

Abstract
Background
Emerging evidence shows that Sirtuin 3 (SIRT3) can exert an antioxidative effect in various neurodegenerative diseases, but whether and how SIRT3 modulates neuronal death after subarachnoid hemorrhage (SAH) remains to be elucidated.
Materia/Methods
Experimental SAH was induced in adult mice by prechiasmatic cistern injection and primary neurons by OxyHb incubation. The peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and SIRT3 protein levels were examined at different time points after SAH induction. The PGC-1α protein gene knockdown in vivo and in vitro was achieved by transfection of lentivirus (LV) vectors expressing shPGC-1α or negative control (NC). Western blot, oxidative stress index, histopathology, neurological function, and cell viability analysis was performed.
Results
Results showed that the PGC-1α/SIRT3 pathway was remarkably activated in vivo and in vitro after SAH. LV-shPGC-1α treatment significantly inhibited the activation of this pathway after SAH, accompanied by deteriorated neurologic function, aggravated oxidative stress, increased neuronal apoptosis, and enhanced cytotoxicity compared with the mice or primary neurons treated with LV-NC only.
Conclusions
The present results highlight the detrimental PGC-1α/SIRT3 pathway, involving regulation of the endogenous antioxidant activity against neuronal damage, which may provide a potential therapeutic target in SAH.
MeSH Keywords: Apoptosis, Brain Injuries, Oxidative Stress, Subarachnoid Hemorrhage
Background
Although most studies considered early brain injury (EBI) as the principal cause of poor prognosis of subarachnoid hemorrhage (SAH) patients, the exact molecular mechanisms of EBI have not yet been fully elucidated. Notably, most therapeutics research has shown that oxidative stress greatly promote the progression of EBI, and antioxidant therapy could be an effective way to ameliorate neuronal damage after SAH [1,2].
It has been well established that neurons are vulnerable to oxidative stress due to high levels of reactive oxygen species (ROS) after SAH insult [3]. Thus, endogenous antioxidants are particularly important in the treatment of neuronal oxidative stress resulting from the damage of SAH. Previous studies have shown that Sirtuin 3 (SIRT3) can effectively modulate mitochondrial ROS levels in neurons by activating superoxide dismutase (SOD) after ischemic stroke [4]. SIRT3, a type of NAD-dependent deacetylase, primarily resides in the mitochondria of neurons. It has been identified as a responsive deacetylase, controlling metabolism and oxidative stress. In several neurological disorders, SIRT3 is considered to be a neuroprotective effect in enhancing cellular defense and promoting cell survival; however, the exact molecular mechanism is still incompletely understood [5,6].
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a transcriptional coactivator, can regulate mitochondrial function, oxygen consumption, and oxidative phosphorylation in the brain [7]. Importantly, it has been demonstrated that upregulation of PGC-1α level significantly protects neural cells against oxidative stress, while loss of its activity caused an enhanced level of mitochondrial-derived ROS [8]. Meanwhile, in-depth studies have shown that in ischemic neurons, PGC-1α level is obviously increased by oxidative stress damage, which further upregulates mitochondrial ROS-detoxifying genes [9,10].
Notably, a recent study has found that PGC-1α can interact with SIRT3, protecting against DAergic neuronal death in the pathogenesis of Parkinson’s disease (PD) [11]. However, little is known about the function of the PGC-1α/SIRT3 pathway, which is involved in regulation of oxidative stress and neuronal death in the pathogenesis of SAH. In this study, we effectively measured the dynamic changes of PGC-1α and SIRT3 expression induced by SAH insult, and further investigated the potential neuroprotective effects of PGC-1α/SIRT3 pathway against oxidative stress resulting from SAH damage.
Material and methods
Animal preparation
All experimental procedures were authorized by Anhui Medical University Animal Care and Use Committee and fully complied with the guidelines of the Care and Use of Laboratory Animals of the National Institute of Health. The C57BL/6 mice (male, 8–10 weeks old) needed for the experiment were provided by the Animal Center of Anhui Medical University (Hefei, China).
SAH model in vivo
SAH was effectively performed by prechiasmatic injection of autologous blood [12]. Following deep anesthesia, the mouse head was appropriately set up in a stereotactic apparatus. The skin was sterilely opened along the sagittal suture, and then a cranial hole was drilled 4.5 mm in front of the bregma. The heart of a blood-donor mouse was sterilely exposed after deep anesthesia, and the arterial blood was collected from the left ventricle with a 27-gauge needle. Immediately, 60 μl nonheparinized arterial blood was slowly injected into the prechiasmatic cistern through a drilled cranial hole of another mouse modelling SAH. To avoid backflow, the needle was left in the prechiasmatic cistern for 5 min. The animals in the Sham group were injected with 60 μl of physiologic saline following the same procedure. After the models were established, brain tissues were collected at different timepoints according to the experimental design. In this study, we used temporal cortex for all detection, which was always adjacent to the clotted blood after the SAH model was induced.
Primary culture of cortical neurons and SAH model in vitro
Primary cortical neurons were cultured following a modified method [13]. Briefly, cerebral cortices from C57BL/6 mice were removed from embryos at 16–18 days. Following the protocol step by step, we stripped the leptomeninges and white matter, and then adequately digested the cerebral cortices with trypsin. After filtering and centrifuging, the rest of the cell pellets were resuspended with DMEM containing serum and dual antibiotics. The cells were plated on culture dish at a density of 3×105 cells/cm2. After 2-h incubation, the DMEM was displaced by neurobasal medium containing GlutaMax-I, B27 supplement, and dual antibiotics. Neurons were placed in a humidified CO2 (5%) incubator at 37°C, and the culture medium was replaced every other day. Primary neurons are cultured until day 8, and then used in vitro. To mimic the experimental SAH model in vitro, primary neurons were replaced in the culture medium containing OxyHb (20 mM, MilliporeSigma, Burlington, MA, USA) [14].
Lentivirus delivery in vivo and in vitro
The PGC-1α gene knockdown in vivo and in vitro was achieved by transfection of lentivirus (LV) vectors expressing shPGC-1α (Hanbio, Shanghai, China). In brief, mice were deeply anesthetized and placed in a stereotaxic frame. The shPGC-1α or negative control (NC) at 1×109 Tu/ml was stereotaxically injected into the left ventricle (bregma: −0.4 mm, lateral: 1.2 mm, depth: 2.5 mm; 3 ul; 0.5 ul/min) by a 10-ul Hamilton syringe, as described previously [15]. After the lentivirus was transfected in the brain, the distribution of transduction in the brain was confirmed (Supplementary Figure 1). Mice were returned to their home cages for 3 days before receiving experimental SAH injury.
Neurons were effectively transfected with LV at a multiplicity of infection (MOI) of 30 following the results of our preliminary experiments. Following the operating instructions, the cultured neurons were transfected with LV-shPGC-1α or LV-NC at 1×109 Tu/ml. After 72-h transfection, the neurons were successfully transfected and used for the following experiments [13]. The sequences of shRNA were:
PGC-1α, 5′-UUUCUGGGUGGAUUGAAGUGGUGUA-3− and
NC, 5−-UUUGGUGGGUAGUAAUGGGUUCGUA-3− [16].
Study design
In experiment 1, 60 mice were randomly assigned to the Sham group (n=12) or 4 SAH groups (6 h, 12 h, 1 day, and 3 days, n=12/group). The mice in the SAH groups were used in the SAH model and were killed at 6 h, 12 h, 1 day, and 3 days after blood injection. Postmortem assessments included Western blot and histopathology study.
In experiment 2, the primary neurons were randomly arranged as: Control group and SAH group (6 h, 12 h, and 1 day). The primary neurons were collected at 6 h, 12 h, and 1 day after OxyHb incubation. Postmortem assessments included Western blot and histopathology study.
In experiment 3, 72 mice were randomly assigned to the Sham group, SAH group, SAH+LV-NC group, and SAH+LV-shPGC-1α group (n=18/group). All the mice were euthanized 1 day after SAH model establishment, and the temporal cortex tissues were immediately collected for further detection. Postmortem assessments included Western blot, biochemical assessment, histopathology detection, and behavioral analysis. Western blot analysis, biochemical assessment, histopathology detection, and behavioral analysis were performed before the mice were euthanized.
In experiment 4, the primary neurons were randomly assigned: Control group, SAH group, SAH+ LV-NC group, and SAH+LV-shPGC-1α group. The primary neurons were incubated OxyHb for 12 h before they were collected. Postmortem assessments included Western blot, histopathology detection, biochemical estimation, and cell viability analysis.
Western blot analysis
The samples from primary neurons in vitro and cerebral cortices in vivo were lysed with RIPA buffer (Beyotime, Jiangsu, China). After protein concentrations were measured, the same amount of protein from every sample was separated and transferred. After blocking with defatted milk, the membranes were hatched with primary antibodies overnight at 4°C against PGC-1α (1: 2000, ab54481; Abcam, Cambridge, MA, USA), SIRT3 (1: 1000, ab86671; Abcam), and β-actin (1: 5000, AP0060; Bioworld Technology, Minneapolis, MN, USA). After washing with PBS containing Tween-20 (PBST), the membranes were incubated with appropriate secondary antibodies at room temperature. After incubation of the chemiluminescence solution, the protein bands were visualized. Band density was quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Immunofluorescence and TUNEL staining
In brief, brain sections or primary neurons were incubated with Triton X-100, FBS, and primary antibodies against PGC-1α (1: 200, ab54481; Abcam), SIRT3 (1: 200, ab86671; Abcam), microtubule-associated protein 2 (MAP2, 1: 300, 8707T; Cell Signaling Technology), and NeuN (1: 300, MAB377; EMD Millipore, USA). After washing with PBST, coverslips and sections were incubated with corresponding secondary antibodies. After PBST washing again, coverslips and sections were counterstained by DAPI (1: 2000; MilliporeSigma). Negative controls were not incubated the primary antibodies.
Using the operating instructions, the terminal-deoxynucleotidyl transferase -mediated dUTP nick-end labeling (TUNEL) reagent kit (Roche, Inc., Indianapolis, USA) was used for apoptosis assay. In brief, the brain sections or primary neurons were incubated with anti-NeuN (1: 300) at 4°C overnight. After washing with PBS, the TUNEL reaction mixture was added to the brain sections and coverslips. After washing again, they were stained with DAPI.
Immunofluorescence and TUNEL staining were examined with a ZEISS (Scope A1; Oberkochen, Germany) microscope system. In vivo, 3 slides (at least 300 um apart) of each mouse were detected. The investigator, who was blinded to the experimental design, identified, counted, and analyzed the positive cells of IF and TUNEL staining. On each slide, 6 fields were randomly selected. The analytical approach was calculated as the percentages of total neurons that was TUNEL positive in each slide and each group. Based on the mean percentage of positive cells on each slide, the final average value of each group was determined. Image J software was used to analyze and quantify the difference in fluorescence staining intensity.
Real-time PCR detection
Briefly, the temporal cortex of each mouse was lysed with TRIzol reagent (Invitrogen). Then, the isolated RNA was reverse transcribed into cDNA with a Reverse Transcriptase Reagent kit (Invitrogen). Real-time PCR detection was performed with the Real-Time PCR System (Roche). Table 1 shows the primer sequences used [4,16].
Table 1.
Primer sequences for real-time PCR detection.
| Gene | Forward primer (5−-3−) | Reverse primer (5−-3−) |
|---|---|---|
| PGC-1α | GACCACAAACGATGACCCTCC | GCCTCCAAATCTCTCAGG |
| SIRT3 | GCCTGCAAGGTTCCTACTCC | TCGAGGACTCAGAACGAACG |
| β-actin | ACTGACTACCTCATGAAGAT | CGTCATACTCCTGCTTGCTGAT |
Biochemical detection
Following to the operating instructions of commercial kits (Beyotime, China), the levels of malondialdehyde (MDA) and superoxide dismutase (SOD) in the fresh brain samples of mice were assessed.
Primary neurons were cultured in 6-well plates for 8 days, then the supernatant medium in each group was collected. The MDA level and SOD activity were assessed using commercial kits following the manufacturer’s instructions (Jiancheng, China) [17].
LDH assay
According to the manufacturer’s protocol, we used lactate dehydrogenase (LDH) assay (Beyotime, China) to evaluate the viability of primary neurons. Briefly, culture medium in each group was incubated with the reaction mixture. After incubation, the supernatants were collected and measured by a spectrophotometer.
Behavioral analysis
Neurologic scoring was performed 1 h before the mice were euthanized in experiment 3. Neurologic functions were examined in accordance with a 18-point scoring system reported previously [18]. Supplementary Table 1 shows the 18-point neurologic scoring system used.
Morris water maze for assessment of spatial learning
A circular water tank with a diameter of 1.2 m and a height of 50 cm was filled with nontransparent water [12]. The tank was divided into 4 quadrants, and each quadrant was pasted with different shapes of cardboard. A round platform with a diameter of 12 cm, placed in a special quadrant, was set 1 cm below the water surface. The spatial memory acquisition of each mouse continued for 7 days (3 trials/mouse/day) before the SAH model was induced. Each mouse was started from 3 different quadrants, excluding the quadrant with the platform. The test time for each mouse was 1 min, and the interval between each test was 5 min. The quadrants of 2 adjacent tests must be inconsistent. If the mouse failed to find the platform within 1 min, we guided the mouse to find the platform and allowed it to rest on the platform for 15 s. After 7 days of continuous trials, SAH model and LV administrations were performed. Following SAH model induction, spatial memory testing (1 trial/mouse/day) continued for 7 days. On the 4th day of the test, the platform was moved to the contralateral quadrant, which was used to assess the spatial learning ability of mice. The time required to successfully find the platform was recorded and analyzed as an index to evaluate the spatial learning ability of each mouse. The investigator was blinded to the study design.
Statistical analysis
Data were recorded as mean±SEM. The statistical differences among multiple groups were analyzed by one-way analysis of variance (ANOVA) and the Bonferroni post hoc test. The difference was statistically significant at P<0.05.
Results
General observations
The overall mortality rate in experiment 1 and experiment 2 was 16.5% (26/158), and no mice died in the Sham group. The total mortality rate in experiment 1 was 16.7% (12/72). The total mortality rate in experiment 2 was 16.3% (14/86).
Expression of endogenous PGC-1α/SIRT3 pathway after SAH
We used Western blot analysis to assess PGC-1α and SIRT3 protein expressions in vivo and in vitro. In experiment 1, the protein levels of PGC-1α and SIRT3 were remarkably increased at 24 h after SAH in comparison with levels in the Sham group (* P<0.05; Figure 1A–1C). Similarly, we also found that compared with levels in the Sham group, the mRNA levels of PGC-1α and SIRT3 were obviously increased and peaked at 24 h after SAH (** P<0.01, * P<0.05; Figure 1D, 1E). It had been reported that PGC-1α is mainly localized in the neuronal nucleus, while SIRT3 is localized in the neuronal cytoplasm of the mouse brain. Similarly, in the Sham group, we found that low PGC-1α-positive and SIRT3-positive staining overlapped with NeuN-positive neurons in the cortex (Figure 1F, 1G). However, strong immunoreactivity of nuclear PGC-1α and cytoplasmic SIRT3 were both found in the SAH group, consistent with the enhanced levels of PGC-1α and SIRT3 found by Western blot analysis (Figure 1F, 1G).
Figure 1.

The expression of PGC-1α/SIRT3 pathway in the cortex of mice following SAH. (A–C) the protein levels of PGC-1α and SIRT3 in different groups. (D, E) the mRNA levels of PGC-1α and SIRT3 in different groups. (F) Colocalization of PGC-1α and NeuN was revealed in different groups. (G) Colocalization of SIRT3 and NeuN was revealed in different groups. (* P<0.05; n=12/group; SAH group: 24 h after SAH model induced in vivo; Scale bar=25 μm).
In experiment 2, we found that the PGC-1α and SIRT3 protein levels in primary neurons clearly increased and peaked at 12 h following SAH in comparison with the levels in the Control group (** P<0.01, * P<0.05; Figure 2A–2C). Similarly, we also found that compared with the levels in Control group, the mRNA levels of PGC-1α and SIRT3 were obviously increased and peaked at 12 h after SAH (* P<0.05; Figure 2D, 2E). In primary neurons, there was low PGC-1α-positive and SIRT1-positive staining in the Control group. However, the fluorescence intensity of PGC-1α and SIRT3 were both highly expressed in primary neurons of the SAH group in comparison with the intensities in the Control group (** P<0.01, * P<0.05; Figure 2F–2I).
Figure 2.

The expression of PGC-1α/SIRT3 pathway in primary neurons after SAH. (A–C) the protein levels of PGC-1α and SIRT3 in different groups. (D, E) the mRNA levels of PGC-1α and SIRT3 in different groups. (F, G) the fluorescence intensity of PGC-1α and SIRT3 in different groups. (H) Colocalization of PGC-1α and MAP2 was revealed in different groups. (I) Colocalization of SIRT3 and MAP2 was revealed in different groups. (** P<0.01, * P<0.05; SAH group: 12 h after SAH model induced in vitro; Scale bar=50 μm).
The PGC-1α/SIRT3 pathway is involved in neuronal injury in vivo after SAH
Next, we examined PGC-1α and SIRT3 expressions in response to LV treatment both in vivo and in vitro. In experiment 3, in the cortex of mice, PGC-1α protein and mRNA levels were significantly decreased after LV-shPGC-1α treatment in comparison with the level in the SAH+LV-NC group (** P<0.01, * P<0.05; Figure 3A, 3C, 3D). Similarly, in the cortex, SIRT3 protein and mRNA levels were remarkably decreased after LV-shPGC-1α treatment in comparison with the level in the SAH+LV-NC group (* P<0.05; Figure 3A, 3B, 3E).
Figure 3.

Changes of the PGC-1α/SIRT3 pathway after LV-shRNA treatment. (A–E) The changes of PGC-1α/SIRT3 pathway after LV-shPGC-1α treatment in vivo. (F–J) The changes of PGC-1α/SIRT3 pathway after LV-shPGC-1α treatment in vitro. (** P<0.01, * P<0.05; n=6/group).
Notably, LV-shPGC-1α treatment significantly inhibited activation of the PGC-1α/SIRT3 pathway in vivo following SAH injury, accompanied by worse neurologic score, oxidative stress aggravation, and increased neuronal apoptosis compared with the mice treated with LV-NC only (**** P<0.001, ** P<0.01, * P<0.05; Figure 4A–4E). The Morris water maze test was performed to evaluate the spatial learning of mice in different groups. On the 1st to 2nd day after bleeding, the time for mice in the SAH+LV-shPGC-1α group to find the platform was significantly longer than that in the SAH+LV-NC group. This indicated that spatial learning abilities of mice were significantly weakened after inhibition of the PGC-1α/SIRT3 pathway (* P<0.05; Figure 4F). There was no significant difference in the time needed to find the platform in the SAH+LV-shPGC-1α group compared with the SAH+LV-NC group on the 3rd day after bleeding. Nonetheless, when the platform was moved to the contralateral quadrant on the 4th day after bleeding, the spatial learning ability of mice in the SAH+LV-shPGC-1α group was significantly lower than that in the SAH+LV-NC group (* P<0.05; Figure 4F). This indicated that when the platform position changed, the spatial learning abilities of mice were significantly reduced after inhibition of the PGC-1α/SIRT3 pathway.
Figure 4.

The inhibition effects of the PGC-1α/SIRT3 pathway on brain injury after SAH. (A, B) The changes of MDA and SOD in different groups after LV-shPGC-1α administration in vivo. (C, D) The number of apoptotic neurons of cortex in different groups at 24 h post-SAH. (E) The neurologic scores of mice in different groups at 24 h post-SAH. (F) The spatial learning ability of mice in different groups after SAH: spatial reversal was performed on day 4 after SAH. (**** P<0.001, ** P<0.01, * P<0.05; n=6/group; Scale bar=25 μm).
The PGC-1α/SIRT3 pathway is involved in neuronal injury in vitro after SAH
In experiment 4, after the LV-shPGC-1α treatment in vitro, the PGC-1α protein and mRNA levels were obviously decreased in comparison with the SAH 12 h group (* P<0.05; Figure 3F, 3H, 3I). Accompanied by the change of PGC-1α in vitro, the level of SIRT3 and mRNA levels were also significantly lower after LV-shPGC-1α treatment in comparison with that in the SAH+LV-NC group (* P<0.05; Figure 3F, 3G, 3J).
Significantly, LV-shPGC-1α treatment obviously inhibited the activation of PGC-1α/SIRT3 pathway in vitro after SAH insult, accompanied by deteriorated cytotoxicity, oxidative stress aggravation, and increased neuronal apoptosis compared with the primary neurons treated with LV-NC only (*** P<0.001, ** P<0.01, * P<0.05; Figure 5A–5E). Nevertheless, no significant difference was detected between the SAH+LV-NC group and the SAH group (P>0.05; Figure 5A–5E).
Figure 5.

The inhibition effects of the PGC-1α/SIRT3 pathway on neuronal damage after SAH in vitro. (A, B) The changes of MDA and SOD in different groups after LV-shPGC-1α administration in primary neurons. (C, D) The number of apoptotic neurons in different groups at 12 h post-SAH. (E) The cytotoxicity of primary neurons in different groups at 12 h post-SAH. (*** P<0.001, ** P<0.01, * P<0.05; Scale bar=25 μm).
Discussion
In this study, we identified the main presence of PGC-1α and SIRT3 in neurons after experimental SAH both in vivo and in vitro, and demonstrated that PGC-1α is responsible for the modulation of SIRT3 and is implicated in alleviating the oxidative stress after SAH (Figure 6). Therefore, inhibition of the PGC-1α/SIRT3 pathway aggravated the damage from oxidative stress after experimental SAH, which suggests a potential therapeutic strategy and target for SAH therapy.
Figure 6.
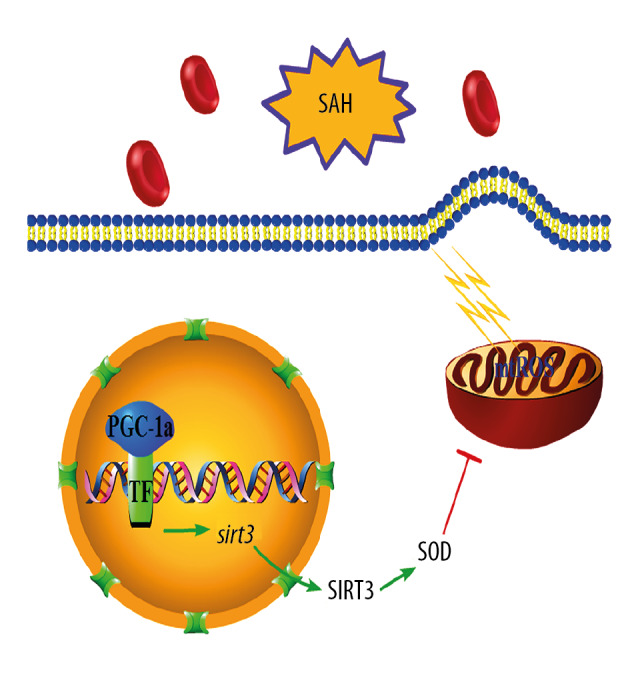
A schematic of the proposed mechanism for PGC-1α/SIRT3 pathway activation on oxidative stress after SAH: PGC-1α, acting as a coactivator of transcriptional factor (TF) in nuclear of neurons, induces the expressions of SIRT3 and a series of antioxidant enzymes, such as SOD1 and SOD2.
The expression of PGC-1α/SIRT3 pathway after experimental SAH
After SAH, intrinsic antioxidant activities can be exhausted by excessive free radicals, resulting in lipid peroxidation, protein breakdown, and DNA damage [3]. As an endogenous antioxidant, SIRT3 is mainly localized in the cytoplasm of neurons at a high level, and regulates almost every major aspect of mitochondrial function. It has been found that H2O2 administration increases SIRT3 expression at mRNA and protein levels of cortical neurons [19]. In addition, elevated level of SIRT3 significantly promotes neuron survival from oxidative stress-mediated stress, while loss of its activity generates an aggravated level of mitochondrial-derived ROS. The antioxidative effects of SIRT3 have been demonstrated in a variety of neurological diseases, such as ischemic stroke, Huntington disease, and Alzheimer’s disease [19]. Substantial evidence indicates that PGC-1α is involved in the regulation of intrinsic antioxidant activity against ROS. Elevated levels of PGC-1α can be obviously induced in response to oxidative stress in ischemic neurons, which further triggers the increased expression of ROS-detoxifying genes [20].
Consistently, we found that PGC-1α and SIRT3 expression in neurons was obviously increased in vivo and in vitro after SAH. It has been well demonstrated that neurons are particularly vulnerable to insult from oxidative stress after SAH. Hence, we speculated here that the increased PGC-1α and SIRT3 expression may be induced after SAH by stimulation from ROS production, but this requires further experimental verification. We also speculated that the predominantly nuclear localization of PGC-1α gives promotes the transcription of ROS-detoxifying genes, thereby contributing to neuroprotection after SAH.
To date, because of conflicting data in the literature, the exact mechanisms by which SIRT3 processes and transports into the cytoplasm remains ambiguous [4]. For instance, it has been reported that SIRT3 exists mainly in the nucleus before translocation into the cytoplasm, while others found that SIRT3 primarily exists in the cytoplasm, not in the nucleus. Similarly, wherever SIRT3 may be processed, our data showed that SAH not only led to the increased expression, but also triggered the translocation of SIRT3 into cytoplasm of neurons both in vivo and in vitro. Therefore, we speculate that the predominantly cytoplasmic localization of SIRT3 allows it to better counter further pathological ROS formation, which will significantly ameliorate SAH-induced injury.
Additionally, it had been reported that there is a reduced level of SIRT3 following SAH [21,22]. In comparison with our study, others used different experimental methods, such as the SAH model and the location of the cortex being detected. Firstly, both of these 2 studies used the puncture model. However, the volumes of hemorrhage in puncture models vary greatly among individuals, which might lead to the need for large sample sizes. In order to make the volume of hemorrhage in each mouse relatively consistent, we used the blood injection model in our study. Therefore, use of different models might show different trends of SIRT3 after SAH. Secondly, the samples from different locations influence the detection results. While Yang et al. reported a reduced level of SIRT3 after SAH in human glioma cell lines U87 and U251 [22], Huang et al. did not specify the location for detection [21]. In our study, we explored the role of the PGC-1α/SIRT3 pathway in the temporal cortex, which directly contacted the initial blood, leading to aggravated cytotoxic injury and ischemic injury after SAH. Hence, we suggest that different experimental designs may produce different experimental results, which needs further study in the future.
Possible neuroprotection by PGC-1α via modulating the SIRT3 pathway in the pathogenesis of oxidative stress after SAH
It has been well demonstrated that ROS accumulation caused by mitochondrial dysfunction is one of the leading causes of neuronal damage after SAH [3]. Thus, endogenous antioxidant activity is an important protective mechanism for neurons after SAH, which is always considered as a potential target for SAH treatment. Significantly, SIRT3 has been demonstrated to enhance the antioxidant activity of SOD2 in nervous system injury [5]. Here, we similarly found increased levels of SIRT3 after SAH, which might also exert an antioxidative effect after SAH. However, how SIRT3 is mediated in oxidative stress after SAH remains to be further elucidated.
It has been reported that PGC-1α regulates the expression of SIRT3 in mitochondria, resulting in activated SOD2 to alleviate ROS [23]. Meanwhile, another study found that increased PGC-1α could interact with estrogen-related receptor alpha (ERRα), which regulated SIRT3 expression by combining with SIRT3 promoter and neuronal death in vitro [11,24]. In this study, we similarly found an increased level of SIRT3 at 24 h after stimulation from the initial bleeding of SAH. Subsequently, LV-shPGC-1α administration decreased endogenous levels of PGC-1α and SIRT3, worsening of neurological function and aggravating levels of oxidative stress, as well as increasing numbers of apoptotic neurons following SAH. This significantly illustrates that PGC-1α regulates the antioxidant activity of SIRT3 after hemorrhage, thereby increasing the endogenous antioxidant effect after SAH. Therefore, therapies targeting the PGC-1α/SIRT3 pathway may help restore the expression of antioxidant enzymes and alleviating oxidative damage after SAH.
Conclusions and Future Perspectives
We found that activation of the PGC-1α/SIRT3 pathway plays a neuroprotective role in preventing oxidative stress after SAH. Small-molecule compound (ZLN005), pharmacologically upregulating the level of PGC-1α, may exert a neuroprotective effect in nervous system diseases [20]. Further studies are required to determine the therapeutic effects of pharmacological activation of the PGC-1α/SIRT3 pathway after SAH.
Supplementary Data
Distribution of lentivirus transfected in the brain. The staining of green fluorescent protein (GFP) in the negative control group (NC-GFP). (A) Distribution of lentivirus in the deep brain tissue, scale bar=200 μm; (B) Distribution of lentivirus in the temporal cortex tissue, scale bar=25 μm.
Supplementary Table 1.
Description of the 18-point neurologic scoring system.
| Test | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Spontaneous activity (in cage for 5 min) | No movement | Barely moves position | Moves but does not approach at least 3 sides of cage | Moves and approaches at least 3 sides of cage |
| Movements of forelimbs (outstretching while held by tail) | No outreaching | Slight outreaching | Outreach is limited and less than pre-SAH | Outreach is the same as pre-SAH |
| Spontaneous movements of all limbs | No movement | Slight movement of limbs | Moves all limbs but slowly | Moves all limbs the same as pre-SAH |
| Climbing wall of wire cage | Not available | Fails to climb | Climbs weakly | Normal climbing |
| Reaction to touch on both sides of trunk | Not available | No response | Weak response | Normal response |
| Response to vibrissae touch | Not available | No response | Weak response | Normal response |
Footnotes
Availability of data and materials
The datasets exhibited and analyzed in the present study are available from the corresponding author on reasonable request.
Conflicts of interest
None.
Source of support: Departmental sources
References
- 1.Sehba FA, Hou J, Pluta RM, Zhang JH. The importance of early brain injury after subarachnoid hemorrhage. Prog Neurobiol. 2012;97(1):14–37. doi: 10.1016/j.pneurobio.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcolini E, Hine J. Approach to the diagnosis and management of subarachnoid hemorrhage. West J Emerg Med. 2019;20(2):203–11. doi: 10.5811/westjem.2019.1.37352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, Li Z, Feng D, et al. Involvement of Nox2 and Nox4 NADPH oxidases in early brain injury after subarachnoid hemorrhage. Free Radic Res. 2017;51(3):316–28. doi: 10.1080/10715762.2017.1311015. [DOI] [PubMed] [Google Scholar]
- 4.Kim SH, Lu HF, Alano CC, et al. Alano, Neuronal Sirt3 protects against excitotoxic injury in mouse cortical neuron culture. PLoS One. 2011;6(3):e14731. doi: 10.1371/journal.pone.0014731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao H, Luo Y, Chen L, et al. Sirt3 inhibits cerebral ischemia-reperfusion injury through normalizing Wnt/beta-catenin pathway and blocking mitochondrial fission. Cell Stress Chaperones. 2018;23(5):1079–92. doi: 10.1007/s12192-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q, Li L, Li CY, et al. SIRT3 protects cells from hypoxia via PGC-1alpha- and MnSOD-dependent pathways. Neuroscience. 2015;286:109–21. doi: 10.1016/j.neuroscience.2014.11.045. [DOI] [PubMed] [Google Scholar]
- 7.Chen SD, Yang DI, Lin TK, et al. Roles of oxidative stress, apoptosis, PGC-1alpha and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci. 2011;12(10):7199–215. doi: 10.3390/ijms12107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.St-Pierre J, Drori S, Uldry M, et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127(2):397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 9.García-Quintans N, Sánchez-Ramos C, Prieto I, et al. Oxidative stress induces loss of pericyte coverage and vascular instability in PGC-1alpha-deficient mice. Angiogenesis. 2016;19(2):217–28. doi: 10.1007/s10456-016-9502-0. [DOI] [PubMed] [Google Scholar]
- 10.Singh SP, Schragenheim J, Cao J, et al. PGC-1 alpha regulates HO-1 expression, mitochondrial dynamics and biogenesis: Role of epoxyeicosatrienoic acid. Prostaglandins Other Lipid Mediat. 2016;125:8–18. doi: 10.1016/j.prostaglandins.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Ren X, Zhang Q, et al. PGC-1alpha/ERRalpha-Sirt3 Pathway Regulates DAergic Neuronal Death by Directly Deacetylating SOD2 and ATP Synthase beta. Antioxid Redox Signal. 2016;24(6):312–28. doi: 10.1089/ars.2015.6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schallner N, Pandit R, LeBlanc R, 3rd, et al. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J Clin Invest. 2015;125(7):2609–25. doi: 10.1172/JCI78443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou X, Wu Q, Lu Y, et al. Crosstalk between soluble PDGF-BB and PDGFRbeta promotes astrocytic activation and synaptic recovery in the hippocampus after subarachnoid hemorrhage. FASEB J. 2019;33(8):9588–601. doi: 10.1096/fj.201900195R. [DOI] [PubMed] [Google Scholar]
- 14.Lu Y, Zhang XS, Zhou XM, et al. Peroxiredoxin 1/2 protects brain against H2O2-induced apoptosis after subarachnoid hemorrhage. FASEB J. 2019;33(2):3051–62. doi: 10.1096/fj.201801150R. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Zhou XM, Xu WD, et al. Inhibition of elevated hippocampal CD24 reduces neurogenesis in mice with traumatic brain injury. J Surg Res. 2020;245:321–29. doi: 10.1016/j.jss.2019.07.082. [DOI] [PubMed] [Google Scholar]
- 16.Uguccioni G, Hood DA. The importance of PGC-1alpha in contractile activity-induced mitochondrial adaptations. Am J Physiol Endocrinol Metab. 2011;300(2):E361–71. doi: 10.1152/ajpendo.00292.2010. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Qiu Z, Lu Y, et al. Edaravone protects primary-cultured rat cortical neurons from ketamine-induced apoptosis via reducing oxidative stress and activating PI3K/Akt signal pathway. Mol Cell Neurosci. 2019;100:103399. doi: 10.1016/j.mcn.2019.103399. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Wu Q, Lu Y, et al. Cerebroprotection by salvianolic acid B after experimental subarachnoid hemorrhage occurs via Nrf2- and SIRT1-dependent pathways. Free Radic Biol Med. 2018;124:504–16. doi: 10.1016/j.freeradbiomed.2018.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai SH, Chen T, Wang YH, et al. Sirt3 protects cortical neurons against oxidative stress via regulating mitochondrial Ca2+ and mitochondrial biogenesis. Int J Mol Sci. 2014;15(8):14591–609. doi: 10.3390/ijms150814591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Y, Kabba JA, Ruan W, et al. The PGC-1alpha activator ZLN005 ameliorates ischemia-induced neuronal injury in vitro and in vivo. Cell Mol Neurobiol. 2018;38(4):929–39. doi: 10.1007/s10571-017-0567-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang W, Huang Y, Huang RQ, et al. SIRT3 expression decreases with reactive oxygen species generation in rat cortical neurons during early brain injury induced by experimental subarachnoid hemorrhage. Biomed Res Int. 2016;2016:8263926. doi: 10.1155/2016/8263926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang S, Chen X, Li S, et al. Melatonin treatment regulates SIRT3 expression in early brain injury (EBI) due to reactive oxygen species (ROS) in a mouse model of subarachnoid hemorrhage (SAH) Med Sci Monit. 2018;24:3804–14. doi: 10.12659/MSM.907734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feng H, Wang JY, Yu B, et al. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha inhibits vascular calcification through Sirtuin 3-mediated reduction of mitochondrial oxidative stress. Antioxid Redox Signal. 2019;31(1):75–91. doi: 10.1089/ars.2018.7620. [DOI] [PubMed] [Google Scholar]
- 24.Giralt A, Hondares E, Villena JA, et al. Peroxisome proliferator-activated receptor-gamma coactivator-1alpha controls transcription of the Sirt3 gene, an essential component of the thermogenic brown adipocyte phenotype. J Biol Chem. 2011;286(19):16958–66. doi: 10.1074/jbc.M110.202390. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Distribution of lentivirus transfected in the brain. The staining of green fluorescent protein (GFP) in the negative control group (NC-GFP). (A) Distribution of lentivirus in the deep brain tissue, scale bar=200 μm; (B) Distribution of lentivirus in the temporal cortex tissue, scale bar=25 μm.
Supplementary Table 1.
Description of the 18-point neurologic scoring system.
| Test | Score | |||
|---|---|---|---|---|
| 0 | 1 | 2 | 3 | |
| Spontaneous activity (in cage for 5 min) | No movement | Barely moves position | Moves but does not approach at least 3 sides of cage | Moves and approaches at least 3 sides of cage |
| Movements of forelimbs (outstretching while held by tail) | No outreaching | Slight outreaching | Outreach is limited and less than pre-SAH | Outreach is the same as pre-SAH |
| Spontaneous movements of all limbs | No movement | Slight movement of limbs | Moves all limbs but slowly | Moves all limbs the same as pre-SAH |
| Climbing wall of wire cage | Not available | Fails to climb | Climbs weakly | Normal climbing |
| Reaction to touch on both sides of trunk | Not available | No response | Weak response | Normal response |
| Response to vibrissae touch | Not available | No response | Weak response | Normal response |