Akt/Akt and Bcl/Bax were reduced, as shown in Figure 3. Rosuvastatin treatment for 2 months decreased the expression of cleaved caspase-3 (P-value < 0.01) but increased the ratio of Bcl-2/Bax (P-value < 0.01). A high dose of rosuvastatin further enhanced the changes mentioned above in a dose-dependent manner (P-value < 0.05). A low dose of rosuvastatin did not markedly increase the ratio of p-Akt/Akt, but a high dose of rosuvastatin increased the ratio of p-Akt/Akt significantly compared with the SHR group and SHR + LD groups (both P-value < 0.05). The TUNEL staining result of 4 groups is shown in Figure 4. The percentage of TUNEL-positive cells was significantly higher in the SHR group than those of the WKY group. However, rosuvastatin treatment decreased the apoptosis of cells, as reflected by the reduced number of TUNEL-positive cells. Overall, these changes in apoptosis markers and TUNEL staining showed that rosuvastatin decreased the apoptosis in the aorta of SHRs.
FIGURE 3.
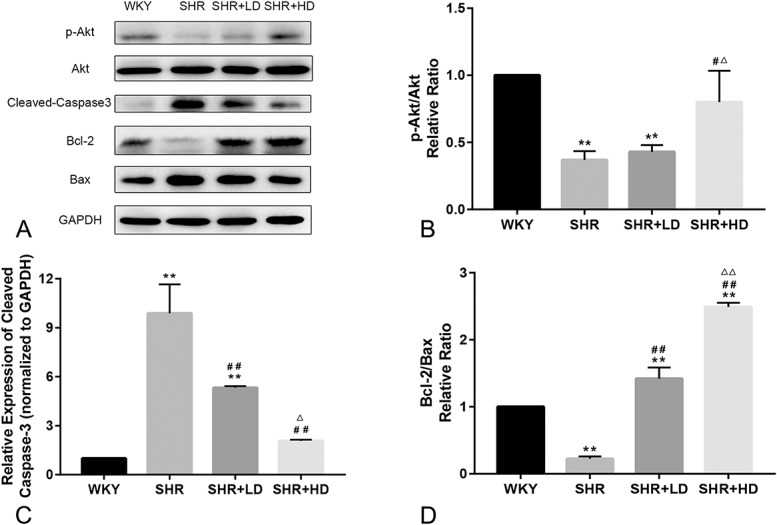
The antiapoptosis effect of rosuvastatin on the aorta of SHRs. A, Representative Western blotting image of protein expression of apoptosis-related biomarkers. B–D, Quantification results of p-Akt/Akt, cleaved Caspase3, and Bcl-2/Bax by Western blotting, respectively (n = 4 for each group). **P < 0.01 versus WKY group, #P < 0.05 versus SHR group, ##P < 0.01 versus SHR group. △P < 0.05 versus SHR + LD group, △△P < 0.01 versus SHR + LD group.
FIGURE 4.
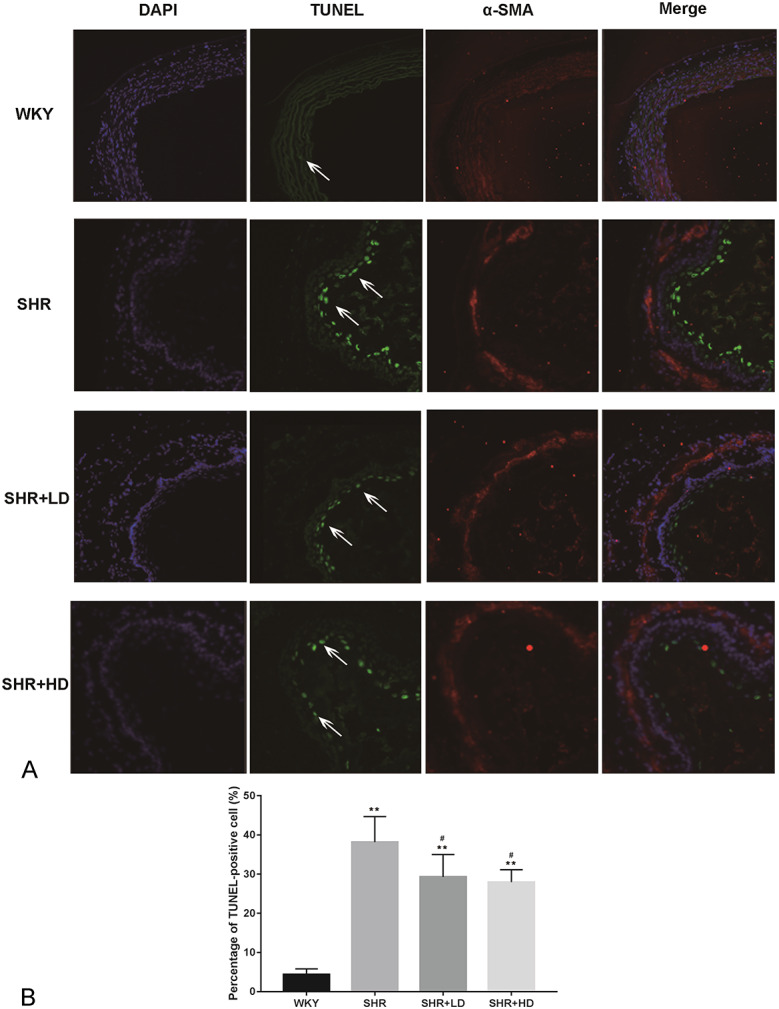
Effect of rosuvastatin on the apoptosis of aorta determined by TUNEL assay. A, Representative image of TUNEL result for 4 groups. The arrows indicate the TUNEL-positive endothelial cells. The α-SMA-positive cells (red dots) refer to vascular SMCs. B, Quantification results of TUNEL-positive cell percentage. Data are expressed as the mean ± SD (n = 5 each group). **P < 0.01 versus WKY group, #P < 0.05 versus SHR group.
Rosuvastatin Increased Cx43 Distribution in Media and Endothelium in SHRs
Further experiments were conducted to explore the distribution of Cx43 in the media and endothelium of the aorta. As shown in Figure 5, Cx43 was significantly lower in terms of intensity and number in SHRs as compared to WKYs in media and endothelium (Figs. 5A, B, respectively). However, treatment of rosuvastatin increased the Cx43 immunofluorescence in a dose-dependent manner in media and endothelium as compared to the SHR group, as shown in Figure 5C.
FIGURE 5.
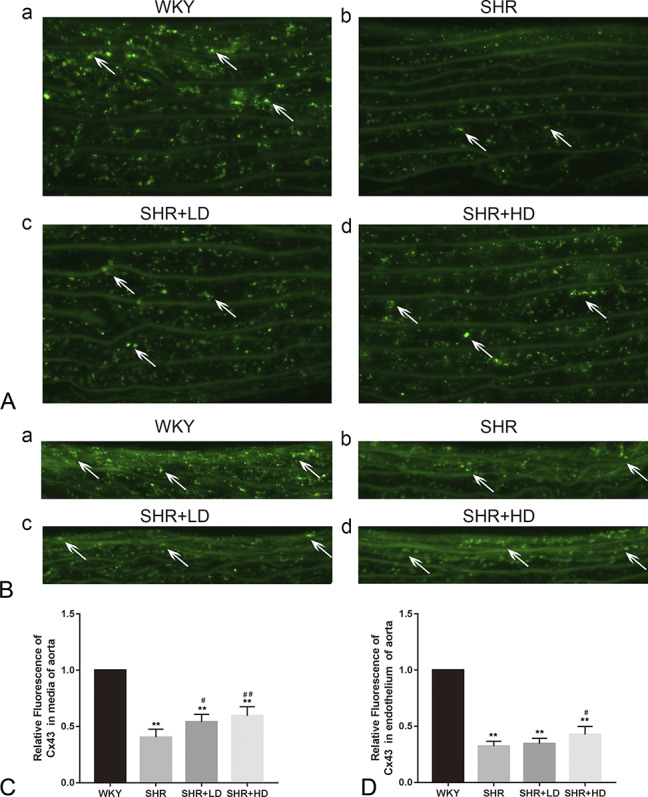
Effect of rosuvastatin on the distribution of Cx43 in the aorta in SHRs. A, Representative image showing Cx43 distribution indicated by white arrows in the media of the aorta. B, Representative image showing Cx43 distribution indicated by white arrows in the endothelium of aorta. C, Quantification results of relative Cx43 expression between 4 groups. Data are expressed as the mean ± SD (n = 5 each group). **P < 0.01 versus WKY group, #P < 0.05 versus SHR group, ##P < 0.01 versus SHR group.
Rosuvastatin Exerted an Anti-Inflammatory Effect of the Aorta in SHRs
We also examined the anti-inflammatory effect of rosuvastatin by detecting the presence of major inflammatory cytokines at the mRNA level (Figs. 6A–E). As shown in Figure 6A, the mRNA levels of C-X-C motif chemokine receptor (CXCR), including CXCR1 (IL8RA) and CXCR2 (IL8RB), were much higher in SHRs as compared to WKYs, as well as interleukin 12a (IL12a). Similar changes in mRNA levels of IL6R and MCP-1 were observed between SHRs and WKYs. Treatment with rosuvastatin decreased the mRNA levels of CXCR2, IL12a, IL6R, and MCP-1 in SHRs (P-value < 0.05). The expression level of ICAM-1 was not significantly increased in SHRs as compared to the WKY group. However, rosuvastatin treatment significantly decreased the expression of ICAM-1 at the mRNA level in a dose-dependent manner (Fig. 6B). The expressions of VCAM-1 was considerably higher in the SHR group than that of the WKY group at mRNA levels (Fig. 6C). Two different doses of rosuvastatin were found to decrease its expression. Moreover, the mRNA levels of MMP10 and TGF-β1 were found to be higher in the SHR group than the WKY group (Figs. 6D, E), and rosuvastatin partly reversed this increase (P-value < 0.05).
FIGURE 6.

The anti-inflammatory effect of rosuvastatin on the aorta of SHRs. A, The mRNA expression of CXCR1, CXCR2, IL12a, and MCP-1 in the 4 groups. B and C, The mRNA expression of the adhesion molecule ICAM-1 and VCAM-1 in the 4 groups. D and E, The mRNA expression of MMP10 and TGF-β1 in the 4 groups (n = 4 for each group). *P < 0.05 versus WKY group, **P < 0.01 versus WKY group, #P < 0.05 versus SHR group, ##P < 0.01 versus SHR group, △P < 0.05 versus SHR + LD group, △△P < 0.01 versus SHR + LD group.
Rosuvastatin Altered the Expression of AT1R-PKCα-HSP70 Pathway
We further determined whether the AT1R-PKCα-HSP70 pathway mediated the antivascular remodeling effect of rosuvastatin. Western blotting was performed to measure the expression of proteins associated with the AT1R-PKCα-HSP70 axis. In the SHR group, the expression of AT1R and HSP70, as well as the activity of PKCα, were higher as compared to the WKY group (P-value < 0.01), as shown in Figure 7. The total protein level of PKCα were unchanged. After treatment with a high dose of rosuvastatin, the expression of AT1R and HSP70 decreased significantly as compared to the SHR group. However, a low dose had no such effect. The phosphorylation level of PKCα was decreased by either a low or high dose of rosuvastatin.
FIGURE 7.
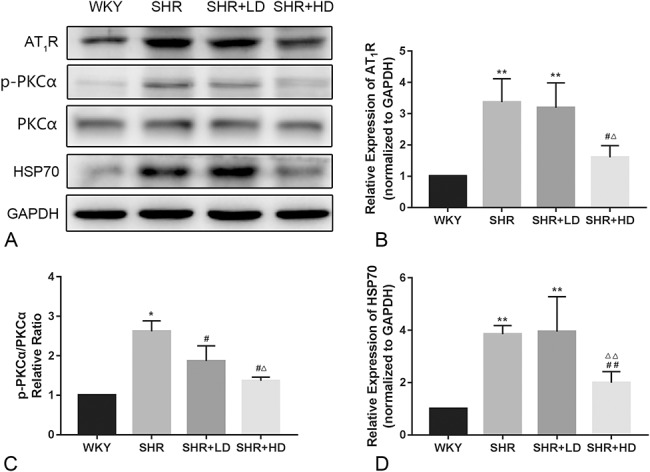
Effect of rosuvastatin on the protein expression related to the AT1R-PKCα-HSP70 axis in the aorta of SHRs. A, Representative Western blotting image of AT1R-PKCα-HSP70 axis protein expression. B–D, The quantification results of AT1R-PKCα-HSP70 axis protein expression normalized to GAPDH (n = 4 for each group). *P < 0.05 versus WKY group, **P < 0.01 versus WKY group, #P < 0.05 versus SHR group, ##P < 0.01 versus SHR group, △P < 0.05 versus SHR + LD group, △△P < 0.01 versus SHR + LD group.
DISCUSSION
This study demonstrated that a two-month treatment with rosuvastatin of SHR could effectively reverse the thickening media and decrease the MT/LD ratio of the thoracic aorta. Also, rosuvastatin improved the endothelium-dependent relaxation of the aorta in SHR. These improvements were associated with increased eNOS expression of the aorta and NO content in plasma. Besides, we also verified that rosuvastatin treatment suppressed the apoptosis and inflammation of the aorta and decreased the plasma LDL-C and TC levels. Furthermore, we reported for the first time that rosuvastatin treatment could upregulate the expression of Cx43 and inhibit the AT1R-PKCα-HSP70 pathway in SHR. Collectively, our finding proved that rosuvastatin exerted a protective role in the hypertension-induced aortic remodeling of SHR.
In this study, we first explored whether such improvements were mediated by the antihypertensive effect of rosuvastatin. Although many studies have confirmed the protective role of statins, either alone or combined with another agent, in attenuating blood pressure of several hypertensive animal models,15,16 studies such as those by Yamashita et al17 and Park et al18 also reported that the blood pressure was not affected in hypertensive rats by statins. So, it is still not entirely clear whether statin alone has an antihypertensive effect. Interestingly, the blood pressure of SHRs was not significantly decreased in our study, whereas the expression and activation of eNOS were both increased by rosuvastatin treatment, accompanied by the increased plasma NO content. There are two possible reasons to explain that rosuvastatin has a limited antihypertensive effect in SHRs. First, the mechanism of blood pressure regulation is complex, and several vasoconstricting agents can counteract the vasodilating effect of NO, such as ET-1 and Ang II. The blood pressure of SHRs was dependent on the balance of vasoconstricting agents and vasodilating agents. Second, the extents of eNOS and NO increase were not so remarkable, so the anti-hypertensive effect by NO increase was not statistically significant. It reminded us that the underlying mechanisms should be explained other than the improvement of blood pressure.
A previous study revealed that rosuvastatin increased the endothelial NO production in a myocardial ischemia/reperfusion mice model.19 So, we also examined the expression and activation of eNOS in the aorta and plasma NO contents. Studies have shown that eNOS is a protective gene in vascular-related diseases, which regulates NO production. NO can diffuse through the VSMCs and catalyze the conversion of GTP into cGMP. The activated PKG induced by increased cGMP further triggers the phosphorylation of downstream target protein to lower the Ca2+ concentration and thereby resulting in relaxation of the vessels. In our study, treatment with different doses of rosuvastatin increased the mRNA and protein levels of eNOS in SHRs, as well as the plasma NO contents. Such improvements might be mediated by its lipid-lowering effect because studies suggested that increased LDL could downregulate the expression of eNOS, inhibit the endothelial-derived NO activity, and decrease receptor-mediated NO release.20,21 In addition, another study found that rosuvastatin promoted a sustained increase of eNOS and endothelial NO production in treating human umbilical vein endothelial cells.22 To explain this, they hypothesized that the increased S-nitrosylation of mt-HSP70 and tropomyosin benefited endothelial integrity and regulated the endothelial migration. Their study provided a new outlook on the regulatory mechanism by rosuvastatin in endothelial cells at the posttranslational level.
As one of the most effective models of studying hypertension, SHRs with sustained high blood pressure often exhibit excessive apoptosis in the arteries, stimulated by high levels of Ang II.23 Apoptosis of the vascular SMC modulates the inward eutrophic remodeling of vessels. Normally, apoptosis is localized to the outer periphery, along with a growth in the inner membrane. Such pathological changes can cause a decrease in the LD and an increase in the MT. To determine whether rosuvastatin has an antiapoptosis effect, we measured the expression of apoptosis biomarkers such as Bcl-2/Bax, cleaved caspase-3, and p-Akt/Ak, and performed the TUNEL staining experiment. Our findings suggested that treatment with rosuvastatin increased the expression of Bcl-2 and reduced the expression of Bax. Meanwhile, as a negative regulator of cell death gene, activated Akt was increased by rosuvastatin and thereby suppressing apoptosis. Furthermore, rosuvastatin decreased the cleaved caspase-3 expression, a promoter of apoptosis. These results, combined with the result of TUNEL assay, confirmed that rosuvastatin had an antiapoptosis effect, which could be a reason for the decreased MT and MT/LD ratio of the aorta in SHRs.
The gap junctions are the key players in cell-to-cell communication and are considered to be crucial for coordinating and synchronizing the contractions of vessels in response to electrotonic signals. Cx43 is one of the most notable gap junction channel proteins expressed between vascular SMCs (VSMC) and endothelial cells (EC) as a connector.24,25 It was reported that Cx43 is a key factor in regulating vascular tone and reactivity.26 Knockdown of Cx43 inhibited the Ang II-induced enhancement of vascular reactivity and contractility.27,28 Previous studies have demonstrated low expression of Cx43 in the media and endothelial in SHRs,14 which is in agreement with our results. Rosuvastatin significantly enhanced the immunofluorescent spots of Cx43 in the media and endothelium in SHRs. It showed that rosuvastatin might improve vascular reactivity and contractility.
Vascular inflammation is one of the most common complications of hypertension. The participation of several inflammatory factors stimulated by Ang II and ox-LDL, including interleukin and intercellular adhesion molecule, normally acts as a contributor to vascular remodeling. They can recruit monocytes/macrophages to the vascular wall and involve in the progression of atherosclerosis.29 In addition to this, hemodynamic abnormalities induced by hypertension can also cause endothelial dysfunction and endothelial macrophage adhesion due to an upregulated expression of ICAM-1 and VCAM-1. So, the decreased ICAM-1 and VCAM-1 expressions by rosuvastatin might be mediated by reduced LDL levels in this study. The formation and degradation of extracellular matrix (ECM) is another key process for vascular remodeling. TGF-β1 is an important profibrotic mediator that works by inhibiting the degradation of ECM. It has been reported that the TGF-β1 level was increased significantly both in the kidney and heart of SHRs mediating renal fibrosis and myocardial fibrosis.30,31 And statins have been reported to decrease the serum TGF-β levels significantly, suggesting that statin might have a suppressive effect on the expression of TGF-β.32 In our study, we found that rosuvastatin can suppress the expression of TGF-β1 in the aortic tissues of SHRs, indicating its antifibrotic effect on SHRs. MMP10 is an enzyme that breaks down the ECM and regulates the migration of inflammatory cytokines across the membranes, eg, neutrophil. It also blocks the activation of macrophage and proliferation of lymphocytes. Meanwhile, MMP10 was found to be regulated by TGF-β1.33 In this study, rosuvastatin significantly suppressed the MMP10 levels in SHRs, which could be attributed to the decreased levels of TGF-β1. These findings suggested that rosuvastatin inhibited the invasion of inflammatory cytokines into the inner membrane, thus improving the endothelium-dependent relaxation function and reducing the MT.
Further experiments were performed to determine the ATR1 expression and relevant signal transduction pathway in vascular remodeling by rosuvastatin. Several studies have elicited the relationship between RAAS and vascular remodeling,34,35 including some of them majoring in the effect of statin in the inhibition of AT1R expression. The expression of AT1R in both heart and aorta was increased by overactivation of the RAAS system in SHRs. Binding with Ang II, AT1R triggers multiple intracellular signaling pathways, such as the coupling of G proteins, which activates vasoconstriction during hypertension.36 A study by Tian et al37 has investigated the effect of rosuvastatin in the endothelial function of db/db mice. They found that rosuvastatin participated in favorable modulation of the RAAS cascade and attenuated vasoconstrictions induced by elevated Ang II concentration through the AT1R inhibition in db/db mice. So, their principal conclusion was that rosuvastatin was attributed to an increase of NO bioavailability by the ROS inhibition from the AT1R-NAD(P)H oxidase cascade. In this study, we only investigated the expression of AT1R expression in the aorta. The expression of AT1R was decreased significantly by a high dose of rosuvastatin, suggesting that the RAAS was attenuated. Another benefit due to RAAS inhibition in our study was reduced vascular inflammation. Blocking RAAS was found to negatively regulate inflammatory factors,38 such as ICAM-1, VCAM-1, IL-6R, and IL-12a.
Several studies have revealed the role of the PKC family in hypertension. The high expression and activation of PKC subtypes such as PKCα, PKCβ, PKCθ, and PKCδ can increase the blood pressure and vasoconstriction.39–41 Previous studies have demonstrated that activation of PKCβ2 and PKCθ is enhanced in the aorta of SHRs.42 This further increases the phosphorylation of the Ca2+-sensitization pathway, such as CPI-17-MYPT1-MLC, causing the contraction of vascular smooth muscles.43 The findings of our study indicated that a high dose of rosuvastatin could decrease the phosphorylation of PKCα in SHRs, suggesting that it might be mediated by the inhibition of PKC.
HSP70 is a molecular chaperone that is found to be enhanced in the vessels of SHRs. Previous studies have demonstrated that HSP70 can not only mediate protein folding and transport during stress but also prevent apoptosis.44,45 Although apoptosis was increased in SHRs, upregulated HSP70 might participate in protecting VSMC and EC against apoptosis of SHRs. However, rosuvastatin treatment decreased the expression of HSP70 in SHRs, which attenuated apoptosis in the aorta.
There are still several limitations to our study. First, given the structural and functional complexity of the adaptive response of the aorta to hypertension, the benefits of rosuvastatin in SHRs shown in this study are only observational. Second, in-depth investigations are required to explore the direct mechanisms on how changes in apoptosis and inflammation, as well as AT1R/PKCα/HSP70 signaling transduction pathways by rosuvastatin, are involved in the vascular remodeling of the aorta during hypertension. Third, our study only revealed the benefits of rosuvastatin in the remodeling of the aorta, but its role for the whole vascular system, particularly the coronary arteries and cerebrovascular vessels, is not determined. Furthermore, the number of observations for each group was quite limited. Only 3 to 5 samples were analyzed for some experiments. Finally, whether those benefits existed in patients with a clinical dose still need clinical trials for confirmation.
CONCLUSIONS
This study provided an observational investigation of the protective role of rosuvastatin in improving vascular structure and function. Mechanistically, we found that rosuvastatin increased the expression of eNOS, enhanced the plasma NO content, and prevented apoptosis of aortic cells. Moreover, it upregulated the expression of Cx43 in media and endothelium and suppressed the inflammation by reducing proinflammation cytokines in the aorta. Inhibition of AT1R/PKCα/HSP70 signaling transduction pathway in SHRs was also discovered by rosuvastatin treatment.
Footnotes
Supported by Natural Science Foundation of Zhejiang Province (LY18H020007 and LQ16H020001) and National Natural Science Foundation of China (81800212 and 81500212).
The authors report no conflicts of interest.
Q. Lv and Y. Wang contributed equally to this article.
REFERENCES
- 1.Arima H, Barzi F, Chalmers J. Mortality patterns in hypertension. J Hypertens. 2011;29(suppl 1):S3–S7. [DOI] [PubMed] [Google Scholar]
- 2.van der Veen PH, Geerlings MI, Visseren FL, et al. Hypertensive target organ damage and longitudinal changes in brain structure and function: the second manifestations of arterial disease-magnetic resonance study. Hypertension. 2015;66:1152–1158. [DOI] [PubMed] [Google Scholar]
- 3.Drawz PE, Alper AB, Anderson AH, et al. Masked hypertension and elevated nighttime blood pressure in CKD: prevalence and association with target organ damage. Clin J Am Soc Nephrol. 2016;11:642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dai X, Hua L, Chen Y, et al. Mechanisms in hypertension and target organ damage: is the role of the thymus key? (review). Int J Mol Med. 2018;42:3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redon J, Tellez-Plaza M, Orozco-Beltran D, et al. Impact of hypertension on mortality and cardiovascular disease burden in patients with cardiovascular risk factors from a general practice setting: the escarval-risk study. J Hypertens. 2016;34:1075–1083. [DOI] [PubMed] [Google Scholar]
- 6.Alloubani A, Saleh A, Abdelhafiz I. Hypertension and diabetes mellitus as a predictive risk factors for stroke. Diabetes Metab Syndr. 2018;12:577–584. [DOI] [PubMed] [Google Scholar]
- 7.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension. 2001;38:581–587. [DOI] [PubMed] [Google Scholar]
- 8.Wang P, Luo L, Shen Q, et al. Rosuvastatin improves myocardial hypertrophy after hemodynamic pressure overload via regulating the crosstalk of Nrf2/ARE and TGF-beta/smads pathways in rat heart. Eur J Pharmacol. 2018;820:173–182. [DOI] [PubMed] [Google Scholar]
- 9.Xu X, Zhang L, Liang J. Rosuvastatin prevents pressure overloadinduced myocardial hypertrophy via inactivation of the Akt, ERK1/2 and GATA4 signaling pathways in rats. Mol Med Rep. 2013;8:385–392. [DOI] [PubMed] [Google Scholar]
- 10.Qiu Z, Zhang W, Fan F, et al. Rosuvastatin-attenuated heart failure in aged spontaneously hypertensive rats via PKCalpha/beta2 signal pathway. J Cell Mol Med. 2012;16:3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colucci R, Fornai M, Duranti E, et al. Rosuvastatin prevents angiotensin II-induced vascular changes by inhibition of NAD(P)H oxidase and COX-1. Br J Pharmacol. 2013;169:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jing L, Zhang J, Sun J, et al. Inhibition of extracellular signal-regulated kinases ameliorates hypertension-induced renal vascular remodeling in rat models. Int J Mol Sci. 2011;12:8333–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Potje SR, Grando MD, Chignalia AZ, et al. Reduced caveolae density in arteries of SHR contributes to endothelial dysfunction and ROS production. Sci Rep. 2019;9:6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dlugosova K, Okruhlicova L, Mitasikova M, et al. Modulation of connexin-43 by omega-3 fatty acids in the aorta of old spontaneously hypertensive rats. J Physiol Pharmacol. 2009;60:63–69. [PubMed] [Google Scholar]
- 15.Li DB, Xu HW, Yang GJ, et al. Effects of rosuvastatin correlated with the down-regulation of CYP4A1 in spontaneously hypertensive rats. Microvasc Res. 2015;98:88–93. [DOI] [PubMed] [Google Scholar]
- 16.Wassmann S, Laufs U, Baumer AT, et al. HMG-CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. 2001;37:1450–1457. [DOI] [PubMed] [Google Scholar]
- 17.Yamashita T, Kawashima S, Miwa Y, et al. A 3-hydroxy-3-methylglutaryl co-enzyme A reductase inhibitor reduces hypertensive nephrosclerosis in stroke-prone spontaneously hypertensive rats. J Hypertens. 2002;20:2465–2473. [DOI] [PubMed] [Google Scholar]
- 18.Park JK, Mervaala EM, Muller DN, et al. Rosuvastatin protects against angiotensin II-induced renal injury in a dose-dependent fashion. J Hypertens. 2009;27:599–605. [DOI] [PubMed] [Google Scholar]
- 19.Jones SP, Gibson MF, Rimmer DM, III, et al. Direct vascular and cardioprotective effects of rosuvastatin, a new HMG-CoA reductase inhibitor. J Am Coll Cardiol. 2002;40:1172–1178. [DOI] [PubMed] [Google Scholar]
- 20.Liao JK, Shin WS, Lee WY, et al. Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J Biol Chem. 1995;270:319–324. [DOI] [PubMed] [Google Scholar]
- 21.Liao JK. Inhibition of Gi proteins by low density lipoprotein attenuates bradykinin-stimulated release of endothelial-derived nitric oxide. J Biol Chem. 1994;269:12987–12992. [PubMed] [Google Scholar]
- 22.Huang B, Li FA, Wu CH, et al. The role of nitric oxide on rosuvastatin-mediated S-nitrosylation and translational proteomes in human umbilical vein endothelial cells. Proteome Sci. 2012;10:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vega F, Panizo A, Pardo-Mindan J, et al. Susceptibility to apoptosis measured by MYC, BCL-2, and BAX expression in arterioles and capillaries of adult spontaneously hypertensive rats. Am J Hypertens. 1999;12:815–820. [DOI] [PubMed] [Google Scholar]
- 24.Haefliger JA, Nicod P, Meda P. Contribution of connexins to the function of the vascular wall. Cardiovasc Res. 2004;62:345–356. [DOI] [PubMed] [Google Scholar]
- 25.Johnstone S, Isakson B, Locke D. Biological and biophysical properties of vascular connexin channels. Int Rev Cell Mol Biol. 2009;278:69–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ampey BC, Morschauser TJ, Lampe PD, et al. Gap junction regulation of vascular tone: implications of modulatory intercellular communication during gestation. Adv Exp Med Biol. 2014;814:117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu H, Li XZ, Peng M, et al. Role of gap junctions in the contractile response to agonists in the mesenteric resistance artery of rats with acute hypoxia. Mol Med Rep. 2017;15:1823–1831. [DOI] [PubMed] [Google Scholar]
- 28.Yang G, Peng X, Wu Y, et al. Involvement of connexin 43 phosphorylation and gap junctional communication between smooth muscle cells in vasopressin-induced ROCK-dependent vasoconstriction after hemorrhagic shock. Am J Physiol Cell Physiol. 2017;313:C362–C370. [DOI] [PubMed] [Google Scholar]
- 29.Weiss D, Kools JJ, Taylor WR. Angiotensin II-induced hypertension accelerates the development of atherosclerosis in apoE-deficient mice. Circulation. 2001;103:448–454. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Martinez E, Ibarrola J, Fernandez-Celis A, et al. Galectin-3 pharmacological inhibition attenuates early renal damage in spontaneously hypertensive rats. J Hypertens. 2018;36:368–376. [DOI] [PubMed] [Google Scholar]
- 31.Fu S, Li YL, Wu YT, et al. Icariside II attenuates myocardial fibrosis by inhibiting nuclear factor-kappaB and the TGF-beta1/Smad2 signalling pathway in spontaneously hypertensive rats. Biomed Pharmacother. 2018;100:64–71. [DOI] [PubMed] [Google Scholar]
- 32.Zhao J, Cheng Q, Liu Y, et al. Atorvastatin alleviates early hypertensive renal damage in spontaneously hypertensive rats. Biomed Pharmacother. 2019;109:602–609. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa F, Miyoshi H, Nose K, et al. Transcriptional induction of MMP-10 by TGF-beta, mediated by activation of MEF2A and downregulation of class IIa HDACs. Oncogene. 2010;29:909–919. [DOI] [PubMed] [Google Scholar]
- 34.Baumann M, Megens R, Bartholome R, et al. Prehypertensive renin-angiotensin-aldosterone system blockade in spontaneously hypertensive rats ameliorates the loss of long-term vascular function. Hypertens Res. 2007;30:853–861. [DOI] [PubMed] [Google Scholar]
- 35.Morishita R, Gibbons GH, Tomita N, et al. Antisense oligodeoxynucleotide inhibition of vascular angiotensin-converting enzyme expression attenuates neointimal formation: evidence for tissue angiotensin-converting enzyme function. Arterioscler Thromb Vasc Biol. 2000;20:915–922. [DOI] [PubMed] [Google Scholar]
- 36.Montezano AC, Nguyen D, Cat A, Rios FJ, et al. Angiotensin II and vascular injury. Curr Hypertens Rep. 2014;16:431. [DOI] [PubMed] [Google Scholar]
- 37.Tian XY, Wong WT, Xu A, et al. Rosuvastatin improves endothelial function in db/db mice: role of angiotensin II type 1 receptors and oxidative stress. Br J Pharmacol. 2011;164:598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pacurari M, Kafoury R, Tchounwou PB, et al. The renin-angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflam. 2014;2014:689360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhao Y, Vanhoutte PM, Leung SW. alpha1-Adrenoceptor activation of PKC-epsilon causes heterologous desensitization of thromboxane receptors in the aorta of spontaneously hypertensive rats. Br J Pharmacol. 2015;172:3687–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Novokhatska T, Tishkin S, Dosenko V, et al. Correction of vascular hypercontractility in spontaneously hypertensive rats using shRNAs-induced delta protein kinase C gene silencing. Eur J Pharmacol. 2013;718:401–407. [DOI] [PubMed] [Google Scholar]
- 41.Liu L, Liu J, Gao Y, et al. Protein kinase Cbeta mediates downregulated expression of glucagon-like peptide-1 receptor in hypertensive rat renal arteries. J Hypertens. 2015;33:784–790. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Zhou Q, Wu B, et al. Propofol induces excessive vasodilation of aortic rings by inhibiting protein kinase Cbeta2 and theta in spontaneously hypertensive rats. Br J Pharmacol. 2017;174:1984–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moreno-Dominguez A, Colinas O, El-Yazbi A, et al. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol. 2013;591:1235–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tremblay J, Hadrava V, Kruppa U, et al. Enhanced growth-dependent expression of TGF beta 1 and hsp70 genes in aortic smooth muscle cells from spontaneously hypertensive rats. Can J Physiol Pharmacol. 1992;70:565–572. [DOI] [PubMed] [Google Scholar]
- 45.Stankiewicz AR, Lachapelle G, Foo CP, et al. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J Biol Chem. 2005;280:38729–38739. [DOI] [PubMed] [Google Scholar]