

Abstract
Nitrogen mustards (NM) are an important class of chemotherapeutic drugs used in the treatment of malignant tumors. The accepted mechanism of action of NM is through the alkylation of DNA bases. NM-adducts block DNA replication in cancer cells by forming cytotoxic DNA interstrand cross-links. We previously characterized several adducts formed by reaction of bis(2-chloroethyl)ethylamine (NM) with calf thymus (CT) DNA and the MDA-MB-231 mammary tumor cell line. The mono-alkylated N7-guanine (NM-G) adduct, and its cross-link (G-NM-G) were major lesions. The cationic NM-G undergoes a secondary reaction through depurination to form an apurinic (AP) site or reacts with hydroxide to yield the stable ring-opened N5-substituted formamidopyrimidine (NM-Fapy-G) adduct. Both of these lesions are mutagenic and may contribute to secondary tumor development, a major clinical limitation of NM chemotherapy. We established a kinetic model with NM-treated female mice and measured the rates of formation and removal of NM-DNA adducts and AP sites. We employed liquid chromatography-mass spectrometry (LC-MS) to measure NM-G, G-NM-G, and NM–Fapy-G adducts in liver, lung, and spleen over 168 hours. NM-G reached a maximum level within 6 h in all organs and then rapidly declined. The G-NM-G cross-link and NM-FapyG were more persistent with half-lives over three times longer than NM-G. We quantified AP site lesions in the liver and showed that NM treatment increased AP site levels by 3.7-fold over the basal levels at 6 h. The kinetics of AP site repair closely followed the rate of removal of NM-G; however, AP sites remained 1.3-fold above basal levels 168 hours post-treatment with NM. Our data provide new insights into NM-induced DNA damage and biological processing in vivo. The quantitative measurement of the spectrum of NM adducts and AP sites can serve as biomarkers in the design and assessment of the efficacy of novel chemotherapeutic regimens.
Keywords: Nitrogen mustard, chemotherapeutics, DNA adduct, abasic/apurinic site
Graphical Absttract
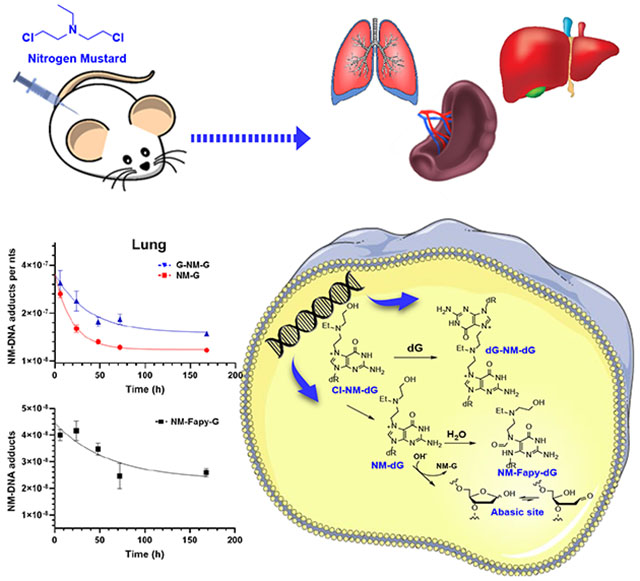
Introduction
Nitrogen mustards (NMs) possess a common bis(2-chloroethyl)amino group and represent the earliest employed anticancer chemotherapeutic agents.1–3 NMs undergo intramolecular cyclization to a highly electrophilic aziridinium ion, which alkylates DNA, most abundantly at the N7-position of deoxyguanosines (Scheme 1). NMs are bis-alkylating agents and can also react with a second deoxyguanosine to form inter- and intrastrand cross-links. Interstrand cross-links preferentially form in GNC sequences and are generally considered the most cytotoxic DNA adduct of NMs even though they are formed in low abundance.4,5 DNA interstrand cross-links interfere with DNA replication, leading to cytotoxicity.6,7 The third substituent of the nitrogen atom can attenuate the reactivity of the nitrogen mustard.1 Nitrogen mustard derivatives, such as melphalan, chlorambucil, and cyclophosphamide (CPA) are currently used as clinical therapeutics for the treatment of many malignant diseases such as ovarian cancer, leukemia, lymphoma, and breast cancer.1
Scheme 1.
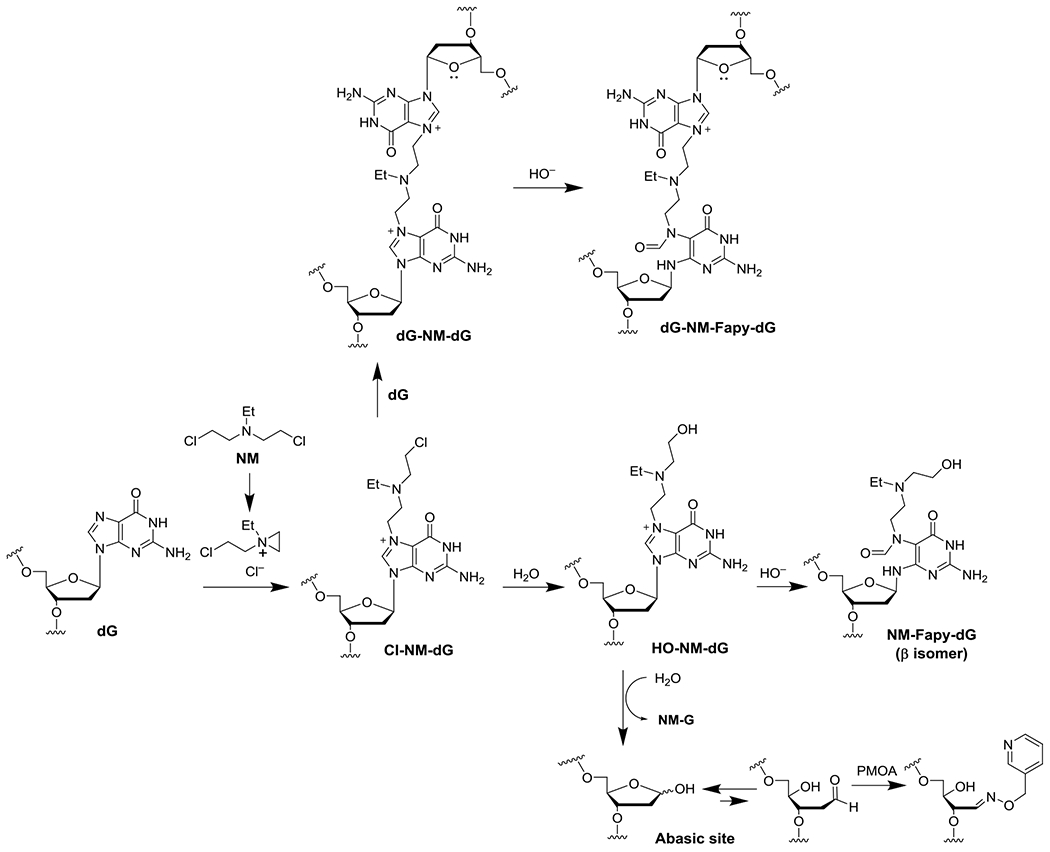
Mechanism of NM-DNA adduct and AP site formation.
Cationic N7-dG adducts can undergo secondary reactions, most commonly depurination to an apurinic (AP) site (Scheme 1).8 The nature of the N7-group affects the rate of depurination. Masta et al. reported the t1/2 of 9.1 hr for NM-dG depurination from calf thymus DNA (pH 7.4, 37 °C) and we subsequently confirmed this result.8–10 AP sites are strong blocks to replication, which can cause double-stranded DNA breaks if not efficiently repaired.11–13 The AP site is non-coding and mutagenic when replicated.14 Some N7-dG adducts add hydroxide at the C8 atom to form the ring-opened N5-substituted formamidopyrimidine (Fapy) adduct.15 The Fapy-dG adduct derived from aflatoxin B1 (AFB1-Fapy-dG) has been observed in vivo, while nitrogen mustard,10 mitomycin C,16 and butadiene epoxide Fapy-dG adducts17 were observed in vitro. The N5-methyl-Fapy-dG (Me-Fapy-dG) adduct was observed from rodents treated with methylating agents;18,19 however, a related study did not observe the formation of Me-Fapy-dG adducts from treated rats.20 The unsubstituted Fapy-dG adducts is a product of oxidative damage and has been observed in vivo.21,22
The methyl, aflatoxin B1, and NM Fapy-dG lesions are mutagenic in cultured cells.23,24 For example, NM-Fapy-dG gave a complex mutation spectrum in cultured COS7 (primate kidney) cells with a frequency of 11–12%; the predominant mutation was G to T transversions.25 An unusual feature of Fapy-dG lesions is they can isomerize to the non-canonical α-anomer.26 Interestingly, the frequency of G to T transversion mutations decreased by ~50% when the α-anomer was selectively removed by pre-treatment with E. coli Endonuclease IV. The mutagenic effects of NM-Fapy-dG and AP sites may increase the risk of secondary malignancies, such as acute non-lymphocytic leukemia.27
AP sites have been exploited to increase drug cytotoxicity and therapeutic efficacy. AP sites exist partially as the ring-opened aldehyde and react with nucleophiles such as methoxyamine (TRC102), to form a stable oxime adduct. The methoxyamine-AP site oxime inhibits the base excision repair (BER) pathway28,29 and enhanced the cytotoxicity of temozolomide30,31 in cultured human breast,32–34 ovarian,35 and colon cancer cells.28,30 Indeed, methoxyamine is part of seven ongoing or recently completed clinical trials.36–42 The reactivity of the ring-opened aldehyde form has also been exploited to quantitate AP sites in DNA (Scheme 1) employing the aldehyde reactive probe43 or O-(pyridin-3-yl-methyl)hydroxylamine (PMOA).44
The extent to which NMs cause DNA adducts and AP sites in vivo are unknown. In the present study, we measure the tissue distribution and concentration-time profile of NM-DNA adducts, and AP sites in female C57BL/6NJ mice by quantitative mass spectrometry-based methods. We measured the NM-adducts as their deglycosylated guanine derivatives. Abbreviations with dG refer to adducts still within DNA.
Experimental Section
Caution:
N,N-bis(2-chloroethyl)ethylamine (NM) is a carcinogen and must be handled in a well-ventilated fume hood with proper use of gloves and protective clothing.
Materials.
O-(Pyridin-3-yl-methyl)hydroxylamine (PMOA), butyraldehyde, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), tris(hydroxymethyl)aminomethane (Tris), EDTA, DNase I (type IV, bovine pancreas), alkaline phosphatase (Escherichia coli), nuclease P1 (Penicillium citrinum), RNase A (bovine pancreas), RNase T1 (Aspergillus oryzae), and proteinase K (Tritirachium album) were purchased from Sigma-Aldrich (St. Louis, MO). Phosphodiesterase I (Crotalus adamanteus venom) and adenosine deaminase were purchased from Worthington Biochemical Corp. (Lakewood, NJ). LC-MS grade solvents were purchased from Fisher Chemical Co. (Pittsburgh, PA). Puregene protein precipitation (PP) solution and DNeasy Blood & Tissue kit were purchased from Qiagen (Germantown, MD). N,N-Bis(2-chloroethyl)ethylamine hydrochloride was purchased from Aaron Chemistry Gmbh (Mittenwald, Germany). Pall 10 kDa filter with omega membrane, MWCO 10 kDa, was purchased from Sigma Aldrich (St. Louis, MO) All other chemicals were ACS grade and purchased from Sigma-Aldrich unless otherwise stated. Strata-X 33 μm Polymeric Reversed Phase 30 mg solid-phase extraction (SPE) cartridge was purchased from Phenomenex (Torrance, CA). Sola SCX cartridges were purchased from Thermo Scientific (Bellafonte, PA). MicroLiter autosampler vials with silanized glass inserts were purchased from Wheaton (Millville, NJ).
Synthesis of standards.
NM-G, G-NM-G, NM-Fapy-G, and PMOA-dR (derivatized abasic site), and their isotopically labeled internal standards [2H5]-NM-G, [2H5]-G-NM-G, [15N5]-NM-FapyG, and [13C5]PMOA-dR, were synthesized according to reported protocols.10,44 Isotopic purities of the internal standards of the NM-DNA adducts exceeded 99.5%,10 and the isotopic purity of [13C5]PMOA-dR exceeded 99.8%.44
Animal studies.
All studies were reviewed and approved by the University of Minnesota Institutional Animal Care and Use Committee. Female C57BL/6NJ mice were 13 to 14 weeks old and a body weight ranging from 18−26 g (The Jackson Laboratories, Bar Harbor, ME). The animals were housed in groups of five per cage, acclimated for one week under standard housing conditions and given an AIN-76A diet and water ad libitum. Thereafter, the mice were injected intraperitoneally with NM (1 μg/μL in sterile 0.9% saline at a dose of 3 mg/kg body weight). The vehicle (100 µL saline) served as the negative control group (n = 5 per group). NM-treated mice were anesthetized with CO2, and sacrificed at 6, 24, 48, 72, and 168 h after dosing. Control mice were sacrificed T0 h after dosing. The liver, lung, and spleen tissues were immediately put on dry ice and frozen at –80 ºC until sample workup.
DNA preparation for quantitation of NM-DNA adducts.
Tissues were thawed on ice and placed in a Petri dish, and cut into small pieces with a scalpel (60 – 100 mg of tissue per tube), followed by homogenization in 4 volumes of TE lysis buffer [Tris buffer (50 mM), EDTA (10 mM), freshly added 2-mercaptoethanol (10 mM), pH 8.0] on ice using a PRO 200 homogenizer (PRO Scientific, Oxford, CT) with a sawtooth type generator (5 mm/7 mm).
DNA was processed as previously reported.10 Briefly, the liver, lung, and spleen tissue homogenates were centrifuged at 3000 g and the nuclear pellet was resuspended in 300 µL of TE buffer and treated with 15 µL RNase A (10 mg/mL) and 1 µL RNase T1 (1000 U/mL) for 90 min at 37 °C. Afterward, proteinase K (20 μL of 20 mg/mL in water) and 10% SDS (0.1 volume) were added and digestion proceeded for 2 h at 37 °C. Nuclear DNA was then extracted using a Qiagen DNeasy Blood & Tissue kit and quantitated with UV to obtain the concentration (assuming that 50 µg/mL of double-stranded DNA gave an absorbance of 1.0 at 260 nm).
A portion of the DNA (10 μg) was digested with DNase I, nuclease P1, alkaline phosphatase, and phosphodiesterase as previously described to determine the extent of RNA contamination.45 The DNA hydrolysate (2.5 µg) was assayed with an Agilent 1260 Infinity HPLC system equipped with a diode array detector DAD (Palo Alto, CA), monitoring UV absorbance at 260 and 280 nm. The chromatographic conditions are reported with a representative HPLC-UV chromatogram of mouse liver DNA in Supporting Information (Figure S1). The % RNA contamination, based on Guo and Ado peak area relative to dG and dA, was less than 2% in all organs.
The DNA (220 µg on average) was assayed independently in duplicate for NM-DNA adducts in each organ. The isolated DNA was subjected to neutral thermal hydrolysis in 3 mL of phosphate buffer (50 mM, pH 7.0) at 37 °C for 72 h in the presence of 350 pg of each internal standard [2H5]-NM-G (1.3 pmol), [15N5]-NM-Fapy-G (1.2 pmol), and [2H5]-G-NM-G (0.88 pmol) corresponding to 2.2, 2.0, and 1.4 adducts per 106 nts, respectively, followed by heating at 95 °C for 4 h.10 The residual DNA backbone was removed with a Pall 10 kDa molecular weight cutoff filter pre-washed with 0.5 mL of H2O, 0.05 M NaOH, followed by H2O. The adducts were enriched by SPE using Sola SCX cartridges and reconstituted in 2 mM ammonium acetate containing 5% CH3CN (50 μL) for LC-MS3 analysis.10
DNA preparation for quantitation of AP sites as the PMOA-dR derivative.
Sample preparation was conducted according to our reported protocol.44 Briefly, chilled HE buffer [1 mL, containing HEPES buffer (50 mM) and EDTA (10 mM), pH 8.0] was added to the liver tissue of each mouse (250 μL, 50 mg wet weight equivalent), homogenized, and centrifuged at 3,000 g at 4 ºC for 10 min. The nuclear pellet was resuspended in HE buffer (500 μL). Proteinase K (20 μL of 20 mg/mL in water) and 10% SDS (60 μL) were added and incubated at 37 ºC for 1.5 h. The AP sites were derivatized by incubation with PMOA (32 μL of 100 mM stock solution) at 37 ºC for 1.5 h. Excess PMOA was quenched by adding butyraldehyde (30 μL, 1 M in 50% isopropanol/water), and the suspension was cooled on ice. Proteins were precipitated with Puregene PP solution (250 μL), and nucleic acids in the supernatant were isolated by precipitation with NaCl/cold isopropanol, washed with 70% ethanol in 5 mM HEPES buffer (pH 8.0) and dried. The pellets were reconstituted with HE buffer (300 μL) and incubated with RNase A (150 μg) and RNase T1 (0.5 μg) at 37 ºC for 1.5 h to remove RNA. The enzymes were precipitated with Puregene PP solution, and the supernatant was treated with NaCl/isopropanol to precipitate the DNA. The DNA pellet was dried and reconstituted in HEPES buffer (5 mM, pH 8.0), the concentration determined by UV, and digested with a cocktail of DNase I, nuclease P1, phosphodiesterase I, alkaline phosphatase, and adenosine deaminase in the presence of [13C5]-PMOA-dR spiked at 100 fmol/30 μg of DNA (1.1 /106 nts) as the internal standard. The derivatized AP sites were enriched by SPE with a Strata-X polymeric cartridge and reconstituted in 2 mM ammonium bicarbonate for LC-MS2 analysis.44
Quantitation of NM-DNA adducts and AP sites by LC-MSn.
Quantitation was conducted on an LTQ Velos Pro ion-trap mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with an UltiMate™ 3000 RSLCnano System (Thermo Fisher) and an Advance CaptiveSpray source (Michrom Bioresource Inc., Auburn, CA). Electrospray ionization (ESI) in the positive ion mode at the MS3 stage was used to detect NM-DNA adducts and the MS2 stage to detect the AP site derivative (PMOA-dR). Detailed MS parameters and MSn transitions were reported,10,44 and provided in Table S1 of the supporting information.
An Acclaim PA II C18 column (300 µm × 15 cm, 3 μm particle size, Thermo Scientific, Bellefonte, PA) was employed for chromatographic separation of NM-DNA adducts at a flow rate of 5 μL/min. The solvents of the mobile phases were (A) 2 mM ammonium acetate in 95% H2O and 5% CH3CN and (B) 2 mM ammonium acetate in 95% CH3CN. A linear gradient was employed to resolve the DNA adducts. The solvent was held at 95:5 A/B for 1 min, and then increased to 5:95 A/B over 11 min and held at this solvent composition for 2 min. The column was heated at 55 °C to optimize the peak shape and chromatographic resolution.
The analysis of AP sites was performed with a Magic C18AQ reversed-phase column (300 µm × 15 cm, 3 μm particle size, NanoLC Solutions, Sacramento, CA) for chromatographic separation. The mobile phases were (A) 2 mM ammonium acetate in H2O and (B) 2 mM ammonium acetate in 95% CH3CN. The following gradient elution profile was used: 5 μL/min, 1% B from 0 to 2 min; 5 – 2 μL/min, 1% B from 2 to 2.5 min; 2 μL/min, 1 to 95% B from 2.5 to 20 min; 2 − 5 μL/min, 95% B from 20 to 22 min; 5 μL/min, 95 to 1% B from 22 to 24 min; 5 μL/min, 1% B from 24 to 30 min.
Data and statistical analyses.
The calibration curves of NM-DNA adducts and AP site derivatives (PMOA-dR) were constructed using seven calibrant levels for NM-DNA adducts and six calibrant levels of AP sites. The amount of NM internal standards added was 350 pg and the levels of unlabeled standard ranged from 0.0029 – 0.86 for NM-G; 0.0029 – 4.29 for G-NM-G; and 0.0029 – 0.29 for NM-Fapy-G (pmol adduct/pmol internal standard), which were added to a matrix of calf thymus DNA (220 µg) and then subjected to neutral thermal hydrolysis, and enriched by SPE.10 [13C5]-PMOA-dR was spiked at a level of 100 fmol in 30 μg DNA (1.1 per 106 nts), together with the synthesized PMOA-dR standard (1:1 isomeric mixture) at calibrant levels of 0, 0.022, 0.11, 0.55, 2.8, 11, 28 PMOA-dR per 106 nts.44 All calibrant levels were conducted in triplicate.
The limit of detection (LOD) and limit of quantification (LOQ) values were set at 3.3σ and 10σ SD units above the background level signal of the DNA hydrolysate of each organ for NM-treated samples.46 Calf thymus DNA served as the background matrix for AP sites.44 NM adducts were expressed as adducts per nucleotides (nts). The average LOD and LOQ values were NM-G (7.9 × 10−8 and 1.7 × 10−7 adducts/nts), G-NM-G (1.1 × 10−8 and 2.3 × 10−8 adducts/nts), and NM-Fapy-G (2.5 × 10−8 and 5.4 × 10−8 adducts/nts), when 220 µg DNA was processed. The LOQ value of the AP site was 2.2 PMOA-dR sites/108 nts in 13.5 μg of enzymatically digested calf thymus DNA. The calibration curves for NM-DNA adducts (amount ratio versus area of response ratio) and AP sites (AP site ratio versus area of response ratio) were fitted with linear least-squares regression analysis and the goodness-of-fit linear regression values r2 are reported: NM-G (y = 0.8360x + 0.003944, r2 = 0.996); G-NM-G (y = 1.139x + 0.0124, r2 = 0.996); NM-Fapy-G (y = 1.151x + 0.006, r2 = 0.993); AP sites (y = 0.9590x + 0.03324, r2 = 0.998).
The kinetics of NM-DNA adduct removal and half-lives of the NM-DNA adducts and AP sites were estimated with a one-phase decay model for each lesion in each organ with the following equation: Y = (Y0 - Plateau)*exp(-K*t) + Plateau, in which Y is the level of the lesion. The plateau was constrained to the background level signals of the MS3 transitions of the NM-DNA adducts in the corresponding organs of the control mice. The one-phase decay model was fitted using the replicate assays (n = 5 animals per time point, 5 time points), with passing tests for normality (Shapiro-Wilk) and the replicates test for homogeneity. The extra sum-of-squares F test was employed to determine whether a one-phase decay rate curve or individual curves fitted all NM-DNA adducts. A Welch’s one-way ANOVA, followed by Dunnet’s T3 multiple comparisons post hoc test was employed to determine statistically significant differences between half-lives (t1/2’s) of the DNA adducts and the AP site. A Welch’s unpaired t-test was used to test the significance between time dependent AP site level abundances, and the Pearson correlation was employed to determine the strength (r) and significance (p value) of the relationship between NM-DNA adducts and AP site levels. A p < 0.05 was deemed significant for all tests. Data are presented as the mean ± standard error of the mean (SEM) or the standard deviation (SD). Statistical analysis was performed with the Prism software (Version 8.3, GraphPad, La Jolla, CA), and Microsoft Excel 2017 (Redmond, WA).
Results
NM-DNA adducts and AP site measurements.
We measured NM-DNA adducts and AP sites using two validated analytical methods. The NM-DNA base adducts (NM-G, NM-Fapy-G, G-NM-G) were recovered by a two-stage neutral thermal hydrolysis of the isolated DNA, which mitigates artifactual formation of the ring-opened NM-Fapy-G adduct.10 The creation of artifactual AP sites was mitigated by directly derivatizing DNA in the nuclear pellet as an oxime derivative with PMOA before DNA isolation, followed by digestion of the DNA and enrichment of the AP site derivative (PMOA-dR) by SPE.44 The DNA adducts were analyzed by LC-MSn employing ESI in the positive ion mode at the MS3 scan stage for NM-DNA adducts, and the MS2 scan stage for PMOA-dR.10,44
NM-DNA adducts and kinetics of adduct removal.
We conducted a preliminary study to find a dose range where NM adducts could be identified and characterized in mouse livers. Female mice were dosed with NM at 0.1, 0.3, and 3 mg /kg body weight. The overall level of NM-DNA adduct formation was low, and the minor ring-opened NM-Fapy-G was detected above the LOD value only at the dose of 3 mg/kg. We did not examine NM-DNA adduct formation at higher doses because a 3 mg/kg dose is near the LD50 value of NM reported in Swiss female mice.47 All treated animals survived the NM treatment at 3 mg/kg; however, the average body weight decreased by 14 and 24% at 72 and 168 h, respectively.
A representative extracted ion chromatogram (EIC) of NM-DNA adducts in the liver of untreated and NM-treated mice (3 mg/kg) at 6 h post-treatment is shown in Figure 1. The impurities of the labeled adducts in the internal standards were responsible for a significant portion of the background signals. The signal to noise (S/N) of the response for NM-DNA adducts over the background signals of the untreated animals was 21 for NM-G, 173 for G-NM-G, and 6.6 for NM-FapyG. The signals for NM-G and G-NM-G were above the LOQ values (background signal + 10σ SD units), whereas the signal of the response of NM-Fapy-G hovered between the LOD (background signal + 3.3σ SD units) and the LOQ. The signal for the second NM cross-linked adduct containing a ring-opened Fapy moiety, G-NM-Fapy-G (Scheme 1), was below the LOD and not reported. A guideline for quantitative measurements of analytes recommends that the internal standard amount be within the lower one-third range of the calibration curve.48 The amounts of [2H5]-NM-G and [2H5]-G-NM-G used for measurement fall within this working range (signal ratios of NM-G (0.27) and G-NM-G (0.52) to their internal standards); however, the amount of NM-Fapy-G formed in the liver (and other organs) is ⁓10-folder lower than the amounts of NM-G and G-NM-G. Thus, the level [15N5]-NM-Fapy-G used for quantitation is elevated, and the signal ratio of the NM-Fapy-G to [15N5]-NM-Fapy-G (0.034) falls below the optimal working range of the calibration curve. The amount of [15N5]-NM-Fapy-G should be decreased by about 5 to 10-fold, resulting in a lower background signal of the unlabeled NM-Fapy-G and improved LOD and LOQ values. However, multiple, broad peaks are observed when lower amounts of [15N5]-NM-Fapy-G are assayed on the column, even when heating the column at 55 °C (possibly due to interaction with active sites within the UPLC system). As a result, the integration of the peaks is inaccurate (R. Turesky, unpublished observations). The calibration curve of NM-Fapy-G is shown in Figure S2 of the Supporting Information. The linearity of the calibration curve is shown by the slope (y = 1.151*x + 0.0056), and the goodness-of-fit linear regression value (r2 = 0.993) over the calibrant range of 0.0029 – 0.29 pmol NM-Fapy-G/pmol [15N5]-NM-Fapy-G used for quantitation. The lowest calibrant level is 5 to 10-fold lower than the amounts of NM-Fapy-G measured over the time course of the study. Thus, we maintained the level of internal standard at 350 pg per DNA sample. The calibration curve permitted a satisfactory estimate of NM-Fapy-G over the 168 h time course of the kinetic study.
Figure 1.
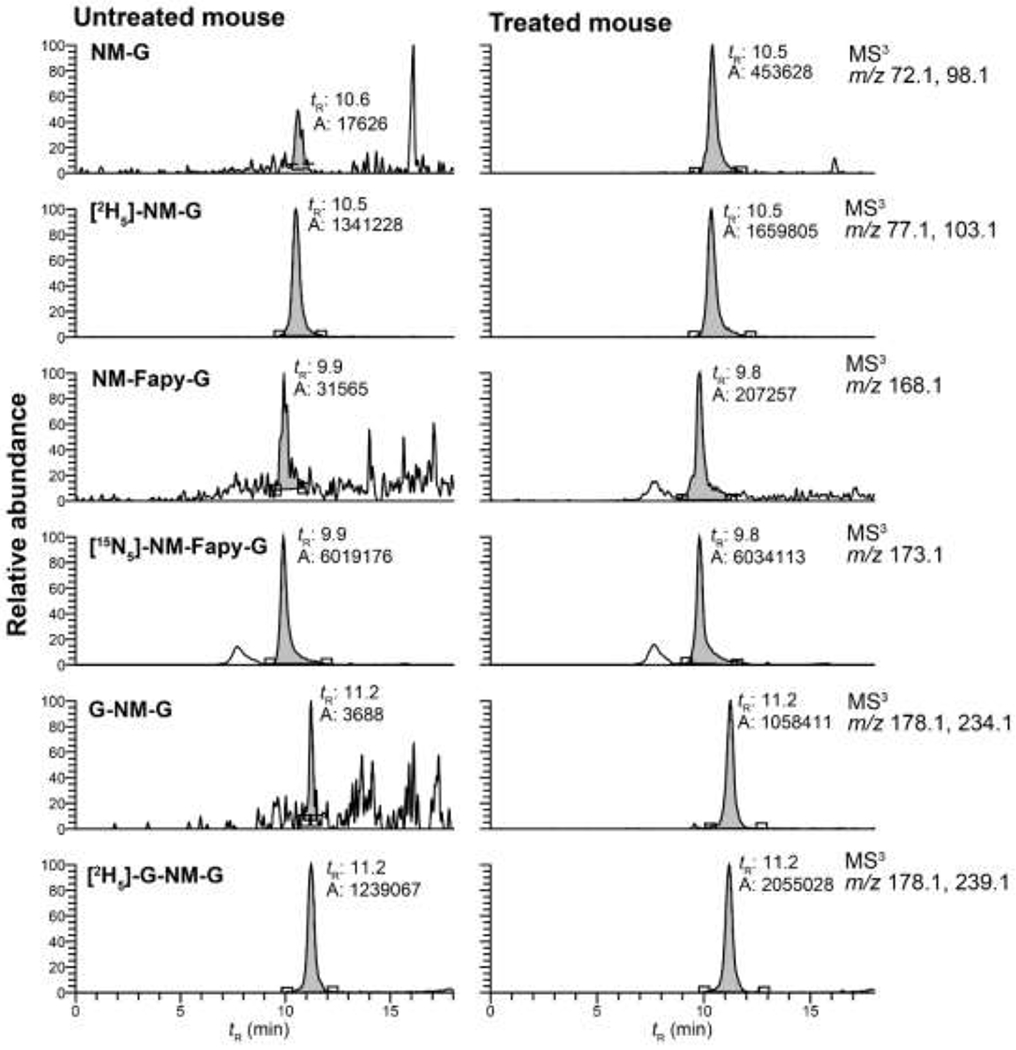
EIC of NM-DNA adduct formation in the liver of female C57BL/6NJ mice injected i.p. with saline or NM (3 mg/kg). Animals were sacrified 6 h post-treatment.
Kinetics of NM-DNA adduct formation and removal.
The kinetics of NM-DNA adduct formation and removal were measured in the liver, lung, and spleen of female mice (Table 1 and Figure 2). The EICs of NM-DNA adducts from the lung at T 0, 6, and 168 h are shown in Figure S3 of the Supporting Information. The signals of NM-Fapy-G and NM-G-NM remained above the LOD and LOQ, respectively, throughout the study, whereas the signal of NM-G declined to basal levels at 168 h. The high trapping efficiency and sensitivity of the ion trap permitted identification of all NM-DNA adducts at the MS3 scan stage.10 The product ion spectra of NM-DNA adducts at the MS3 scan stage are shown in Figure S4 of the Supporting Information.
Table 1.
The levels and ranges of NM-DNA adducts per 108 nts in mice tissues (liver, spleen, and lung) after administration of NM.a
| Time (h) | NM-G | G-NM-G | NM-Fapy-G |
|---|---|---|---|
| 6 | 26.9-40.4 | 33.1-71.8 | 2.81-5.10 |
| 24 | 12.5-20.2 | 25.6-43.1 | 2.97-4.22 |
| 48 | 6.94-12.1 | 16.9-43.3 | 2.28-3.87 |
| 72 | 4.66-5.48 | 18.0-39.7 | 0.82-2.20 |
| 168 | 0.00-3.44 | 12.0-20.8 | 1.09-1.40 |
Background signals of the MS3 transitions obtained from the organs of the untreated mice were subtracted from the organs of treated mice.
Figure 2.
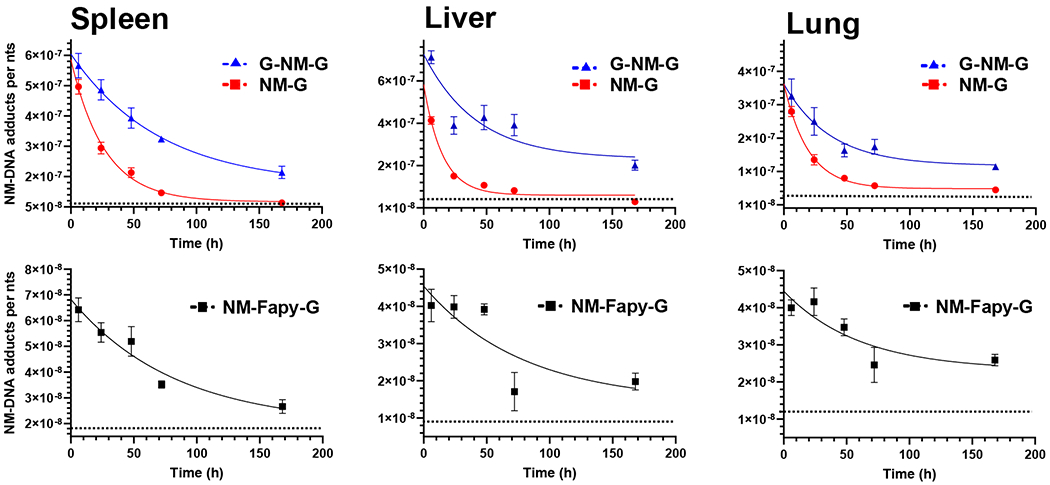
Time course of the NM-DNA adduct levels in mouse DNA after treatment of NM. Female C57BL/6NJ mice were injected i.p. with NM (3 mg/kg) and sacrificed after 6, 24, 48, 72, and 168 h. Control mice were injected i.p. with saline and sacrificed at T0 h. Adduct levels in liver, spleen, and lung DNA are expressed as adducts per nts (mean ± SEM, n = 5). The dashed lines represent the background signals of the MS3 transitions for NM-G and NM-Fapy-G. The background signal for G-NM-G is below the background signal of NM-G and not presented.
A one-phase decay curve was used to characterize adduct removal and the estimates of the t1/2’s of the NM-DNA adducts. The decay plateau was constrained at the basal level of the MS3 transition signal, representing the background signal for each adduct in the corresponding organs of the control mice (Figure 2). The NM-DNA adducts were already at their peak levels in most organs 6 h post-dosing. G-NM-G was the predominant NM-DNA adduct formed at 6 h post-treatment; however, the neutral thermal hydrolysis process does not allow the distinction between interstrand and intrastrand cross-links.49 NM-G was the second most abundant adduct in all organs, occurring at a level ranging between 58 – 94% of the G-NM-G cross-link. Though, the actual level of NM-G is about 30% greater than the level measured due to partial hydrolysis of the labile glycosidic linkage during the DNA isolation process.10 NM-Fapy-G was the least abundant adduct, and present at 10 – 20% of the NM-G level. These relative levels of NM adducts agree with our in vitro studies on NM-DNA adducts formed with NM-treated calf thymus DNA.10
NM-G declined rapidly and was undetectable in all organs at 168 h. In contrast, the G-NM-G cross-link and NM-Fapy-G were persistent, and approximately 30 – 50% of these adducts remained at 168 h relative to levels at 6 h post-treatment with NM (Figure 3 and Table 1). The decay curves for all NM-DNA adducts (Figure 2) were best fitted with independent equations (p < 0.0001). The estimated biological t1/2’s of NM-dG, dG-NM-dG, and NM-Fapy-dG are reported in Table 2. The mean t1/2’s among the adducts were significantly different for spleen (F(2, 34) = 20.9, p < 0.0001), lung (F(2,32) = 7.6, p = 0.0019), and liver (F(3,40) = 8.2, p = 0.0002), when compared using an unequal variance F-test (Welch’s one-way ANOVA). The level of significance between the mean t1/2’s between each adduct pair within each organ were determined Dunnet’s T3 multiple comparisons post hoc test (Table S2, Supporting Iinformation). The t1/2 of NM-dG was at least 3-fold lower than the t1/2’s of NM-Fapy-dG and dG-NM-dG in all organs. In spleen, the t1/2’s were significantly different between NM-G and both G-NM-G (p < 0.0001) and NM-Fapy-G (p = 0.0022), and also significantly different between NM-G and both G-NM-G (p = 0.0351) and NM-Fapy-G (p = 0.0242) in the lung. The t1/2’s were significantly different between NM-G and G-NM-G (p = 0.00311), while the t1/2’s of NM-G and NM-Fapy-G were borderline significantly different in the liver (p = 0.0603). The relatively short t1/2 of NM-dG is attributed to the biochemical processing of the adduct, including chemical depurination, enzymatic repair of the lesion, and ring-opening of NM-dG to NM-Fapy-dG. We did not observe a significant statistical difference between the t1/2’s of NM-Fapy-G and G-NM-G in any of the organs. Additional data points across the time course are required to fit the decay curves more accurately.
Figure 3.
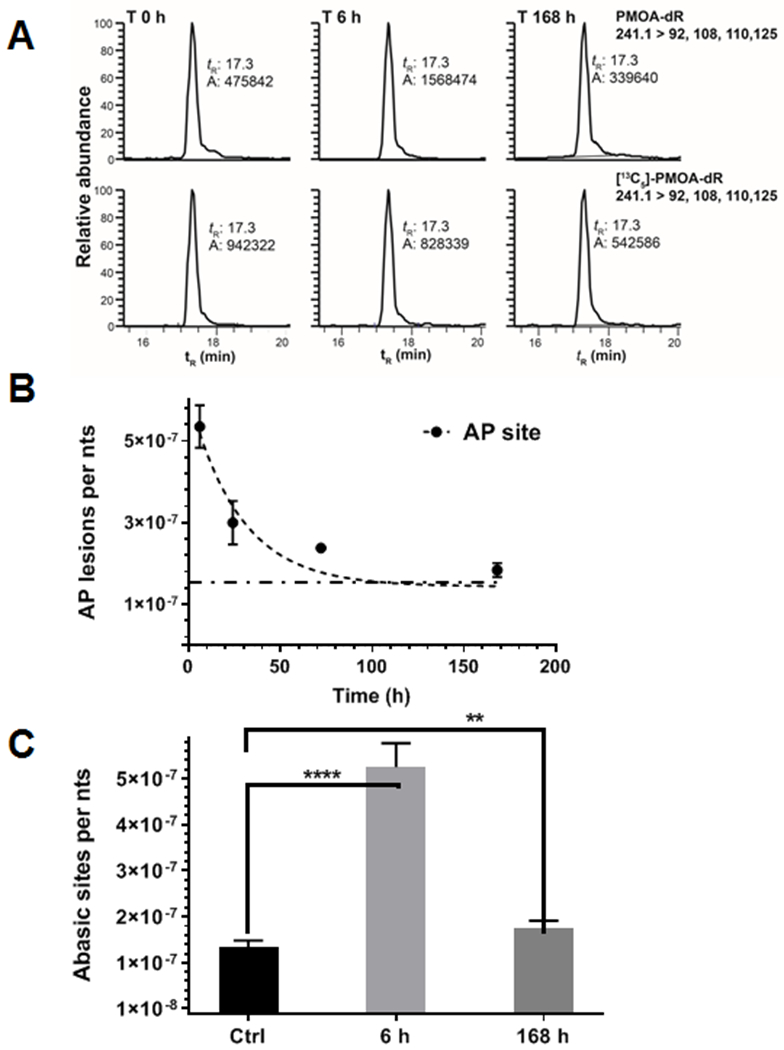
(A) EIC of AP site levels and internal standard in female mouse liver DNA after treatment of NM (3 mg/kg) at 6 h and 168 h. Control mice were injected i.p. with saline and sacrificed at T0 h. (B) One-phase decay model was applied to estimate the half-life of AP site removal (mean ± SD, n = 5 animals per data point). The decay plateau was constrained to the basal level of AP sites and shown as a dashed line. (C) Levels of AP sites in NM-treated mice at 6 and 168 h versus control mice (mean ± SD, n = 5 animals per data point). A Welch’s t-test for unequal variances was applied to compare the significant difference between the treated group at 6 and 168 h and the untreated control group. **** p < 0.0001 and ** p = 0.0039.
Table 2.
Estimated half-lives (h) with standard error (SE) of the mean of NM-DNA adducts in liver, spleen and lung of mice after administration of NM (3 mg/kg).a
| Adduct | Liver | Spleen | Lung |
|---|---|---|---|
| NM-G | 13.7 ± 1.3 | 22.2 ± 2.0 | 21.0 ± 2.3 |
| G-NM-G | 91.4 ± 19.4** | 84.3 ± 11.4**** | 89.3 ± 23.6* |
| NM-FapyG | 76.0 ± 22.4* | 74.0 ± 13.3** | 111 ± 33.1* |
One-way Welch’s ANOVA and Dunnet’s T3 multiple comparisons post hoc test on adduct half-lives in the respective organs; Liver: NM-G vs. NM-Fapy-G (p = 0.0603), NM-G vs. G-NM-G (p = 0.0031); Spleen: NM-G vs. NM-Fapy-G (p = 0.0022), NM-G vs. G-NM-G (p < 0.0001); Lung: NM-G vs. NM-Fapy-G (p = 0.0242), NM-G vs. G-NM-G (p = 0.0351).
p < 0.1,
p < 0.05,
p < 0.001,
p < 0.0001.
Quantification and Kinetics of Abasic Sites of NM-treated mice.
Chemical depurination and glycosylase-catalyzed BER of NM-DNA adducts leads to the generation of AP sites, which are cytotoxic and potentially mutagenic lesions.50,51 AP sites exist as the aldehyde form in equilibrium with the predominant cyclic acetal form (Scheme 1).52 The aldehyde form can lead to strand breaks through β-elimination,53 or can react with primary amines to form covalent adducts. A recent study showed the mechlorethamine-induced AP sites reacts with the N6-amino group of 2′-deoxyadenosine, resulting in a novel interstrand cross-link.54 We measured the level of AP sites in NM-treated mice to gain a better understanding of their kinetics of formation and repair.
AP sites are formed rapidly in liver DNA of mice treated with NM. NM-treatment increased the level of AP sites to 5.35 ± 0.52 per 107 nts (mean ± SD, n = 5), reaching a maximum at 6 h post-treatment with the drug. The level of AP sites in liver was significantly higher than the basal level (1.44 ± 0.14 per 107 nts). The EIC of AP sites at basal level, T 6 and 168 h are shown in Figure 3A. Therefore, NM-DNA adduct formation was accompanied by a concurrent increase of AP sites through spontaneous depurination or base excision repair. The AP site level induced by NM treatment at 6 h (3.91 per 107 nts) is approximately 32% of the total NM-DNA adducts (12.1 per 107 nts) measured at 6 h and accounts for a large portion of total NM-induced DNA damage. The AP site is a BER intermediate, and efficiently repaired.55 The level of AP sites rapidly declined after 6 h with an estimated repair t1/2 of 19.3 ± 3.6 h (mean ± SEM) (Figure 3B). The t1/2 of the AP site was significantly different than the t1/2 of G-NM-G (p = 0.0066) but not significantly different for the t1/2’s of NM-G (p = 0.6208) and NM-Fapy-G (p = 0.1078) (Table S2, Supporting Information). The high variance of the NM-DNA adduct levels at the different time points results in a large standard error in the estimated t1/2’s obtained from the decay curve. However, the percent of each adduct remaining at T 168 h compared to T 6 h reveal signifcant differences in the rates of removal of NM-DNA adducts and AP sites. The relative fractions remaining for NM-Fapy-G (44.8 ± 17.1%) and G-NM-G (29.8 ± 7.8%) are significantly different than fractions remaining for the AP site (10.1 ± 4.3%) and NM-G (0%) ( p < 0.0001) at T 168 h.
The level of AP sites in livers of NM-treated mice remained above the control saline-treated group at T 168 h (Figure 3C). The AP site levels were significantly higher at T 6 h (p < 0.0001 ) and T 168 h ( p = 0.0039) time points compared to the control group. The Pearson correlation (r) and p values (two-tailed) between AP site formation and NM-DNA adducts over the T 6 – 168 h time scale are: NM-G r = 0.99, (p = 0.0001); G-NM-G r = 0.97,( p = 0.0278); and NM-Fapy-G r = 0.76, (p = 0.2370), underscoring the strong correlation between NM-G and G-NM-G with AP site formation. It is worth noting that isolated AP sites are repaired much faster than the clustered lesion, e.g., the ones generated by interstrand cross-links.56 Considering the greater persistence of dG-NM-dG cross-links and NM-Fapy-dG adducts (Figure 2), we surmise that most of the AP sites were derived from NM-dG. Also, the dose of NM approached the LD50 value,47 and oxidative stress-mediated DNA damage probably contributed to AP site formation and may explain the levels of AP sites remaining above the basal levels at 168 h post-treatment with NM.47,57
Discussion
Our findings show that NM forms the mono-alkylated NM-dG adduct and its cross-link dG-NM-dG as major lesions in liver, lung, and spleen of the female mouse. The NM-dG adduct is removed by spontaneous or BER-catalyzed hydrolysis of the glycosidic linkage to produce AP sites. In addition, hydroxide adds to NM-dG to form the ring-opened NM-Fapy-dG as a minor adduct. Both AP sites and NM-Fapy-dG are mutagenic and may contribute to secondary tumor development, a major clinical limitation of NM chemotherapy.58,59 NM-Fapy-dG is a persistent lesion in all organs studied with a t1/2 of 73 – 111 h. Several studies have reported the formation and kinetics of N7-Fapy-dG adducts of genotoxicants in rodents. The AFB1-N7-dG adduct is formed in rats and mice treated with aflatoxin B1 (AFB1) after bioactivation to the reactive exo-8,9-epoxide of AFB1.60 This adduct underwent rapid removal with an apparent t1/2 of 7.5 h in the liver of rats,61 while its ring-opened AFB1-Fapy-dG adduct persisted over 72 h in both species.61,62 Similarly, the ring-opened Me-Fapy-dG, derived from N7-methyl-dG, persisted for three weeks in the bladder of rats treated with N-methylnitrosourea, while N7-methyl-dG, O6-methyl-dG, and other methylated DNA adducts were removed.18,19
The glycosidic linkage of NM-Fapy-dG is relatively stable and slowly undergoes depurination compared to NM-dG under physiological conditions, which contributes to the persistence of NM-Fapy-G in vivo and the toxicity and mutagenicity of NM.10 The reported t1/2’s of the structurally related N7-Me-dG and its ring-opened Me-Fapy-dG at neutral pH and 37 ºC are 105−150 h and 1500 h, respectively.8 DNA repair enzymes such as the 3-methyladenine-DNA glycosylases can remove NM-dG,63 and bacterial formamidopyrimidine DNA glycosylase (FPG) or human NEIL1 and oxoguanine DNA glycosylases repair the N5-substituted Fapy-dG lesions.24,64 Fapy-dG lesions exist in a number of interconverting species. In addition to α- and β-anomers, there are geometric isomers of the formamide group, syn and anti conformations of the glycosidic bond, and atropisomers of C5-N5 bond. Six discrete species of the Me-Fapy-dG lesions in duplex DNA were observed by NMR.65 It is possible that glycosylases recognized and excise these various conformations with varying efficiencies. We also reported that Me-Fapy-dG and NM-Fapy-dG were substrates for FPG.10,26,66 Recently, human NEIL1 was reported to efficiently excise the highly mutagenic AFB1-Fapy-dG adduct.24,62 The β-NM-Fapy-dG anomer was excised by hNeil1 more efficiently than AFB1-Fapy-dG and the reported rates of several oxidative base lesions (5-hydroxycytosine, 5-hydroxyuracil, thymine glycol) in vitro.24 However, the non-canonical α-NM-Fapy-dG anomer was excised 30-fold less efficiently. This latter observation may explain the increased persistence of the more mutagenic NM-Fapy-dG lesion.23,25
The cytotoxic dG-NM-dG interstrand DNA cross-link requires adduct removal from both DNA strands and is more persistent than NM-dG. The cross-link is a mixture of inter- and intrastrand cross-links; however, the neutral thermal hydrolysis treatment does not allow us to distinguish between interstrand and intrastrand cross-links. The interstrand cross-link accounted for 5 – 7% of the total DNA alkylation product from CHO B11 cells treated with the structurally related bis(2-chloroethy1)methylamine (mechlorethamine, HN2) as visualized by a gel electrophoresis assays.67 Mechlorethamine formed intrastrand cross-link in a GGC sequence in both single- (13%) and double-stranded DNA (28%) in vitro.68 The frequency of intrastrand cross-links was ~5.6-fold higher than interstrand cross-links in double-stranded DNA. In our mouse study, the levels of G-NM-G and NM-G are comparable at 6 h post-dosing, when accounting for the ~30% loss of NM-G during isolation and processing of DNA and the portion of the NM-G removed in vivo by hydrolysis and repair. The percent of G-NM-G ranged between 47 to 57% of the total adducts measured at T6 h, the G-NM-G is twice the level of NM-G (Table 1) at 24 h post-dosing and was the most abundant NM-lesion in nuclear DNA.
We previously measured the levels of NM-DNA adducts formed in vitro using calf thymus DNA or human breast cancer cells (MDA-MB-231) treated with NM (100 μM). The level of G-NM-G was 3-fold greater than NM-G in the NM-treated calf thymus DNA, but the level of G-NM-G was 25% of the level of NM-G in MDA-MB-231 24 h after treatment. The relative level of G-NM-G formed in the breast cancer cells occurred at a much lower level relative to the total NM-DNA adducts formed in organs of our mouse model.10 This result suggests a faster enzymatic removal of the G-NM-G cross-links in the tumor cells compared to healthy organs of our mouse study. Consistent with this observation, the structurally related G-NM-G cross-link adduct of mechlorethamine was removed more rapidly than the NM-G adduct in Ehrlich cells, when determined by DNA renaturation assays.69 DNA cross-links formed by the chemotherapeutic NM chlorambucil were also more rapidly removed in a drug-resistant strain of Yoshida ascites sarcoma cells compared to a drug-sensitive strain.70 The accelerated repair of dG-NM-dG cross-links can significantly contribute to drug resistance to NM.71,72 Strategies that downregulate the cross-link repair activity in tumor cells or increase repair in healthy tissue could lead to more effective chemotherapy treatments with fewer side-effects.72
To our knowledge, there are no studies reported on the quantitative measurements and kinetics of NM-DNA adducts formed in vivo by mass spectrometry-based methods. A study conducted by Hemminki in 1985 measured DNA binding of tritium labeled-cyclophosphamide ([3H]CPA) in NIH Swiss mice given a dose of 3.3 µmol (0.8 mg)/kg body weight,73 which is about 4-folder lower than that used in our study conducted with NM (14.5 µmol, 3 mg/kg body weight). CPA is a prodrug and must be metabolized to the reactive phosphoramide nitrogen intermediate to produce a panel of N7 nornitrogen mustard substituted guanine adducts similar to the NM adducts in our work. The level of DNA binding was low and peaked at ~4 adducts per 107 nts at 7 h post-treatment, with highest levels formed in lung, followed by kidney and liver. A portion of the radioactivity bound to lung DNA persisted and about 50% of the activity remained after 72 h, whereas 15 – 30% of the radioactivity remained bound to kidney and liver DNA at 72 h. The low level of binding precluded identification of the respective NM adducts by HPLC with radiometric detection. In comparison, the level of NM adduct formation in our study at 6 h post-dosing ranged from 6.7 – 12 adducts per 107 nts (NM-G + NM-Fapy-G + G-NM-G) in liver, lung, and spleen: the total level of adducts remaining in these organs ranged between 35 – 40% and 19 – 27%, respectively, at 72 and 168 h.
There is one report on the LC-MS measurement of bis-N7-guanine norNM cross-links in white blood cells of human cancer patients receiving CPA therapy. The patients received 50-60 mg/kg of intravenous CPA, and very high levels of the adduct, up to 18 adducts per 106 dG, were formed 4 – 8 h following CPA administration.74 The t1/2 of bis-N7-guanine norNM cross-link was approximately 4 – 6 h.75 The adduct level declined to background levels within 24 h. The short t1/2 of the adduct is due to the rapid turnover of white blood cells,76 in combination with adduct hydrolysis, DNA repair, and cell death. The t1/2 of bis-N7-guanine norNM cross-link in human white blood cells is considerably less than the t1/2 of NM-G-NM reported in the longer-lived cells in liver, lung, and spleen of NM-treated mice (Table 2). The levels and t1/2 of the norNM-G or ring-opened nor-NM-Fapy-G adduct were not reported in human patients.
The structurally related sulfur mustard (SM) analogue bis-(2-chloroethyl) sulphide, used in chemical warfare, is a powerful alkylating agent and also forms adducts at the N7 position of guanine (N7-hydroxyethylthioethylguanine, HETE-G), a cross-link between two guanines (bis(N7-guanine)-ethylthioethyl, G-ETE-G), and at N3 of adenine (N3-hydroxyethylthioethyladenine, HETE-A).77,78 Male euthymic, hairless SKH-1 mice were dosed with SM (2 – 60 mg/kg), and the formation and persistence of SM adducts were measured.79 HETE-G formed the highest levels of adducts in the brain, followed by lung, spleen, and kidney. The levels of G-ETE-G cross-link were about 10-fold lower than HETE-G, followed by lower levels of HETE-A.79 The level of the HETE-A adduct decreased significantly faster than that of HETE-G and G-ETE-G. No significant difference was observed between the half-lives of HETE-G and G-ETE-G, which ranged between 2.8 – 10.8 h in the different organs. SM adducts were most persistent in the lung. A similar study was conducted in male Sprague-Dawley rats dosed percutaneously with SM (1 – 10 mg/kg).80 The levels of three SM adducts peaked (tmax) in the different organs within 3 h. HETE-G was the predominant adduct formed in all organs. The t1/2 of all SM adducts was less than 24 h. To our knowledge, ring-opened SM-Fapy-G adducts have not been identified. Our kinetics data suggest NM-G adducts are removed more slowly in mouse organs than SM adducts.
Conclusions
The risk for secondary malignancies after NM chemotherapies is well established, and is largely dependent on dose intensity and the length of treatment.58,59,81 One study examined over 19,000 patients with non-uroepithelial cancers and estimated that CPA increased the risk of bladder cancer 9-fold compared to those not treated with the drug.82 Our study shows that the secondary NM-induced DNA lesions G-NM-G and NM-Fapy-G are more resistant to DNA removal than NM-G, and they persist for more than 7 days in mouse organs. The dG-NM-dG interstrand cross-link is the putative cytotoxic lesion responsible for therapeutic efficacy of NM drugs. The dG-NM-dG and NM-Fapy-dG lesions may also play a role in the secondary cancer potential of NMs because of their persistence. These adducts could serve as biomarkers to assess secondary cancer risk. The levels of AP sites increased significantly after NM treatment. The high level of AP sites is of interest not only due to its cytotoxicity and mutagenicity, but also for its potential to react with methyoxyamine and enhance NM cytotoxicity. With our quantitative LC-MS-based methods, studies on the biological processing of NM adducts and AP sites, and the potential for synergistic effect of NM with methoxyamine can be investigated in non-cancerous and cancer cell lines.
Supplementary Material
ACKNOWLEDGMENT
The Turesky laboratory gratefully acknowledges the support of the Masonic Chair in Cancer Causation, University of Minnesota.
Funding Sources
This work is funded by the National Cancer Institute (P01 CA160032). Mass spectrometry was conducted in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, University of Minnesota, funded in part by Cancer Center Support Grant CA 077598.
Footnotes
Supporting Information.
The following data are available free of charge. Methods and Instrumentation; DNA digestion and HPLC analyses of nucleosides for DNA purity; MS parameters for NM-DNA adducts and AP sites; Calibration curve for NM-Fapy-G; EIC of NM-DNA adducts in untreated and NM-treated (3 mg/kg) female C57BL/6NJ mouse lung at T 6 and T 168 h post-dosing (PDF).
REFERENCES
- (1).Diethelm-Varela B, Ai Y, Liang DD, and Xue FT (2019) Nitrogen mustards as anticancer chemotherapies: Historic perspective, current developments and future trends. Curr. Top. Med. Chem 19, 691–712. [DOI] [PubMed] [Google Scholar]
- (2).Einhorn J (1985) Nitrogen mustard: The origin of chemotherapy for cancer. Int. J. Radiat. Oncol. Biol. Phys 11, 1375–1378. [DOI] [PubMed] [Google Scholar]
- (3).Emadi A, Jones RJ, and Brodsky RA (2009) Cyclophosphamide and cancer: golden anniversary. Nat. Rev. Clin. Oncol 6, 638–647. [DOI] [PubMed] [Google Scholar]
- (4).Rajski SR, and Williams RM (1998) DNA cross-linking agents as antitumor drugs. Chem. Rev 98, 2723–2796. [DOI] [PubMed] [Google Scholar]
- (5).McHugh PJ, Spanswick VJ, and Hartley JA (2001) Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol. 2, 483–490. [DOI] [PubMed] [Google Scholar]
- (6).Schärer OD (2005) DNA interstrand crosslinks: Natural and drug-induced DNA adducts that induce unique cellular responses. Chembiochem 6, 27–32. [DOI] [PubMed] [Google Scholar]
- (7).Dronkert MLG, and Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat. Res. - DNA Repair 486, 217–247. [DOI] [PubMed] [Google Scholar]
- (8).Gates KS, Nooner T, and Dutta S (2004) Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol 17, 839–856. [DOI] [PubMed] [Google Scholar]
- (9).Masta A, Gray PJ, and Phillips DR (1994) Molecular basis of nitrogen mustard effects on transcription processes: role of depurination. Nucleic Acids Res 22, 3880–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Gruppi F, Hejazi L, Christov PP, Krishnamachari S, Turesky RJ, and Rizzo CJ (2015) Characterization of nitrogen mustard formamidopyrimidine adduct formation of bis(2-chloroethyl)ethylamine with calf thymus DNA and a human mammary cancer cell line. Chem. Res. Toxicol 28, 1850–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kingma PS, Corbett AH, Burcham PC, Marnett LJ, and Osheroff N (1995) Abasic sites stimulate double-stranded DNA cleavage mediated by topoisomerase II: DNA lesions as endogenous topoisomerase II poisons. J. Biol. Chem 270, 21441–21444. [DOI] [PubMed] [Google Scholar]
- (12).Boiteux S, and Guillet M (2004) Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 3, 1–12. [DOI] [PubMed] [Google Scholar]
- (13).Wilson DM, and Barsky D (2001) The major human abasic endonuclease: formation, consequences and repair of abasic lesions in DNA. Mutat. Res 485, 283–307. [DOI] [PubMed] [Google Scholar]
- (14).Kow YW, Bao G, Minesinger B, Jinks-Robertson S, Siede W, Jiang YL, and Greenberg MM (2005) Mutagenic effects of abasic and oxidized abasic lesions in Saccharomyces cerevisiae. Nucleic Acids Res. 33, 6196–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pujari SS, and Tretyakova N (2017) Chemical biology of N5-substituted formamidopyrimidine DNA adducts. Chem. Res. Toxicol 30, 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tomasz M, Lipman R, Lee MS, Verdine GL, and Nakanishi K (1987) Reaction of acid-activated mitomycin C with calf thymus DNA and model guanines: Elucidation of the base-catalyzed degradation of N7-alkylguanine nucleosides. Biochemistry 26, 2010–2027. [DOI] [PubMed] [Google Scholar]
- (17).Groehler AS, Najjar D, Pujari SS, Sangaraju D, and Tretyakova NY (2018) N6-(2-deoxy-D-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-(2-hydroxy-3-buten-1-yl)-formamidopyrimidine adducts of 1,3-butadiene: Synthesis, structural identification, and detection in human cells. Chem. Res. Toxicol 31, 885–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Beranek DT, Weis CC, Evans FE, Chetsanga CJ, and Kadlubar FF (1983) Identification of N5-methyl-N5-formyl-2,5,6-triamino-4-hydroxypyrimidine as a major adduct in rat liver DNA after treatment with the carcinogens, N,N-dimethylnitrosamine or 1,2-dimethylhydrazine. Biochem. Biophys. Res. Commun 110, 625–631. [DOI] [PubMed] [Google Scholar]
- (19).Kadlubar FF, Beranek DT, Weis CC, Evans FE, Cox R, and Irving CC (1984) Characterization of the purine ring-opened 7-methylguanine and its presistence in rat bladder epithelial DNA after treatment with the carcinogen N-methylnitrosourea. Carcinogenesis 5, 587–592. [DOI] [PubMed] [Google Scholar]
- (20).Den Engelse L, Menkveld GJ, De Brij RJ, and Tates AD (1986) Formation and stability of alkylated pyrimidines and purines (including imidazole ring-opened 7-alkylguanine) and alkylphosphotriesters in liver DNA of adult rats treated with ethylnitrosourea or dimethylnitrosamine. Carcinogenesis 7, 393–403. [DOI] [PubMed] [Google Scholar]
- (21).Dizdaroglu M, Kirkali G, and Jaruga P (2008) Formamidopyrimidines in DNA: mechanisms of formation, repair, and biological effects. Free Radic. Biol. Med 45, 1610–1621. [DOI] [PubMed] [Google Scholar]
- (22).Greenberg MM (2012) The formamidopyrimidines: purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res 45, 588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Earley LF, Minko IG, Christov PP, Rizzo CJ, and Lloyd RS (2013) Mutagenic spectra arising from replication bypass of the 2,6-diamino-4-hydroxy-N5-methyl formamidopyrimidine adduct in primate cells. Chem. Res. Toxicol 26, 1108–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Minko IG, Christov PP, Li L, Stone MP, McCullough AK, and Lloyd RS (2019) Processing of N5-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair 73, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Minko IG, Rizzo CJ, and Lloyd RS (2017) Mutagenic potential of nitrogen mustard-induced formamidopyrimidine DNA adduct: Contribution of the non-canonical α-anomer. J. Biol. Chem 292, 18790–18799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Christov PP, Banerjee S, Stone MP, and Rizzo CJ (2010) Selective incision of the a-N5-methyl-formamidopyrimidine anomer by Escherichia coli Endonuclease IV. J. Nucleic Acids 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Povirk LF, and Shuker DE (1994) DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res.- Rev. Genet. Toxicol 318, 205–226. [DOI] [PubMed] [Google Scholar]
- (28).Liu L, Nakatsuru Y, and Gerson SL (2002) Base excision repair as a therapeutic target in colon cancer. Clin. Cancer Res 8, 2985–2991. [PubMed] [Google Scholar]
- (29).Liu L, and Gerson SL (2004) Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr. Opin. Investig. Drugs 5, 623–627. [PubMed] [Google Scholar]
- (30).Taverna P, Liu L, Hwang HS, Hanson AJ, Kinsella TJ, and Gerson SL (2001) Methoxyamine potentiates DNA single strand breaks and double strand breaks induced by temozolomide in colon cancer cells. Mutat. Res. - DNA Repair 485, 269–281. [DOI] [PubMed] [Google Scholar]
- (31).Yan L, Bulgar A, Miao Y, Mahajan V, Donze JR, Gerson SL, and Liu L (2007) Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase IIa. Clin. Cancer Res 13, 1532–1539. [DOI] [PubMed] [Google Scholar]
- (32).Rinne M, Caldwell D, and Kelley MR (2004) Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol. Cancer Ther 3, 955–967. [PubMed] [Google Scholar]
- (33).Rinne ML, He Y, Pachkowski BF, Nakamura J, and Kelley MR (2005) N-methylpurine DNA glycosylase overexpression increases alkylation sensitivity by rapidly removing non-toxic 7-methylguanine adducts. Nucleic Acids Res 33, 2859–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Krishnamurthy N, Liu L, Xiong X, Zhang J, and Montano MM (2015) Downregulation of hPMC2 imparts chemotherapeutic sensitivity to alkylating agents in breast cancer cells. Cancer Biol. Ther 16, 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fishel ML, and Kelley MR (2007) The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol. Aspects Med 28, 375–395. [DOI] [PubMed] [Google Scholar]
- (36).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Identifier: NCT01851369, TRC102 and temozolomide for relapsed solid tumors and lymphomas. https://clinicaltrials.gov/ct2/show/NCT01851369/ [Google Scholar]
- (37).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Methoxyamine and temozolomide in treating patients with advanced solid tumors. https://clinicaltrials.gov/ct2/show/NCT00892385/ [Google Scholar]
- (38).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Methoxyamine and temozolomide in treating patients with recurrent glioblastoma. https://clinicaltrials.gov/ct2/show/NCT02395692/ [Google Scholar]
- (39).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Study of TRC102 in combination With pemetrexed in cancer patients. https://ClinicalTrials.gov/show/NCT00692159 [Google Scholar]
- (40).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Methoxyamine hydrochloride, pemetrexed disodium, cisplatin, and radiation therapy in Treating patients with stage IIIA-IV non-small cell lung cancer. https://ClinicalTrials.gov/show/NCT02535325 [Google Scholar]
- (41).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Methoxyamine and fludarabine phosphate in treating patients with relapsed or refractory hematologic malignancies. https://ClinicalTrials.gov/show/NCT01658319 [Google Scholar]
- (42).ClinicalTrials.gov, Bethesda (MD): National Library of Medicine; Methoxyamine, cisplatin, and pemetrexed disodium in treating patients with advanced solid tumors or mesothelioma that cannot be removed by surgery or mesothelioma that is refractory to pemetrexed disodium and cisplatin or carboplatin https://ClinicalTrials.gov/show/NCT02535312 [Google Scholar]
- (43).Nakamura J, Walker VE, Upton PB, Chiang SY, Kow YW, and Swenberg JA (1998) Highly sensitive apurinic/apyrimidinic site assay can detect spontaneous and chemically induced depurination under physiological conditions. Cancer Res. 58, 222–225. [PubMed] [Google Scholar]
- (44).Chen H, Yao L, Brown C, Rizzo CJ, and Turesky RJ (2019) Quantitation of apurinic/apyrimidinic sites in isolated DNA and in mammalian tissue with a reduced level of artifacts. Anal. Chem 91, 7403–7410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gu D, Turesky RJ, Tao Y, Langouet SA, Nauwelaers GC, Yuan JM, Yee D, and Yu MC (2012) DNA adducts of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 4-aminobiphenyl are infrequently detected in human mammary tissue by liquid chromatography/tandem mass spectrometry. Carcinogenesis 33, 124–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Shrivastava A, and Gupta VB (2011) Methods for the determination of limit of detection and limit of quantitation of the analytical methods. Chron. Young Sci 2, 21–25. [Google Scholar]
- (47).Sharma M, Vijayaraghavan R, and Ganesan K (2008) Comparison of toxicity of selected mustard agents by percutaneous and subcutaneous routes. Indian J. Exp. Biol 46, 822–830. [PubMed] [Google Scholar]
- (48).IonSourcesm The internal standard,https://www.ionsource.com/tutorial/msquan/is.htm
- (49).Douki T, Laporte G, and Cadet J (2003) Inter-strand photoproducts are produced in high yield within A-DNA exposed to UVC radiation. Nucleic Acids Res. 31, 3134–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Chakravarti D, Mailander PC, Li K-M, Higginbotham S, Zhang HL, Gross ML, Meza JL, Cavalieri EL, and Rogan EG (2001) Evidence that a burst of DNA depurination in SENCAR mouse skin induces error-prone repair and forms mutations in the H-ras gene. Oncogene 20, 7945–7953. [DOI] [PubMed] [Google Scholar]
- (51).Loeb LA, and Preston BD (1986) Mutagenesis by apurinic/apyrimidinic sites. Annu. Rev. Genet 20, 201–230. [DOI] [PubMed] [Google Scholar]
- (52).Wilde JA, Bolton PH, Mazumder A, Manoharan M, and Gerlt JA (1989) Characterization of the equilibrating forms of the aldehydic abasic site in duplex DNA by oxygen-17 NMR. J. Am. Chem. Soc 111, 1894–1896. [Google Scholar]
- (53).Lindahl T, and Andersson A (1972) Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry 11, 3618–3623. [DOI] [PubMed] [Google Scholar]
- (54).Nejad MI, Johnson KM, Price NE, and Gates KS (2016) A new cross-link for an old cross-linking drug: The nitrogen mustard anticancer agent mechlorethamine generates cross-links derived from abasic sites in addition to the expected drug-bridged cross-links. Biochemistry 55, 7033–7041. [DOI] [PubMed] [Google Scholar]
- (55).Whitaker AM, and Freudenthal BD (2018) APE1: A skilled nucleic acid surgeon. DNA Repair 71, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Georgakilas AG, Bennett PV, Wilson DM III, and Sutherland BM (2004) Processing of bistranded abasic DNA clusters in γ-irradiated human hematopoietic cells. Nucleic Acids Res. 32, 5609–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Gurtoo HL, Hipkens JH, and Sharma SD (1981) Role of glutathione in the metabolism-dependent toxicity and chemotherapy of cyclophosphamide. Cancer Res. 41, 3584–3591. [PubMed] [Google Scholar]
- (58).Boffetta P, and Kaldor JM (1994) Secondary malignancies following cancer chemotherapy. Acta Oncol. 33, 591–598. [DOI] [PubMed] [Google Scholar]
- (59).Pich O, Muiños F, Lolkema MP, Steeghs N, Gonzalez-Perez A, and Lopez-Bigas N , (2019) The mutational footprints of cancer therapies. Nat. Genet 51, 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Brown KL, Bren U, Stone MP, and Guengerich FP (2009) Inherent stereospecificity in the reaction of aflatoxin B1 8,9-epoxide with deoxyguanosine and efficiency of DNA catalysis. Chem. Res. Toxicol 22, 913–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Croy RG, and Wogan GN (1981) Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res. 41, 197–203. [PubMed] [Google Scholar]
- (62).Vartanian V, Minko IG, Chawanthayatham S, Egner PA, Lin Y-C, Earley LF, Makar R, Eng JR, Camp MT, Li L, Stone MP, Lasarev MR, Groopman JD, Croy RG, Essigmann JM, McCullough AK, and Lloyd RS (2017) NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proceedings of the National Academy of Sciences 114, 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mattes WB, Lee C-S, Laval J, and O’Connor TR (1996) Excision of DNA adducts of nitrogen mustards by bacterial and mammalian 3-methyladenine-DNA glycosylases. Carcinogenesis 17, 643–648. [DOI] [PubMed] [Google Scholar]
- (64).Kobune M, Xu Y, Baum C, Kelley MR, and Williams DA (2001) Retrovirus-mediated expression of the base excision repair proteins, formamidopyrimidine DNA glycosylase or human oxoguanine dna glycosylase, protects hematopoietic cells from N,N′,N-triethylenethiophosphoramide (thioTEPA)-induced toxicity in vitro and in vivo. Cancer Res. 61, 5116–5125. [PubMed] [Google Scholar]
- (65).Bamberger SN, Malik CK, Voehler MW, Brown SK, Pan H, Johnson-Salyard TL, Rizzo CJ, and Stone MP (2018) Configurational and conformational equilibria of N6-(2-deoxy-D-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine (MeFapy-dG) lesion in DNA. Chem. Res. Toxicol 31, 924–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Christov PP, Son KJ, and Rizzo CJ (2014) Synthesis and characterization of oligonucleotides containing a nitrogen mustard formamidopyrimidine monoadduct of deoxyguanosine. Chem. Res. Toxicol 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Larminat F, Zhen W, and Bohr VA (1993) Gene-specific DNA repair of interstrand cross-links induced by chemotherapeutic agents can be preferential. J. Biol. Chem 268, 2649–2654. [PubMed] [Google Scholar]
- (68).Bauer GB, and Povirk LF (1997) Specificity and kinetics of interstrand and intrastrand bifunctional alkylation by nitrogen mustards at a G-G-C sequence. Nucleic Acids Res. 25, 1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Yin L, Chun EH, and Rutman RJ (1973) A comparison of the effects of alkylation on the DNA of sensitive and resistant Lettre-Ehrlich cells following in vivo exposure to nitrogen mustard. Biochim. Biophys. Acta 324, 472–481. [DOI] [PubMed] [Google Scholar]
- (70).Harrap KR, and Gascoigne EW (1976) The interaction of bifunctional alkylating agents with the DNA of tumour cells. Eur. J. Cancer 12, 53–59. [DOI] [PubMed] [Google Scholar]
- (71).Torres-Garcia SJ, Cousineau L, Caplan S, and Panasci L (1989) Correlation of resistance to nitrogen mustards in chronic lymphocytic leukemia with enhanced removal of melphalan-induced DNA cross-links. Biochem. Pharmacol 38, 3122–3123. [DOI] [PubMed] [Google Scholar]
- (72).Deans AJ, and West SC (2011) DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 11, 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Hemminki K (1985) Binding of metabolites of cyclophosphamide to DNA in a rat liver microsomal system and in vivo in mice. Cancer Res. 45, 4237–4243. [PubMed] [Google Scholar]
- (74).Malayappan B, Johnson L, Nie B, Panchal D, Matter B, Jacobson P, and Tretyakova N (2010) Quantitative high-performance liquid chromatography-electrospray ionization tandem mass spectrometry analysis of bis-N7-guanine DNA-DNA cross-links in white blood cells of cancer patients receiving cyclophosphamide therapy. Anal. Chem 82, 3650–3658. [DOI] [PubMed] [Google Scholar]
- (75).Johnson LA, Malayappan B, Tretyakova N, Campbell C, MacMillan ML, Wagner JE, and Jacobson PA (2012) Formation of cyclophosphamide specific DNA adducts in hematological diseases. Pediatr. Blood Cancer 58, 708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Godschalk RW, Van Schooten FJ, and Bartsch H (2003) A critical evaluation of DNA adducts as biological markers for human exposure to polycyclic aromatic compounds. J. Biochem. Mol. Biol 36, 1–11. [DOI] [PubMed] [Google Scholar]
- (77).Lawley PD, Lethbridge JH, Edwards PA, and Shooter KV (1969) Inactivation of bacteriophage T7 by mono- and difunctional sulphur mustards in relation to cross-linking and depurination of bacteriophage DNA. J. Mol. Biol 39, 181–198. [DOI] [PubMed] [Google Scholar]
- (78).Fidder A, Moes GW, Scheffer AG, van der Schans GP, Baan RA, de Jong LP, and Benschop HP (1994) Synthesis, characterization, and quantitation of the major adducts formed between sulfur mustard and DNA of calf thymus and human blood. Chem. Res. Toxicol 7, 199–204. [DOI] [PubMed] [Google Scholar]
- (79).Batal M, Boudry I, Mouret S, Clery-Barraud C, Wartelle J, Berard I, and Douki T (2014) DNA damage in internal organs after cutaneous exposure to sulphur mustard. Toxicol. Appl. Pharmacol 278, 39–44. [DOI] [PubMed] [Google Scholar]
- (80).Yue L, Zhang Y, Chen J, Zhao Z, Liu Q, Wu R, Guo L, He J, Zhao J, Xie J, and Peng S (2015) Distribution of DNA adducts and corresponding tissue damage of Sprague-Dawley rats with percutaneous exposure to sulfur mustard. Chem. Res. Toxicol 28, 532–540. [DOI] [PubMed] [Google Scholar]
- (81).Belitskiy GA, Kirsanov KI, Lesovaya EA, and Yakubovskaya MG (2020) Drug-Related Carcinogenesis: Risk Factors and Approaches for Its Prevention. Biochemistry (Mosc) 85, S79–S107. [DOI] [PubMed] [Google Scholar]
- (82).Fairchild WV, Spence CR, Solomon HD, and Gangai MP (1979) The incidence of bladder cancer after cyclophosphamide therapy. J. Urol 122, 163–164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.