

Abstract
Alzheimer’s disease is the most common cause of dementia. Cellular changes in the brains of the patients suffering from Alzheimer’s disease occur well in advance of the clinical symptoms. At the cellular level, the most dramatic is a demise of neurones. As astroglial cells carry out homeostatic functions of the brain, it is certain that these cells are at least in part a cause of Alzheimer’s disease. Historically, Alois Alzheimer himself has recognised this at the dawn of the disease description. However, the role of astroglia in this disease has been understudied. In this chapter, we summarise the various aspects of glial contribution to this disease and outline the potential of using these cells in prevention (exercise and environmental enrichment) and intervention of this devastating disease.
Keywords: Pathological ageing, Astrocytes, Alzheimer’s disease, Astroglial atrophy, Neurodegeneration, Calcium signaling, Stem cells
11.1. Senile Dementia—The Outcome of Pathological Ageing
Dementia as a medical term was introduced in the first century AD by Aulus Cornelius Celsus in his fundamental discourse De Medicina [50] to characterise major cognitive impairments of the mankind. The term dementia originates from the prefix “de” (meaning “out of”), the stem “ment” (‘mind’) and the suffix “ia” (diseased condition). Historically, this term was used in a very broad sense to indicate chronic cognitive impairments associated with psychotic symptoms such as delusions or hallucinations. However, dementia was not associated with age-dependent cognitive decline, although from the very dawn of medical observations, these impairments were considered as an essential and inevitable part of ageing process. Already in the seventh century BC, Pythagoras defined the advanced ages of human life as “senium” when “the system returns to the imbecility of the first epoch of infancy” [26]. From Aristotle to Lucretius and Galen, the ageing was considered to be associated with mental decline, impossible to arrest or recuperate [26, 38, 121, 302]. This gloomy outlook was not, however, shared by Cicero who believed in selective development of a senescence-dependent cognitive decline: “senile imbecility does not occur in all old men, but only in those of feeble mind” [54].
Over centuries the mental weakness of old people was defined as senility, idiocy, morosis, dotage, etc.; in 1794, the term dementia was formalised by Philippe Pinel and this term was officially recognised by the French Law [291]. At the end of the nineteenth century, the definition of senile dementia became widespread and underlying histopathology became to be scrutinised. The specific lesions, the plaques (then known as miliary foci), were discovered by Block and Marinesco in the brain of old epileptic patient [36], and subsequently these plaques were observed in the post-mortem tissues of patients suffering from senile dementia by Redlich and by Otto Fischer [77, 225]. In 1903, Max Bielschowsky developed an improved version of the Golgi silver stain that allowed visualisation of neurofibrilles [30]. Using this new technique, Alois Alzheimer (in 1906) was able to visualise neurofibrillary tangles in the post-mortem brain of Mrs. Augusta D, whom he first seen in 1901 in Frankfurt with the symptoms of confusion, delusions and dementia. These tangles displayed extraordinary thickness and often merged into dense bundles reaching the surface of a neurone [7, 26]. The brain of Augusta D also contained senile plaques, and thus the case of early dementia associated with appearance of senile plaques and pathological neurofibrillary tangles has been reported in 1907 [7]. This was a rather unique description, which differed from the widespread dementia of the early twentieth century associated predominantly with neurosyphilis or vascular ischemic brain damage. The disease was named “Alzheimersche krankheit” by Emil Kraepelin in the 8th edition of his immensely influential textbook on Psychiatry (Psychiatrie: Ein Lehrbuch fuer Studierende und Arzte). Kraepelin defined this new disease as a rapidly progressive, early-onset dementia distinct from the senile dementia [136]; for the history, people involved, histological details and controversies see [27, 109, 172].
Alzheimer’s disease (AD) was rarely diagnosed in the first half of the twentieth century and was generally regarded as a rare pathology that affected relatively young persons. Only in 1960, the AD histopathology was related to the sporadic cases of age-dependent (i.e. senile) dementia and the notion of AD as senescent-associated pathology had been developed [244, 245, 290]. It seems also that the pandemic of the senile dementia observed in recent years has evolved over the last century. Detailed physiological investigation of organs and systems of an extended population (826 subjects) of elderly (80–100 years of age) inhabitants of the UK performed in 1889 revealed surprisingly little changes in their cognitive status. Furthermore “… the brain in many held out as well or better than other organs - which may be regarded one of the bright rays, if not the brightest, in the centenarian landscape” [111]. Indeed, in this study dementia was observed only in 2 out of 74 centenarians. This contrasts remarkably with our times, when >50% of people older than 85 demonstrate signs of severe cognitive impairment [343]. Of course, evolution of epidemiological changes may be interpreted from many angles, and yet it is impossible not to speculate that an increased environmental toxicity, changes in diet and mounting social pressure have contributed to a rise of sporadic AD in the modern population.
Increased prevalence of senile dementia at advanced age parallels increase in life expectancy. Modern definition regards AD as a severe neurodegenerative disorder associated with specific histopathological markers represented by (i) focal extracellular deposits of fibrillar β-amyloid generally known as neuritic or senile plaques in the brain parenchyma and the walls of blood vessels, and (ii) intraneuronal accumulation of neurofibrillary tangles composed of abnormal hyperphosphorylated tau filaments [39, 70]. AD affects specific brain regions associated with learning and memory, including the basal forebrain, the hippocampus and the neocortex. Clinical symptoms of AD are manifested by a progressive decline of cognitive functions including short- and long-term memory [185]. At advanced stages of the disease, clinical presentation of AD is complicated by a variety of behavioural disturbances including agitation, irritability, anxiety, delusions and depression [165].
Conceptually, two forms of AD are defined: (i) early-onset or familial Alzheimer’s disease (FAD) and (ii) late-onset or sporadic AD, or SAD [35]. Epidemiologically, the late-onset SAD accounts for the absolute majority (95–99%) of AD cases in people above 65 years of age. The familial variant of AD is associated with mutations in three genes encoding for amyloid precursor protein (APP), presenilin-1 (PS-1) and presenilin-2 (PS-2), which are inherited in an autosomal dominant manner [23, 257, 294]. In contrast to SAD, which is linked to an old age, the familial AD occurs in much younger group of patients between 40 and 50 years old; the FAD is characterised by a rapid progression and idiosyncratic clinical manifestation. Anatomical and histopathological progression of AD begins from early degeneration of cholinergic neurones in the nucleus basalis of Meynert and septum. In parallel, the accumulation of intraneuronal β-amyloid and formation of neurofibrillary tangles develop, which ultimately leads to an emergence of senile plaques [182]. Deterioration of neuronal networks begins with synaptic damage and malfunction which affects CNS plasticity; these changes occur prior to the formation of senile plaques and neurofibrillary tangles and prior to neuronal death [251, 255]. In addition, this process suppresses neurogenesis, which further impairs neuronal plasticity [231, 237]. Apart from the cholinergic system, AD pathology impairs other major neurotransmitter systems including noradrenergic, serotonergic and dopaminergic [153, 234, 348].
11.2. Experimental Animal Models of Alzheimer’s Disease
Alzheimer’s disease, similar to other neurodegenerative diseases, is a specific disease of humans; animals as a rule do not develop AD-like pathology [289]. Experimental study of AD therefore requires the development of animal disease models which are capable of faithful reproduction of single or multiple subsets of neuropathological, histological, cellular, behavioural or biochemical alterations resembling those seen in classical AD [48, 91].
11.2.1. Old Animals
The very first models of AD were represented by aged animals [28, 29, 289]; several species from rodents to primates have demonstrated atrophy and death of basal forebrain neurones expressing choline acetyltransferase or nerve growth factor [60, 78, 248]. In monkeys, alterations of cholinergic neurones were even associated with β-amyloid depositions [289]. In addition, old animals showed not only a cholinergic dysfunction but also a concomitant alteration of other neurotransmitter systems such as the monoaminergic, peptidergic or serotonergic [15, 176, 198, 234].
11.2.2. Lesions
The global lesion models of AD (Table 11.1) relied on destruction of certain brain areas. At the beginning, the electrolytic lesions were used; these caused diffuse damage of several brain areas and lacked specificity [156, 289]. In the majority of global lesion models, the non-selective excitotoxic toxins such as NMDA, ibotenic acid, quisqualic acid, quinolinic acid, colchicine and other alkaloid substances were employed [289]. Injections of these substances triggered cell death with consequent neurological dysfunctions including impaired cognition. The global lesion models also employed injections of alcohol, which is toxic to cholinergic neurons [13, 289]; or injections of β-amyloid peptides, which produces multiple alterations of corticobasal neurones affecting acetylcholine release and cholinergic receptors [88, 211].
Table 11.1.
Summary of lesion models of Alzheimer’s disease. Modified from [232]
| Lesion | Cholinergic | Non-cholinergic | Neuropathology | References |
|---|---|---|---|---|
| Electrolytic | Yes | Yes | Neuronal death | [156, 293] |
| Excitotoxins (NMDA, Ibotenic acid, Quisalic acid) | Yes | Yes | Neuronal death | [69, 330] |
| Quinolinic acid | Yes | Yes | Neuronal death | [37] |
| Colchicine | Yes | Yes | Neuronal death | [258] |
| Alkaloids | Yes | Yes | Neuronal death | [65] |
| AF64A | Yes | No | Neuronal death | [53, 97, 318] |
| 192Ig-G saporin | Yes | No | Neuronal death | [325, 327] |
| Alcohol | Yes | Yes | Neuronal death | [13] |
| β-Amyloid | No | No | Cholinergic Dysfunction | [88, 211] |
As mentioned above, a loss of cholinergic neurones is a prominent feature of AD [18]. With this in mind, animal models, which employed specific lesioning of cholinergic neurones, were developed. Among these, the most relevant were the rodent models with lesions in the nucleus basalis magnocellularis, which is the equivalent of the nucleus of Meynert in humans [214, 322], in the diagonal band of Broca and the septum [289] (Table 11.1).
Specific cholinergic lesion models used toxins, which affected only cholinergic neurones in the brain regions relevant to AD, including septum, nucleus basalis magnocellularis and the diagonal band of Broca, but did not impair non-cholinergic neurones [289, 325]. For example, the AF64A cholinotoxin, which binds to the high-affinity choline uptake system, was injected. Alternatively, the immunotoxin 192 IgG-saporin that binds selectively and irreversibly to low-affinity nerve growth factor receptor interrupting cholinergic neuronal protein synthesis was employed. Both techniques lead to selective impairment and death of cholinergic neurones [289].
Similarly, the noradrenergic system can be lesioned in rats by the injection of the construct, consisting of antibody against dopamine-β-hydroxylase, the enzyme converting dopamine to noradrenaline, and saporin [215], a ribosome-inactivating plant lectin extracted from Saponaria officinalis (Caryophyllaceae) [17, 151]. This technology allows a selective and gradual lesioning of noradrenergic neurones in the brain stem nucleus locus coeruleus, the primary site of noradrenaline production in the CNS [75]. Upon injection into the LC, this immunotoxin binds dopamine-β-hydroxylase, which is not only localised mainly in the cytosol, but also at the plasma membrane surface of noradrenergic neurones [271, 321]. Due to its structure, saporin cannot enter the cell [56], but when coupled to a carrier molecule (for example, an antibody), is able to specifically bind a surface antigen protein (such as dopamine-β-hydroxylase, in this case), the toxin gains access to the cytosol and binds to the ribosomal 60S subunit, interfering with protein synthesis, and soon leading to cell death [326]. In initial anatomical investigations, the immunotoxin, infused into the lateral ventricles of either adult [331] or developing rats [57], produced specific and dose-dependent depletions of locus coeruleus neurones, with no effects on other cholinergic, dopaminergic or serotonergic neuronal populations [153]. The possibility to induce a partial or total noradrenergic loss (by varying the injected dose) makes this immunotoxic approach an ideal model to study events within the noradrenergic projection system, as they occur during age-related demise of locus coeruleus in humans [329].
11.2.3. Transgenic Animals
The AD models described above, although triggering neuronal death with consequent cognitive impairments, did not mimic histopathology and temporal progression of the disease. In the last two decades, an alternative and much more effective approach of using transgenic technologies have produced numerous models of familial forms of AD, which have been widely employed in experimental research. These transgenic animal models replicate several neuropathological features of AD (Table 11.2 and [91, 142, 232]) and they are based on mutated genes isolated from patients with various forms of familial AD. The very first transgenic animal carrying mutant APP and showing an AD-like pathology was developed in 1995 [83]. In this model, known as PDAPP, several pathological hallmarks of AD have been identified, including extracellular β-amyloid deposits, dystrophy of neurites, astrogliosis and memory impairments. Memory impairments, however, did not show any correlation with β-amyloid load [83]. The next transgenic AD mouse model, designated as Tg2576 mice, harboured APPswe (Swedish K670 N/M671L) mutation; this model developed numerous senile plaques in parallel with learning and memory impairments, which begun to develop from 9 months of age onwards [110]. The next generation of transgenic models carried double mutation of APP gene; such a model known as APP23 demonstrated some (~14%) neuronal death in the hippocampus [72, 272]. The next step was to combine mutated APP and PS genes; co-expression of PS1dE9 with APPSwe resulted in an AD mouse model characterised by accelerated β-amyloid deposition and memory deficits but without tangle formation [250]. These developments culminated in creation of 5xTG AD mice, designated as Tg6799; these animals carry a single human APP Swedish K670N/M671L double mutation as well as the Florida I716V mutation, and the London V717I mutation, along with PS with double M146L and L286V mutations. These mice develop amyloid depositions as early as 2 months of age [200].
Table 11.2.
Transgenic mouse and rat models of Alzheimer’s disease
| Transgenic mouse and rat models | Neuropathology | References |
|---|---|---|
| APP751SL | Plaques | [32] |
| APP/Ld/2 | Plaques | [183] |
| APPSwe | Plaques | [72] |
| APP Swedish, 695 K670N M671L | Plaques | [272] |
| PS1M146L | Diffused plaques | [32] |
| APP751SL/PS1M146L | Plaques | [32] |
| APPSWE/PS1dE9 | Plaques | [250] |
| APPSwedish and PS1m146L | Plaques | [115] |
| APP695SWE | Plaques | [110] |
| APPV717F | Plaques | [67] |
| K670N/M671L and V717F | Plaques | [115] |
| APP Swedish, 695 K670N-M671L and Indiana V717F | Plaques | [72] |
| APPSwedish and V717F | Plaques | [51] |
| V337 M | Tangles | [280] |
| 4R/2 N | Tangles | [281] |
| TauP301L (4R,2-,3-) | Tangles | [159] |
| P301L | Tangles | [89] |
| TauP301L | Tangles | [12] |
| P301S/G272V | Tangles | [252] |
| P301S | Tangles | [6] |
| G272V, P301L, R406W | Tangles | [72] |
| Endogenous tau KO | Tangles | [9] |
| P301L TET-off | Tangles | [224] |
| 7TauTg | Tangles | [113] |
| Tg2576 × JNPL3 (APPSWE) | Plaques and Tangles | [158] |
| Tg2576 and VLW | Plaques and Tangles | [228] |
| 3xTg-AD | Plaques and Tangles | [202] |
| Tg478 | None | [79] |
| Tg1116 | None | [79] |
| Tg478/Tg1116 | Plaques | [79] |
| Tg 478/1116/11587 | Plaques | [79] |
| K670M/N671L | Plaques | [132] |
In parallel to the animal models with increased β-amyloid production, the pathological tau models also have been created; the first being produced in 1995. In this model, the hyperphosphorylated tau was accumulated in neuronal somata and dendrites, although neurofibrillary tangles were never developed [90]. The next tauopa thy model, over-expressing TauP301L, did develop neurofibrillary tangles without β-amyloid pathology and neuronal loss [159].
In 2003, the triple transgenic AD mice (3xTg-AD) was created combining the mutants of the three major implicated genes; these animals harbour the mutant genes for APPSwe, for presenilin PS1M146V and for TauP301L [201, 202]. These animals demonstrated temporal- and region-specific Aβ and tau pathology, which resembles that seen in the human AD brain. Additionally, the 3xTg-AD animals also displayed plaques and tangles, and also showed reduced long-term potentiation in the hippocampus along with functional and cognitive impairments seen as deficient spatial and long-term memory [201, 202]. These pathological changes progress in an age-related manner; most importantly functional deficits precede the appearance of histological hallmarks. Cognitive deficits in the 3xTg-AD model correlate with the accumulation of intraneuronal Aβ [44, 177]. Moreover, at the cellular level, changes in astroglial subcellular vesicle traffic contribute to the pathophysiology of AD [268, 269].
11.3. Neurodegenerative Diseases and Neuroglia
Neurodegenerative diseases, which affect almost exclusively humans, are chronic disorders that result in a progressive loss of function, structure and number of neural cells, ultimately resulting in atrophy of the brain and profound cognitive deficit. The aetiology of neurodegeneration is complex and multifaceted. Neurodegeneration can have a genetic background or it can be instigated by acute trauma, by chemical poisoning, by metabolic insufficiencies or by infectious attacks, as well as by vascular abnormalities, or by sporadic accumulation of genetic/biochemical errors of unknown nature. At the early stages, neurodegeneration as a rule affects synaptic contacts in the brain tissue, thus causing early cognitive deficits. The early stages of neurodegeneration are of course of specific significance, because during this early phase the pathological process can be arrested or even reversed, thus offering the hope for preventing the cognitive decline.
Cellular and molecular mechanisms of underlying initiation and progression of neurodegenerative diseases are highly complex, which makes it almost impossible to identify a single leading cause. At the level of cellular biochemistry, neurodegeneration is frequently linked to aberrant handling of proteins, which promotes intra- or extracellular accumulation of abnormal proteins such as, for example, β-amyloid, tau or α-synuclein [116]. At a more systemic level, however, neurodegeneration reflects a generalised failure of brain homeostasis, which results in a functional and structural decline in the connectivity of neural networks, thus ultimately destroying information processing. Neurodegeneration starts from functional impairment of synaptic connectivity and synaptic plasticity which leads to a neurotransmission misbalance; these processes stipulate early cognitive deficiency. With further progression of the neurodegenerative process, the structural abnormalities develop, trigger disappearance of synapses and death of neural cells, ultimately resulting in a generalised atrophy of the brain accompanied with profound cognitive deficiency [133, 207, 254, 282].
The neuroglia provides for the birth, maintenance and demise of synapses, as well as for overall homeostasis of the nerve tissue, these functions being summarised in a concept of the astroglial cradle [303, 304]. Thus, these non-neuronal cells likely represent the main cellular element shaping the progression of neurodegenerative processes. The generally acknowledged and prevailing point of view considers neurones as main substrates of neurodegeneration, and it is generally assumed that failures in neuronal protein synthesis and/or direct neuronal damage caused by various factors constitute the leading mechanism of neurodegenerative pathologies. These neurone-centric doctrine has been challenged in the past decade, with considerable attention re-routed to neuroglia, which being primary cells responsible for the brain homeostasis and defences, fundamentally contribute to an overall homeostatic failure promoting neurodegeneration [40, 45, 106, 212, 235, 238, 242, 300, 307, 310, 311, 315].
11.4. Astroglial Atrophy and Astrogliosis in Neurodegenerative Diseases
Astrogliopathology in neurodegenerative diseases includes reactivity and astroglial atrophy, asthenia and loss of function. These processes develop in a stage-specific manner and contribute to pathological progression; frequently astroglial asthenia develops at early stages of the disease, whereas at the advanced stages an emergence of disease-specific lesions (for example, senile plaques) and death of neurones instigates astroglial reactivity [14, 306, 308, 317]. Pathological changes in astroglia evolve in parallel with microglial responses. Microglial reactions, at least in the context of human disease, are represented by either activation (that may contribute to neuroinflammatory progression) or microglial paralysis with loss of neuroprotective capabilities, which all contribute to brain atrophy. Cells of oligodendroglial lineage are also affected, which leads to a failure in myelination and atrophy of brain connectome.
11.4.1. Neurodegeneration Following Toxic Brain Injury
Astrocytes play the leading role in chronic neurodegeneration following the brain poisoning by toxic agents. The core mechanism underlying this astroglial-dependent neurotoxicity, which leads to a substantial neuronal death, is linked to a failure of astroglial glutamate uptake. Glutamate clearance from the extracellular space is mainly accomplished by astroglial Na+-dependent plasmalemmal glutamate transporters; astrocytes specifically express two types of these glutamate transporters, the excitatory amino acid transporters 1 and 2 (EAAT1 and 2). This glutamate uptake is fundamental for astroglia-mediated neuroprotection against glutamate excitotoxicity; suppressing of astroglial glutamate uptake greatly increases neuronal damage following exposure to glutamate [58]. Astroglial glutamate uptake is usually impaired in neurodegeneration and can be considered as one of the common mechanisms of this process [126].
Exposure to heavy metals triggers neuronal death underlying condition known as heavy metal toxic encephalopathy, which manifests itself by impaired cognition and psychotic symptoms. This neurotoxicity results from astroglial homeostatic failure; heavy metals are accumulated by astroglial cells, thus damaging pathways responsible for glutamate homeostasis and catabolism. In methylmercury-induced encephalopathy known as Minamata disease (the name derives from the Japanese city of Minamata where massive poisoning with methylmercury occurred in 1950s, see [174]). When in astrocytes, methylmercury inhibits glutamate, glutamine and cystine transporters which compromises glutamate homoeostasis [194, 339]. Resulting increase in extracellular glutamate concentration triggers neuronal death underlying clinical symptoms that include cognitive decline, impaired vision and hearing, as well as motor symptoms. Similarly, astrocytes, endowed with capacity manganese transport system, emerge as a main target for manganese toxicity. Again, increased manganese in astroglial cells suppresses astroglial glutamate uptake with subsequent excitotoxic neuronal damage [260]. Similarly, astrocytes appear as a primary target for other heavy metals, such as arsenic, lead and cadmium, which all reduce expression of glial fibrillary acidic protein (GFAP) and trigger astroglial apoptosis, thus reducing astroglial homeostatic presence [223]. In aluminium toxic encephalopathy (symptoms of which include cognitive deficits and speech alterations), astrocytes are again the main targets. Aluminium accumulated by astrocytes impairs plasmalemmal glutamate transporters as well as gap junctions and causes astrocytic death [275]. Likewise, astroglial loss through apoptotic death plays a leading role in the enchephalotoxic damage caused by cypermethrin, a synthetic II pyrethroid insecticide [173].
11.4.2. Wernicke Encephalopathy
Wernicke encephalopathy is a pathoanatomical substrate for Korsakoff syndrome, symptoms of which include ante- and retrograde amnesia, apathy and confabulation [135, 323]. This type of encephalopathy is essentially rapidly progressing malignant thalamo-cortical neurodegeneration. The pathological mechanism of Korsakoff–W-ernicke syndrome is primarily associated with acute failure in astroglial glutamate uptake resulting from ~60 to 70% decrease in expression of EAAT1 and EAAT2 glutamate transporters. This remarkable decrease in plasmalemmal glutamate transporters expression has been identified in post-mortem human samples, as well as in the rat thiamine deficiency model of the disease [104, 105]. In addition to decrease in EAAT1/2 expression, astrocytes demonstrated signs of atrophy including decrease in GFAP morphological profiles, as well as decrease in expression of glutamine synthetase (GS) and GAT-3 GABA transporter.
11.4.3. The Human Immunodeficiency Virus-1 (HIV-1)-Associated Dementia (HAD)
In the nervous system, the HIV-1 virus primarily infects and propagates in microglial cells. Microglia contributes to neuronal death by releasing various neurotoxic factors [123, 171], including Nef protein [267]. In HAD, astroglial cells develop both astrodegeneration and reactive astrogliosis. In the basal ganglia, astrocytes undergo a serious loss with the degree of astroglial death correlated with the degree of cognitive impairments [283]. In the entorhinal cortex and in the hippocampus, astrocytes show prominent reactivity [295].
11.4.4. Non-AD Dementia
The non-AD dementia is represented by many disorders including fronto-temporal lobar degeneration, Pick’s disease, Cockayne syndrome, juvenile neuronal ceroid lipofuscinosis (JNCL) or Niemann-Pick type C disease. Astroglial contribution to these disorders is complex with signs for astroglial atrophy and astroglial apoptotic death [42, 320] as well as for astroglial reactivity, which is particularly prominent in the frontal and temporal cortices of patients with fronto-temporal dementia [124]. In thalamic dementia, a profound astrogliosis likely represents a key pathophysiological factor [221]. There is evidence indicating uncoupling of astroglial syncytium and aberrant activity of astroglial connexin hemichannels in JNCL [43], whereas early astroglial reactivation was reported in animal models of Niemann-Pick type C disease [222].
11.4.5. Amyotrophic Lateral Sclerosis (ALS)
Astrocytes play fundamental role in the pathogenesis of hereditary familial ALS associated with the mutation of the human superoxide dismutase 1 (hSOD1) gene. In hSOD1/G93A, associated animal models of ALS astrocytes undergo atrophy, pathological remodelling loss of function and cell death. These astroglial changes precede neuronal abnormalities and the emergence of clinical symptoms [241, 242]. The key pathogenetic factor linked to the neurotoxicity is represented by deficient astroglial glutamate uptake. Selective silencing of hSOD1 gene in astrocytes in animal model delays ALS progression [337].
11.4.6. Parkinson’s Disease
The role of neuroglia in emergence and progression of Parkinson’s disease remains to be fully elucidated. There are some indications for microglial activation in relevant brain regions; this activation being possibly linked to neurotoxicity [63]. This microglial response, however, can be secondary, being triggered by neuronal death [108]. In 6-hydroxydopamine (6-OHDA) animal model of Parkinson’s disease, inhibition of microglial activation was found to be neuroprotective [152]. Astrocytes have been considered to provide neuroprotection to dopaminergic neurones, based on in vitro experiments [180, 181]. In primary neuronal–glial co-cultures, astrocytes were shown to convert L-DOPA, the immediate precursor of dopamine, from neurotoxic to neurotrophic substance, and hence astroglia can be crucial for L-DOPA substitute therapy [179].
11.5. Astrocytes in Alzheimer’s Disease
Alzheimer’s disease is characterised by progressive neurodegeneration and an occurrence of specific histopathological markers represented by (i) focal extracellular deposits of fibrillar β-amyloid (also called neuritic or senile plaques) in the brain parenchyma and in the walls of blood vessels, and by (ii) intraneuronal accumulation of neurofibrillary tangles composed from abnormal hyperphosphorylated tau filaments. The initial neurodegenerative events in AD appear in the transentorhinal cortex, which subsequently spread to the entorhinal cortex and hippocampus. At later stages of the disease, the neurodegenerative process disseminates through the temporal, frontal, and parietal lobes [284, 285]. At these late stages, the grey matter undergoes severe damage with a profound loss of neurones and synaptic contacts and generalised atrophy of the brain parenchyma; this atrophy includes both white and grey matters. Contribution of neuroglia to the histopathology of Alzheimer’s disease has been initially suggested by Alois Alzheimer himself; Fig. 11.1 shows original drawings of Alzheimer depicting pathologically modified glial cells of a senile plaque [8]. The role of astrocytes in the pathogenesis and progression of AD remains to be fully characterised, primarily because of the lack of longitudinal studies assessing the status of astroglia at different stages of the disease. From analyses of human post-mortem tissues, there has been generally agreed that at the late stages of the disease there are prominent reactive astrogliosis and inclusion of astrocytes into senile plaques [106, 193, 235].
Fig. 11.1.
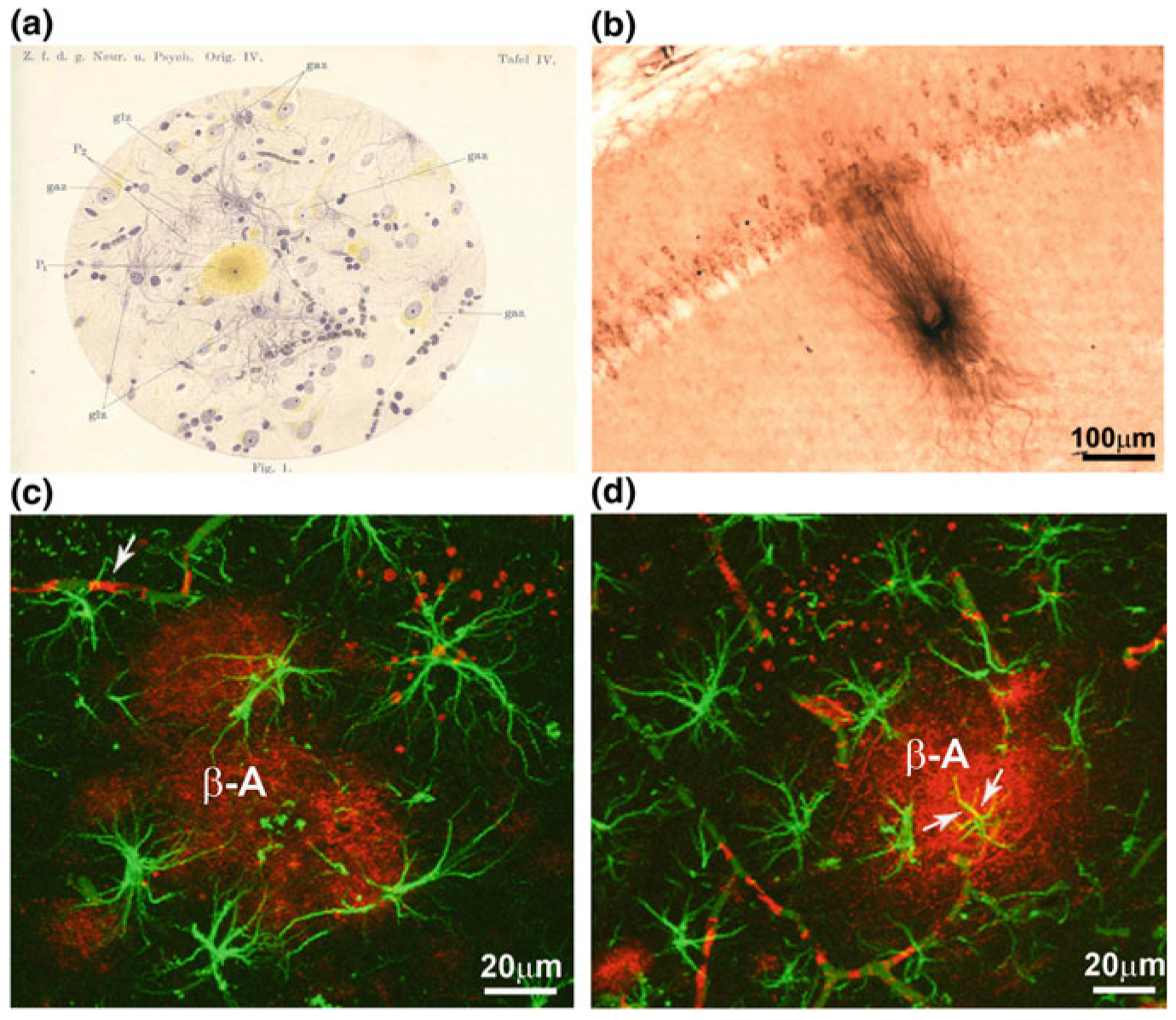
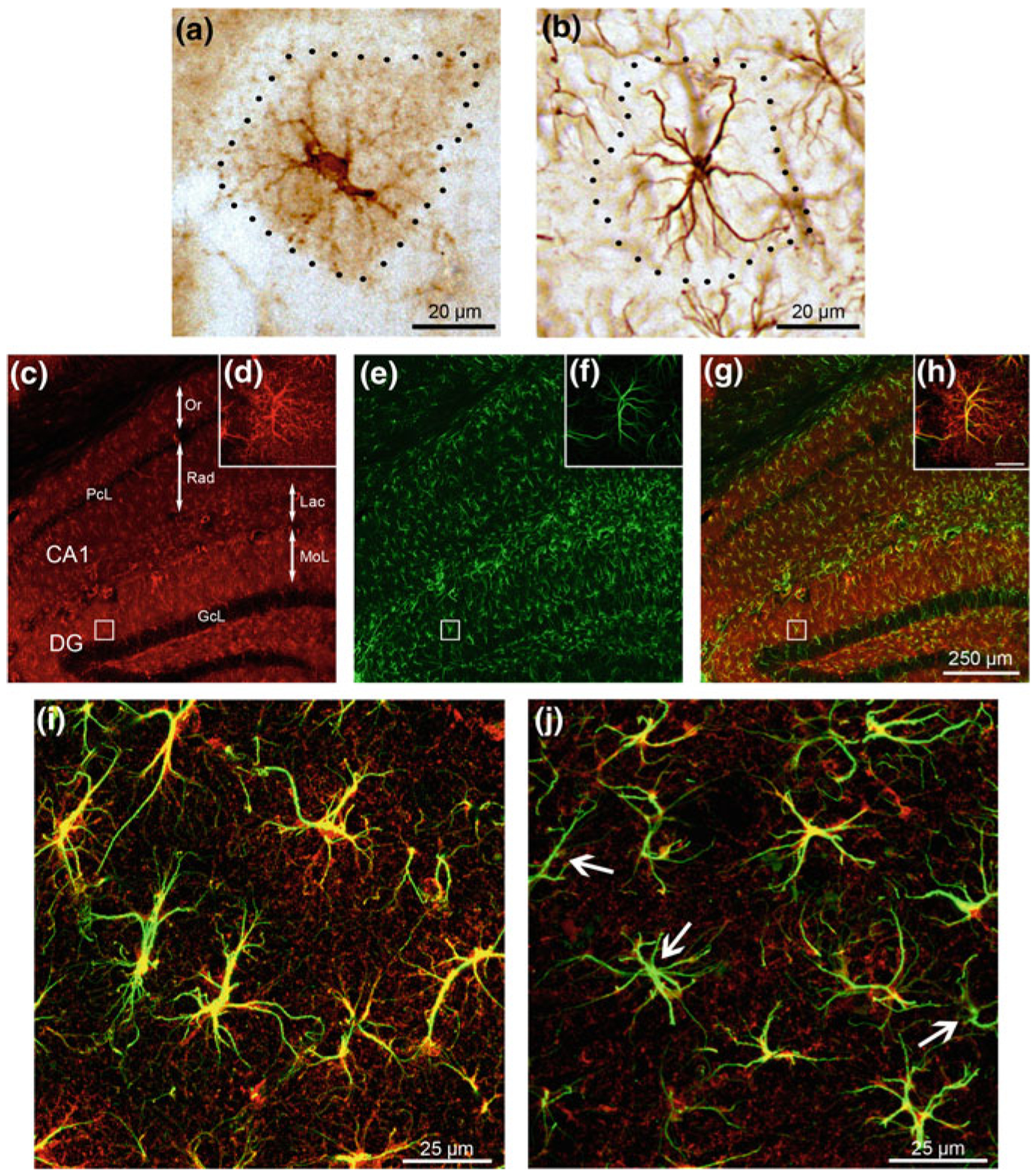
Glial cells in AD a Alois Alzheimeŕs drawing illustrating the glial reaction (astro- and/or micro-gliosis and hypertrophy) in a pathological brain containing senile plaques. Abbreviations: gaz, ganglionic cell—i.e. neurone; glz, glial cell, P, central part of the plaque; P2, peripheral part of the plaque. From [8]. b Photomicrograph showing the presence of β-amyloid within the pyramidal neurones of the hippocampal CA1 area as well as the presence of a plaque in 12 months 3xTg-AD mice. c, d Confocal images showing GFAP-positive (green) reactive astrocytes surrounding β-amyloid plaques (β-A red; c). d Reactive astrocytes (green) and an astrocyte showing cytoplasmic β-amyloid accumulation (indicated by arrows; co-localisation is in yellow) near a neuritic plaque (red). Modified and adapted with permission from [235]
11.5.1. Astrocytes and β-Amyloid
The prevailing views on AD pathogenesis associate disease evolution with progressive accumulation of β-amyloid protein in the brain parenchyma and formation of senile plaques [87, 100, 122, 134]. Recently, however, the β-amyloid hypothesis became the subject of extensive criticism [49, 98, 99, 226]. Production of β-amyloid is mostly associated with neurones although there are several reports indicating the role of astrocytes in this process through either direct β-amyloid production or through deficient clearance.
Astroglial contribution to the clearance and degradation of β-amyloid has been suggested some 15 years ago [96, 195]. Reactive astrocytes associated with senile plaques in the transgenic AD mouse model expressing mutant APP were found to express a zinc-dependent metalloendopeptidase neprilysin, an enzyme capable of degrading β-amyloid [11]. In experiments in vitro, cultured astroglial cells obtained from healthy mice were shown to accumulate exogenous β-amyloid. In contrast, this ability was absent in astrocytes isolated from the brains of APP AD model [333]. Similarly, astroglial β-amyloid accumulation was detected in cells from the entorhinal cortex of AD patients [192]. Conversely, β-amyloid was almost never detected in astrocytes from 3xTg-AD mice (Fig. 11.1d, [203]).
Astroglial contribution to β-amyloid production is not fully characterised. Neurones, which express β-amyloid producing enzymes β- and γ-secretases, were for a long time considered to be the main source for β-amyloid [143]. Indeed, healthy astrocytes seem not to express β-secretase; nonetheless, its expression can be induced by conditions of chronic stress or neuroinflammatory environment, thus adding astroglia to amyloidogenesis [33, 82, 117, 157, 205, 243, 342]. Expression of β-secretase was detected in reactive astrocytes emerging following immune lesion of cholinergic septohippocampal afferents or an occlusion of the middle cerebral artery [243]. Similarly, expression of β-secretase was found in reactive astrocytes in AD mice models expressing mutant human APP; these models, for example, included Tg2576, K670N-M671L APP or APP V717I mutations [101, 107, 243]. Of note, an increase in production of APP was characterised in a rat model of chronic neocortical astrogliosis, induced by grafting a foetal cortical tissue in the midbrain of neonatal animals; these chronically activated astrocytes were immunopositive for APP, as well as for another AD-related marker apolipoprotein E4 [166].
11.5.2. Astrogliosis in AD
Astroglial reactivity, generally characterised by an increase in expressions of GFAP, vimentin or s100β protein, has been detected in post-mortem tissues from AD patients [19, 92, 178, 189]. No obvious correlation between GFAP levels, degree of astrogliosis and β-amyloid load was detected [261]. Similarly, no differences in GFAP expression were found between the brains obtained from cognitively sound and demented patients [324]. Reactive, hypertrophic astrocytes, associated with senile plaques and perivascular β-amyloid deposits, are also observed in the brains of AD mice models (Fig. 11.1, [204, 235, 306]). It is important to highlight that astrogliosis in AD is never associated with the scar formation and it does not hamper the physiological non-overlap of astroglial territorial domains. It can be classified therefore as isomorphic or mild astrogliotic response. In the context of AD, the astrogliotic response can be triggered by various molecules, such as β-amyloid, molecules released from damaged cells or certain cytokines and chemokines. Soluble β-amyloid was found to initiate reactive astrogliosis in astrocytes in vitro [64]. This may be associated with certain intracellular signalling events, including, for example, Ca2+ signals. Such signals are indeed generated by exposure of cultured astrocytes to β-amyloid [2, 3, 94]. Treatment of cultured astrocytes with β-amyloid also resulted in inhibition of glutamate uptake, which can contribute to pathological progression [168].
11.5.3. Astroglial Atrophy in AD
Pathological changes of astrocytes in the AD pathology are not limited to astrogliotic response; it seems that astrogliosis occurs at later stages of the disease, with reactive astrocytes being mainly associated with senile plaques. Recent studies of transgenic AD mice models revealed a profound astrodegeneration that occurs at the early stages of AD progression [20, 203, 306].
Total number of astrocytes (labelled with antibodies against GFAP, s100β or GS) did not show any age-dependent variations in 3xTg-AD mice of 3–24 months of age [203, 204]. There are complex region- and disease stage-specific morphological changes in astrocytes in 3xTg-AD mice (Figs. 11.2, 11.3 and 11.4). At the early (i.e. pre-plaque) stages of the AD, astrocytes in the entorhinal cortex, prefrontal cortex and hippocampus demonstrate signs of morphological atrophy [139, 203, 204, 338]. Astroglial atrophy develops first in the entorhinal cortex (from 1 month of age, Fig. 11.2); next, it occurs in the prefrontal cortex (3–4 months of age, Fig. 11.3) and finally in the hippocampus (6–9 months of age, Fig. 11.4). Atrophy of GFAP-positive profiles preceded β-amyloid deposition and formation of senile plaques. The reduction in GFAP profiles coincided with the reduced morphological presence of astroglial cells labelled with GS antibodies in the hippocampus and in the prefrontal cortex, but not in the entorhinal cortex. Morphological atrophy of astrocytes was manifested by reduced expression of GFAP-rich cytoskeleton (surface and volume coverage) and decreased somata volume, as well as number and branching of cell processes. Very similar atrophic changes were observed in hippocampal astrocytes from another AD animal model, the mutant APP (PDAPP-J20) mice carrying the Swedish and Indiana APP human mutations [20, 21]. Astroglial atrophy was subsequently confirmed in human material, in astrocytes derived from pluripotent stem cells isolated from patients with family and sporadic AD (Fig. 11.5, [119, 184]).
Fig. 11.2.
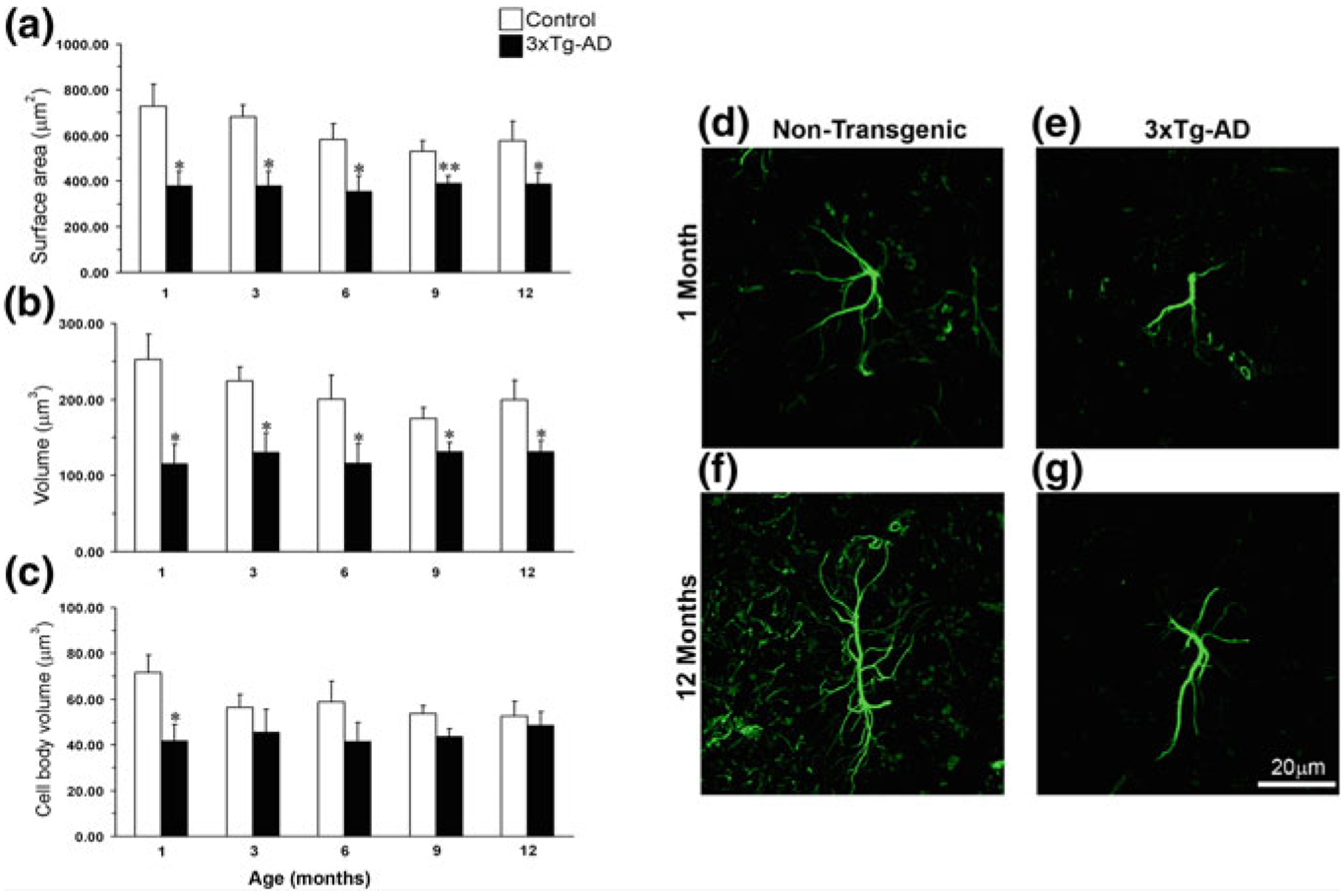
Astroglial atrophy in the entorhinal cortex (EC) of 3xTg-AD mice. Comparison of astrocytic GFAP surface area and volume in the EC of non-Tg and 3xTg-AD animals of different ages. The histograms show a comparison of a surface area, b total cell volume and c somata volume in the EC at the ages of 1, 3, 6, 9 and 12 months between 3xTg-AD and non-Tg animals. Results are means ± S.E.M. (*p < 0.05 compared with the age-matched non-Tg control). Confocal micrographs show astrocytic atrophy in 3xTg-AD at 1 month (e) and 12 months (g) compared with the control animals (d, f). Reproduced with permission from [338]
Fig. 11.3.
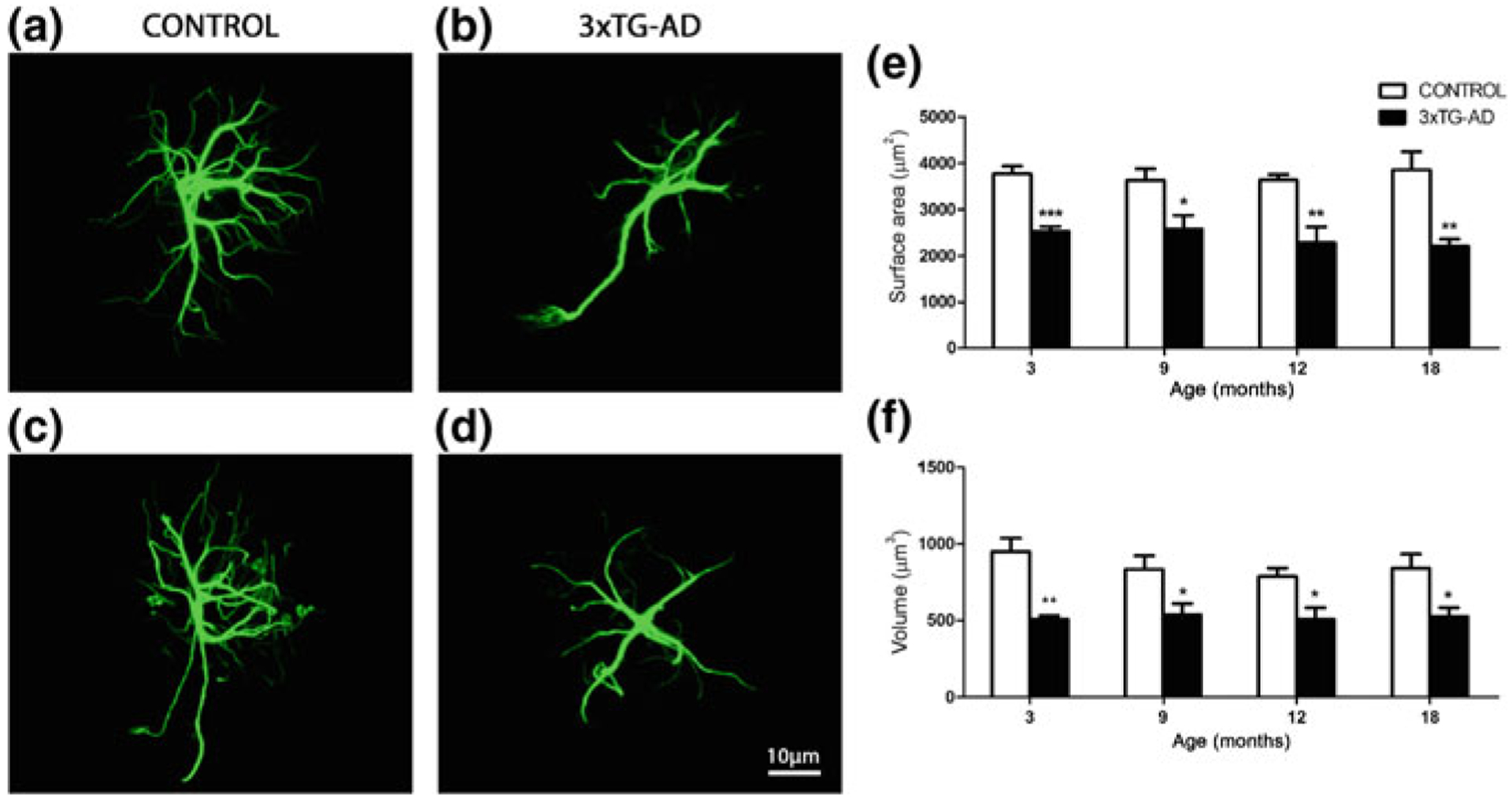
Astroglial atrophy in the prefrontal cortex of 3xTg-AD mice. Confocal images showing morphology of GFAP-positive astrocytes in control non-Tg animals and astrocytic atrophy in the 3xTg-AD animals at 3 months (a and b, respectively) and 18 months (c and d, respectively) in the prefrontal cortex. Bar graphs showing the decreases in the surface area and volume (e, f) in 3xTg-AD mice when compared with control animals. Bars represent mean ±SEM. Reproduced with permission from [139]
Fig. 11.4.
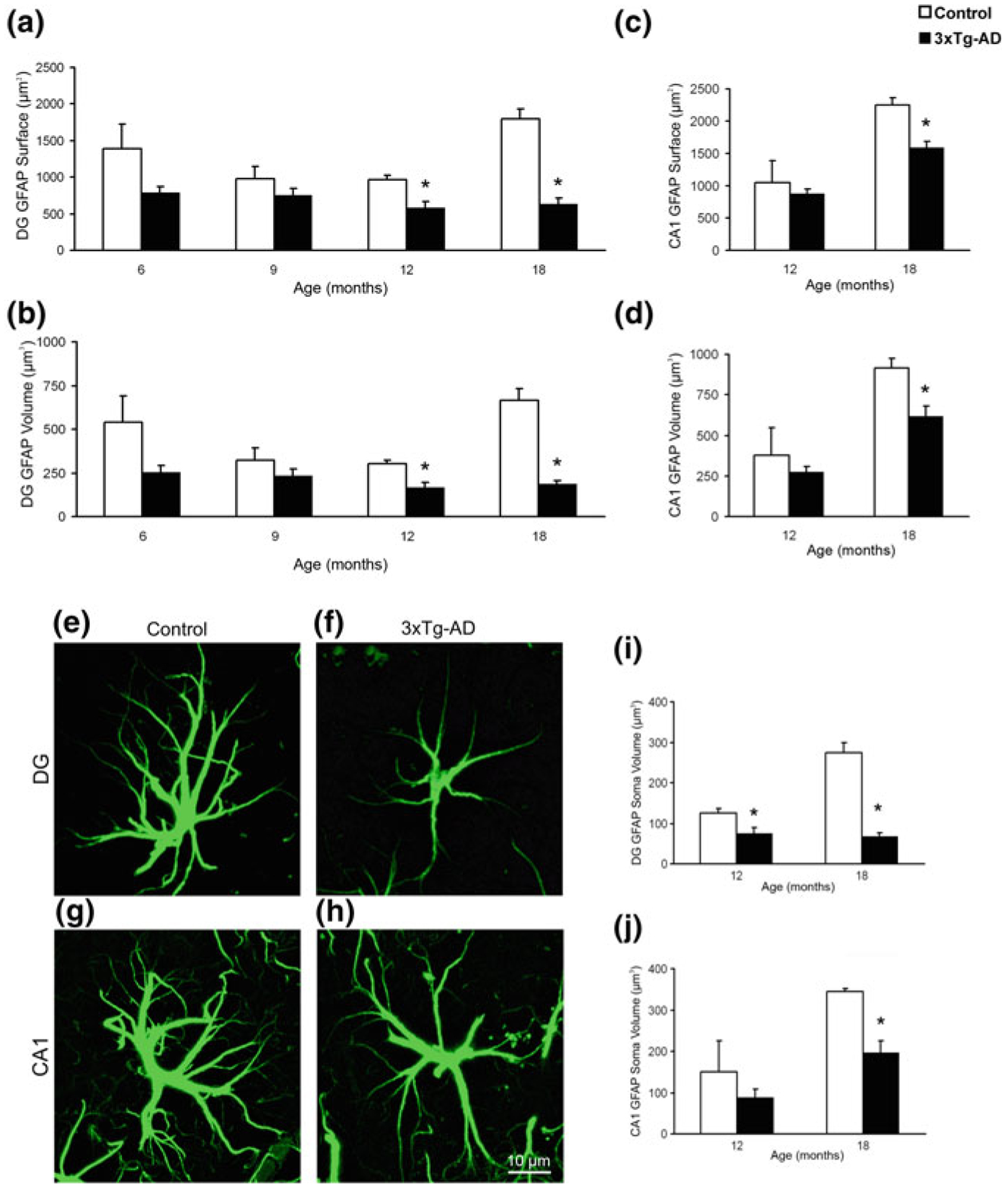
Astroglial atrophy in the hippocampal areas of 3xTg-AD mice. Bar graphs showing the significant decrease in surface area, volume, and soma volume of GFAP-positive astrocytes in the dentate gyrus (DG) (a, b, i) and the CA1 region (c, d, j) of the hippocampus of the 3xTg-AD mice when compared with control animals. Bars represent mean ± SEM (p < 0.05). (g–j). Confocal micrographs illustrating the astrocytic atrophy in 3xTg-AD mice in the DG (f) and CA1 (h) compared to control animals (e and g). Reproduced with permission from [203]
Fig. 11.5.
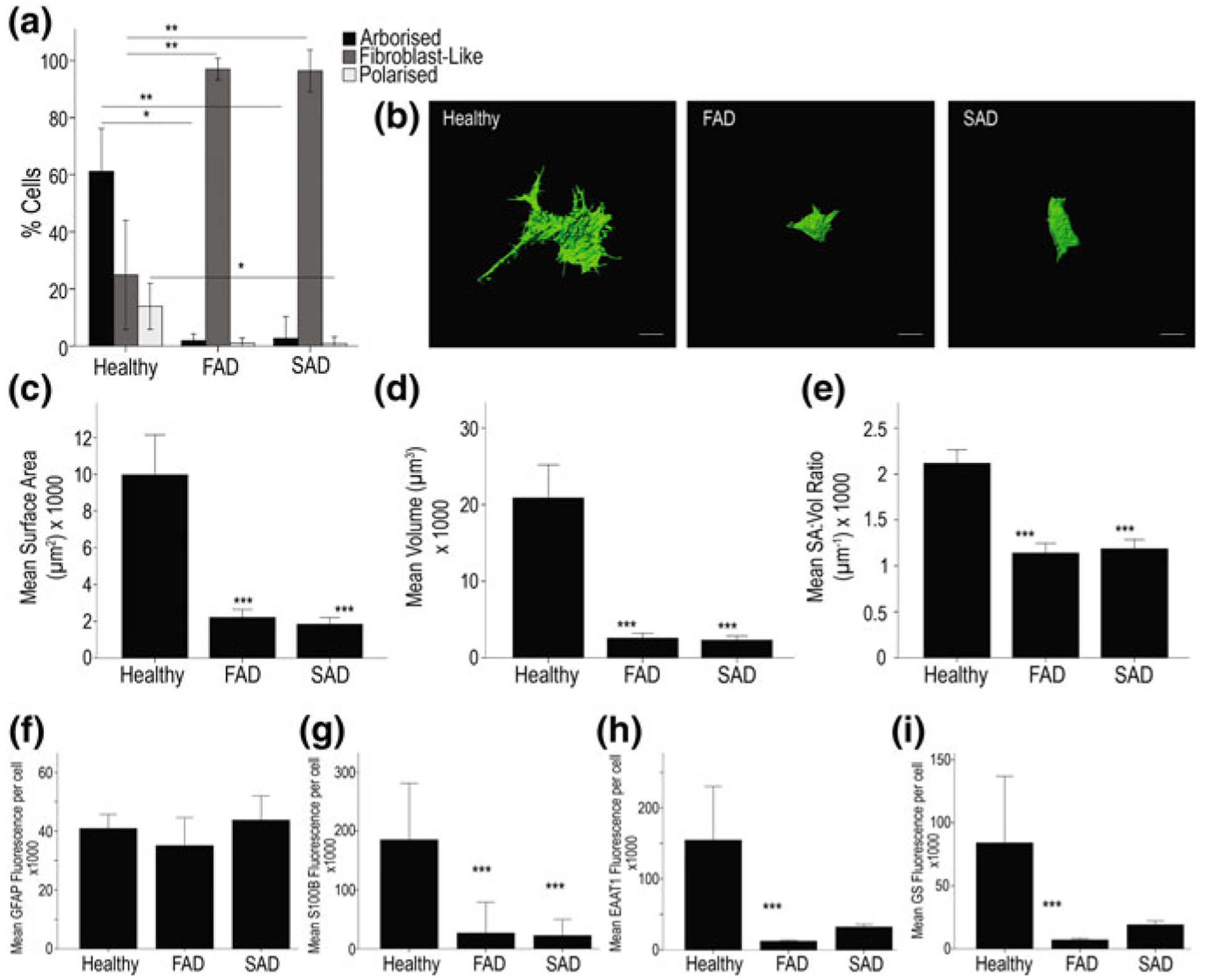
Astrocytes derived from PSEN1 M146L FAD and ApoE4+/+ SAD patients exhibit significant atrophy when compared to those from healthy patients. a Exemplar 3D isosurface renders constructed from serial confocal z-stacks display clear differences in cell size and overall morphology (b). Scale bar = 10 μm. Quantification of cells using these renders by way of surface area (c), cell volume (d) and SA:Vol ratio (e) reveal significant differences in all aspects of cellular morphology between healthy and diseased astrocytes. Quantification of mean fluorescence intensity per immunoreactive cell reveals no significant difference in GFAP staining intensities between AD and control astrocytes (f) but S100B, EAAT1 and GS intensities are reduced in both FAD and SAD cells (g, h and i, respectively). Asterisks on graph; ***p < 0.001, **p < 0.005, *p < 0.05. Reproduced from [119]
At the later stages of AD pathology in hippocampi of 3xTG-AD animals (12–18 months; at this time neurofibrillary tangles also start to develop in neurones), formation of plaques and accumulation of extracellular β-amyloid initiates reactive astrogliosis. Numerous hypertrophic astrocytes accumulate exclusively around senile plaques and β-amyloid inundated blood vessels (Figs. 11.6 and 11.7; [203, 230, 316]). This astroglial hypertrophy is characterised by an increased volume and surface of both astrocyte somata and processes, which can increase their size up to 70% (Fig. 11.3). At the same time, astrocytes positioned away from the senile plaques retain their atrophic morphology (Fig. 11.6). In contrast to the hippocampus, accu mulation of β-amyloid and formation of senile plaques do not induce reactivity of astrocytes neither in the entorhinal nor in the prefrontal cortex (Fig. 11.7, [139, 338]). Deficient reactivity of astrocytes may determine the specific vulnerability of different brain regions to AD-type pathology. Atrophy of astrocytes at the early stages of AD may have important functional consequences. The decrease in astroglial complexity may affect synaptic coverage and homoeostatic support as well as functional performance of the neuronal–glial–vascular unit. This in turn can affect connectivity in neural network, reduce synaptic strength and disturb synaptic plasticity thus contributing to cognitive deficits.
Fig. 11.6.
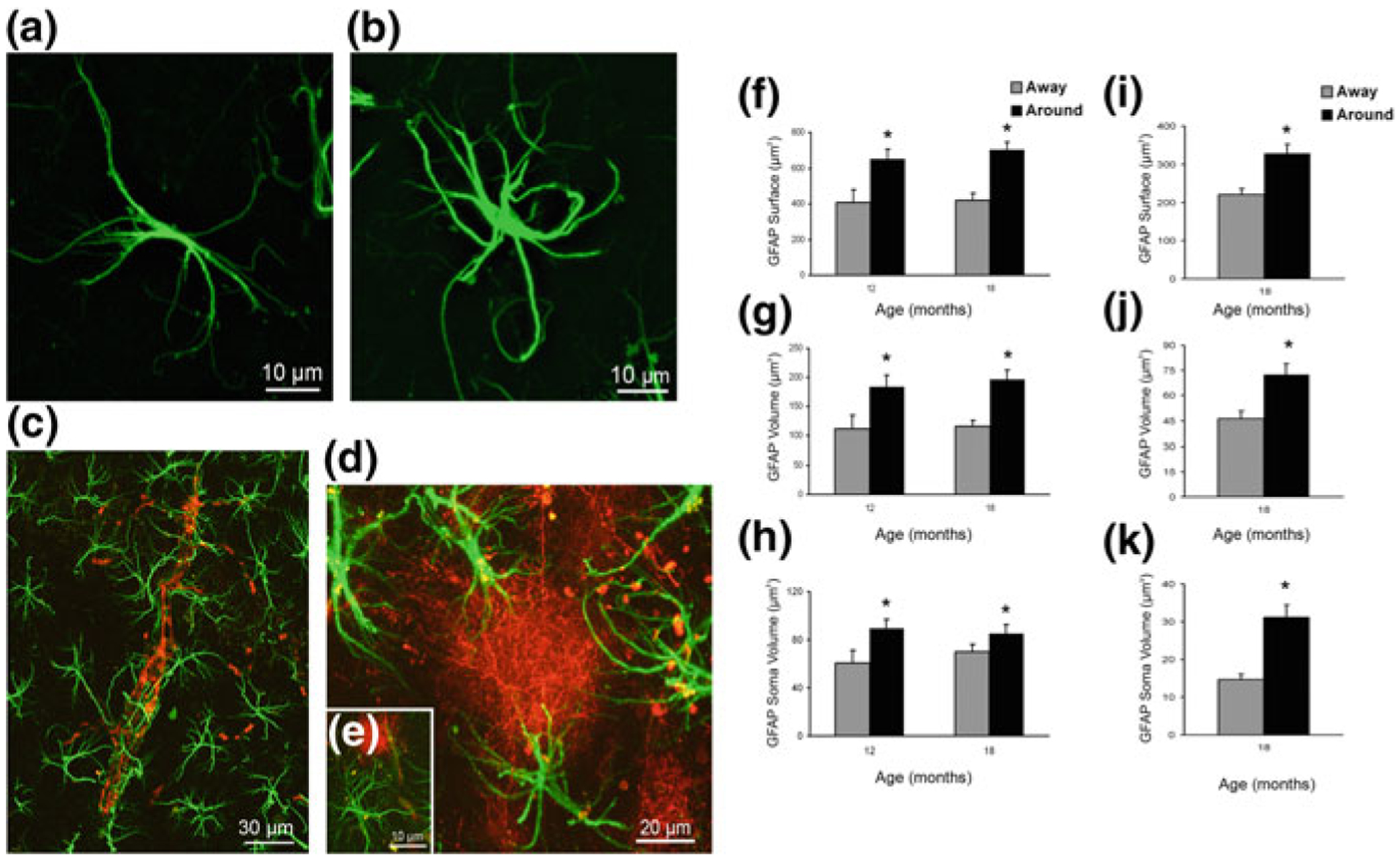
Concomitant astroglial atrophy and astrogliosis at the advanced stages of AD-like pathology in 3xTg-Ad mice. a, b Confocal images of hippocampal preparations dually labelled by GFAP and by anti-β amyloid monoclonal antibody illustrating differential changes in GFAP profiles in astrocytes distant to the plaques (a) and associated with the β-amyloid plaques (b). c–e Confocal dual labelling images (GFAP in green and β-amyloid in red) in 3xTg-AD mice showing the accumulation of astrocytes around the β-amyloid plaques and vascular β-amyloid deposits. Astrocytes surrounding β-amyloid plaques (d, e) and β-amyloid deposits around a blood vessel (c), undergo astrogliosis. f–k Bar graphs showing GFAP-positive astrocytic surface area (f), volume (g) and somata volume (h) differences between astrocytes located around the β-amyloid plaques (Aβ) and those distant to the plaques in the CA1 of 3xTg-AD animals. i–k Similar astrocytic surface area (i), volume (j) and somata volume (k) differences are observed in the DG at 18 months of age. Bars represent mean 6 SEM (p < 0.05). Reproduced with permission from [203]
Fig. 11.7.
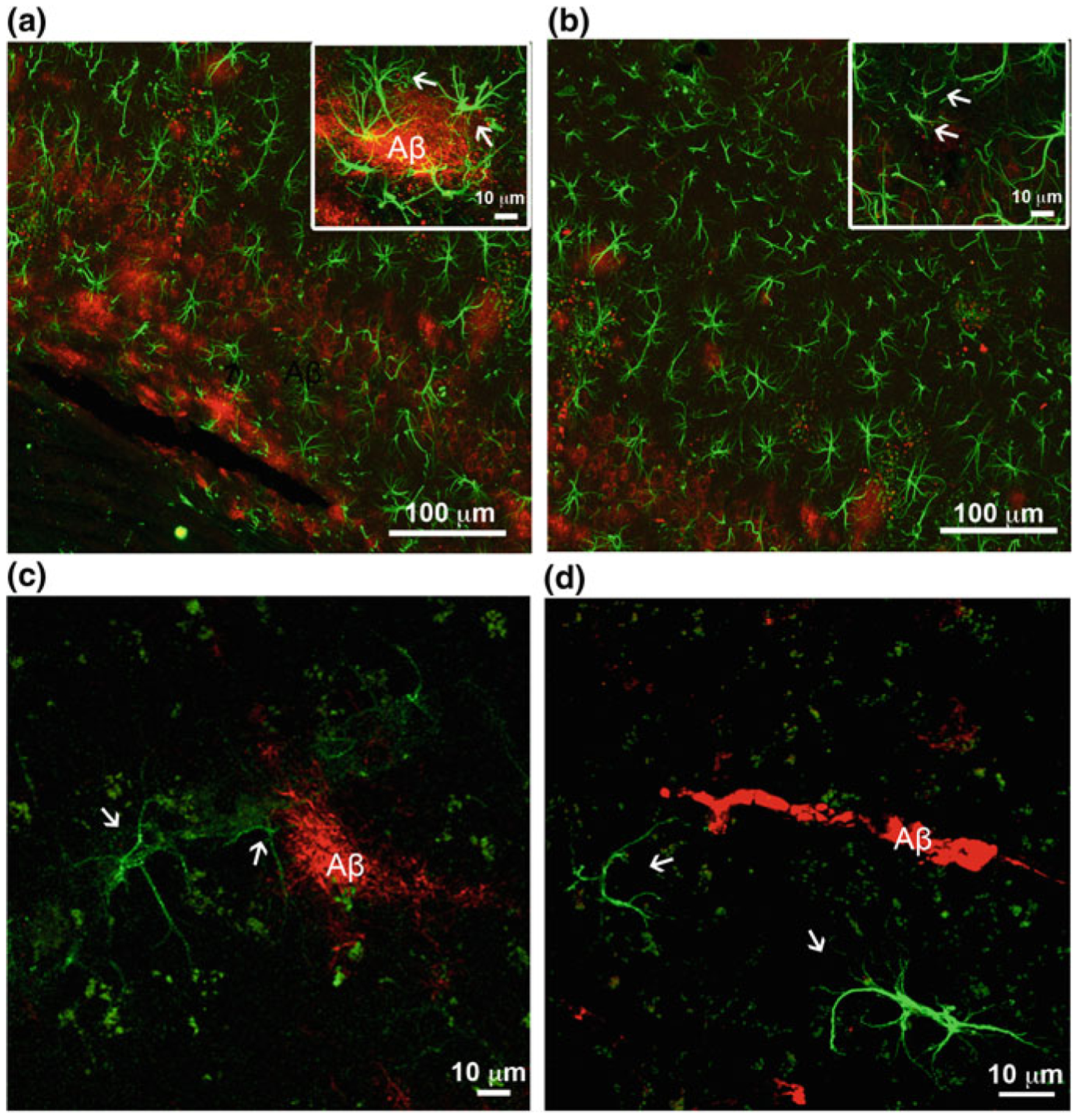
β-Amyloid depositions trigger gliotic response in associated astrocytes in the hippocampus but not in the entorhinal cortex. a, b Confocal images of hippocampal preparations labelled by GFAP (green) and β-amyloid (red) illustrating differential changes in GFAP profiles in astrocytes in close association with Aβ plaques (a) and atrophic profiles of astrocytes (arrows) distant from β-amyloid deposits (b) in 3xTg-AD mice. c, d Confocal dual labelling images (GFAP in green and β-amyloid in red) showing the absence of reactive response of astrocytes in the entorhinal cortex of 3xTg-AD mice around perivascular vascular β-amyloid deposits (c) and β-amyloid plaques (d). Modified and reproduced with permission from [300, 338]
11.5.4. Loss of Astroglial Homeostatic Support Contributes to Early Cognitive Impairments
Atrophic changes in astrocytes, characterised in several AD animal models as well as in stem cells derived astrocytes appear as general diminution of astroglial territories, of astroglial coverage of neuronal membranes and overall decrease in astroglial homeostatic support. Arguably, this atrophy and loss of function of astrocytes, which occur early in the disease progression, may contribute to the disease pathophysiology. Atrophic astrocytes provide less synaptic coverage with deleterious consequences for synaptic transmission associated with compromised ion and neurotransmitter homeostasis or reduced local metabolic support; astroglial asthenia also results in decreased neuroprotection [232, 238, 316, 317]. All these changes are likely to weaken synaptic transmission and affect synaptic plasticity, thereby being responsible for initial cognitive deficiency observed at the early stages of AD.
Early cognitive deficits are the very first symptoms of AD, which start to emerge decades before the occurrence of specific histopathology [55, 282]. Loss or impairment of cognitive capacities reflects reduced synaptic connectivity due to decreased synaptic function and synaptic loss [347]. Decrease in number of synapses indeed was found to be the earliest morphological change in AD [282]; and moreover the degree of synaptic loss correlates with the severity of dementia [61, 247]. Atrophy of astroglial perisynaptic processes may indeed underlie synaptic loss at the early stages of AD. Furthermore, astrocytes are fundamental for synaptogenesis and synaptic maintenance; furthermore, astroglial plasmalemmal transporters control local concentrations of ions and neurotransmitters, most notably glutamate, that may contribute to local excitotoxicity [73, 292, 303]. Astroglial asthenia also impairs metabolic support accomplished by lactate shuttle [213]. Astrocytes are also critical for maintaining normal neurotransmission by supplying neurones with glutamine that is an indispensable precursor for both glutamate and GABA. Impairment of all these fundamental functions associated with astroglial atrophy and loss of function may be considered as a primary cause for distorted synaptic connectivity and early cognitive deficits in AD [300, 306, 316].
11.5.5. Neurovascular Unit in AD
Clinical evolution of AD is almost invariably associated with vascular deficiency and pathologies of the blood–brain barrier [276, 277]. It is well documented that the blood flow is significantly reduced in the brains of patients with AD, with these vascular deficits being prominent already at the early stages of the disease [24, 345]. These functional deficits stem from substantial remodelling of vascularisation in the brains altered by AD pathology [74]. Brain vessels are controlled by both neuronal and astroglial inputs [112, 346]. Astrocytes are central integrating elements of neurovascular units that bridge brain parenchyma with local circulation. By secreting various agents astrocytes target pericytes, vascular smooth muscle cells and endothelial cells, thus contributing to functional hyperaemia and regulating the blood–brain barrier [191, 279, 346]. Astroglial atrophy as well as reactivity may differentially remodel the neurovascular unit, even that can occur at both early and late stages of the disease and can contribute to cognitive abnormalities and neuronal damage.
11.5.6. AD and Astroglial Metabolic Support
Metabolic deficiency of the brain is a common feature of AD. Functional brain imaging demonstrated a progressive loss of utilisation of glucose in patients with different stages of AD; deficits in brain metabolism are present already at the very early stages of the disease, having thus diagnostic significance [187, 229]. Exposure of cultured astrocytes to β-amyloid impairs cellular metabolism, although both decrease [209, 264] and increase [5] of glucose utilisation were detected. Likewise, both decrease [34, 160] and increase [31, 264] of the activity of glucose metabolism enzymes were described in post-mortem AD brains.
11.5.7. Deficient Astroglial Reactivity Defines Susceptibility of Brain Tissue to AD Pathology
Astroglial atrophy and asthenia in AD also lead to a loss of their defensive function [300]. As has been alluded previously, in experiments on 3xTg-AD mice, reactive astrocytes were accumulated around senile plaques and form perivascular β-amyloid deposits [203, 204]. These hypertrophic astrocytes are specifically associated with extracellular β-amyloid deposits, whereas astrocytes distant to the plaques remain atrophic (so in this sense astroglial atrophy emerges at the early stages of AD and is complimented by astrogliosis at later stages, when specific lesions develop). In contrast, in entorhinal and prefrontal cortices, extracellular β-amyloid accumulation does not trigger astrogliotic response (Fig. 11.7, [139, 338]) indicating failure of astroglial neuroprotection.
There are several lines of evidence demonstrating that reactive astrocytes are neuroprotective in the context of AD. For example, the Tg2576 mice (that harbour the APPSwe mutation—see [110]) demonstrate early and prominent astroglial reactivity which correlates with relatively slow development of AD. Furthermore, senile plaques in these animals are resembling human β-amyloid deposit being represented by fleecy, granular, cored and diffused amyloid plaques [336], Incidentally, the Tg2576 mice display certain similarities with the prodromal stage of AD known in humans as mild cognitive impairment [16]. Astroglial capabilities to mount astrogliotic response change with age. The density of reactive astrocytes changes with age. In old Tg2576 mice, GFAP staining demonstrated prevalence of atrophic astrocytes with fewer reactive astroglial cells, which may be related with increased AD pathology in ageing [300]. Inhibition of reactive astrogliosis in the AD mouse model significantly increased β-amyloid load and exacerbated pathological progression [137].
The in vivo brain imaging of astrocytes uses PET detection of 11C-deuterium-L-deprenyl (11C-DED) that binds to MAO-B in the astrocytes [81]. When using a multi-tracer PET detecting 11C-PIB (marker of fibrillar β-amyloid), 18F-FDG (marker cerebral glucose metabolism) and 11C-DED (marker of astrogliosis), the highest binding of 11C-DED (which reflects maximal reactivity of astrocytes) was observed in patients with mild cognitive impairment (MCI) and high levels of fibrillar amyloid plaques in the brain (PIB+) reflecting prodromal AD [46]. Decrease in astroglial reactivity parallels the switch from MCI to full-blown AD with senile dementia again demonstrating the neuroprotective role of astrogliotic remodelling (Fig. 11.8 and [300]).
Fig. 11.8.
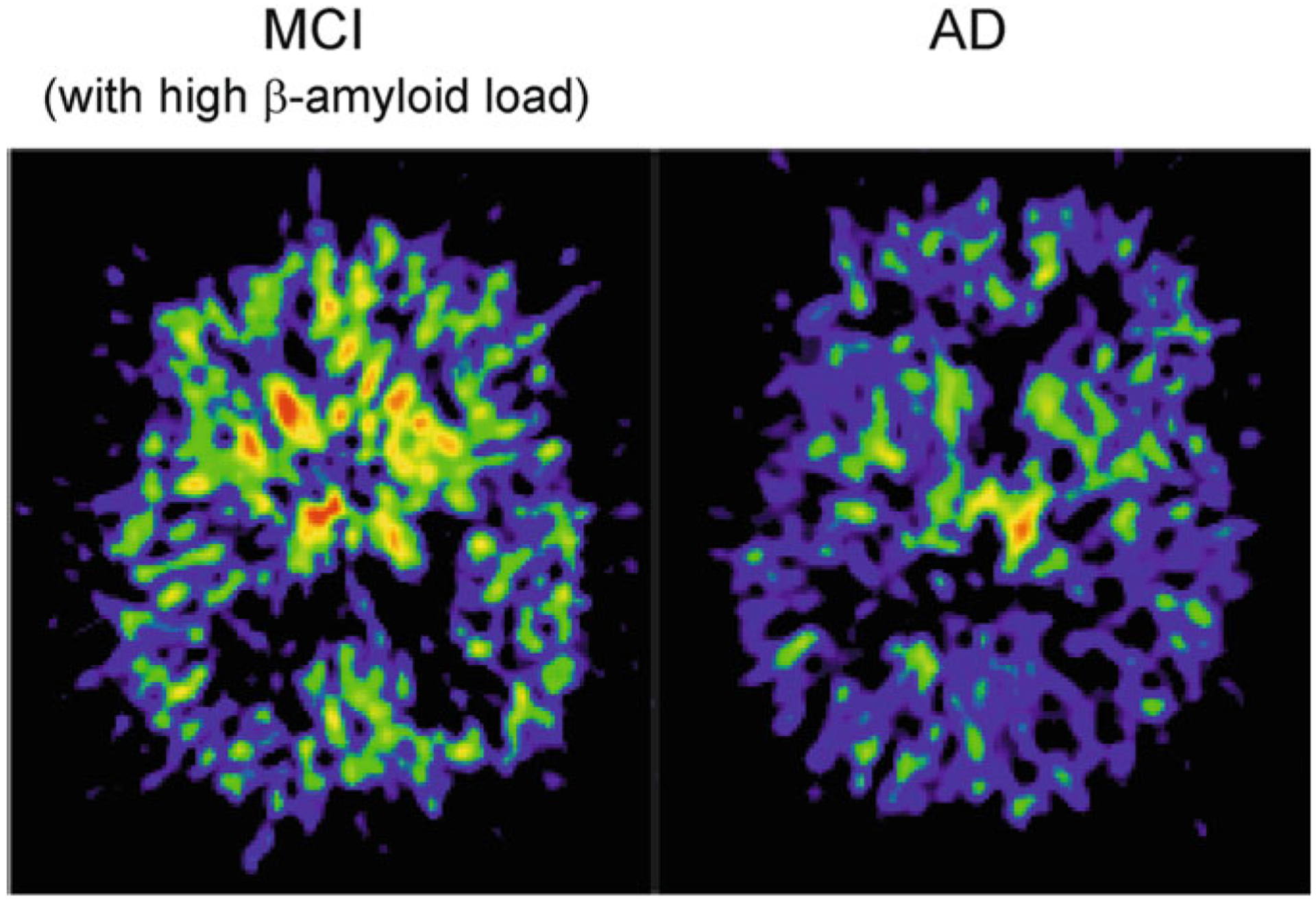
Failure in astroglial reactivity defines the switch between mild cognitive impairment and senility in AD. Prominent astrogliosis in the brain of patient with mild cognitive impairment associated with high β-amyloid load (Left panel) in comparison with patient with Alzheimer’s disease (Right panel). Representative images of 11C-d-deprenyl binding (that reflects MAO-B expression in astrocytes) were obtained by position emission tomography. The MCI patient also showed high presence of fibrillar amyloid plaque as measured with 11C-PIB (the status that could be identified as a prodromal AD). The PET scans show sagittal sections of the brain at the level of basal ganglia. Colour scale indicates red = very high, yellow = moderately high, green = high, blue = low 11C-d-deprenyl binding. Photo courtesy of A. Nordberg, Karolinska institutet. Reproduced with permission from [300]
11.6. Astroglial Calcium Signalling in AD
11.6.1. Ionic Signalling as a Substrate of Astroglial Excitability
Astroglial excitability is based on spatially and temporally controlled fluctuations of intracellular concentration of ions, most notably of Ca2+ and Na+, although recently the signalling role for K+ and Cl− begun to be considered [130, 240, 246, 256, 313, 314, 328]. Astroglial Ca2+ signalling is the most studied; astroglial Ca2+ responses have been discovered in the late 1980s [71, 299], and are implicated in various signalling functions.
Physiological stimulation has been demonstrated to trigger Ca2+ signals in astrocytes in vitro, in situ and in vivo [22, 66, 129, 131, 256]. Astroglial calcium signalling has a spatio-temporal hierarchical organisation: at the cellular level, astrocytic Ca2+ signals are classified into local Ca2+ microdomains, intracellular propagating waves, global Ca2+ signals and Ca2+ oscillations [95, 196, 259, 265]. These distinct forms of Ca2+ signals reflect operation of different mechanisms. Global Ca2+ signals and propagating Ca2+ waves originate from Ca2+ release from the endoplasmic reticulum Ca2+ store; this release is primarily mediated by inositol 1,4,5 trisphosphate (InsP3) receptor type 2, (InsP3R2). Local Ca2+ microdomains in contrast are often generated by Ca2+ entry through ionotropic receptors, transient receptor potential channels, store-operated Ca2+ entry (SOCE) or reversed Na+/Ca2+ exchanger [305]. Astroglial Ca2+ signals regulate several cellular processes, including secretion, metabolism and astroglial reactivity. Astroglial Na+ signalling is much less characterised, although basic parameters of Na+ transients evoked by physiological stimulation have been described in experiments in cultured cells and in astrocytes in brain slices [86, 127, 128, 148–150, 227, 344]. Astroglial Na+ signals regulate many homeostatic plasmalemmal transporters, thus coordinating neuronal activity with astroglial support [130, 240].
11.6.2. Aberrant Calcium Signalling in AD
The fundamental role of Ca2+ in regulation of cellular survival and cell death inspired the “calcium hypothesis of ageing and neurodegeneration” formulated by Zaven Khachatirian [125] who based this hypothesis on experimental studies of Philipp W. Landfield [146, 147]. This Ca2+ hypothesis posits that ageing neurones experience increased Ca2+ influx during depolarisation, which elevates cytosolic Ca2+ concentration ([Ca2+]i), thus triggering excitotoxicity. Subsequent studies revealed that physiological neuronal ageing is associated with much subtle alterations of neuronal Ca2+ extrusion, which, although capable of handling normal Ca2+ loads, fail to clear excessive Ca2+ influx. This deficit in Ca2+ handling stipulates higher vulnerability of old neurones to the periods of high activity [286, 287, 312, 334]. In neurodegenerative diseases (including AD), Ca2+ homeostatic machinery is, however, seriously compromised, and hence these disorders have been regarded as “chronic calciumopathies” [273, 274]. Almost nothing is known about changes in Ca2+ homeostatic machinery, resting Ca2+ handling, and Ca2+ signalling in aged astrocytes. There are several isolated reports demonstrating a decrease in evoked astrocytic Ca2+ signals in mice aged 16–21 months, when compared to adult animals [144, 145].
11.6.3. Exposure to β-Amyloid Disturbs Astroglial Ca2+ Signalling
Whether β-amyloid is indeed a causal factor in AD or a mere epiphenomenon, exposure to it affects astroglial Ca2+ dynamics. Experimental studies in vitro in primary astroglial cultures demonstrated acute effects of β-amyloid on Ca2+ signalling. Resting [Ca2+]i significantly (2–3 times) increased in astrocytes exposed to β-amyloid (in concentrations ranging between 100 nM and 5 μM) for several hours [103, 161]. These findings, however, have not been universally confirmed; several investigations found that incubations of cultured astrocytes with 100–200 nM of β-amyloid (or its toxic fragment β-amyloid25–35) for 48–72 h did not significantly change resting [Ca2+]i [47, 288].
Acute exposure to β-amyloid triggered oscillations of [Ca2+]i transients in cultured astrocytes and in astrocytes in organotypic slices [2–4, 52, 114, 163]. These acute effects, however, were not always observed and several studies have not noticed such acute effects [47, 288]. Treatment of cultured astrocytes with 1 μM of β-amyloid1 – 40 induced [Ca2+]i elevations only in 17% of all the cells, whereas application of β-amyloid25–35 triggered Ca2+ signals in 36% of all astrocytes [114]. In primary cultured rat newborn astrocytes, application of 1 μM of β-amyloid25–35 induced [Ca2+]i transients in 27% of primary cultured rat newborn astrocytes; at 2–5 μM ~60% of astrocytes responded with [Ca2+]i transients [270]. Of note, low concentrations of β-amyloid apparently stimulate astroglial Ca2+-permeable α7 nicotinic cholinoreceptors, which resulted in Ca2+ influx and generation of Ca2+ responses [154, 216].
11.6.4. Pathological Ca2+ Signalling in AD Astrocytes In Vitro
Analysis of [Ca2+]i dynamics in astrocytes isolated from several mouse models of AD also demonstrated aberrant Ca2+ signalling [162, 163]. Abnormally large Ca2+ signals have been detected in astrocytes isolated from newborn 3xTg-AD mice, indicating intrinsic alterations of Ca2+ homeostatic cascades [239]. Astrocytes isolated from 3xTg-AD mice in particular showed increased store-operated calcium entry (SOCE) [239]. Cultures of astrocytes isolated from 3xTg-AD animals also demonstrated aberrant kinetics of ATP-induced Ca2+ signals and Ca2+ oscillations [269]. Further analysis revealed that these aberrations are most likely associated with expression of mutant PS1 presenilins residing in the endoplasmic reticulum [269]. In the Tg5469 AD mouse which over-expressed APP, the SOCE was not changed, whereas deletion of APP caused an inhibition of store-operated Ca2+ entry [164]. This inhibition may be associated with down-regulation of expression of either TRPC1 or Orai 1 channels.
11.6.5. Pathological Ca2+ Signalling in Astrocytes In Vivo
Imaging astroglial [Ca2+]i dynamics in vivo in the brains of AD animal models reliably demonstrated aberrant, hyperactive [Ca2+]i dynamics, which is fundamentally similar to neuronal hyperexcitability routinely observed in AD-like experimental pathology [350]. Aberrant hyperactive [Ca2+]i oscillations have been observed in reactive astrocytes associated with senile plaques. High levels of resting [Ca2+]i, pathological Ca2+ oscillations and long-projecting propagating Ca2+ waves have been identified in plaque-associated astrocytes in the brains of APP/PS1 mice [138]. Emergence of astroglial Ca2+ hyperactivity was also suggested to be linked with abnormal purinergic signalling in reactive astrocytes. There are claims that reactive astrocytes release excessive amounts of ATP through connexin hemichannels. This ATP, acting in autocrine manner, activates astroglial P2Y purinoceptors, which mediate pathological Ca2+ signalling [62]. An increased frequency of astroglial Ca2+ oscillations was also observed in AD animals in the pre-plaque stage, and these abnormal [Ca2+]i dynamics coincided with the instability of vascular tone probably indicating that astrocytes in their ability to regulate local blood flow [278].
11.6.6. Astroglial Ca2+ Signalling Toolkit Is Remodelled in AD
The AD as a chronic pathology leads to a substantial remodelling of astroglial Ca2+ signalling toolkit. Chronic exposure of cultured astrocytes to β-amyloid as well in vivo AD pathology (in model animals) changes expression of various components of Ca2+ homeostatic/signalling system; these molecules include, for example, metabotropic and ionotropic receptors, intracellular Ca2+ channels, store-operated Ca2+ channels and Ca2+ sensors [162, 163, 309].
Exposure of astroglial cultures to 10–30 μM β-amyloid1–40 for 48–72 h resulted in an increase of the amplitude of [Ca2+]i transients in response to stimulation of metabotropic glutamate receptor mGluR5. This augmentation of metabotropic Ca2+ signalling was a consequence of an up-regulated expression of mGluR5 detected at both mRNA and protein levels [47]. This was corroborated in another series of experiments which demonstrated that 24–72 h exposure of cultured astrocytes to 100 nM–20 μM of oligomeric β-amyloid increased expression of mGluR5 [93, 94, 161]. This up-regulation of mGluR5 expression was suppressed by the inhibitors of calcineurin and Nf-κB (nuclear factor κ-light-chain-enhancer of activated B cells) [161]. Similar increase in expression of mGluR5 was detected in astrocytes in the animal AD models and in post-mortem human tissues. Increased levels of mGluR5 protein were found in the post-mortem hippocampal preparations obtained from AD patients at advanced (Braak V–VI) stages of the disease [47, 161]. Incubation of astrocytes with nanomolar (0.1–100 nM) concentrations of β-amyloid1–42 for 24–72 h increased the expression of several subunits of nicotinic cholinoreceptors including α7nAChR, α 4nAChR and β2nAChR [335]. Similarly, increased levels of α7nAChR were identified in the post-mortem brain tissue of patients with sporadic AD and familial AD associated with the Swedish APP mutation [341].
Another important class of molecules affected by AD progression is represented by intracellular Ca2+ release channels. Exposure of cultured astrocytes to 125 nM of Tat-ProADAM10709–729 peptide (this peptide inhibits production of β-amyloid1–40 and β-amyloid1–42) for 72 h leads to an increased expression of InsP3R1 [93]. Sim ilarly, up-regulation of expression of InsP3R1 and InsP3R2 mRNA was detected in astrocytes in vitro which were exposed to 100 nM oligomeric β-amyloid1–42 [161]. Pathological remodelling of Ca2+ homeostatic and signalling cascades differ between different brain regions. Expression of InsP3R1 is increased in healthy hippocampal astrocytes exposed to β-amyloid, but remains unchanged in astrocytes from the entorhinal cortex [94]. However, β-amyloid did not affect expression of InsP3R1 in astrocytes isolated from 3xTg-AD animals, indicating that exogenous β-amyloid and over-expression of mutated AD-related genes share common molecular pathways that cause deregulation of Ca2+ homeostasis. In post-mortem studies, however, generalised decrease in the expression of InsP3Rs was detected in all brain regions including frontal, parietal and entorhinal cortices and the hippocampus [102, 141, 340]. These studies did not, however, discriminate between cell types. Many other components of Ca2+ signalling system are affected by AD; these include components calpain-10 [85], NFAT (Nuclear factor of activated T-cells) [1], NF-κB [93], calcineurin [93, 199], L-type calcium channels [59] and store-operated Ca2+ channels [239]. All in all 32 genes associated with Ca2+ signalling were found to be affected in the transcriptome of astrocytes microdissected from patients with different Braak stages of AD. It appeared that expression of several isoforms of calmodulin kinase CaMKII, two isoforms of calmodulin, plasma membrane Ca2+-ATPases, ryanodine receptors and InsP3Rs, was decreased at advanced (Braak V–VI) stage when compared with early (Braak I–II) stage of the disease [262].
11.6.7. Ca2+ Release and Astroglial Reactivity
As has been alluded before, astrogliosis is a prominent component in certain brain regions in the context of AD; reactive astrocytes associate themselves with senile plaques in human tissue and in the brains of AD animal models, arguably forming a defensive barrier protecting neural networks [106, 306]. Suppression of astrogliotic response (for instance, by genetic deletion of GFAP and vimentin) exacerbates β-amyloid load and facilitates plaques dissemination [137]. Astroglial reactivity, however, is different in different regions of the brain. Prominent astroglial reactivity is observed in the hippocampus, whereas the emergence of senile plaques and β-amyloid depositions does not trigger astrogliosis in entorhinal and prefrontal cortices of AD mice models. Underlying molecular mechanisms might be linked to a deficient Ca2+ signalling in astrocytes from different brain regions.
In the AD context, one of the most relevant signals instigating astroglial reactivity is β-amyloid, and indeed exposure of astroglia to β-amyloid in vitro or in situ triggers astrogliosis [4, 306]. As has been described above, β-amyloid also evokes [Ca2+]i elevation. It appears that β-amyloid-induced Ca2+ signals originate from Ca2+ release from the endoplasmic reticulum Ca2+ store and these Ca2+ signals are directly linked to the initiation of astrogliotic response. Suppression of Ca2+ release from the ER with pharmacological tools effectively inhibits astrogliosis induced by β-amyloid in both cultured astrocytes and astroglia in organotypic slices [4]. The causal role of Ca2+ release in astroglial reactivity was directly demonstrated: deletion of InsP3R2 effectively suppressed astrogliotic activation [120]. Sensitivity of astrocytes from different brain regions to β-amyloid is different: β-amyloid up-regulates expression of molecules providing for ER Ca2+ release in hippocampal but not in entorhinal astrocytes [94]. This may explain the absence of astrogliotic defensive response in astrocytes from cortical regions, which renders these parts of the brain vulnerable to the AD pathology [162, 300].
11.7. AD Pathology Affects Astroglial Vesicular Trafficking and Secretion
Astrocytes are secretory cells, being a part of CNS-wide “gliocrine” system [301]. Astrocytes are known to secrete ~200 molecules, many of which are released through exocytosis of secretory vesicles [349]. Intracellular astroglial vesicles are also fundamental for delivery of various molecules [298] such as ion channels, membrane receptors and transporters, as well as major histocompatibility complex II (MHC-II, [296]) and EAAT 2 [266], to the plasma membrane. Vesicular traffic is controlled by sophisticated molecular cascades, which in turn are regulated by increases in [Ca2+]i [220, 266]. Changes in [Ca2+]i differentially regulate motility of distinct vesicles types. Increases in [Ca2+]i reduce the motility of vesicles carrying peptides, such as atrial natriuretic peptide, while accelerating motility of vesicles containing vesicular L-glutamate transporter VGLUT1 [218–220]. Proteolytic enzymes stored in endolysosomes may contribute to the development of AD. One of these proteases is represented by the insulin-degrading enzyme (IDE), which, when secreted into the extracellular space, may degrade β-amyloid. Astrocytes are the main cell type which produces and releases IDE [68, 263]. It has been hypothesised that in AD the capacity of secreting IDE is reduced, leading to an increase in β-amyloid, which involves a reduction in autophagy-based lysosomal secretion of IDE [263].
Astrocytes from 3xTg-AD mice demonstrated an aberrant vesicular traffic. Spontaneous mobility of peptidergic and endolysosomal vesicles as well as the ATP-evoked, Ca2+-dependent, vesicle mobility was all diminished in AD astrocytes (Fig. 11.9). Transfection of healthy rat astrocytes to express familial AD-associated mutated presenilin 1 (PS1M146V) caused very similar impairment of peptidergic vesicle trafficking. The stimulation-dependent peptide secretion from single vesicles was less efficient in 3xTg-AD and PS1M146V-expressing astrocytes than in healthy controls. The impaired vesicle dynamics and reduced evoked secretion of the signalling peptides both may contribute to the development of AD [269].
Fig. 11.9.
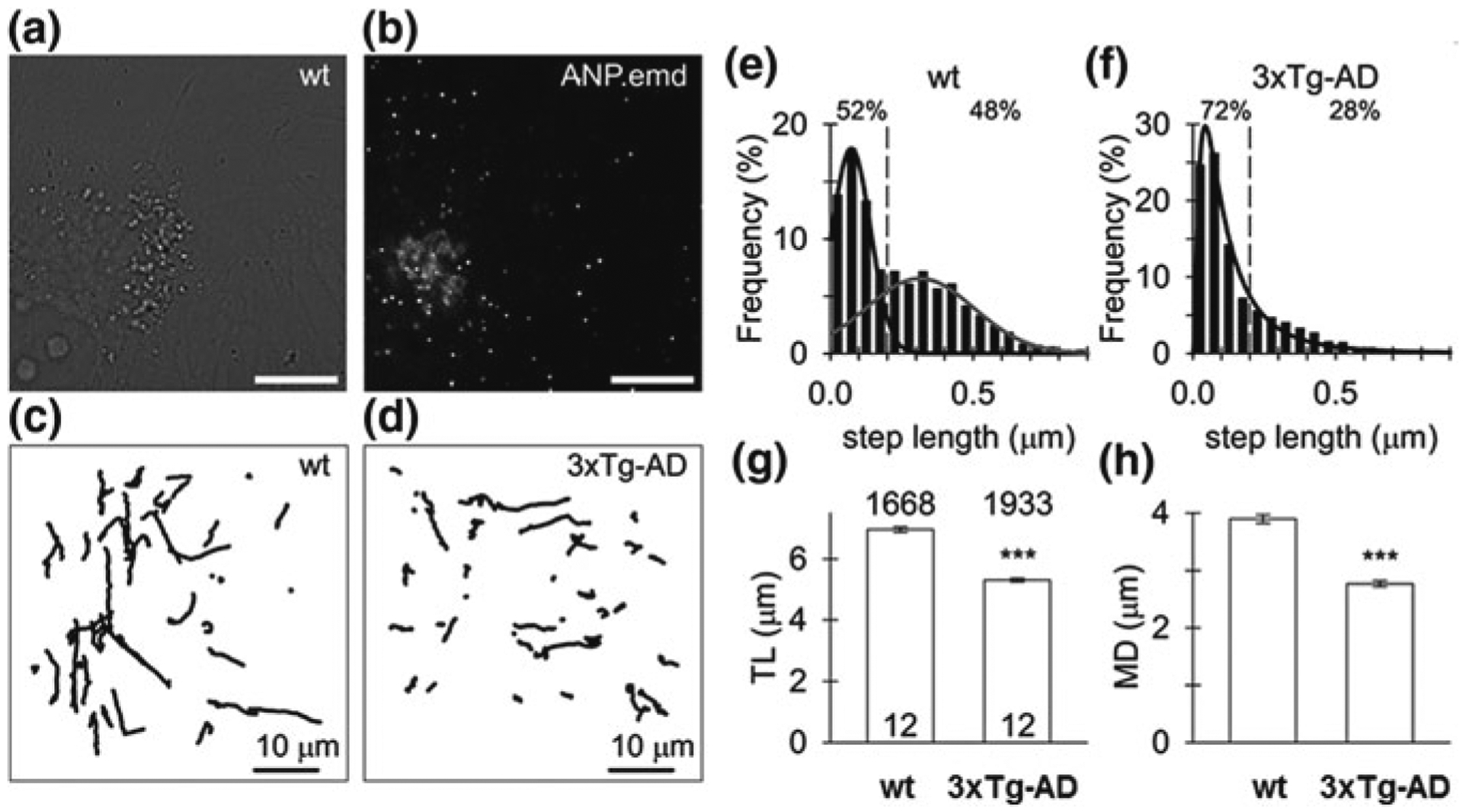
Decreased spontaneous mobility of peptidergic vesicles in 3xTg-AD astrocytes. a Live cultured wild-type (wt) astrocyte under DIC optics and b the confocal image of the same cell expressing fluorescent peptide atrial natriuretic peptide-emerald green (ANP.emd), stored in individual vesicles, observed as bright fluorescent puncta; scale bars, 10 μm. c Vesicle tracks (N = 50) obtained in a 15-s epoch of imaging representative control (wt) and d 3xTg-AD astrocytes expressing ANP.emd, respectively. Note less elongated vesicle tracks in the 3xTg-AD astrocyte. e, f Frequency histogram of the step length in spontaneously moving vesicles in wt (N = 5025, e) and 3xTg-AD (N = 5072, f) astrocytes. The data were fitted with the function f = a × exp(− 0.5 × (x/x0)/b)2/x, where a = 17.88±0.00 μm−0.5, x0 = 0.07±0.00 μm(black curve) and a=6.53±0.13, b=0.19±0.01 μm−0.5, x0 = 0.31±0.01 μm (grey curve) in wt astrocyte, and with the function f = a × exp(−0.5 × (lnx/x0)/b)2/x, where a = 1.96 ± 0.04, b=0.92 ± 0.02 μm−0.5, x0 = 0.10± 0.00 μm (black curve) in 3xTg-AD astrocyte. The indicates the step length of 0.2 μm obtained close to the intersection of distributions (black and grey curve) in wt astrocytes to discriminate small (<0.2 μm) from large (≥ 0.2 μm) steps. Note the higher proportion (%) of smaller steps lengths in the 3xTg-AD astrocyte indicated by the absence of the second mode distribution seen in wt astrocytes. g Track length (TL), h maximal displacement (MD),note substantially diminished TL, MD in 3xTg-AD astrocytes. The numbers above the top of the bars (mean ± SEM) indicate the number of vesicles analysed; the numbers at the bottom of the bars indicate the number of cells analysed; “***”—indicates p values < 0.001. Modified with permission from [269]
11.8. GABAergic Astrocytes in AD
In the healthy young CNS, astrocytes contribute to GABAergic transmission through (i) supplying glutamine, needed for GABA biosynthesis in neuronal terminals and (ii) removing ~20% of all released GABA by dedicated plasmalemmal transporters GAT-1 and GAT-3. After being transported into the astrocytes, most of GABA is catabolised by GABA transaminase (GABA-T) to succinate, which is subsequently utilised for production of ATP [253, 305, 319]. Due to this energy-oriented catabolism, the concentration of GABA in the cytosol of astrocytes is rather low. Ageing and neurodegeneration, however, significantly affect astroglial GABA metabolism; concentration of GABA in astrocytes in elderly [155], in patients with AD [118, 332] and in transgenic AD models [41, 118, 332], is significantly higher. This increase is particularly prominent in reactive astrocytes associated with senile plaques in AD animal models; intracellular GABA concentration in these AD reactive astrocytes is several times higher than in age-matched controls and is very similar to neuronal GABA content [41, 118, 332]. These changes in astrocytic GABA content in reactive astrocytes are accompanied with an up-regulation of expression of GABA producing enzyme glutamic acid decarboxylase GAD67 as well as with an increase in expression of astroglia-specific monoaminoxidase-B (MAO-B) [118]. At the same time, expression of glutamine synthetase is specifically down-regulated in reactive astrocytes surrounding senile plaques in the hippocampus and prefrontal cortex of 3xTg-AD mice (Fig. 11.10 and [204]). Thus reactive astrocytes acquire machinery to synthetase GABA either from glutamate (through GAD67 and increased glutamate availability due to the loss of glutamine synthetase) or from putrescine through MAO-B pathway [84]. Furthermore, there is an increased glutamatergic neuronal activity around senile plaques [206]; this conceivably increases astroglial glutamate uptake and availability of cytosolic glutamate for conversion to GABA cytosolic glutamate concentration glutamate transport into astrocytes Astroglial GABA may potentially be released from astrocytes by diffusion through Bestrophin-1 Cl− channels or through reversed GAT3 transporters (Fig. 11.11, [84]). The emergence of GABAergic astrocytes may represent yet another defensive response; as GABA release from astroglia may counteract neuronal hyperexcitability by an increase of tonic inhibition [84].
Fig. 11.10.
Down-regulation of glutamine synthetase (GS) expression in hippocampal astrocytes in 3xTg-AD mice. a, b Light microscopy images of GS—(a) and GFAP—(b) positive astrocytes. c, e, g Confocal images of hippocampal preparation labelled for GS (c, red), GFAP (e, green) and their co-localisation (g, yellow). d, f, h High magnification confocal images illustrating the co-expression of GS and GFAP. i, j Ubiquitous co-expression of GS and GFAP in wild-type control mice (i) and down-regulation of GS expression (astrocytes lacking GS are indicated by arrows) in 3xTg-AD mice (j). DG, dentate gyrus; GcL, granule cell layer; MoL, molecular layer; Lac, stratum lacunosum moleculare; Or, stratum oriens; PcL, pyramidal layer; Rad, stratum radiatum. Modified and reproduced with permission from [204]
Fig. 11.11.
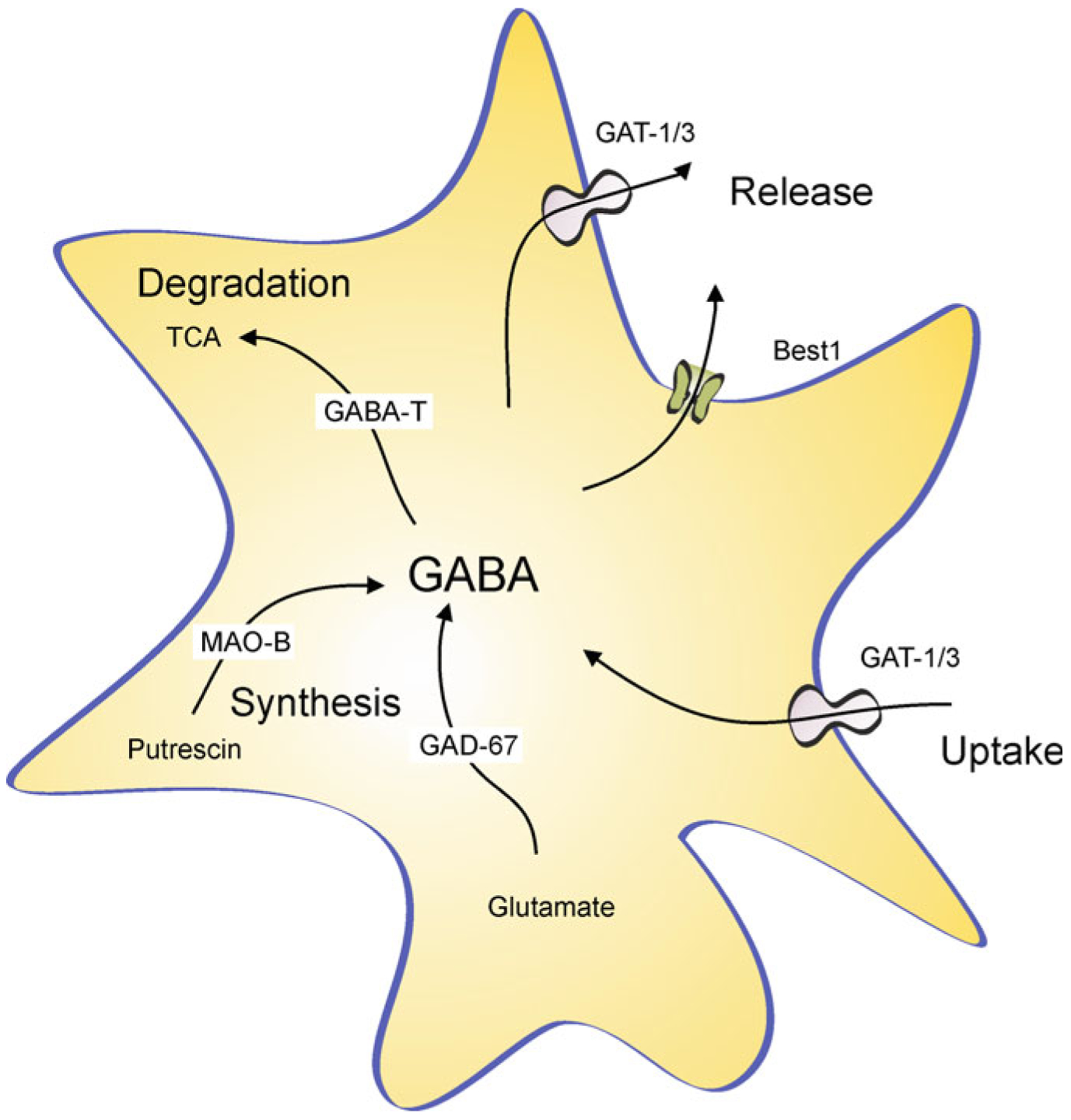
GABAergic reactive astrocytes in AD. See text for explanation. Abbreviations: GAT1/3 GABA transporters 1 (SLC6A1) and 3 (SLC6A11); Best1—bestrophin 1 anion channel 1; GABAT—GABA transaminase; TCA—tricarboxylic acid (Krebs) cycle; MAO-B—Monoamine oxidase B; GAD67—glutamate decarboxylase. Modified from [84]
11.9. Astrocytes as Therapeutic Targets in AD
Neuroglia is yet to be considered as a fundamental target for novel therapeutic agents for neurological disorders and neurodegenerative diseases in particular. It is conceivable that by modulating the status of astrocytes, by reversing or halting astrocytes degeneration and asthenia or by modulating astroglial reactivity, the course of AD can be altered and the disease can be delayed or cognitive alterations reversed. Several possible strategies that may affect astroglial pathology have recently emerged.
11.9.1. Lifestyle Changes May Reverse Astrodegeneration
Recent experiments have demonstrated that environmental modifications such as sensory stimulation, dieting or usage of food supplements may affect AD progression and at the same time change astroglia morphology and revert astroglial atrophy. Experiments on APP and 3xTg-AD mice models revealed that chronic exposure of these animals to physical activity and/or to enriched environment reversed morphological atrophy of astrocytes, increased GFAP expression and normalised GFAP-positive astroglial profiles (Fig. 11.12); most importantly, these astroglia-specific changes developed in parallel to a decrease in β-amyloid load [20, 236]. Incidentally, environmental stimulation also improved neurogenesis which is impaired in the AD [231, 233]. Another AD model, the 5xFAD mice chronically treated with polyunsatu-rated fatty acid 2-hydroxy-docosahexaenoic acid similarly rescued astroglial atrophy, restored adult neurogenesis and improved cognitive performance [76]. Treatment with specific diets may also affect ageing and AD progression. It is well appreciated that caloric restriction exerts prominent positive effect of lifespan of several species and may boost cognitive resilience of the brain [80, 169, 170, 175]. It appeared that caloric restriction induces growth of astroglial perisynaptic processes, thus extending synaptic coverage, preventing glutamate spillover, improving K+ buffering and glu tamate uptake from the synaptic cleft, thus ultimately enhancing synaptic plasticity [217].
Fig. 11.12.
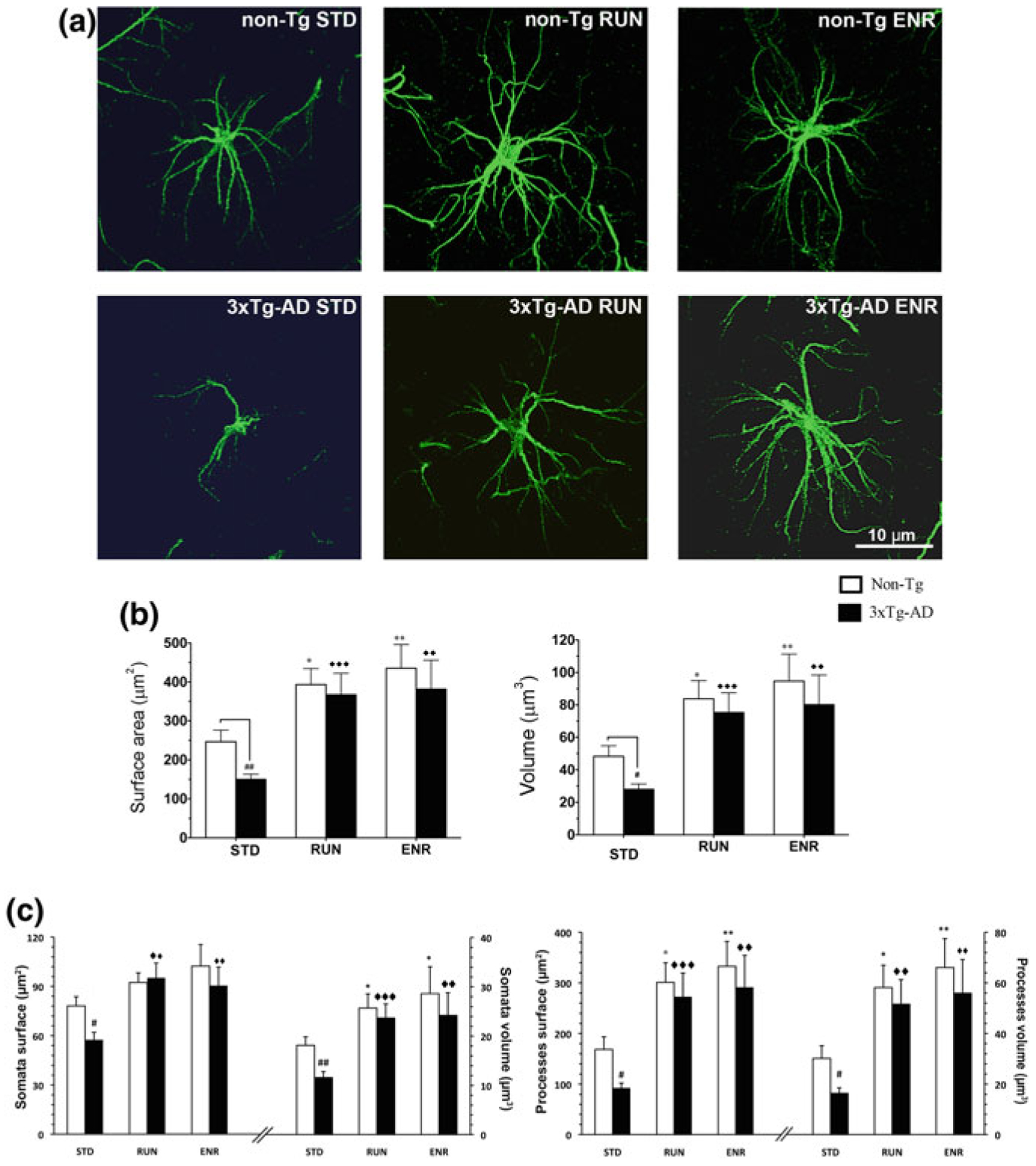
Environmental stimulation (enriched environment, ENR and physical activity, RUN) reverse morphological atrophy of astrocytes seen in the dentate gyrus isolates from 3xTg-AD mice. GFAP-immunoreactivity of astrocytes in the DG of non-Tg and 3xTg-AD animals housed in different conditions. a High magnification of representative confocal micrographs showing the astrocytic morphology in mice housed in standard conditions (STD), RUN and ENR. Scale bars, 10 μm. Note the morphological changes of the astrocytes from both genotypes induced by the different living conditions. b Histograms showing difference of surface area and volume of GFAP-positive astrocytes in the DG of non-Tg and 3xTg-AD mice housed under different housing conditions. c Histograms showing differences in surface area and volume of GFAP-immunoreactivity of astrocytic cell bodies and processes detected between non-Tg and 3xTg-AD mice housed under different housing conditions. Bars represent means ± S.E.M., #p < 0.05, ##p < 0.01 compared with non-Tg animals in same housing environment; *p < 0.05, **p < 0.01 compared with non-Tg mice housed under STD; ◆◆p < 0.01 and ◆◆◆p < 0.001 compared with 3xTg-AD mice housed under STD. Reproduced with permission from [236]
11.9.2. Preventing Neurodegeneration by Adrenergic Astroglial Excitation
Noradrenergic innervation of the CNS is provided by projections of adrenergic neurones localised in the brainstem nucleus locus coeruleus. This small nucleus is located near the fourth ventricle and, in humans, comprises around 50,000 neurones [188]. Diffuse innervation by projections of locus coeruleus neurones reaches practically all parts of the brain and the spinal cord [25]. The locus coeruleus neurones are vulnerable to oxidative stress; apparently, they are lost in ageing and they are first to die during neurodegeneration including AD [75, 167, 190, 249]. Astrocytes, being universally sensitive to noradrenaline, represent the major target for deficient noradrenergic innervation and interfering with astroglial adrenergic mechanisms may be therapeutically relevant [348].
Astroglial sensitivity to noradrenaline, released from locus coeruleus neuronal projections, is mediated by both α- and β-adrenoceptors linked, respectively, to cytosolic Ca2+ signalling [66, 131] and cyclic AMP (cAMP) cascades [297]. In the in vivo experiments in the awake mice, the vast majority of astrocytes generated synchronous [Ca2+]i signals in response to noradrenaline released from locus coeruleus projections [22, 66, 210]; of note neurones did not generate Ca2+ responses to the same stimulation [208]. This difference reflects upon much higher density of adrenoceptors in astrocytes when compared to neurones [10]. Degeneration of locus coeruleus neurones associated with ageing most certainly impairs adrenoceptors–medicated astroglial excitability, which may be linked to the cognitive decline [348]. Consequently, preventing death of locus coeruleus neurones or boosting astroglial adrenergic excitability may represent a valid therapeutic strategy [348]. Alternative possibilities may involve drugs, such as deprenyl, that limit noradrenaline catabolism in astrocytes.
Transcranial direct current stimulation (tDCS) was used with positive effects including memory enhancements, accelerated motor function rehabilitation, alleviation of depressive symptoms and decelerated progression of cognitive impairments in AD patients [140, 197]. The mechanism of action of tDCS is astroglial and noradrenergic. It has been revealed that tDCS induces a massive increase in astroglial [Ca2+]i which has been suppressed by the ablation of noradrenergic neurones or by the inhibition of α1-adrenoceptors [186]. Alternative possibilities may involve drugs that limit noradrenaline catabolism in astrocytes such as, for example, deprenil.
11.10. Conclusions
Astroglial contributions to the pathophysiology of AD are complex and range from early astroglial atrophy, which limits homeostatic support and may cause synaptic weakness and early cognitive decline, to astroglial reactivity, which seems to protect the CNS against AD-associated pathology and limits the spread of β-amyloid load. Astroglia may also undergo pathological remodelling, in which astrocytes may acquire new functions such as, for example, secreting GABA. Specific manipulations with astroglia may represent a valid therapeutic approach for treating neurodegenerative disorders including AD.
Acknowledgments
VP’s work is supported by a grant from the National Institute of General Medical Sciences of the National Institutes of Health (R01GM123971). VP is an Honorary Professor at University of Rijeka, Croatia.
References
- 1.Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H 3rd, Kraner SD, Norris CM (2009) Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci 29:12957–12969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abramov AY, Canevari L, Duchen MR (2003) Changes in intracellular calcium and glutathione in astrocytes as the primary mechanism of amyloid neurotoxicity. J Neurosci 23:5088–5095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abramov AY, Canevari L, Duchen MR (2004) β-Amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24:565–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alberdi E, Wyssenbach A, Alberdi M, Sanchez-Gomez MV, Cavaliere F, Rodriguez JJ, Verkhratsky A, Matute C (2013) Ca2+-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid β-treated astrocytes and in a model of Alzheimer’s disease. Aging Cell 12:292–302 [DOI] [PubMed] [Google Scholar]
- 5.Allaman I, Gavillet M, Belanger M, Laroche T, Viertl D, Lashuel HA, Magistretti PJ (2010) Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. J Neurosci 30:3326–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M (2002) Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22:9340–9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alzheimer A (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg Z Psychiat Psych-Gericht Med 64:146–148 [Google Scholar]
- 8.Alzheimer A (1910) Beiträge zur Kenntnis der pathologischen Neuroglia und ihrer Beziehungen zu den Abbauvorgängen im Nervengewebe In: Nissl F, Alzheimer A (eds) Histologische und histopathologische Arbeiten über die Grosshirnrinde mit besonderer Berücksichtigung der pathologischen Anatomie der Geisteskrankheiten. Jena, Gustav Fischer, pp 401–562 [Google Scholar]
- 9.Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K, Davies P (2003) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86:582–590 [DOI] [PubMed] [Google Scholar]
- 10.Aoki C (1992) β-adrenergic receptors: astrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. J Neurosci 12:781–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Apelt J, Ach K, Schliebs R (2003) Aging-related down-regulation of neprilysin, a putative β-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of β-amyloid plaques. Neurosci Lett 339:183–186 [DOI] [PubMed] [Google Scholar]
- 12.Arendash GW, Lewis J, Leighty RE, McGowan E, Cracchiolo JR, Hutton M, Garcia MF (2004) Multi-metric behavioral comparison of APPsw and P301L models for Alzheimer’s disease: linkage of poorer cognitive performance to tau pathology in forebrain. Brain Res 1012:29–41 [DOI] [PubMed] [Google Scholar]
- 13.Arendt T (1994) Impairment in memory function and neurodegenerative changes in the cholinergic basal forebrain system induced by chronic intake of ethanol. J Neural Transm Suppl 44:173–187 [DOI] [PubMed] [Google Scholar]
- 14.Arranz AM, De Strooper B (2019) The role of astroglia in Alzheimer’s disease: pathophysiology and clinical implications. Lancet Neurol 18:406–414 [DOI] [PubMed] [Google Scholar]
- 15.Arranz B, Blennow K, Ekman R, Eriksson A, Mansson JE, Marcusson J (1996) Brain monoaminergic and neuropeptidergic variations in human aging. J Neural Transm (Vienna) 103:101–115 [DOI] [PubMed] [Google Scholar]
- 16.Ashe KH, Zahs KR (2010) Probing the biology of Alzheimer’s disease in mice. Neuron 66:631–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barthelemy I, Martineau D, Ong M, Matsunami R, Ling N, Benatti L, Cavallaro U, Soria M, Lappi DA (1993) The expression of saporin, a ribosome-inactivating protein from the plant Saponaria officinalis, in Escherichia coli. J Biol Chem 268:6541–6548 [PubMed] [Google Scholar]
- 18.Bartus RT, Dean RL 3rd, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408–414 [DOI] [PubMed] [Google Scholar]
- 19.Beach TG, McGeer EG (1988) Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res 463:357–361 [DOI] [PubMed] [Google Scholar]
- 20.Beauquis J, Pavia P, Pomilio C, Vinuesa A, Podlutskaya N, Galvan V, Saravia F (2013) Environmental enrichment prevents astroglial pathological changes in the hippocampus of APP transgenic mice, model of Alzheimer’s disease. Exp Neurol 239:28–37 [DOI] [PubMed] [Google Scholar]
- 21.Beauquis J, Vinuesa A, Pomilio C, Pavia P, Galvan V, Saravia F (2014) Neuronal and glial alterations, increased anxiety, and cognitive impairment before hippocampal amyloid deposition in PDAPP mice, model of Alzheimer’s disease. Hippocampus 24:257–269 [DOI] [PubMed] [Google Scholar]
- 22.Bekar LK, He W, Nedergaard M (2008) Locus coeruleus α-adrenergic-mediated activation of cortical astrocytes in vivo. Cereb Cortex 18:2789–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bekris LM, Yu CE, Bird TD, Tsuang DW (2010) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23:213–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bell RD, Zlokovic BV (2009) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benarroch EE (2009) The locus ceruleus norepinephrine system: functional organization and potential clinical significance. Neurology 73:1699–1704 [DOI] [PubMed] [Google Scholar]
- 26.Berchtold NC, Cotman CW (1998) Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960s. Neurobiol Aging 19:173–189 [DOI] [PubMed] [Google Scholar]
- 27.Berrios GE (1990) Alzheimer’s disease: A conceptual history. Int J Geriatric Psychiat 5:355–365 [Google Scholar]
- 28.Bertoni-Freddari C, Giuli C, Pieri C, Paci D (1986) Quantitative investigation of the morphological plasticity of synaptic junctions in rat dentate gyrus during aging. Brain Res 366:187–192 [DOI] [PubMed] [Google Scholar]
- 29.Biegon A, Greenberger V, Segal M (1986) Quantitative histochemistry of brain acetylcholinesterase and learning rate in the aged rat. Neurobiol Aging 7:215–217 [DOI] [PubMed] [Google Scholar]
- 30.Bielschowsky M (1903) Die Ziele bei Impregnation der Neurofibrillen. Neurol Centralbl 22:997–1006 [Google Scholar]
- 31.Bigl M, Bruckner MK, Arendt T, Bigl V, Eschrich K (1999) Activities of key glycolytic enzymes in the brains of patients with Alzheimer’s disease. J Neural Transm 106:499–511 [DOI] [PubMed] [Google Scholar]
- 32.Blanchard V, Moussaoui S, Czech C, Touchet N, Bonici B, Planche M, Canton T, Jedidi I, Gohin M, Wirths O, Bayer TA, Langui D, Duyckaerts C, Tremp G, Pradier L (2003) Time sequence of maturation of dystrophic neurites associated with Abeta deposits in APP/PS1 transgenic mice. Exp Neurol 184:247–263 [DOI] [PubMed] [Google Scholar]
- 33.Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B (2000) Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1–40 and Abeta1–42 by human astrocytes. Neurobiol Dis 7:682–689 [DOI] [PubMed] [Google Scholar]
- 34.Blass JP, Sheu RK, Gibson GE (2000) Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann NY Acad Sci 903:204–221 [DOI] [PubMed] [Google Scholar]
- 35.Blennow K, de Leon MJ, Zetterberg H (2006) Alzheimer’s disease. Lancet 368:387–403 [DOI] [PubMed] [Google Scholar]
- 36.Blocq P, Marinesco G (1892) Sur les lesions et la pathogenie de l’epilepsie dite essentielle. Semaine Medical 12:445–446 [Google Scholar]
- 37.Boegman RJ, el-Defrawy SR, Jhamandas K, Beninger RJ, Ludwin SK (1985) Quinolinic acid neurotoxicity in the nucleus basalis antagonized by kynurenic acid. Neurobiol Aging 6:331–336 [DOI] [PubMed] [Google Scholar]
- 38.Boller F, Forbes MM (1998) History of dementia and dementia in history: an overview. J Neurol Sci 158:125–133 [DOI] [PubMed] [Google Scholar]
- 39.Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259 [DOI] [PubMed] [Google Scholar]
- 40.Brambilla L, Martorana F, Rossi D (2013) Astrocyte signaling and neurodegeneration: new insights into CNS disorders. Prion 7:28–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brawek B, Chesters R, Klement D, Muller J, Lerdkrai C, Hermes M, Garaschuk O (2018) A bell-shaped dependence between amyloidosis and GABA accumulation in astrocytes in a mouse model of Alzheimer’s disease. Neurobiol Aging 61:187–197 [DOI] [PubMed] [Google Scholar]
- 42.Broe M, Kril J, Halliday GM (2004) Astrocytic degeneration relates to the severity of disease in frontotemporal dementia. Brain 127:2214–2220 [DOI] [PubMed] [Google Scholar]
- 43.Burkovetskaya M, Karpuk N, Xiong J, Bosch M, Boska MD, Takeuchi H, Suzumura A, Kielian T (2014) Evidence for aberrant astrocyte hemichannel activity in Juvenile Neuronal Ceroid Lipofuscinosis (JNCL). PLoS ONE 9:e95023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ (2007) Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci 27:13357–13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, Zimmer ER (2019) Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med 25:77–95 [DOI] [PubMed] [Google Scholar]
- 46.Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, Nordberg A (2012) Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med 53:37–46 [DOI] [PubMed] [Google Scholar]
- 47.Casley CS, Lakics V, Lee HG, Broad LM, Day TA, Cluett T, Smith MA, O’Neill MJ, Kingston AE (2009) Up-regulation of astrocyte metabotropic glutamate receptor 5 by amyloid-β peptide. Brain Res [DOI] [PubMed] [Google Scholar]
- 48.Cassel JC, Mathis C, Majchrzak M, Moreau PH, Dalrymple-Alford JC (2008) Coexisting cholinergic and parahippocampal degeneration: a key to memory loss in dementia and a challenge for transgenic models? Neurodegener Dis 5:304–317 [DOI] [PubMed] [Google Scholar]
- 49.Castellani RJ, Lee HG, Siedlak SL, Nunomura A, Hayashi T, Nakamura M, Zhu X, Perry G, Smith MA (2009) Reexamining Alzheimer’s disease: evidence for a protective role for amyloid-beta protein precursor and amyloid-beta. J Alzheimers Dis 18:447–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Celsus AC (1935–1938) De Medicina With an english translation by Spencer WG. William Heinemann Ltd., London [Google Scholar]
- 51.Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, Pearson J, Strome R, Zuker N, Loukides J, French J, Turner S, Lozza G, Grilli M, Kunicki S, Morissette C, Paquette J, Gervais F, Bergeron C, Fraser PE, Carlson GA, George-Hyslop PS, Westaway D (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276:21562–21570 [DOI] [PubMed] [Google Scholar]
- 52.Chow SK, Yu D, Macdonald CL, Buibas M, Silva GA (2010) Amyloid β-peptide directly induces spontaneous calcium transients, delayed intercellular calcium waves and gliosis in rat cortical astrocytes. ASN Neuro 2:e00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chrobak JJ, Hanin I, Schmechel DE, Walsh TJ (1988) AF64A-induced working memory impairment: behavioral, neurochemical and histological correlates. Brain Res 463:107–117 [DOI] [PubMed] [Google Scholar]
- 54.Cicero (2003) On old age. In: On the good life, pp 160–194. Folio Society, London [Google Scholar]
- 55.Coleman P, Federoff H, Kurlan R (2004) A focus on the synapse for neuroprotection in Alzheimer disease and other dementias. Neurology 63:1155–1162 [DOI] [PubMed] [Google Scholar]
- 56.Contestabile A, Stirpe F (1993) Ribosome-inactivating proteins from plants as agents for suicide transport and immunolesioning in the nervous system. Eur J Neurosci 5:1292–1301 [DOI] [PubMed] [Google Scholar]
- 57.Coradazzi M, Gulino R, Garozzo S, Leanza G (2010) Selective lesion of the developing central noradrenergic system: short- and long-term effects and reinnervation by noradrenergic-rich tissue grafts. J Neurochem 114:761–771 [DOI] [PubMed] [Google Scholar]
- 58.Danbolt NC (2001) Glutamate uptake. Progr Neurobiol 65:1–105 [DOI] [PubMed] [Google Scholar]
- 59.Daschil N, Geisler S, Obermair GJ, Humpel C (2014) Short- and long-term treatment of mouse cortical primary astrocytes with beta-amyloid differentially regulates the mRNA expression of L-type calcium channels. Pharmacology 93:24–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Decker MW (1987) The effects of aging on hippocampal and cortical projections of the forebrain cholinergic system. Brain Res 434:423–438 [DOI] [PubMed] [Google Scholar]
- 61.DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457–464 [DOI] [PubMed] [Google Scholar]
- 62.Delekate A, Fuchtemeier M, Schumacher T, Ulbrich C, Foddis M, Petzold GC (2014) Metabotropic P2Y1 receptor signalling mediates astrocytic hyperactivity in vivo in an Alzheimer’s disease mouse model. Nat Commun 5:5422. [DOI] [PubMed] [Google Scholar]
- 63.Depboylu C, Stricker S, Ghobril JP, Oertel WH, Priller J, Hoglinger GU (2012) Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Exp Neurol 238:183–191 [DOI] [PubMed] [Google Scholar]
- 64.DeWitt DA, Perry G, Cohen M, Doller C, Silver J (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 149:329–340 [DOI] [PubMed] [Google Scholar]
- 65.Di Patre PL, Abbamondi A, Bartolini L, Pepeu G (1989) GM1 ganglioside counteracts cholinergic and behavioral deficits induced in the rat by intracerebral injection of vincristine. Eur J Pharmacol 162:43–50 [DOI] [PubMed] [Google Scholar]
- 66.Ding F, O’Donnell J, Thrane AS, Zeppenfeld D, Kang H, Xie L, Wang F, Nedergaard M (2013) α1-Adrenergic receptors mediate coordinated Ca2+ signaling of cortical astrocytes in awake, behaving mice. Cell Calcium 54:387–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dodart JC, Mathis C, Saura J, Bales KR, Paul SM, Ungerer A (2000) Neuroanatomical abnormalities in behaviorally characterized APP(V717F) transgenic mice. Neurobiol Dis 7:71–85 [DOI] [PubMed] [Google Scholar]
- 68.Dorfman VB, Pasquini L, Riudavets M, Lopez-Costa JJ, Villegas A, Troncoso JC, Lopera F, Castano EM, Morelli L (2010) Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer’s disease. Neurobiol Aging 31:1743–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dunnett SB, Everitt BJ, Robbins TW (1991) The basal forebrain-cortical cholinergic system: interpreting the functional consequences of excitotoxic lesions. Trends Neurosci 14:494–501 [DOI] [PubMed] [Google Scholar]
- 70.Duyckaerts C, Delatour B, Potier MC (2009) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118:5–36 [DOI] [PubMed] [Google Scholar]
- 71.Enkvist MO, Holopainen I, Akerman KE (1989) Glutamate receptor-linked changes in membrane potential and intracellular Ca2+ in primary rat astrocytes. Glia 2:397–402 [DOI] [PubMed] [Google Scholar]
- 72.Eriksen JL, Janus CG (2007) Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet 37:79–100 [DOI] [PubMed] [Google Scholar]
- 73.Eroglu C, Barres BA (2010) Regulation of synaptic connectivity by glia. Nature 468:223–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Farkas E, Luiten PG (2001) Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol 64:575–611 [DOI] [PubMed] [Google Scholar]
- 75.Feinstein DL, Kalinin S, Braun D (2016) Causes, consequences, and cures for neuroinflammation mediated via the locus coeruleus: noradrenergic signaling system. J Neurochem 139(Suppl 2):154–178 [DOI] [PubMed] [Google Scholar]
- 76.Fiol-deRoque MA, Gutierrez-Lanza R, Torres M, Terés S, Barceló P, Rial RV, Verkhratsky A, Escribá PV, Busquets X, Rodríguez JJ (2013) Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology (in press) [DOI] [PubMed] [Google Scholar]
- 77.Fischer O (1907) Miliäre Nekrosen mit drusigen wucherungen der neurofibrillen, eine regelmässige veränderung der hirnrinde bei seniler demenz. Monatsschr Psychiatr Neurol 22:361–372 [Google Scholar]
- 78.Fischer W, Chen KS, Gage FH, Bjorklund A (1992) Progressive decline in spatial learning and integrity of forebrain cholinergic neurons in rats during aging. Neurobiol Aging 13:9–23 [DOI] [PubMed] [Google Scholar]
- 79.Flood DG, Lin YG, Lang DM, Trusko SP, Hirsch JD, Savage MJ, Scott RW, Howland DS (2009) A transgenic rat model of Alzheimer’s disease with extracellular Aβ deposition. Neurobiol Aging 30:1078–1090 [DOI] [PubMed] [Google Scholar]
- 80.Fontana L, Partridge L, Longo VD (2010) Extending healthy life span–from yeast to humans. Science 328:321–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, MacGregor R (1997) Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging 18:431–435 [DOI] [PubMed] [Google Scholar]
- 82.Frost GR, Li YM (2017) The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F et al. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature 373:523–527 [DOI] [PubMed] [Google Scholar]
- 84.Garaschuk O, Verkhratsky A (2019) GABAergic astrocytes in Alzheimer’s disease. Aging (Albany NY) 11:1602–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Garwood C, Faizullabhoy A, Wharton SB, Ince PG, Heath PR, Shaw PJ, Baxter L, Gelsthorpe C, Forster G, Matthews FE, Brayne C, Simpson JE (2013) Calcium dysregulation in relation to Alzheimer-type pathology in the ageing brain. Neuropathol Appl Neurobiol 39:788–799 [DOI] [PubMed] [Google Scholar]
- 86.Gerkau NJ, Kafitz KW, Rose CR (2019) Imaging of local and global sodium signals in astrocytes. Methods Mol Biol 1938:187–202 [DOI] [PubMed] [Google Scholar]
- 87.Gerlai R (2001) Alzheimer’s disease: beta-amyloid hypothesis strengthened! Trends Neurosci 24:199. [DOI] [PubMed] [Google Scholar]
- 88.Giovannini MG, Scali C, Prosperi C, Bellucci A, Vannucchi MG, Rosi S, Pepeu G, Casamenti F (2002) Beta-amyloid-induced inflammation and cholinergic hypofunction in the rat brain in vivo: involvement of the p38MAPK pathway. Neurobiol Dis 11:257–274 [DOI] [PubMed] [Google Scholar]
- 89.Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science 293:1491–1495 [DOI] [PubMed] [Google Scholar]
- 90.Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M (1995) Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J 14:1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gotz J, Streffer JR, David D, Schild A, Hoerndli F, Pennanen L, Kurosinski P, Chen F (2004) Transgenic animal models of Alzheimer’s disease and related disorders: histopathology, behavior and therapy. Mol Psychiatry 9:664–683 [DOI] [PubMed] [Google Scholar]
- 92.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL 3rd, Araoz C (1989) Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci USA 86:7611–7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grolla AA, Fakhfouri G, Balzaretti G, Marcello E, Gardoni F, Canonico PL, DiLuca M, Genazzani AA, Lim D (2013) Aβ leads to Ca2+ signaling alterations and transcriptional changes in glial cells. Neurobiol Aging 34:511–522 [DOI] [PubMed] [Google Scholar]
- 94.Grolla AA, Sim JA, Lim D, Rodriguez JJ, Genazzani AA, Verkhratsky A (2013) Amyloid-β and Alzheimer’s disease type pathology differentially affects the calcium signalling toolkit in astrocytes from different brain regions. Cell Death Dis 4:e623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Grosche J, Matyash V, Moller T, Verkhratsky A, Reichenbach A, Kettenmann H (1999) Microdomains for neuron-glia interaction: parallel fiber signaling to Bergmann glial cells. Nat Neurosci 2:139–143 [DOI] [PubMed] [Google Scholar]
- 96.Guenette SY (2003) Astrocytes: a cellular player in Aβ clearance and degradation. Trends Mol Med 9:279–280 [DOI] [PubMed] [Google Scholar]
- 97.Hanin I (1996) The AF64A model of cholinergic hypofunction: an update. Life Sci 58:1955–1964 [DOI] [PubMed] [Google Scholar]
- 98.Hardy J (2006) Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr Alzheimer Res 3:71–73 [DOI] [PubMed] [Google Scholar]
- 99.Hardy J (2009) The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J Neurochem 110:1129–1134 [DOI] [PubMed] [Google Scholar]
- 100.Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297:353–356 [DOI] [PubMed] [Google Scholar]
- 101.Hartlage-Rubsamen M, Zeitschel U, Apelt J, Gartner U, Franke H, Stahl T, Gunther A, Schliebs R, Penkowa M, Bigl V, Rossner S (2003) Astrocytic expression of the Alzheimer’s disease β-secretase (BACE1) is stimulus-dependent. Glia 41:169–179 [DOI] [PubMed] [Google Scholar]
- 102.Haug H, Eggers R (1991) Morphometry of the human cortex cerebri and corpus striatum during aging. Neurobiol Aging 12:336–338; discussion 352–335 [DOI] [PubMed] [Google Scholar]
- 103.Haughey NJ, Mattson MP (2003) Alzheimer’s amyloid β-peptide enhances ATP/gap junction-mediated calcium-wave propagation in astrocytes. Neuromolecular Med 3:173–180 [DOI] [PubMed] [Google Scholar]
- 104.Hazell AS (2009) Astrocytes are a major target in thiamine deficiency and Wernicke’s encephalopathy. Neurochem Int 55:129–135 [DOI] [PubMed] [Google Scholar]
- 105.Hazell AS, Sheedy D, Oanea R, Aghourian M, Sun S, Jung JY, Wang D, Wang C (2009) Loss of astrocytic glutamate transporters in Wernicke encephalopathy. Glia 58:148–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Heneka MT, Rodriguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration. Brain Res Rev (in press) [DOI] [PubMed] [Google Scholar]
- 107.Heneka MT, Sastre M, Dumitrescu-Ozimek L, Dewachter I, Walter J, Klockgether T, Van Leuven F (2005) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APPV717I transgenic mice. J Neuroinflam 2:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Henry V, Paille V, Lelan F, Brachet P, Damier P (2009) Kinetics of microglial activation and degeneration of dopamine-containing neurons in a rat model of Parkinson disease induced by 6-hydroxydopamine. J Neuropathol Exp Neurol 68:1092–1102 [DOI] [PubMed] [Google Scholar]
- 109.Hodges JR (2006) Alzheimer’s centennial legacy: origins, landmarks and the current status of knowledge concerning cognitive aspects. Brain 129:2811–2822 [DOI] [PubMed] [Google Scholar]
- 110.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274:99–102 [DOI] [PubMed] [Google Scholar]
- 111.Humphry GM (1889) Old age. Macmillan & Bowes, Cambridge [Google Scholar]
- 112.Iadecola C, Nedergaard M (2007) Glial regulation of the cerebral microvasculature. Nat Neurosci 10:1369–1376 [DOI] [PubMed] [Google Scholar]
- 113.Ishihara T, Higuchi M, Zhang B, Yoshiyama Y, Hong M, Trojanowski JQ, Lee VM (2001) Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J Neurosci 21:6026–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jalonen TO, Charniga CJ, Wielt DB (1997) β-Amyloid peptide-induced morphological changes coincide with increased K+ and Cl- channel activity in rat cortical astrocytes. Brain Res 746:85–97 [DOI] [PubMed] [Google Scholar]
- 115.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, St George-Hyslop P, Westaway D (2000) A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408:979–982 [DOI] [PubMed] [Google Scholar]
- 116.Jellinger KA (2008) Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis 5:118–121 [DOI] [PubMed] [Google Scholar]
- 117.Jin SM, Cho HJ, Kim YW, Hwang JY, Mook-Jung I (2012) Abeta-induced Ca(2+) influx regulates astrocytic BACE1 expression via calcineurin/NFAT4 signals. Biochem Biophys Res Commun 425:649–655 [DOI] [PubMed] [Google Scholar]
- 118.Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee DY, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ (2014) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20:886–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jones VC, Atkinson-Dell R, Verkhratsky A, Mohamet L (2017) Aberrant iPSC-derived human astrocytes in Alzheimer’s disease. Cell Death Dis 8:e2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kanemaru K, Kubota J, Sekiya H, Hirose K, Okubo Y, Iino M (2013) Calcium-dependent N-cadherin up-regulation mediates reactive astrogliosis and neuroprotection after brain injury. Proc Natl Acad Sci USA 110:11612–11617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Karenberg A, Forstl H (2006) Dementia in the Greco-Roman world. J Neurol Sci 244:5–9 [DOI] [PubMed] [Google Scholar]
- 122.Karran E, Mercken M, De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10:698–712 [DOI] [PubMed] [Google Scholar]
- 123.Kaul M, Lipton SA (2006) Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr HIV Res 4:307–318 [DOI] [PubMed] [Google Scholar]
- 124.Kersaitis C, Halliday GM, Kril JJ (2004) Regional and cellular pathology in frontotemporal dementia: relationship to stage of disease in cases with and without Pick bodies. Acta Neuropathol 108:515–523 [DOI] [PubMed] [Google Scholar]
- 125.Khachaturian ZS (1987) Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging 8:345–346 [DOI] [PubMed] [Google Scholar]
- 126.Kim K, Lee SG, Kegelman TP, Su ZZ, Das SK, Dash R, Dasgupta S, Barral PM, Hedvat M, Diaz P, Reed JC, Stebbins JL, Pellecchia M, Sarkar D, Fisher PB (2011) Role of excitatory amino acid transporter-2 (EAAT2) and glutamate in neurodegeneration: opportunities for developing novel therapeutics. J Cell Physiol 226:2484–2493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kirischuk S, Kettenmann H, Verkhratsky A (1997) Na+/Ca2+ exchanger modulates kainate-triggered Ca2+ signaling in Bergmann glial cells in situ. FASEB J 11:566–572 [DOI] [PubMed] [Google Scholar]
- 128.Kirischuk S, Kettenmann H, Verkhratsky A (2007) Membrane currents and cytoplasmic sodium transients generated by glutamate transport in Bergmann glial cells. Pflugers Arch 454:245–252 [DOI] [PubMed] [Google Scholar]
- 129.Kirischuk S, Moller T, Voitenko N, Kettenmann H, Verkhratsky A (1995) ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosci 15:7861–7871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kirischuk S, Parpura V, Verkhratsky A (2012) Sodium dynamics: another key to astroglial excitability? Trends Neurosci 35:497–506 [DOI] [PubMed] [Google Scholar]
- 131.Kirischuk S, Tuschick S, Verkhratsky A, Kettenmann H (1996) Calcium signalling in mouse Bergmann glial cells mediated by α1-adrenoreceptors and H1 histamine receptors. Eur J Neurosci 8:1198–1208 [DOI] [PubMed] [Google Scholar]
- 132.Kloskowska E, Pham TM, Nilsson T, Zhu S, Oberg J, Codita A, Pedersen LA, Pedersen JT, Malkiewicz K, Winblad B, Folkesson R, Benedikz E (2010) Cognitive impairment in the Tg6590 transgenic rat model of Alzheimer’s disease. J Cell Mol Med 14:1816–1823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Knight RA, Verkhratsky A (2010) Neurodegenerative diseases: failures in brain connectivity? Cell Death Differ 17:1069–1070 [DOI] [PubMed] [Google Scholar]
- 134.Korczyn AD (2008) The amyloid cascade hypothesis. Alzheimers Dement 4:176–178 [DOI] [PubMed] [Google Scholar]
- 135.Korsakoff SS (1889) Kopcakov, C.C. Pcixiqeckoe pacctpo ctvo v coqetanii c mno ectvennym nevpitom (psychosis polineuritica, s. cerebropathia psychica toxaemica). English translation: Korsakoff SS. Psychic disorder in conjunction with multiple neuritis, Translated from Russian by M. Victor and P. Yakovlev, Neurology (1955), 5:394–406. Med obozp 32:3–18 [Google Scholar]
- 136.Kraepelin E (1910) Psychiatrie: Ein Lehrbuch fuer Studierende und Arzte. Johann Ambrosius Barth, Leipzig [Google Scholar]
- 137.Kraft AW, Hu X, Yoon H, Yan P, Xiao Q, Wang Y, Gil SC, Brown J, Wilhelmsson U, Restivo JL, Cirrito JR, Holtzman DM, Kim J, Pekny M, Lee JM (2013) Attenuating astrocyte activation accelerates plaque pathogenesis in APP/PS1 mice. FASEB J 27:187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ (2009) Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science 323:1211–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kulijewicz-Nawrot M, Verkhratsky A, Chvatal A, Sykova E, Rodriguez JJ (2012) Astrocytic cytoskeletal atrophy in the medial prefrontal cortex of a triple transgenic mouse model of Alzheimer’s disease. J Anat 221:252–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kuo MF, Paulus W, Nitsche MA (2014) Therapeutic effects of non-invasive brain stimulation with direct currents (tDCS) in neuropsychiatric diseases. Neuroimage 85(Pt 3):948–960 [DOI] [PubMed] [Google Scholar]
- 141.Kurumatani T, Fastbom J, Bonkale WL, Bogdanovic N, Winblad B, Ohm TG, Cowburn RF (1998) Loss of inositol 1,4,5-trisphosphate receptor sites and decreased PKC levels correlate with staging of Alzheimer’s disease neurofibrillary pathology. Brain Res 796:209–221 [DOI] [PubMed] [Google Scholar]
- 142.LaFerla FM, Green KN (2012) Animal models of Alzheimer disease. Cold Spring Harb Perspect Med 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25:11693–11709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lalo U, Palygin O, North RA, Verkhratsky A, Pankratov Y (2011) Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell 10:392–402 [DOI] [PubMed] [Google Scholar]
- 145.Lalo U, Rasooli-Nejad S, Pankratov Y (2014) Exocytosis of gliotransmitters from cortical astrocytes: implications for synaptic plasticity and aging. Biochem Soc Trans 42:1275–1281 [DOI] [PubMed] [Google Scholar]
- 146.Landfield PW (1987) ‘Increased calcium-current’ hypothesis of brain aging. Neurobiol Aging 8:346–347 [DOI] [PubMed] [Google Scholar]
- 147.Landfield PW, Pitler TA (1984) Prolonged Ca2+ -dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science 226:1089–1092 [DOI] [PubMed] [Google Scholar]
- 148.Langer J, Gerkau NJ, Derouiche A, Kleinhans C, Moshrefi-Ravasdjani B, Fredrich M, Kafitz KW, Seifert G, Steinhauser C, Rose CR (2017) Rapid sodium signaling couples glutamate uptake to breakdown of ATP in perivascular astrocyte endfeet. Glia 65:293–308 [DOI] [PubMed] [Google Scholar]
- 149.Langer J, Rose CR (2009) Synaptically induced sodium signals in hippocampal astrocytes in situ. J Physiol 587:5859–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Langer J, Stephan J, Theis M, Rose CR (2012) Gap junctions mediate intercellular spread of sodium between hippocampal astrocytes in situ. Glia 60:239–252 [DOI] [PubMed] [Google Scholar]
- 151.Lappi DA, Esch FS, Barbieri L, Stirpe F, Soria M (1985) Characterization of a Saponaria officinalis seed ribosome-inactivating protein: immunoreactivity and sequence homologies. Biochem Biophys Res Commun 129:934–942 [DOI] [PubMed] [Google Scholar]
- 152.Lazzarini M, Martin S, Mitkovski M, Vozari RR, Stuhmer W, Bel ED (2013) Doxycycline restrains glia and confers neuroprotection in a 6-OHDA Parkinson model. Glia 61:1084–1100 [DOI] [PubMed] [Google Scholar]
- 153.Leanza G, Gulino R, Zorec R (2018) Noradrenergic hypothesis linking neurodegeneration-based cognitive decline and astroglia. Front Mol Neurosci 11:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lee L, Kosuri P, Arancio O (2014) Picomolar amyloid-β peptides enhance spontaneous astrocyte calcium transients. J Alzheimers Dis 38:49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lee M, Schwab C, McGeer PL (2011) Astrocytes are GABAergic cells that modulate microglial activity. Glia 59:152–165 [DOI] [PubMed] [Google Scholar]
- 156.Lescaudron L, Stein DG (1999) Differences in memory impairment and response to GM1 ganglioside treatment following electrolytic or ibotenic acid lesions of the nucleus basalis magnocellularis. Restor Neurol Neurosci 15:25–37 [PubMed] [Google Scholar]
- 157.Leuba G, Wernli G, Vernay A, Kraftsik R, Mohajeri MH, Saini KD (2005) Neuronal and non-neuronal quantitative BACE immunocytochemical expression in the entorhinohippocampal and frontal regions in Alzheimer’s disease. Dement Geriatr Cogn Disord 19:171–183 [DOI] [PubMed] [Google Scholar]
- 158.Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293:1487–1491 [DOI] [PubMed] [Google Scholar]
- 159.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25:402–405 [DOI] [PubMed] [Google Scholar]
- 160.Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, Niedzielko TL, Schneider LE, Mastroeni D, Caselli R, Kukull W, Morris JC, Hulette CM, Schmechel D, Rogers J, Stephan DA (2008) Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A 105:4441–4446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lim D, Iyer A, Ronco V, Grolla AA, Canonico PL, Aronica E, Genazzani AA (2013) Amyloid β deregulates astroglial mGluR5-mediated calcium signaling via calcineurin and Nf-κB. Glia 61:1134–1145 [DOI] [PubMed] [Google Scholar]
- 162.Lim D, Rodriguez-Arellano JJ, Parpura V, Zorec R, Zeidan-Chulia F, Genazzani AA, Verkhratsky A (2016) Calcium signalling toolkits in astrocytes and spatio-temporal progression of Alzheimer’s disease. Curr Alzheimer Res 13:359–369 [DOI] [PubMed] [Google Scholar]
- 163.Lim D, Ronco V, Grolla AA, Verkhratsky A, Genazzani AA (2014) Glial calcium signalling in Alzheimer’s disease. Rev Physiol Biochem Pharmacol 167:45–65 [DOI] [PubMed] [Google Scholar]
- 164.Linde CI, Baryshnikov SG, Mazzocco-Spezzia A, Golovina VA (2011) Dysregulation of Ca2+ signaling in astrocytes from mice lacking amyloid precursor protein. Am J Physiol Cell Physiol 300:C1502–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lyketsos CG, Olin J (2002) Depression in Alzheimer’s disease: overview and treatment. Biol Psychiatry 52:243–252 [DOI] [PubMed] [Google Scholar]
- 166.Martins RN, Taddei K, Kendall C, Evin G, Bates KA, Harvey AR (2001) Altered expression of apolipoprotein E, amyloid precursor protein and presenilin-1 is associated with chronic reactive gliosis in rat cortical tissue. Neuroscience 106:557–569 [DOI] [PubMed] [Google Scholar]
- 167.Mather M, Harley CW (2016) The locus coeruleus: essential for maintaining cognitive function and the aging brain. Trends Cogn Sci 20:214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Matos M, Augusto E, Oliveira CR, Agostinho P (2008) Amyloid-β peptide decreases glutamate uptake in cultured astrocytes: involvement of oxidative stress and mitogen-activated protein kinase cascades. Neuroscience 156:898–910 [DOI] [PubMed] [Google Scholar]
- 169.Mattison JA, Colman RJ, Beasley TM, Allison DB, Kemnitz JW, Roth GS, Ingram DK, Weindruch R, de Cabo R, Anderson RM (2017) Caloric restriction improves health and survival of rhesus monkeys. Nat Commun 8:14063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mattson MP (2012) Energy intake and exercise as determinants of brain health and vulnerability to injury and disease. Cell Metab 16:706–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mattson MP, Haughey NJ, Nath A (2005) Cell death in HIV dementia. Cell Death Differ 12(Suppl 1):893–904 [DOI] [PubMed] [Google Scholar]
- 172.Maurer K, Volk S, Gerbaldo H (1997) Auguste D and Alzheimer’s disease. Lancet 349:1546–1549 [DOI] [PubMed] [Google Scholar]
- 173.Maurya SK, Rai A, Rai NK, Deshpande S, Jain R, Mudiam MK, Prabhakar YS, Bandy-opadhyay S (2012) Cypermethrin induces astrocyte apoptosis by the disruption of the autocrine/paracrine mode of epidermal growth factor receptor signaling. Toxicol Sci 125:473–487 [DOI] [PubMed] [Google Scholar]
- 174.McAlpine D, Araki S (1958) Minamata disease: an unusual neurological disorder caused by contaminated fish. Lancet 2:629–631 [DOI] [PubMed] [Google Scholar]
- 175.McCay CM, Crowell MF, Maynard LA (1935) The effect of retarded growth upon the length of life span and upon the ultimate body size. J Nutr 10:63–79 [PubMed] [Google Scholar]
- 176.McEntee WJ, Crook TH (1991) Serotonin, memory, and the aging brain. Psychopharmacology 103:143–149 [DOI] [PubMed] [Google Scholar]
- 177.McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A (2008) Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res 1207:225–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Meda L, Baron P, Scarlato G (2001) Glial activation in Alzheimer’s disease: the role of Abeta and its associated proteins. Neurobiol Aging 22:885–893 [DOI] [PubMed] [Google Scholar]
- 179.Mena MA, Casarejos MJ, Carazo A, Paino CL, Garcia de Yebenes J (1996) Glia conditioned medium protects fetal rat midbrain neurones in culture from L-DOPA toxicity. NeuroReport 7:441–445 [DOI] [PubMed] [Google Scholar]
- 180.Mena MA, de Bernardo S, Casarejos MJ, Canals S, Rodriguez-Martin E (2002) The role of astroglia on the survival of dopamine neurons. Mol Neurobiol 25:245–263 [DOI] [PubMed] [Google Scholar]
- 181.Mena MA, Garcia de Yebenes J (2008) Glial cells as players in parkinsonism: the “good,” the “bad,” and the “mysterious” glia. Neuroscientist 14:544–560 [DOI] [PubMed] [Google Scholar]
- 182.Mesulam M, Shaw P, Mash D, Weintraub S (2004) Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol 55:815–828 [DOI] [PubMed] [Google Scholar]
- 183.Moechars D, Lorent K, De Strooper B, Dewachter I, Van Leuven F (1996) Expression in brain of amyloid precursor protein mutated in the alpha-secretase site causes disturbed behavior, neuronal degeneration and premature death in transgenic mice. EMBO J 15:1265–1274 [PMC free article] [PubMed] [Google Scholar]
- 184.Mohamet L, Jones VC, Dayanithi G, Verkhratsky A (2018) Pathological human astroglia in Alzheimer’s disease: opening new horizons with stem cell technology. Future Neurol 13:87–99 [Google Scholar]
- 185.Mohs RC (2005) The clinical syndrome of Alzheimer’s disease: aspects particularly relevant to clinical trials. Genes Brain Behav 4:129–133 [DOI] [PubMed] [Google Scholar]
- 186.Monai H, Ohkura M, Tanaka M, Oe Y, Konno A, Hirai H, Mikoshiba K, Itohara S, Nakai J, Iwai Y, Hirase H (2016) Calcium imaging reveals glial involvement in transcranial direct current stimulation-induced plasticity in mouse brain. Nat Commun 7:11100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann NY Acad Sci 1147:180–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Mouton PR, Pakkenberg B, Gundersen HJ, Price DL (1994) Absolute number and size of pigmented locus coeruleus neurons in young and aged individuals. J Chem Neuroanat 7:185–190 [DOI] [PubMed] [Google Scholar]
- 189.Mrak RE, Griffin WS (2005) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26:349–354 [DOI] [PubMed] [Google Scholar]
- 190.Mravec B, Lejavova K, Cubinkova V (2014) Locus (coeruleus) minoris resistentiae in pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 11:992–1001 [DOI] [PubMed] [Google Scholar]
- 191.Mulligan SJ, MacVicar BA (2004) Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature 431:195–199 [DOI] [PubMed] [Google Scholar]
- 192.Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A β 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209 [DOI] [PubMed] [Google Scholar]
- 193.Nagele RG, Wegiel J, Venkataraman V, Imaki H, Wang KC, Wegiel J (2004) Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiol Aging 25:663–674 [DOI] [PubMed] [Google Scholar]
- 194.Ni M, Li X, Rocha JB, Farina M, Aschner M (2012) Glia and methylmercury neurotoxicity. J Toxicol Environ Health A 75:1091–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Nicoll JA, Weller RO (2003) A new role for astrocytes: β-amyloid homeostasis and degradation. Trends Mol Med 9:281–282 [DOI] [PubMed] [Google Scholar]
- 196.Nimmerjahn A, Mukamel EA, Schnitzer MJ (2009) Motor behavior activates Bergmann glial networks. Neuron 62:400–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nitsche MA, Cohen LG, Wassermann EM, Priori A, Lang N, Antal A, Paulus W, Hummel F, Boggio PS, Fregni F, Pascual-Leone A (2008) Transcranial direct current stimulation: state of the art 2008. Brain Stimul 1:206–223 [DOI] [PubMed] [Google Scholar]
- 198.Noristani HN, Olabarria M, Verkhratsky A, Rodriguez JJ (2010) Serotonin fibre sprouting and increase in serotonin transporter immunoreactivity in the CA1 area of hippocampus in a triple transgenic mouse model of Alzheimer’s disease. Eur J Neurosci 32:71–79 [DOI] [PubMed] [Google Scholar]
- 199.Norris CM, Kadish I, Blalock EM, Chen KC, Thibault V, Porter NM, Landfield PW, Kraner SD (2005) Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J Neurosci 25:4649–4658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Oddo S, Caccamo A, Kitazawa M, Tseng BP, LaFerla FM (2003) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease. Neurobiol Aging 24:1063–1070 [DOI] [PubMed] [Google Scholar]
- 202.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 39:409–421 [DOI] [PubMed] [Google Scholar]
- 203.Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2010) Concomitant astroglial atrophy and astrogliosis in a triple transgenic animal model of Alzheimer’s disease. Glia 58:831–838 [DOI] [PubMed] [Google Scholar]
- 204.Olabarria M, Noristani HN, Verkhratsky A, Rodriguez JJ (2011) Age-dependent decrease in glutamine synthetase expression in the hippocampal astroglia of the triple transgenic Alzheimer’s disease mouse model: mechanism for deficient glutamatergic transmission? Mol Neurodegener 6:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Orre M, Kamphuis W, Osborn LM, Jansen AHP, Kooijman L, Bossers K, Hol EM (2014) Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol Aging 35:2746–2760 [DOI] [PubMed] [Google Scholar]
- 206.Ovsepian SV, O’Leary VB, Zaborszky L, Ntziachristos V, Dolly JO (2018) Amyloid plaques of Alzheimer’s disease as hotspots of glutamatergic activity. Neuroscientist. 10.1177/1073858418791128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Palop JJ, Mucke L (2010) Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci 13:812–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Pankratov Y, Lalo U (2015) Role for astroglial α1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front Cell Neurosci 9:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Parpura-Gill A, Beitz D, Uemura E (1997) The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res 754:65–71 [DOI] [PubMed] [Google Scholar]
- 210.Paukert M, Agarwal A, Cha J, Doze VA, Kang JU, Bergles DE (2014) Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 82:1263–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Pavia J, Alberch J, Alvarez I, Toledano A, de Ceballos ML (2000) Repeated intracerebroventricular administration of beta-amyloid(25–35) to rats decreases muscarinic receptors in cerebral cortex. Neurosci Lett 278:69–72 [DOI] [PubMed] [Google Scholar]
- 212.Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, Hol EM, Sofroniew MV, Verkhratsky A (2016) Astrocytes: a central element in neurological diseases. Acta Neuropathol 131:323–345 [DOI] [PubMed] [Google Scholar]
- 213.Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32:1152–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Pepeu G, Marconcini Pepeu I (1994) Dysfunction of the brain cholinergic system during aging and after lesions of the nucleus basalis of Meynert. J Neural Transm Suppl 44:189–194 [DOI] [PubMed] [Google Scholar]
- 215.Picklo MJ, Wiley RG, Lappi DA, Robertson D (1994) Noradrenergic lesioning with an anti-dopamine beta-hydroxylase immunotoxin. Brain Res 666:195–200 [DOI] [PubMed] [Google Scholar]
- 216.Pirttimaki TM, Codadu NK, Awni A, Pratik P, Nagel DA, Hill EJ, Dineley KT, Parri HR (2013) α7 Nicotinic receptor-mediated astrocytic gliotransmitter release: Aβ effects in a preclinical Alzheimer’s mouse model. PLoS ONE 8:e81828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Plata A, Popov A, Denisov P, Bychkov M, Brazhe A, Lyukmanova E, Natalia Lazareva N, Verkhratsky A, Semyanov A (2019) An astrocytic basis of caloric restriction action on the brain plasticity. BioRxiv [Google Scholar]
- 218.Potokar M, Kreft M, Pangrsic T, Zorec R (2005) Vesicle mobility studied in cultured astrocytes. Biochem Biophys Res Commun 329:678–683 [DOI] [PubMed] [Google Scholar]
- 219.Potokar M, Stenovec M, Gabrijel M, Li L, Kreft M, Grilc S, Pekny M, Zorec R (2010) Intermediate filaments attenuate stimulation-dependent mobility of endosomes/lysosomes in astrocytes. Glia 58:1208–1219 [DOI] [PubMed] [Google Scholar]
- 220.Potokar M, Vardjan N, Stenovec M, Gabrijel M, Trkov S, Jorgacevski J, Kreft M, Zorec R (2013) Astrocytic vesicle mobility in health and disease. Int J Mol Sci 14:11238–11258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Potts R, Leech RW (2005) Thalamic dementia: an example of primary astroglial dystrophy of Seitelberger. Clin Neuropathol 24:271–275 [PubMed] [Google Scholar]
- 222.Pressey SN, Smith DA, Wong AM, Platt FM, Cooper JD (2012) Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol Dis 45:1086–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Rai A, Maurya SK, Sharma R, Ali S (2013) Down-regulated GFAPα: a major player in heavy metal induced astrocyte damage. Toxicol Mech Methods 23:99–107 [DOI] [PubMed] [Google Scholar]
- 224.Ramsden M, Kotilinek L, Forster C, Paulson J, McGowan E, SantaCruz K, Guimaraes A, Yue M, Lewis J, Carlson G, Hutton M, Ashe KH (2005) Age-dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci 25:10637–10647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Redlich E (1898) Ueber miliare Sklerose der Hirnrinde bei seniler Atrophie. J Psychiat Neurol 17:208–216 [Google Scholar]
- 226.Reitz C (2012) Alzheimer’s disease and the amyloid cascade hypothesis: a critical review. Int J Alzheimers Dis 2012:369808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Reyes RC, Verkhratsky A, Parpura V (2013) TRPC1-mediated Ca2+ and Na+ signalling in astroglia: differential filtering of extracellular cations. Cell Calcium 54:120–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ribe EM, Perez M, Puig B, Gich I, Lim F, Cuadrado M, Sesma T, Catena S, Sanchez B, Nieto M, Gomez-Ramos P, Moran MA, Cabodevilla F, Samaranch L, Ortiz L, Perez A, Ferrer I, Avila J, Gomez-Isla T (2005) Accelerated amyloid deposition, neurofibrillary degeneration and neuronal loss in double mutant APP/tau transgenic mice. Neurobiol Dis 20:814–822 [DOI] [PubMed] [Google Scholar]
- 229.Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Scholl M, Chiotis K, Thordardottir S, Graff C, Wall A, Langstrom B, Nordberg A (2016) Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 139:922–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Rodriguez JJ, Butt AM, Gardenal E, Parpura V, Verkhratsky A (2016) Complex and differential glial responses in Alzheimer’s disease and ageing. Curr Alzheimer Res 13:343–358 [DOI] [PubMed] [Google Scholar]
- 231.Rodriguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, Verkhratsky A (2008) Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3:e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rodríguez JJ, Matute C, Verkhratsky AI(ed) (2011) Neuroglia in Alzheimer’s disease In:Schemes E, Spray DC (eds) Astrocytes: wiring the brain, pp 311–337. Taylor & Francis Inc [Google Scholar]
- 233.Rodriguez JJ, Noristani HN, Olabarria M, Fletcher J, Somerville TD, Yeh CY, Verkhratsky A (2011) Voluntary running and environmental enrichment restores impaired hippocampal neurogenesis in a triple transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res 8:707–717 [DOI] [PubMed] [Google Scholar]
- 234.Rodriguez JJ, Noristani HN, Verkhratsky A (2012) The serotonergic system in ageing and Alzheimer’s disease. Prog Neurobiol 99:15–41 [DOI] [PubMed] [Google Scholar]
- 235.Rodriguez JJ, Olabarria M, Chvatal A, Verkhratsky A (2009) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16:378–385 [DOI] [PubMed] [Google Scholar]
- 236.Rodriguez JJ, Terzieva S, Olabarria M, Lanza RG, Verkhratsky A (2013) Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis 4:e678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rodriguez JJ, Verkhratsky A (2011) Neurogenesis in Alzheimer’s disease. J Anat 219:78–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Rodriguez JJ, Verkhratsky A (2011) Neuroglial roots of neurodegenerative diseases? Mol Neurobiol 43:87–96 [DOI] [PubMed] [Google Scholar]
- 239.Ronco V, Grolla AA, Glasnov TN, Canonico PL, Verkhratsky A, Genazzani AA, Lim D (2014) Differential deregulation of astrocytic calcium signalling by amyloid-β, TNFα, IL-1β and LPS. Cell Calcium 55:219–229 [DOI] [PubMed] [Google Scholar]
- 240.Rose CR, Verkhratsky A (2016) Principles of sodium homeostasis and sodium signalling in astroglia. Glia [DOI] [PubMed] [Google Scholar]
- 241.Rossi D, Brambilla L, Valori CF, Roncoroni C, Crugnola A, Yokota T, Bredesen DE, Volterra A (2008) Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ 15:1691–1700 [DOI] [PubMed] [Google Scholar]
- 242.Rossi D, Volterra A (2009) Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull 80:224–232 [DOI] [PubMed] [Google Scholar]
- 243.Rossner S, Lange-Dohna C, Zeitschel U, Perez-Polo JR (2005) Alzheimer’s disease beta-secretase BACE1 is not a neuron-specific enzyme. J Neurochem 92:226–234 [DOI] [PubMed] [Google Scholar]
- 244.Roth M, Tomlinson BE, Blessed G (1966) Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature 209:109–110 [DOI] [PubMed] [Google Scholar]
- 245.Roth M, Tomlinson BE, Blessed G (1967) The relationship between quantitative measures of dementia and of degenerative changes in the cerebral grey matter of elderly subjects. Proc R Soc Med 60:254–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Rusakov DA (2015) Disentangling calcium-driven astrocyte physiology. Nat Rev Neurosci 16:226–233 [DOI] [PubMed] [Google Scholar]
- 247.Samuel W, Masliah E, Hill LR, Butters N, Terry R (1994) Hippocampal connectivity and Alzheimer’s dementia: effects of synapse loss and tangle frequency in a two-component model. Neurology 44:2081–2088 [DOI] [PubMed] [Google Scholar]
- 248.Sani S, Traul D, Klink A, Niaraki N, Gonzalo-Ruiz A, Wu CK, Geula C (2003) Distribution, progression and chemical composition of cortical amyloid-beta deposits in aged rhesus monkeys: similarities to the human. Acta Neuropathol 105:145–156 [DOI] [PubMed] [Google Scholar]
- 249.Satoh A, Iijima KM (2019) Roles of tau pathology in the locus coeruleus (LC) in age-associated pathophysiology and Alzheimer’s disease pathogenesis: potential strategies to protect the LC against aging. Brain Res 1702:17–28 [DOI] [PubMed] [Google Scholar]
- 250.Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR (2005) Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 18:602–617 [DOI] [PubMed] [Google Scholar]
- 251.Scheff SW, Price DA, Schmitt FA, Mufson EJ (2006) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27:1372–1384 [DOI] [PubMed] [Google Scholar]
- 252.Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L (2006) Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol 169:599–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Schousboe A, Wellendorph P, Frolund B, Clausen RP, Krogsgaard-Larsen P (2017) Astrocytic GABA transporters: pharmacological properties and targets for antiepileptic drugs. Adv Neurobiol 16:283–296 [DOI] [PubMed] [Google Scholar]
- 254.Selkoe DJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:741–766 [DOI] [PubMed] [Google Scholar]
- 255.Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298:789–791 [DOI] [PubMed] [Google Scholar]
- 256.Semyanov A (2019) Spatiotemporal pattern of calcium activity in astrocytic network. Cell Calcium 78:15–25 [DOI] [PubMed] [Google Scholar]
- 257.Shastry BS, Giblin FJ (1999) Genes and susceptible loci of Alzheimer’s disease. Brain Res Bull 48:121–127 [DOI] [PubMed] [Google Scholar]
- 258.Shaughnessy LW, Barone S Jr, Mundy WR, Herr DW, Tilson HA (1994) Comparison of intracranial infusions of colchicine and ibotenic acid as models of neurodegeneration in the basal forebrain. Brain Res 637:15–26 [DOI] [PubMed] [Google Scholar]
- 259.Shigetomi E, Kracun S, Sofroniew MV, Khakh BS (2010) A genetically targeted optical sensor to monitor calcium signals in astrocyte processes. Nat Neurosci 13:759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sidoryk-Wegrzynowicz M, Aschner M (2013) Role of astrocytes in manganese mediated neurotoxicity. BMC Pharmacol Toxicol 14:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Simpson JE, Ince PG, Lace G, Forster G, Shaw PJ, Matthews F, Savva G, Brayne C, Wharton SB (2010) Astrocyte phenotype in relation to Alzheimer-type pathology in the ageing brain. Neurobiol Aging 31:578–590 [DOI] [PubMed] [Google Scholar]
- 262.Simpson JE, Ince PG, Shaw PJ, Heath PR, Raman R, Garwood CJ, Gelsthorpe C, Baxter L, Forster G, Matthews FE, Brayne C, Wharton SB (2011) Microarray analysis of the astrocyte transcriptome in the aging brain: relationship to Alzheimer’s pathology and APOE genotype. Neurobiol Aging 32:1795–1807 [DOI] [PubMed] [Google Scholar]
- 263.Son SM, Cha MY, Choi H, Kang S, Choi H, Lee MS, Park SA, Mook-Jung I (2016) Insulin-degrading enzyme secretion from astrocytes is mediated by an autophagy-based unconventional secretory pathway in Alzheimer disease. Autophagy 12:784–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Soucek T, Cumming R, Dargusch R, Maher P, Schubert D (2003) The regulation of glucose metabolism by HIF-1 mediates a neuroprotective response to amyloid beta peptide. Neuron 39:43–56 [DOI] [PubMed] [Google Scholar]
- 265.Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, Khakh BS (2015) Ca2+ signaling in astrocytes from Ip3r2−/− mice in brain slices and during startle responses in vivo. Nat Neurosci 18:708–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Stenovec M, Kreft M, Grilc S, Pangrsic T, Zorec R (2008) EAAT2 density at the astrocyte plasma membrane and Ca2+-regulated exocytosis. Mol Membr Biol 25:203–215 [DOI] [PubMed] [Google Scholar]
- 267.Stenovec M, Lasic E, Dominkus PP, Bobnar ST, Zorec R, Lenassi M, Kreft M (2019) Slow release of HIV-1 protein nef from vesicle-like structures is inhibited by cytosolic calcium elevation in single human microglia. Mol Neurobiol 56:102–118 [DOI] [PubMed] [Google Scholar]
- 268.Stenovec M, Trkov Bobnar S, Smolic T, Kreft M, Parpura V, Zorec R (2018) Presenilin PS1E9 disrupts mobility of secretory organelles in rat astrocytes. Acta Physiol (Oxf) 223:e13046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Stenovec M, Trkov S, Lasic E, Terzieva S, Kreft M, Rodriguez Arellano JJ, Parpura V, Verkhratsky A, Zorec R (2016) Expression of familial Alzheimer disease presenilin 1 gene attenuates vesicle traffic and reduces peptide secretion in cultured astrocytes devoid of pathologic tissue environment. Glia 64:317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Stix B, Reiser G (1998) β-amyloid peptide 25–35 regulates basal and hormone-stimulated Ca2+ levels in cultured rat astrocytes. Neurosci Lett 243:121–124 [DOI] [PubMed] [Google Scholar]
- 271.Studelska DR, Brimijoin S (1989) Partial isolation of two classes of dopamine beta-hydroxylase-containing particles undergoing rapid axonal transport in rat sciatic nerve. J Neurochem 53:622–631 [DOI] [PubMed] [Google Scholar]
- 272.Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94:13287–13292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Stutzmann GE (2007) The pathogenesis of Alzheimers disease is it a lifelong “calciumopathy”? Neuroscientist 13:546–559 [DOI] [PubMed] [Google Scholar]
- 274.Stutzmann GE, Mattson MP (2011) Endoplasmic reticulum Ca2+ handling in excitable cells in health and disease. Pharmacol Rev 63:700–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Suarez-Fernandez MB, Soldado AB, Sanz-Medel A, Vega JA, Novelli A, Fernandez-Sanchez MT (1999) Aluminum-induced degeneration of astrocytes occurs via apoptosis and results in neuronal death. Brain Res 835:125–136 [DOI] [PubMed] [Google Scholar]
- 276.Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, Markus HS, Salman RA, Allan SM, Quinn TJ, Kalaria RN, Werring DJ, Carare RO, Touyz RM, Williams SCR, Moskowitz MA, Katusic ZS, Lutz SE, Lazarov O, Minshall RD, Rehman J, Davis TP, Wellington CL, Gonzalez HM, Yuan C, Lockhart SN, Hughes TM, Chen CLH, Sachdev P, O’Brien JT, Skoog I, Pantoni L, Gustafson DR, Biessels GJ, Wallin A, Smith EE, Mok V, Wong A, Passmore P, Barkof F, Muller M, Breteler MMB, Roman GC, Hamel E, Seshadri S, Gottesman RF, van Buchem MA, Arvanitakis Z, Schneider JA, Drewes LR, Hachinski V, Finch CE, Toga AW, Wardlaw JM, Zlokovic BV (2019) Vascular dysfunction-The disregarded partner of Alzheimer’s disease. Alzheimers Dement 15:158–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV (2019) Blood-brain barrier: from physiology to disease and back. Physiol Rev 99:21–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Takano T, Han X, Deane R, Zlokovic B, Nedergaard M (2007) Two-photon imaging of astrocytic Ca2+ signaling and the microvasculature in experimental mice models of Alzheimer’s disease. Ann N Y Acad Sci 1097:40–50 [DOI] [PubMed] [Google Scholar]
- 279.Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M (2006) Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–267 [DOI] [PubMed] [Google Scholar]
- 280.Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22:133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A (2002) Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci USA 99:13896–13901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Terry RD (2000) Cell death or synaptic loss in Alzheimer disease. J Neuropathol Exp Neurol 59:1118–1119 [DOI] [PubMed] [Google Scholar]
- 283.Thompson KA, McArthur JC, Wesselingh SL (2001) Correlation between neurological progression and astrocyte apoptosis in HIV-associated dementia. Ann Neurol 49:745–752 [DOI] [PubMed] [Google Scholar]
- 284.Thompson PM, Hayashi KM, de Zubicaray G, Janke AL, Rose SE, Semple J, Herman D, Hong MS, Dittmer SS, Doddrell DM, Toga AW (2003) Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci 23:994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Thompson PM, Hayashi KM, Dutton RA, Chiang MC, Leow AD, Sowell ER, De Zubicaray G, Becker JT, Lopez OL, Aizenstein HJ, Toga AW (2007) Tracking Alzheimer’s disease. Ann NY Acad Sci 1097:183–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Toescu EC, Verkhratsky A (2007) The importance of being subtle: small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 6:267–273 [DOI] [PubMed] [Google Scholar]
- 287.Toescu EC, Verkhratsky A, Landfield PW (2004) Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci 27:614–620 [DOI] [PubMed] [Google Scholar]
- 288.Toivari E, Manninen T, Nahata AK, Jalonen TO, Linne ML (2011) Effects of transmitters and amyloid-beta peptide on calcium signals in rat cortical astrocytes: Fura-2AM measurements and stochastic model simulations. PLoS ONE 6:e17914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Toledano A, Alvarez MI (2004) Lesions and dysfunctions of the nucleus basalis as Alzheimer’s disease models: general and critical overview and analysis of the long-term changes in several excitotoxic models. Curr Alzheimer Res 189–214 [DOI] [PubMed] [Google Scholar]
- 290.Tomlinson BE, Blessed G, Roth M (1970) Observations on the brains of demented old people. J Neurol Sci 11:205–242 [DOI] [PubMed] [Google Scholar]
- 291.Torack R (1996) The early history of senile dementia In: Reisberg B (ed) Alzheimer’s disease: the standard reference. The Free Press, New York, pp 23–28 [Google Scholar]
- 292.Ullian EM, Christopherson KS, Barres BA (2004) Role for glia in synaptogenesis. Glia 47:209–216 [DOI] [PubMed] [Google Scholar]
- 293.Vale-Martinez A, Guillazo-Blanch G, Marti-Nicolovius M, Nadal R, Arevalo-Garcia R, Morgado-Bernal I (2002) Electrolytic and ibotenic acid lesions of the nucleus basalis magnocellularis interrupt long-term retention, but not acquisition of two-way active avoidance, in rats. Exp Brain Res 142:52–66 [DOI] [PubMed] [Google Scholar]
- 294.Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med 18:421–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Vanzani MC, Iacono RF, Caccuri RL, Troncoso AR, Berria MI (2006) Regional differences in astrocyte activation in HIV-associated dementia. Medicina (B Aires) 66:108–112 [PubMed] [Google Scholar]
- 296.Vardjan N, Gabrijel M, Potokar M, Svajger U, Kreft M, Jeras M, de Pablo Y, Faiz M, Pekny M, Zorec R (2012) IFN-γ-induced increase in the mobility of MHC class II compartments in astrocytes depends on intermediate filaments. J Neuroinflammation 9:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Vardjan N, Kreft M, Zorec R (2014) Dynamics of β-adrenergic/cAMP signaling and morphological changes in cultured astrocytes. Glia 62:566–579 [DOI] [PubMed] [Google Scholar]
- 298.Vardjan N, Verkhratsky A, Zorec R (2015) Pathologic potential of astrocytic vesicle traffic: new targets to treat neurologic diseases? Cell Transplant 24:599–612 [DOI] [PubMed] [Google Scholar]
- 299.Verkhratsky A, Kettenmann H (1996) Calcium signalling in glial cells. Trends Neurosci 19:346–352 [DOI] [PubMed] [Google Scholar]
- 300.Verkhratsky A, Marutle A, Rodriguez-Arellano JJ, Nordberg A (2015) Glial asthenia and functional paralysis: a new perspective on neurodegeneration and Alzheimer’s disease. Neuroscientist 21:552–568 [DOI] [PubMed] [Google Scholar]
- 301.Verkhratsky A, Matteoli M, Parpura V, Mothet JP, Zorec R (2016) Astrocytes as secretory cells of the central nervous system: idiosyncrasies of vesicular secretion. EMBO J 35:239–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Verkhratsky A, Mattson MP, Toescu EC (2004) Aging in the mind. Trends Neurosci 27:577–578 [DOI] [PubMed] [Google Scholar]
- 303.Verkhratsky A, Nedergaard M (2014) Astroglial cradle in the life of the synapse. Philos Trans R Soc Lond B Biol Sci 369:20130595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Verkhratsky A, Nedergaard M (2016) The homeostatic astroglia emerges from evolutionary specialization of neural cells. Philos Trans R Soc Lond B Biol Sci 371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Verkhratsky A, Nedergaard M (2018) Physiology of astroglia. Physiol Rev 98:239–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ (2010) Astrocytes in Alzheimer’s disease. Neurotherapeutics 7:399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Verkhratsky A, Parpura V (2016) Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol Dis 85:254–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Verkhratsky A, Parpura V, Rodriguez JJ (2014) Neurodegeneration and neuroglia: emphasis on astroglia in Alzheimer’s disease In: Parpura V, Verkhratsky A (eds) Pathological potential of neuroglia: possible new targets for medical intervention. Springer, Heidelberg, pp 264–291 [Google Scholar]
- 309.Verkhratsky A, Rodriguez-Arellano JJ, Parpura V, Zorec R (2017) Astroglial calcium signalling in Alzheimer’s disease. Biochem Biophys Res Commun 483:1005–1012 [DOI] [PubMed] [Google Scholar]
- 310.Verkhratsky A, Rodriguez JJ, Parpura V (2013) Astroglia in neurological diseases. Future Neurol 8:149–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Verkhratsky A, Sofroniew MV, Messing A, deLanerolle NC, Rempe D, Rodriguez JJ, Nedergaard M (2012) Neurological diseases as primary gliopathies: a reassessment of neurocentrism. ASN Neuro 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Verkhratsky A, Toescu EC (1998) Calcium and neuronal ageing. Trends Neurosci 21:2–7 [DOI] [PubMed] [Google Scholar]
- 313.Verkhratsky A, Trebak M, Perocchi F, Khananshvili D, Sekler I (2018) Crosslink between calcium and sodium signalling. Exp Physiol 103:157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Verkhratsky A, Untiet V, Rose CR (2019) Ionic signalling in astroglia beyond calcium. J Physiol [DOI] [PubMed] [Google Scholar]
- 315.Verkhratsky A, Zorec R, Parpura V (2017) Stratification of astrocytes in healthy and diseased brain. Brain Pathol 27:629–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Verkhratsky A, Zorec R, Rodriguez JJ, Parpura V (2016) Astroglia dynamics in ageing and Alzheimer’s disease. Curr Opin Pharmacol 26:74–79 [DOI] [PubMed] [Google Scholar]
- 317.Verkhratsky A, Zorec R, Rodriguez JJ, Parpura V (2017) Neuroglia: functional paralysis and reactivity in Alzheimer’s disease and other neurodegenerative pathologies. Adv Neurobiol 15:427–449 [DOI] [PubMed] [Google Scholar]
- 318.Waite JJ, Chen AD, Wardlow ML, Wiley RG, Lappi DA, Thal LJ (1995) 192 immunoglobulin G-saporin produces graded behavioral and biochemical changes accompanying the loss of cholinergic neurons of the basal forebrain and cerebellar Purkinje cells. Neuroscience 65:463–476 [DOI] [PubMed] [Google Scholar]
- 319.Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U (2015) The glutamineglutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res 40:402–409 [DOI] [PubMed] [Google Scholar]
- 320.Weidenheim KM, Dickson DW, Rapin I (2009) Neuropathology of Cockayne syndrome: evidence for impaired development, premature aging, and neurodegeneration. Mech Ageing Dev 130:619–636 [DOI] [PubMed] [Google Scholar]
- 321.Weinshilboum RM (1978) Serum dopamine beta-hydroxylase. Pharmacol Rev 30:133–166 [PubMed] [Google Scholar]
- 322.Wellman CL, Pelleymounter MA (1999) Differential effects of nucleus basalis lesions in young adult and aging rats. Neurobiol Aging 20:381–393 [DOI] [PubMed] [Google Scholar]
- 323.Wernicke C (1881–1883) Lehrbuch der Gehirnkrankheiten für Aerzte und Studirende. Theodor Fischer, Kassel und Berlin [Google Scholar]
- 324.Wharton SB, O’Callaghan JP, Savva GM, Nicoll JA, Matthews F, Simpson JE, Forster G, Shaw PJ, Brayne C, Ince PG (2009) Population variation in glial fibrillary acidic protein levels in brain ageing: relationship to Alzheimer-type pathology and dementia. Dement Geriatr Cogn Disord 27:465–473 [DOI] [PubMed] [Google Scholar]
- 325.Wiley RG (1992) Neural lesioning with ribosome-inactivating proteins: suicide transport and immunolesioning. Trends Neurosci 15:285–290 [DOI] [PubMed] [Google Scholar]
- 326.Wiley RG, Kline IR (2000) Neuronal lesioning with axonally transported toxins. J Neurosci Methods 103:73–82 [DOI] [PubMed] [Google Scholar]
- 327.Wiley RG, Oeltmann TN, Lappi DA (1991) Immunolesioning: selective destruction of neurons using immunotoxin to rat NGF receptor. Brain Res 562:149–153 [DOI] [PubMed] [Google Scholar]
- 328.Wilson CS, Mongin AA (2019) The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci Lett 689:33–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Wilson RS, Nag S, Boyle PA, Hizel LP, Yu L, Buchman AS, Schneider JA, Bennett DA (2013) Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 80:1202–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Winkler J, Thal LJ (1995) Effects of nerve growth factor treatment on rats with lesions of the nucleus basalis magnocellularis produced by ibotenic acid, quisqualic acid, and AMPA. Exp Neurol 136:234–250 [DOI] [PubMed] [Google Scholar]
- 331.Wrenn CC, Picklo MJ, Lappi DA, Robertson D, Wiley RG (1996) Central noradrenergic lesioning using anti-DBH-saporin: anatomical findings. Brain Res 740:175–184 [DOI] [PubMed] [Google Scholar]
- 332.Wu Z, Guo Z, Gearing M, Chen G (2014) Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat Commun 5:4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Wyss-Coray T, Loike JD, Brionne TC, Lu E, Anankov R, Yan F, Silverstein SC, Husemann J (2003) Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat Med 9:453–457 [DOI] [PubMed] [Google Scholar]
- 334.Xiong J, Verkhratsky A, Toescu EC (2002) Changes in mitochondrial status associated with altered Ca2+ homeostasis in aged cerebellar granule neurons in brain slices. J Neurosci 22:10761–10771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Xiu J, Nordberg A, Zhang JT, Guan ZZ (2005) Expression of nicotinic receptors on primary cultures of rat astrocytes and up-regulation of the α7, α4 and β2 subunits in response to nanomolar concentrations of the β-amyloid peptide1–42. Neurochem Int 47:281–290 [DOI] [PubMed] [Google Scholar]
- 336.Yamaguchi H, Sugihara S, Ogawa A, Saido TC, Ihara Y (1998) Diffuse plaques associated with astroglial amyloid beta protein, possibly showing a disappearing stage of senile plaques. Acta Neuropathol 95:217–222 [DOI] [PubMed] [Google Scholar]
- 337.Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW (2008) Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci 11:251–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Yeh CY, Vadhwana B, Verkhratsky A, Rodriguez JJ (2011) Early astrocytic atrophy in the entorhinal cortex of a triple transgenic animal model of Alzheimer’s disease. ASN Neuro 3:271–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Yin Z, Milatovic D, Aschner JL, Syversen T, Rocha JB, Souza DO, Sidoryk M, Albrecht J, Aschner M (2007) Methylmercury induces oxidative injury, alterations in permeability and glutamine transport in cultured astrocytes. Brain Res 1131:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Young LT, Kish SJ, Li PP, Warsh JJ (1988) Decreased brain [3H]inositol 1,4,5-trisphosphate binding in Alzheimer’s disease. Neurosci Lett 94:198–202 [DOI] [PubMed] [Google Scholar]
- 341.Yu WF, Guan ZZ, Bogdanovic N, Nordberg A (2005) High selective expression of α7 nicotinic receptors on astrocytes in the brains of patients with sporadic Alzheimer’s disease and patients carrying Swedish APP 670/671 mutation: a possible association with neuritic plaques. Exp Neurol 192:215–225 [DOI] [PubMed] [Google Scholar]
- 342.Zhao J, O’Connor T, Vassar R (2011) The contribution of activated astrocytes to Abeta production: implications for Alzheimer’s disease pathogenesis. J Neuroinflammation 8:150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhu CW, Sano M (2006) Economic considerations in the management of Alzheimer’s disease. Clin Interv Aging 1:143–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Ziemens D, Oschmann F, Gerkau NJ, Rose CR (2019) Heterogeneity of activity-induced sodium transients between astrocytes of the mouse hippocampus and neocortex: mechanisms and consequences. J Neurosci 39:2620–2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zlokovic BV (2008) The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 57:178–201 [DOI] [PubMed] [Google Scholar]
- 346.Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G (2003) Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50 [DOI] [PubMed] [Google Scholar]
- 347.Zorec R, Parpura V, Vardjan N, Verkhratsky A (2017) Astrocytic face of Alzheimer’s disease. Behav Brain Res 322:250–257 [DOI] [PubMed] [Google Scholar]
- 348.Zorec R, Parpura V, Verkhratsky A (2018) Preventing neurodegeneration by adrenergic astroglial excitation. FEBS J 285:3645–3656 [DOI] [PubMed] [Google Scholar]
- 349.Zorec R, Verkhratsky A, Rodriguez JJ, Parpura V (2016) Astrocytic vesicles and gliotransmitters: Slowness of vesicular release and synaptobrevin2-laden vesicle nanoarchitecture. Neuroscience 323:67–75 [DOI] [PubMed] [Google Scholar]
- 350.Zott B, Busche MA, Sperling RA, Konnerth A (2018) What happens with the circuit in Alzheimer’s disease in mice and humans? Annu Rev Neurosci 41:277–297 [DOI] [PMC free article] [PubMed] [Google Scholar]