

Abstract
Seizures can be evident within minutes of exposure to an organophosphorus (OP) agent and often progress to status epilepticus (SE) resulting in a high mortality if left untreated. Effective medical countermeasures are necessary to sustain patients suffering from OP poisoning and to mitigate the ensuing brain injury. Here, the hypothesis was tested that a single subanesthetic dose of urethane prevents neuropathology measured 24 hours following diisopropylfluorophosphate (DFP)-induced SE. Adult Sprague-Dawley rats were injected with DFP to induce SE. During SE rats displayed increased neuronal activity in the hippocampus and an upregulation of immediate early genes as well as pro-inflammatory mediators. In additional experiments rats were administered diazepam (10 mg/kg, ip) or urethane (0.8 g/kg, sc) one hour after DFP-induced SE and compared to rats that experienced uninterrupted SE. Cortical electroencephalography (EEG) and power analysis strengthen the conclusion that urethane effectively terminates SE and prevents the overnight return of seizure activity. Neurodegeneration in limbic brain regions and the seizure-induced upregulation of key inflammatory mediators present 24 hours after DFP-induced SE were strongly attenuated by administration of urethane. A trivial explanation for these beneficial effects, that urethane simply reactivates acetylcholinesterase, has been ruled out. These findings indicate that, by contrast to rats administered diazepam or rats that experience uninterrupted SE, the early neuropathology after SE is prevented by subanesthetic urethane, which terminates rather than interrupts, SE.
Keywords: DFP, urethane, diazepam, EEG, organophosphate, hippocampus, neurodegeneration, inflammation, status epilepticus, FluoroJade B, RT-PCR, ELISA, IBA1, COX-2
INTRODUCTION
Status epilepticus (SE) is a medical emergency resulting from multiple seizures occurring consecutively without regain of consciousness between them, or a continuous seizure that lasts longer than five minutes in man. If left untreated SE results in a high mortality. Exposure to high levels of organophosphorus (OP) based compounds can induce SE. In rodents, SE induced by diisopropyl fluorophosphate (DFP), a mimic of the nerve agent sarin, usually persists for more than five hours if uninterrupted pharmacologically (Pouliot et al., 2016; Rojas et al., 2015, 2016). Rats that survive SE tend to display neurological damage (Cherian & Thomas, 2009) and develop epilepsy (Rojas et al., 2018). Like many other OP based agents, DFP is a potent inhibitor of acetylcholinesterase (AChE), the major enzyme that breaks down acetylcholine at cholinergic synapses. Inhibition of AChE leads to accumulation of acetylcholine, which causes prolonged stimulation of cholinergic receptors, inducing a cholinergic crisis that includes enhanced seizure activity. Rats that experience DFP-induced SE tend to display neurodegeneration and a robust inflammatory response in the brain that is evident days after the SE experience (Deshpande & DeLorenzo, 2019; Ferchmin et al., 2014; Flannery et al., 2016; Guignet et al., 2019; Hobson et al., 2018; Kadriu et al., 2009; Kuruba et al., 2018; Li et al., 2015; Liang et al., 2018; Rojas et al., 2015, 2018, 2020; Wu et al., 2018; Zaja-Milatovic et al., 2009).
Currently benzodiazepines, in particular diazepam and midazolam, are still the initial standard of care for managing patients experiencing SE (Isojarvi & Tokola, 1998). Benzodiazepines are a class of drugs that promote the binding of gamma-amino-butyric acid (GABA) to its receptors. Therefore, the facilitation of GABA receptor activation in the central nervous system (CNS) is a potential mechanism by which seizure activity terminates. Like benzodiazepines, the general anesthetic urethane (ethyl carbamate) also potentiates the activation of GABA and glycine receptors, which are the main inhibitory neurotransmitter receptors in the CNS. However, in addition to potentiation of these inhibitory neurotransmitter receptors, urethane also inhibits the excitatory N-methyl-D-aspartate (NMDA) and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors (Hara & Harris, 2002). The proposed method by which a sub-anesthetic dose of urethane terminates seizures involves a dual mechanism of action that promotes net neuronal network inhibition. Recently, urethane was shown to be superior to diazepam at blocking the return of high power seizure activity that returned following DFP-induced SE in adult rats (Rojas et al., 2018). Although seizure activity returned 6–10 hours later in diazepam treated rats following 1 hour of DFP-induced SE, all rats administered diazepam survived the following four days (Rojas et al., 2018), supporting a benefit of administering benzodiazepines to patients experiencing SE.
Administration of a single sub-anesthetic dose of urethane attenuated hippocampal neurodegeneration, neuroinflammation and gliosis while improving functional recovery of rats 4 days after DFP-induced SE (Rojas et al., 2018). The efficacy of urethane to attenuate seizure-induced neuropathology is attributed to the ability of urethane to block the overnight return of high-intensity seizures. Considering the neuropathology was measured 4 days following SE, it remains unclear whether urethane has an immediate or early beneficial effect following termination of SE and whether urethane attenuates neuropathology by simply reactivating AChE. The hypothesis that a single subanesthetic dose of urethane, which aborts electrographic seizures following DFP exposure in adult rats and attenuates SE, prevents neuropathology measured 24 hours after SE was investigated. The use of continuous electroencephalography (EEG) recordings and investigation of neuropathology early after SE allows us to ask the questions: Does the duration of SE correlate to the degree of neuropathology? Does neuroinflammation precede neurodegeneration and gliosis following DFP exposure? A rat organophosphate toxicity model that involves administration of DFP was used to induce SE. The results provide important information regarding the early consequences of OP poisoning and further support a new strategy for the complete termination of OP-induced SE.
MATERIALS & METHODS
Ethics Statement
All procedures concerning animals were approved by the Emory University Animal Care and Use Committee and conformed to the guidelines of the National Institutes of Health.
Electrode implantation and EEG recording
Adult male Sprague-Dawley rats 8–12 weeks old (240–420 g body weight) were implanted with monopolar electrodes on the surface of the cortex under general anesthesia as previously reported (Rojas et al., 2018). Briefly, the screw electrodes were positioned through burr holes above the left and right parietal cortices with a third surface electrode positioned at the base of the frontal as the reference. Rats were injected with yohimbine to reverse the effects of the anesthesia, meloxicam for pain management and allowed to recover from surgery 7–10 days before experimentation. EEG signals were recorded using a Nicolet Endeavor CR system (Viasys Healthcare, Madison, WI) with a Tornado v32 amplifier (Natus Medical Inc, San Carlos, CA) and the NicoletOne recorder software (Viasys Healthcare) as previously reported (Rojas et al., 2018). A script written in Python was used to obtain a running mean of EEG power in the 20–70 Hz band, defined as the square of the EEG amplitude signal, integrated over 300 sec periods. EEG power was plotted as a function of time. The mean power during the final 4 h (20–24 hours) was obtained for each rat in the diazepam and urethane treatment groups. The final 4 hours was chosen for power analysis in an attempt to identify differences in the seizure return phase of the EEG and to correlate seizure activity towards the end of the EEG recording with neuropathology and the activity of immediate early genes such as cFos.
DFP-induced SE
Adult male Sprague-Dawley rats (200–240 g body weight) were purchased from Charles River Labs (Wilmington, MA, USA) and housed in standard plastic cages (2 rats/cage) in a temperature-controlled room (22 ± 2° C) on a 12-hour reverse light-dark cycle. Food and water were provided ad libitum. On the day of organophosphorus exposure, the rats were weighed, placed individually into a plastic cage and moved into a ventilation hood. To induce SE awake rats were injected with pyridostigmine bromide followed twenty minutes later with ethylatropine bromide and subsequently ten minutes later with diisopropyl fluorophosphate (DFP) (D0879, Sigma) diluted in sterile distilled water (5 mg/kg, sc) as previously described (Rojas et al., 2018). This dose of DFP with the given route of administration results in prolonged SE lasting >5h without pharmacological intervention. Control (non-seizure) rats were treated similarly except they were given sterile water instead of DFP. Rats were administered diazepam (Hospira, Lake Forest, IL) (10 mg/kg, ip) 1h after SE onset to interrupt SE. This dose and route of diazepam was chosen as it interrupts electrographic seizures and stops behavioral convulsions in all rats, as well as improves survival of rats that experience DFP-induced SE suggesting that the dose is not too high (Rojas et al., 2018). It has not been determined whether diazepam produces any beneficial effects on neuropathology 24 hrs after SE.
One hour after SE onset additional groups of rats were exposed for 5–7 min to isoflurane (Piramal Enterprises, Andhra Pradesh, India) by inhalation followed by administration of a sub-anesthetic dose of urethane (U2500, Sigma) (0.8 g/kg, sc) dissolved in physiological saline. Isoflurane was used to mimic the rapid behavioral sedative effect of diazepam. This dose of urethane effectively terminates electrographic SE without producing lung tumors (Rojas et al., 2018). Status epilepticus was not interrupted pharmacologically in an additional subset of rats (termed “uninterrupted”), whose seizures eventually waned. By combining cortical EEG recordings with the modified Racine scoring we were able to determine that SE persists for more than 5 hours in rats that experience uninterrupted SE although it varied considerably. Each rat received a volume of injected compound based on measured body weight (1 ml/kg).
Behavioral scoring of seizure activity and Modified Irwin test
Following DFP exposure rats presenting distinct motor behaviors that include tail extension, forelimb clonus, and whole body clonic seizures with increasing seizure intensity, duration, and frequency were in SE characterized by non-intermittent whole body clonic seizures that persist. A small subset of rats injected with DFP experienced occasional seizures but did not experience SE and so were grouped in the category “DFP no SE”. The seizure activity was scored and recorded every 5 min for at least 1h using a modified Racine scale (Racine, 1972) that is appropriate for scoring rat seizure behavior in our DFP model as previously reported (Rojas et al., 2018). All rats were monitored for at least 6 h and persistent non-intermittent seizure activity was detected shortly after DFP exposure that lasted until pharmacological interruption post SE onset, or until the seizures waned on their own. The rats were then placed individually into clean plastic cages with fresh bedding, soft food and water and allowed to recover overnight. To maintain hydration, a single injection of lactated Ringer’s solution (2 ml, ip) was administered when the rats were placed into cages. All rats were sacrificed at 2 h or 24 h. A modified Irwin test (Irwin, 1968) was performed to access the health of rats prior to and after DFP-induced SE as previously described (Rojas et al., 2018). The test was given twice (once prior to DFP and again just prior to euthanization 24 h after SE onset). Rats were observed for ptosis, exophthalmia, lacrimation, body posture, running and walking, hypoactivity, hypothermia and vocalization when handled. Each observation item was scored as: 0=normal; 1=mild to moderate impairment; 2=severe impairment.
Quantification of proteins by ELISA
Enzyme-linked immunosorbent assays (ELISA) were performed to quantify brain levels of interleukin 1β (IL-1β), tumor necrosis factor α (TNFα), cyclooxygenase-1 and −2 (COX-1, COX-2). COX-1 was included as a control as it is constitutively expressed in tissues and is not induced following seizures, whereas COX-2 is induced following DFP exposure (Rojas et al., 2015, 2018). Protein lysates were generated from half brains (hemisphere without cerebellum) and skeletal muscle (Vastus lateralis) taken 2h after DFP exposure from rats that experienced DFP-induced SE, DFP no SE rats, and non-seizure control rats that received no drugs, by homogenizing the tissue in RIPA lysis and extraction buffer (25 mM Tris-HCl, pH7.6, 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS) (Thermo Scientific, Rockford, IL) with proteinase and phosphatase inhibitors (Thermo Scientific). The lysates were incubated on ice for 2 h and then cleared by centrifugation at 14,000 × g for 15 min. The soluble fraction of the lysates was stored at −80 °C until further use. The total protein level was measured by spectrophotometry using a SmartSpec 3000 (Biorad, Hercules, CA) and using the Pierce protein assay (1861426, Thermo Scientific, Rockford, IL). ELISA was performed according to the manufacturer’s protocol provided with the kit.
Quantification of acetylcholinesterase activity
Protein lysates from hemi-brains and skeletal muscle mentioned above were used to measure AChE activity using an acetylcholinesterase colorimetric assay kit (ab138871, Abcam) according to the manufacturer’s protocol as previously described (Rojas et al., 2018). The data from the samples were normalized using the acetylcholinesterase standard curve according to the manufacturer’s protocol provided with the kit and compared to homogenates obtained from non-seizure control rats.
FluoroJade B histochemistry
Twenty-four hours following DFP-induced status epilepticus onset a subset of rats was deeply anesthetized under isoflurane and decapitated. The brains were removed rapidly and longitudinally bisected. One hemisphere lacking the cerebellum of each brain was rapidly frozen on dry ice and kept for RNA isolation (below). The other half brain was fixed in 4% paraformaldehyde, transferred to sucrose and 40 μm coronal sections were prepared for immunohistochemistry and FluoroJade B (FJB) histochemistry (Rojas et al., 2018). For FJB staining the sections were mounted onto slides prior to staining. FluoroJade B staining was performed according to the manufacturer protocol (Histo-chem Inc., Jefferson, AR) as described by Rojas et al. (2018). Briefly, hippocampal sections were immersed in 100% ethyl alcohol for 3 min followed by a 1 min change in 70% alcohol and a 1 min change in distilled water. The sections were transferred to a solution of 0.06% potassium permanganate for 15 min, rinsed for 1 min in distilled water and then transferred to a FJB staining solution (0.0004% FJB) for 30 min. Sections were rinsed following FJB staining with Tris-buffered saline (TBS), mounted onto clean slides and rapidly air dried. The slides were immersed in xylenes and coverslipped with D.P.X. (Aldrich Chem. Co., Milwaukee, WI) mounting media. FJB labeling was visualized using an AxioObserver A1 epifluorescence microscope equipped with an AxioCam MRc5 camera and pictures were obtained with the AxioVision AC 4.7 (Zeiss) software.
Quantification of FluoroJade B labeled cells
Following FJB labeling, images of three hippocampal areas (i.e., hilus, CA1, CA3) were taken with a 5x objective lens using the AxioVision AC 4.1 software (Zeiss) from each of 5 sections in each rat, from the dorsal hippocampus between bregma −2.56 and −4.16 mm (Paxinos & Watson, 1986). An observer blinded to the treatment of the animals and the experimental conditions counted the number of bright FJB positive cells in each area. The cell numbers were recorded and expressed as the mean number of positive FluoroJade B (+FJB) cells/section for each area in each rat. FJB staining was also quantified in the medial amygdala (MEA), piriform cortex (PC), lateral amygdala (LA), the ventral endopiriform nucleus (vEND) and the dorsal endopiriform nucleus (dEND). These regions were chosen for FJB quantification given their implication in temporal lobe epilepsy. All of the regions chosen for FJB quantification are highly interconnected with other limbic nuclei. Quantification of the number of cells was performed using ImageJ software (National Institute of Health, USA). In ImageJ the area of interest was selected and the FJB positive cells were quantified using the “analyze particles” feature. The intensity threshold and the minimum and maximum cell size were initially determined in an empirical manner by an investigator blinded to the treatment conditions. Automatic quantification by ImageJ was carried out on 5 sections obtained from each animal for the treatments. The data are presented as the mean ± standard error of the mean. The number of +FJB cells per section was also graphed for each individual rat.
Immunohistochemistry, Nissl staining and acetylcholinesterase activity staining
Brains were prepared and sectioned as described above for FJB labeling. Immunohistochemistry was performed on free-floating sections as described previously (Rojas et al., 2018). Briefly, for fluorescence immunohistochemistry, the sections were blocked for 4 h in PBS containing 1% BSA, 10% goat serum and 0.3% Triton X-100 at 25°C for 2 h. The section were subsequently incubated in the primary antibody [rabbit anti-COX-2 (1:1000), Abcam; rabbit anti-cFos (1:1000), Abcam; mouse anti-AChE, (1:1000), Thermo Scientific; rabbit anti-Iba1 (1:2000), Wako; mouse anti-NeuN, (1:2000), Millipore; Goat anti-IL-1β (1:500), R&D Systems] diluted in antibody dilution solution (ADS) at 4°C overnight (18 h). After washing with ADS, the sections were incubated for 4 h at 25°C with Alexa Fluor fluorescent conjugated antibodies (all purchased from Molecular Probes, Eugene, OR) diluted to 1:500 in ADS. The sections were washed with PBS and then incubated with the blue-fluorescent Hoechst 33342 dye (Molecular Probes) diluted 1:2000 in ADS for four hours at 25°C. After staining the sections were washed again with PBS, mounted onto slides, rapidly air dried, and cover slipped with FluoroGel mounting media. The fluorescence reactions were visualized using an AxioObserver A1 fluorescence microscope and pictures were obtained using the AxioVision AC 4.7 software. Subsequently, confocal images were taken with a BX51WI microscope equipped with a disk spinning unit (DSU) (Olympus, Center Valley, PA) and SlideBook4.2 software (Intelligent Imaging Innovations, Denver, CO). In control experiments, the sections were treated in a similar manner, except the primary antibodies were omitted. All negative control sections showed no staining (data not shown). All sections used for IHC were obtained from the dorsal hippocampus between bregma −2.56 and −4.16 mm (Paxinos & Watson, 1986).
For Nissl staining free-floating sections were mounted onto slides and allowed to air dry overnight 25°C. The next day the slides were immersed into a 0.1% cresyl violet solution for 15 min at 25°C. The sections were washed with sterile water, dehydrated by exposure to increasing concentrations of ethanol, made transparent with xylenes and then cover slipped in the presence of permount. Acetylcholinesterase activity can be viewed histologically in brain sections using an acetylcholinesterase stain. The method adopted is based on the staining procedure widely used in the rodent brain atlases generated by Paxinos & Watson (1980). Free-floating sections (40 μm) were mounted onto slides and allowed to air dry overnight at 25°C. The slides were placed overnight at 25°C in an incubation solution containing cupric sulfate (4 mM), glycine (16 mM), S-Acetylthiocholine iodide (4 mM) and ethopropazine HCL (86 μM) in sodium acetate buffer (50 mM). The sections were washed with sterile water for 5 min and developed for 10 min by immersion in a 1% sodium sulfide solution. The sections were air dried at 25°C, made transparent in 100% ethanol for 5 min followed by Xylenes for 30 min and cover slipped in the presence of permount. The Nissl and AChE activity reactions were visualized using an AxioObserver A1 microscope (Zeiss, Oberkochen, Germany). Pictures were taken using the AxioVision AC 4.7 software (Zeiss).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using Trizol with the PureLink RNA Mini Kit (Invitrogen, Carlsbad, CA) from the frozen half brains lacking the cerebellum of rats. RNA concentration and purity were measured by an Epoch microplate spectrophotometer (BioTek, Winooski, VT) using the A260 value and the A260/A280 ratio, respectively. The first-strand complementary DNA (cDNA) synthesis was performed with 1 μg of total RNA, using a qScript cDNA superMix kit (Quanta Biosciences, Beverly, MA) in a reaction volume of 20 μl at 42 °C for 30 min. The reaction was terminated by heating at 85 °C for 5 min. The qPCR was performed as previously described (Rojas et al., 2018). Melting curve analysis was used to verify specificity of the primers by single-species PCR product. Real time PCR primer sequences are listed in Table 1. Three housekeeping genes [β-actin, glyceraldehyde-3-phosphate (GAPDH), and hypoxanthine phosphoribosyltransferase 1 (HPRT1)] (Table 2) were used as an internal control. Samples without cDNA template served as the negative controls. The 12 inflammatory mediators investigated are listed with the primer sequences in Table 1. Interleukin 1-beta (IL-1β), interleukin 6 (IL-6), and tumor necrosis factor alpha (TNFα) are produced and secreted by cells involved in both innate and acquired immunity to stimulate inflammation. C-C motif chemokine ligands 2, 3 and 4 (CCL2, CCL3 and CCL4) are chemokines that recruit peripheral leukocytes to sites of inflammation or injury. FBJ murine osteosarcoma viral oncogene homolog (cFos) and nerve growth factor-induced early-response gene C [NGFI-c, encoded by the Early Growth Response 4 (EGR4)] gene are immediate early genes that are activity dependent. Fos is often used as an indicator of neuronal discharge and thus seizure intensity (Harvey & Sloviter, 2005). NGFI-c is a member of the zinc finger transcriptional activators and it is induced following seizures (Mack et al., 1995; Honkaniemi & Sharp, 1999). Also investigated are two growth factors [brain derived neurotrophic factor (BDNF) and transforming growth factor beta-1 (TGFβ1)] that stimulate cell growth. Cyclooxygenase 2 (COX-2) is an intracellular rate-limiting enzyme in the production of prostanoids that contribute to the inflammatory response. Analysis of quantitative real time PCR data for each gene of interest was performed by subtracting the geometric mean of the three internal housekeeping genes from the measured cycle threshold value obtained from the log phase of the amplification curve to yield ΔCT for relative quantification. The fold change of each gene of interest was estimated for each animal 24 h after DFP-induced SE relative to the amount of RNA found in the control animals using the 2−ΔΔCT method (Livak & Schmittgen, 2001). All conditions for qRT-PCR were the same. Although the average fold change is shown in figures of qRT-PCR data the ΔΔCT values, which are more normally distributed, were used for statistical comparison.
Table 1.
Real Time PCR Primer Sequences. The approved human gene nomenclature symbol is in parentheses if different from gene name.
| Genes: | Forward Primer (sequence 5’−3’): | Reverse Primer (sequence 5’−3’): |
|---|---|---|
| HPRT1 | GGTCCATTCCTATGACTGTAGATTTT | CAATCAAGACGTTCTTTCCAGTT |
| β-ACTIN (ACTB) | CCAACCGTGAAAAGATGACC | ACCAGAGGCATACAGGGACA |
| GAPDH | GGTGAAGGTCGGTGTGAAC | CCTTGACTGTGCCGTTGAA |
| CCL2 | CAGAAACCAGCCAACT CTCA | GTGGGGCATTAACTGCATCT |
| CCL3 | T CCACGAAAATTCATTGCTG | AGATCTGCCGG111CTCTTG |
| CCL4 | CATCGGAACTTTGTGATGGA | CACAGATTTGCCTGCCTTTT |
| IL-1β (IL1B) | CAGGAAGGCAGTGTCACTCA | TCCCACGAGTCACAGAGGA |
| IL-6 (IL6) | AACTCCATCTGCCCTTCAGGAACA | AAGGCAGTGGCTGTCAACAACATC |
| TNFα (TNF) | CGTAGCCCACGTCGTAGC | GGTTGTC111GAGATCCATGC |
| COX-2 (PTGS2) | ACCAACGCTGCCACAACT | GGTTGGAACAGCAAGGA111 |
| cFos (FOS) | GGAATTAACCTGGTGCTGGA | T GAACATGGACGCTGAAGAG |
| NGFI-c (EGR4) | AACCTCATGTCTGGCATC | CTCGAACAGGCACTGCGA |
| BDNF | CGGAAACAGAACGAACAGAAAC | TGGCTCTCATACCCACTAAGA |
| TGFβl (TGFB1) | CTGGGCACCATCCATGAC | CAGTTCTTCTCTGTGGAGCTGA |
| GP91Phox (CYBB) | TGTGACAATGCCACCAGTCT | TCTTGCATCTGGGTCTCCA |
| GFAP | CATCTCCACCGTCTTTACCAC | AACCGCATCACCATTCCTG |
| lba1 (AIF1) | TCGATATCTCCATTGCCATTCAG | GATGGGATCAAACAAGCACTTC |
| CD11B (ITGAM) | GAGCATCAGTAGCCAGCAT | CCGTCCATTGTGAGATCCTT |
Table 2.
CT values and geometric means of mRNA for three housekeeping genes obtained from brain hemisphere in groups of rats for 24-hour inflammatory mediator measurement. The number in parentheses represents the number of rats in each group.
| Uninterrupted | β-Actin | GAPDH | HPRT1 | Geomean |
|---|---|---|---|---|
| Control (6) | 22.5 ± 0.3 | 21.3 ± 0.2 | 24.4 ± 0.1 | 22.7 ± 0.2 |
| DFP-SE (7) | 23.5 ± 0.2 | 20.4 ± 0.3 | 24.2 ± 0.1 | 22.6 ± 0.1 |
| Diazepam-1 hr | β-Actin | GAPDH | HPRT1 | Geomean |
| Control (4) | 20.4 ± 0.3 | 19.1 ± 0.3 | 22.5 ± 0.04 | 20.6 ± 0.2 |
| DFP-SE (9) | 18.3 ± 0.6 | 17.9 ± 0.6 | 21.7 ± 0.4 | 19.2 ± 0.5 |
| Urethane-1 hr | β-Actin | GAPDH | HPRT1 | Geomean |
| Control (5) | 22.1 ± 0.5 | 20.7 ± 0.5 | 23.6 ± 0.6 | 22.1 ± 0.5 |
| DFP-SE (11) | 21.7 ± 0.3 | 20.9 ± 0.3 | 23.7 ± 0.3 | 22.1 ± 0.3 |
Data are the mean ± standard error
Western blot
Frozen half brains lacking the cerebellum from rats that experienced SE and non-seizure controls were homogenized on ice in 1.5 ml RIPA buffer (25 mM Tris HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing a mixture of protease and phosphatase inhibitors (Roche Applied Science). The homogenates were placed on ice for 2 h and then centrifuged (14,000 ×g, 15 min, 4 °C). The protein concentration in the supernate was measured by Bradford assay (Thermo Fisher Scientific). The supernates (20 μg protein each) were resolved by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto PVDF membranes (Millipore). Membranes were blocked with 5% (w/v) non-fat milk at 25 °C for 4 h and then incubated overnight at 4 °C with the primary antibody: rabbit anti-COX-2 (1:2000, Abcam), rabbit anti-IBA1 (1:4000, Wako) or mouse anti-β-Actin (1:1000, Abcam). This procedure was followed by incubation with horseradish peroxidase-conjugated secondary antibodies (1:2000, Jackson Immuno) at room temperature for 2 h. The blots were developed by enhanced chemiluminescence (ECL) (Thermo Fisher Scientific) and scanned using a ChemiDoc MP imaging system (BioRad, Hercules, CA). The band intensities were quantified by ImageLab 6.0.1 (BioRad).
Reagents and solutions
The rat COX-2 ELISA kit was purchased from IBL international (Hamburg, Germany). The rat COX-1 ELISA kit was obtained from MyBioSource (San Diego, CA). Rat ELISA kits for IL-1β and TNFα were from R&D Systems (Minneapolis, MN). All plasticware and reagents were sterile and RNase and DNase-free.
Data analysis
Data obtained from ELISA (COX-1, COX-2, TNFα, IL-1β) and the AChE activity assays are presented as box plots with individual points. Data obtained from EEG power analysis, FJB quantification and western blot are presented as means ± standard error with individual points shown. All qRT-PCR data are presented as means with individual points. Statistical analysis was performed with GraphPad Prism version 7 (GraphPad software, San Diego, CA). Student’s t test, Mann-Whitney test, or one-way ANOVA (with Bonferroni or Dunnett’s posthoc tests) were performed as appropriate based on the questions asked and the comparisons made to examine differences of chemical effects. Student’s t test was used for comparisons made with parametric data and the Mann-Whitney test for comparisons made with nonparametric data for only two group data sets. The Dunnett’s posthoc was performed with the one-way ANOVA when two or more treatment groups were compared to a common control group. The Bonferroni posthoc was used to compare multiple selected pairs of treatment groups. The differences were considered to be statistically significant if p < .05. The Shapiro-Wilk test in Origin 9.4.2 (OriginLab, Northampton, MA) was used to test normality of the data. Experimental power analysis was performed with GPower3.1.9.2 (Universitat Kiel, Germany). The observed power values, the statistical test performed and the obtained p-value are presented in Table 3. The superscript following data in the results corresponds to a specific row of statistical information in Table 3. Estimation statistics was performed for qRT-PCR mRNA fold change of inflammatory mediators from brain hemispheres only using an internet application (http://www.estimationstats.com/#/) built for common use by Dr. Adam Claridge-Chang and Mr. Joses Ho. The application performs data analysis with a bootstrap estimation (DABEST) package in python (Ho et al., 2019) with 5000 resampling, bias-corrected and accelerated bootstrap analysis to determine the nonparametric confidence interval of differences between treatment groups. The mean differences between groups, the confidence interval from the bootstrapping and the p-values obtained using the Mann-Whitney test to compare treatments are presented in Table 4.
Table 3.
Statistical table
| Data structure | Type of test | Observed Power |
p-value |
|---|---|---|---|
| a Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.98 | < 0.01 |
| b Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.99 | < 0.01 |
| c Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.99 | < 0.001 |
| d Log-normal distribution | Mann-Whitney test | 0.88 | 0.01 |
| e Log-normal distribution | Mann-Whitney test | 0.84 | 0.0002 |
| f Log-normal distribution | Mann-Whitney test | 0.95 | 0.002 |
| g Normally distributed | Unpaired t test | 0.09 | > 0.05 |
| h Log-normal distribution | Mann-Whitney test | 0.80 | 0.01 |
| i Normally distributed | One-way ANOVA with posthoc Bonferroni | 0.87 | 0.015 |
| j Normally distributed | Unpaired t test | 1 | < 0.0001 |
| k Normally distributed | One-way ANOVA with posthoc Bonferroni | 1 | < 0.0001 |
| l Normally distributed | One-way ANOVA with posthoc Bonferroni | 1 | < 0.0001 |
| m Normally distributed | One-way ANOVA with posthoc Bonferroni | 0.99 | > 0.05 |
| n Normally distributed | One-way ANOVA with posthoc Bonferroni | 0.99 | > 0.05 |
| o Normally distributed | One-way ANOVA with posthoc Bonferroni | 0.85 | 0.006 |
| p Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.99 | < 0.001 |
| q Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.99 | < 0.001 |
| r Normally distributed | One-way ANOVA with posthoc Dunnett’s | 0.58 | < 0.01 |
| s Normally distributed | One-way ANOVA with posthoc Bonferroni | 0.29 | > 0.05 |
Table 4.
Estimation statistics for qRT-PCR mRNA fold change from brain hemispheres with 5000 bootstrap iterations
| Analyte/treatment | Mean ± SEM | Mean difference (compared to urethane) | 95% confidence interval | p-value |
|---|---|---|---|---|
| IL-1β | ||||
| Uninterrupted (7) | 287 ± 53 | 284.9 | 199 – 389 | 0.0006 |
| Diazepam (9) | 10 ± 2 | 8.3 | 5.5 – 13.8 | 0.0003 |
| Urethane (11) | 2 ± 0.4 | 0 | - | - |
| TNFα | ||||
| Uninterrupted (7) | 2 ± 0.5 | −0.7 | −1.5 – 0.7 | 0.15 |
| Diazepam (9) | 2.9 ± 0.6 | 0.4 | −0.6 – 1.7 | 0.94 |
| Urethane (11) | 2.6 ± 0.2 | 0 | − | − |
| IL-6 | ||||
| Uninterrupted | 16 ± 3.3 | 2.8 | −5.4 – 11.9 | 0.5 |
| Diazepam | 31 ± 3.4 | 17.8 | 7 – 25 | 0.006 |
| Urethane | 13 ± 3.2 | 0 | - | - |
| COX-2 | ||||
| Uninterrupted | 8 ± 0.7 | 1.9 | −0.4 – 4.3 | 0.15 |
| Diazepam | 13.4 ± 1.6 | 7.5 | 3.8 – 11 | 0.003 |
| Urethane | 5.9 ± 1 | 0 | - | - |
| cFos | ||||
| Uninterrupted | 8 ± 1.5 | 3.3 | 0.4 – 7.3 | 0.02 |
| Diazepam | 5.6 ± 1.5 | 1.2 | −1.6 – 5.2 | 0.65 |
| Urethane | 4.4 ± 1 | 0 | - | - |
| NGFI-c | ||||
| Uninterrupted | 8 ± 2.4 | 5.7 | 2.7 – 12.9 | 0.004 |
| Diazepam | 5.5 ± 1.5 | 3.3 | 0.9 – 7 | 0.06 |
| Urethane | 2 ± 0.4 | 0 | - | - |
| CCL2 | ||||
| Uninterrupted | 1040±113 | 754 | 453 – 980 | 0.002 |
| Diazepam | 604 ± 69 | 320 | 62 – 490 | 0.003 |
| Urethane | 284 ± 85 | 0 | - | - |
| CCL3 | ||||
| Uninterrupted | 13 ± 1.5 | 11.1 | 8.1 – 13.7 | 0.0006 |
| Diazepam | 13.6 ± 3.2 | 12.1 | 7.7 – 21 | 0.0002 |
| Urethane | 1.5 ± 0.2 | 0 | - | - |
| CCL4 | ||||
| Uninterrupted | 63 ± 21 | 61.2 | 33.5 – 118 | 0.0006 |
| Diazepam | 7 ± 2 | 5.1 | 2.3 – 10.3 | 0.003 |
| Urethane | 1.9 ± 0.4 | 0 | - | - |
| TGFβ1 | ||||
| Uninterrupted | 1.3 ± 0.2 | −0.9 | −1.8 – 0.1 | 0.3 |
| Diazepam | 1.9 ± 0.2 | −0.4 | −1.2 – 0.5 | 0.5 |
| Urethane | 2.2 ± 0.4 | 0 | - | - |
| Gp91phox | ||||
| Uninterrupted | 1.4 ± 0.3 | 0.1 | −0.5 – 0.7 | 0.9 |
| Diazepam | 1 ± 0.1 | −0.3 | −0.8 – 0 | 0.2 |
| Urethane | 1.4 ± 0.2 | 0 | - | - |
| BDNF | ||||
| Uninterrupted | 2.9 ± 0.5 | −2.6 | −4.2 – −0.7 | 0.03 |
| Diazepam | 3.2 ± 0.5 | −2.3 | −4.2 – −0.6 | 0.04 |
| Urethane | 5.5 ± 0.8 | 0 | - | - |
SEM = standard error of mean. P-value is obtained by a Mann-Whitney test comparing diazepam and uninterrupted to urethane.
RESULTS
Early induction of inflammatory mediators and cFos during DFP-induced SE in rats
Adult male Sprague-Dawley rats were injected with a single dose of DFP (9.5 mg/kg in sterile water, ip) to induce SE and sacrificed 2 h after SE onset (Figure 1A). This dose of DFP resulted in a high incidence of SE in adult rats (Rojas et al., 2015, 2016, 2018). In rats, seizure-induced behaviors began within a few minutes of DFP exposure and consisted of distinct motor behaviors that include tail extension, forelimb clonus and whole body clonic seizures. These behaviors were observed and scored using a modified Racine scale.
Figure 1. DFP-induced SE elicits an early inflammatory response and induction of cFos in the brain of rats.
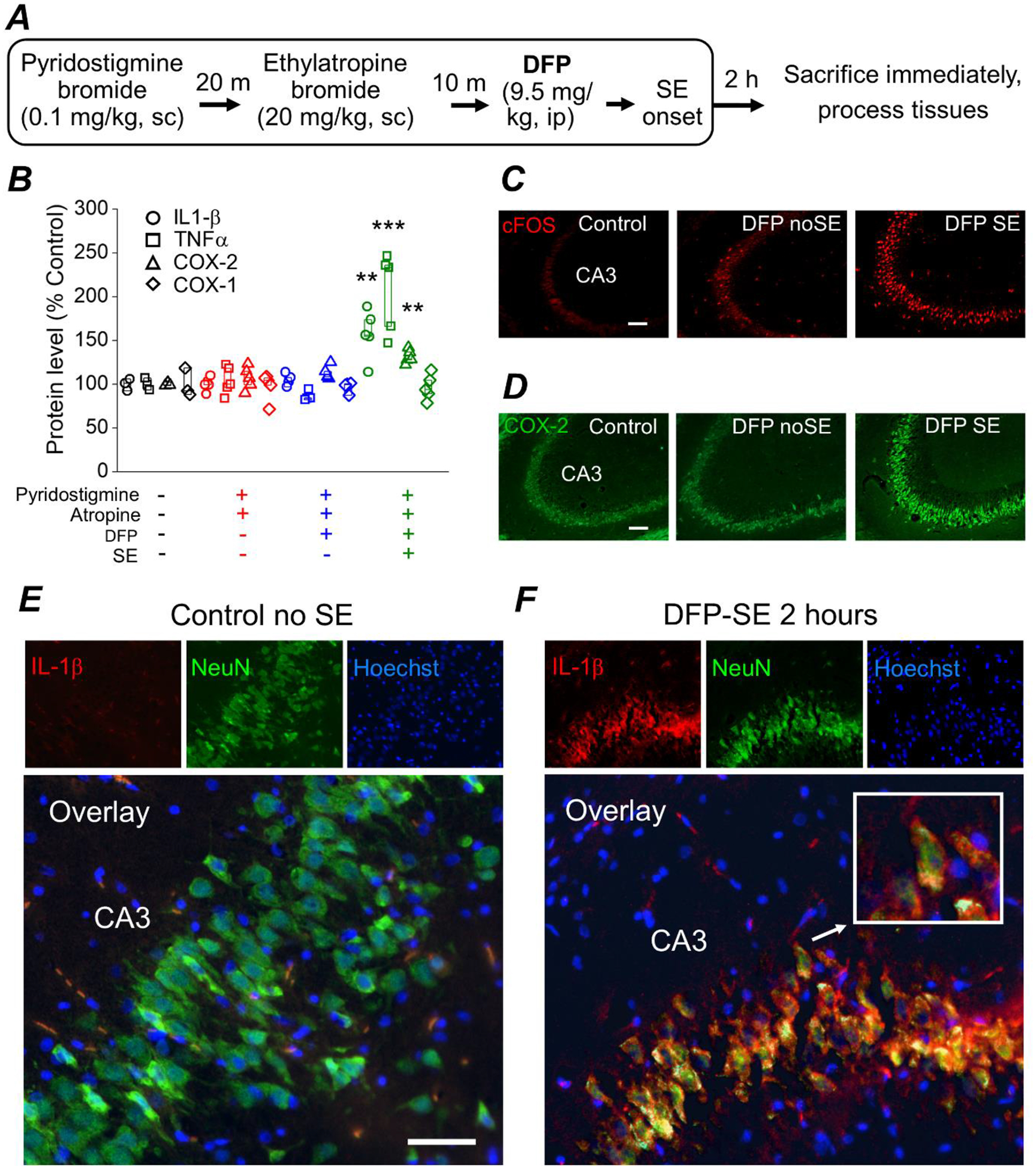
A, experimental paradigm of chemical administration in a rat model of DFP-induced SE. All rats were administered pyridostigmine bromide and ethylatropine bromide followed by DFP to induce status epilepticus. After 2 h of SE all rats were immediately euthanized and the brains were removed and bisected longitudinally. One hemisphere was homogenized in RIPA lysis buffer to obtain protein lysates and the other hemisphere was cut on a crytostat to obtain coronal sections (40 μm) throughout the hippocampus for immunohistochemistry. B, changes in IL-1β, TNFα, COX-2 and COX-1 protein in the brains of rats following DFP exposure measured 2 hours after DFP-induced SE by ELISA. Data are box plots with a 25 and 75 range. The symbols represent each individual rat within the group (** = p < .01, one-way ANOVA with posthoc Dunnett’s compared to the “no treatment” controls). The color of the symbol correspond to the treatment received shown on the bottom of panel B. All four analytes (COX-1, COX-2, IL-1β, TNFα) were measured in all rats regardless of treatment. The protein level is the measured amount of each analyte by ELISA compared to the no treatment controls. C, fluorescence images taken from the cornu ammonis 3 (CA3) region in the hippocampus reveals basal expression of cFos in non-seizure control rats. cFos expression (bright red stain) is induced in rats that experienced 2 h of DFP-induced SE and to a lesser degree in rats that were administered DFP but did not experience SE (“DFP no SE”). The images shown are a single representative of 5 hippocampal sections each from 3 rats in the groups. Scale bar = 100 μm. D, fluorescence images taken from the CA3 region in the hippocampus reveals basal expression of neuronal COX-2 in non-seizure control rats. COX-2 expression is induced slightly in DFP no SE rats and more robustly in rats that experienced 2 h of DFP-induced SE. The images shown are a single representative of 5 hippocampal sections each from 3 rats in the groups. Scale bar = 100 μm. E and F, immunohistochemistry was performed on rat coronal hippocampal sections for IL-1β and NeuN. Fluorescent images taken from the CA3 region in the hippocampus (200x total magnification) revealed little basal expression of neuronal IL-1β in rats that did not experience status epilepticus (panel E; Control no SE, left insert). Neuronal IL-1β in the CA3 region is greatly induced 2 h after DFP-induced SE (panel F; DFP-SE, left insert). Green fluorescent images of the CA3 region in the hippocampus reveals expression of the neuronal marker NeuN (middle insert). The nuclei were labelled by Hoechst staining shown in blue (right insert). Overlaying the red IL-1β stain, the green NeuN stain and the Hoechst revealed IL-1β induction in the same neurons positively stained for NeuN for rats that experienced DFP-induced SE. An example of a neuron with IL-1β induction is magnified in the white outlined box (insert). The images shown are representative of five sections each from three to five rats. Scale bar, 30 μm.
Protein lysates from half-brains lacking the cerebellum were subjected to ELISA for quantification of Interleukin 1-beta (IL-1β), tumor necrosis factor alpha (TNFα), COX-1 (constitutive cyclooxygenase) and COX-2 (inducible cyclooxygenase). The level of COX-2 in the brain was significantly increased 2 h following DFP-SE onset as measured by ELISA (132 ± 4% of non-seizure controls, n = 5 rats; p < .01, one-way ANOVA with posthoc Dunnett’s)a (Figure 1B). On the other hand, COX-1 remained near the baseline in all treatment groups (Figure 1B) demonstrating that exposure of rats to DFP and subsequent SE rapidly induces COX-2. Levels of the pro-inflammatory cytokines IL-1β and TNFα were increased 2 h following DFP-induced SE (158 ± 13% of non-seizure controls for IL-1β, p < .01, one-way ANOVA with posthoc Dunnett’s; 206 ± 21% of non-seizure controls for TNFα, p < .001, one-way ANOVA with posthoc Dunnett’s)b, c (Figure 1B). COX-1, IL-1β, and TNFα remained at the baseline level in rats administered DFP but did not experience SE at the 2 h time point (Figure 1B). Fluorescence immunohistochemistry performed on coronal hippocampal sections (40 μm) revealed induction of COX-2 and cFos in hippocampal pyramidal neurons 2 h after SE onset (Figure 1C, D). cFos was also induced in a small subset of hippocampal pyramidal neurons in rats administered DFP that did not experience SE. Rats that experienced DFP-induced SE, but not non-seizure control rats, displayed positive immunostaining of IL-1β in CA3 pyramidal cells (Figure 1E, F). Taken together, these data reveal increased neuronal activity in the brain and upregulation of inflammatory mediators early during DFP-induced SE.
Urethane blocks the return of seizure activity following DFP-induced SE
In the above studies, a cohort of rats was administered DFP (9.5 mg/kg) intraperitoneally and all were sacrificed 2 h after SE onset. In the following experiments, during further model optimization, we investigated subcutaneous injection of DFP as an alternative route of administration. DFP administered at 5 mg/kg subcutaneously resulted in a similar percent of rats experiencing SE compared to 9.5 mg/kg given intraperitoneally (Rojas et al., 2018). These two doses and routes of DFP resulted in a similar latency to SE onset and prolonged SE lasting >5 h without pharmacological intervention, consistent with a previous study demonstrating that a dose of DFP equal to or greater than 4 mg/kg administered subcutaneously results in consistent SE in adult rats (Pouliot et al., 2016). To minimize sensitization of parasympathetic nerves in the abdomen to DFP, in experiments described below DFP was administered at 5 mg/kg subcutaneously and the rats were sacrificed 24 h after SE onset.
To determine whether termination of SE by urethane produces beneficial effects rats were administered urethane (0.8 g/kg, sc) to interrupt SE 1 h after DFP-induced SE onset and compared to rats administered diazepam (10 mg/kg, ip) (Figure 2A). In a separate cohort of rats, SE was uninterrupted pharmacologically and waned on its own after several hours (Figure 2A). Adult male rats were instrumented with bilateral cortical electrodes prior to EEG recording and DFP exposure. On the day of experiment, rats were connected to the EEG instrument and their baseline brain activity was recorded for 20–30 min prior to DFP. All rats tested displayed normal cortical activity prior to any drug administration as determined by the low amplitude, frequency and shape of the waveforms (Figure 2B). EEG was recorded continuously for 24 h in all rats. Exposure to DFP led to the appearance of seizures defined by the sudden onset of a burst of large amplitude (>2x the baseline prior to drug treatment) and high frequency spikes. All rats received a single injection of DFP and the latency to SE onset was very similar. The onset of SE was defined as the appearance of the initial electrographic seizure that consisted of large amplitude spikes persisting for more than 10 sec followed by a rapid quieting of electrical activity. Recurring large amplitude, high frequency spikes persisted following the first seizure (Figure 2B). Both diazepam and urethane reduced the large amplitude high frequency spikes and thus interrupted SE compared to rats that experienced uninterrupted SE (Figure 3A–B).
Figure 2. Experimental paradigm of chemical administration in a rat model of DFP-induced status epilepticus.
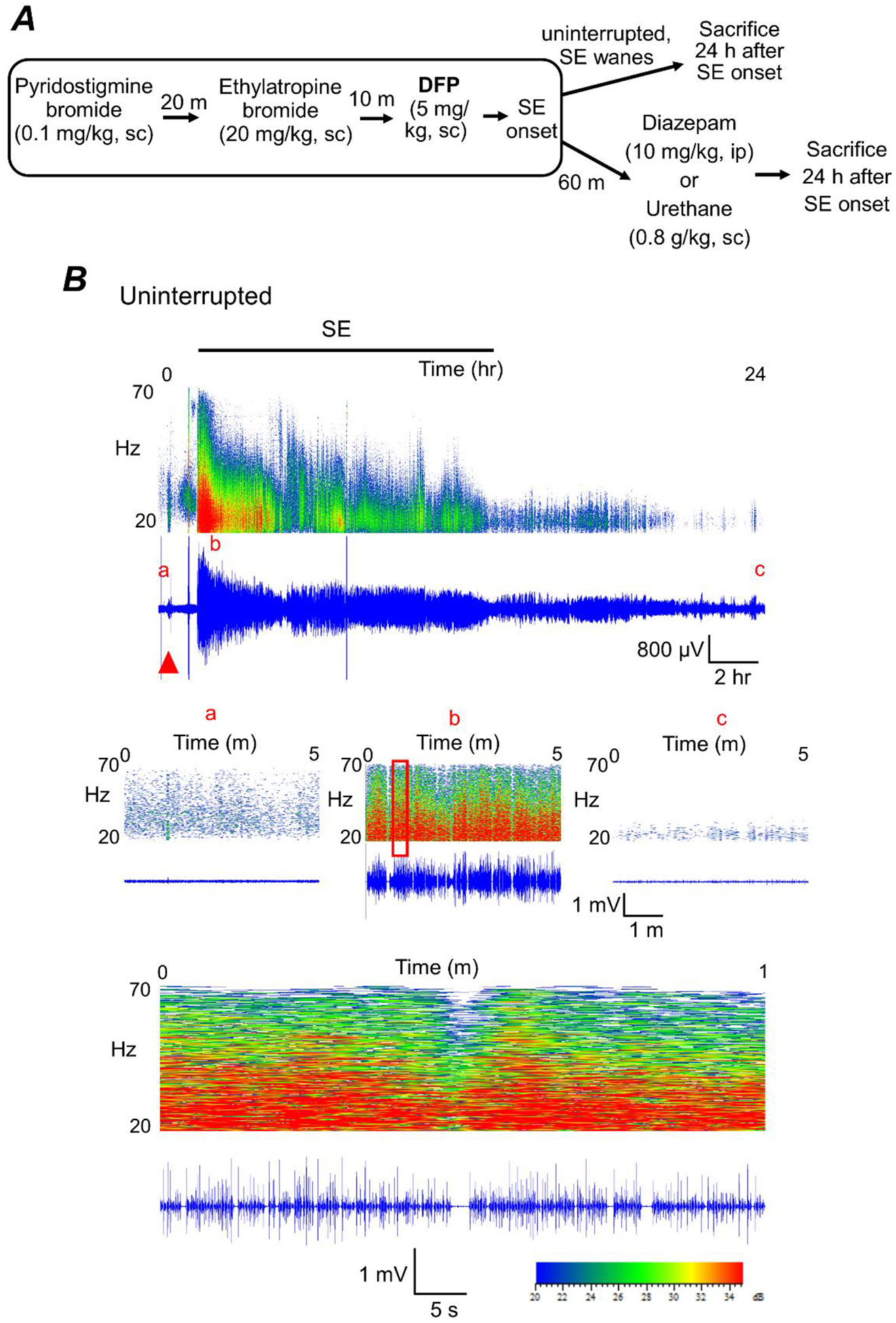
A, all rats were administered pyridostigmine bromide and ethylatropine bromide (Rojas et al., 2017) followed by DFP to induce status epilepticus. For the diazepam cohort, rats were injected with one dose of diazepam 1 h after DFP-induced SE onset. For urethane-treatment following DFP, rats were injected with a single dose of urethane 1 h after the onset of SE. A separate cohort of rats was not administered any drugs after SE onset (uninterrupted SE). All rats were sacrificed 24 h after SE onset. B, cortical electroencephalography (EEG) activity was recorded prior and during SE induced by exposure to DFP for 24 h. A representative EEG trace (in blue) from the cortical recording of an adult male rat showing increased spike activity just after exposure to DFP (red triangle) that develops into SE (denoted by the bar at the top). Above each raw EEG trace is a sonogram of the spike activity obtained in Spike2. The colors of the sonogram indicate the spectral power density in decibels (dB) at the indicated frequency. The middle panels show magnified 5-minute sections of the recording taken from the baseline prior to DFP (a), at the peak intensity of SE (b) and at the end of the recording (c). The area within the red outlined box in panel b was magnified and shown in the bottom panel that represents a 1 min interval of the recording during SE.
Figure 3. The return of seizure activity following 1 hour of DFP-induced SE is suppressed by urethane.
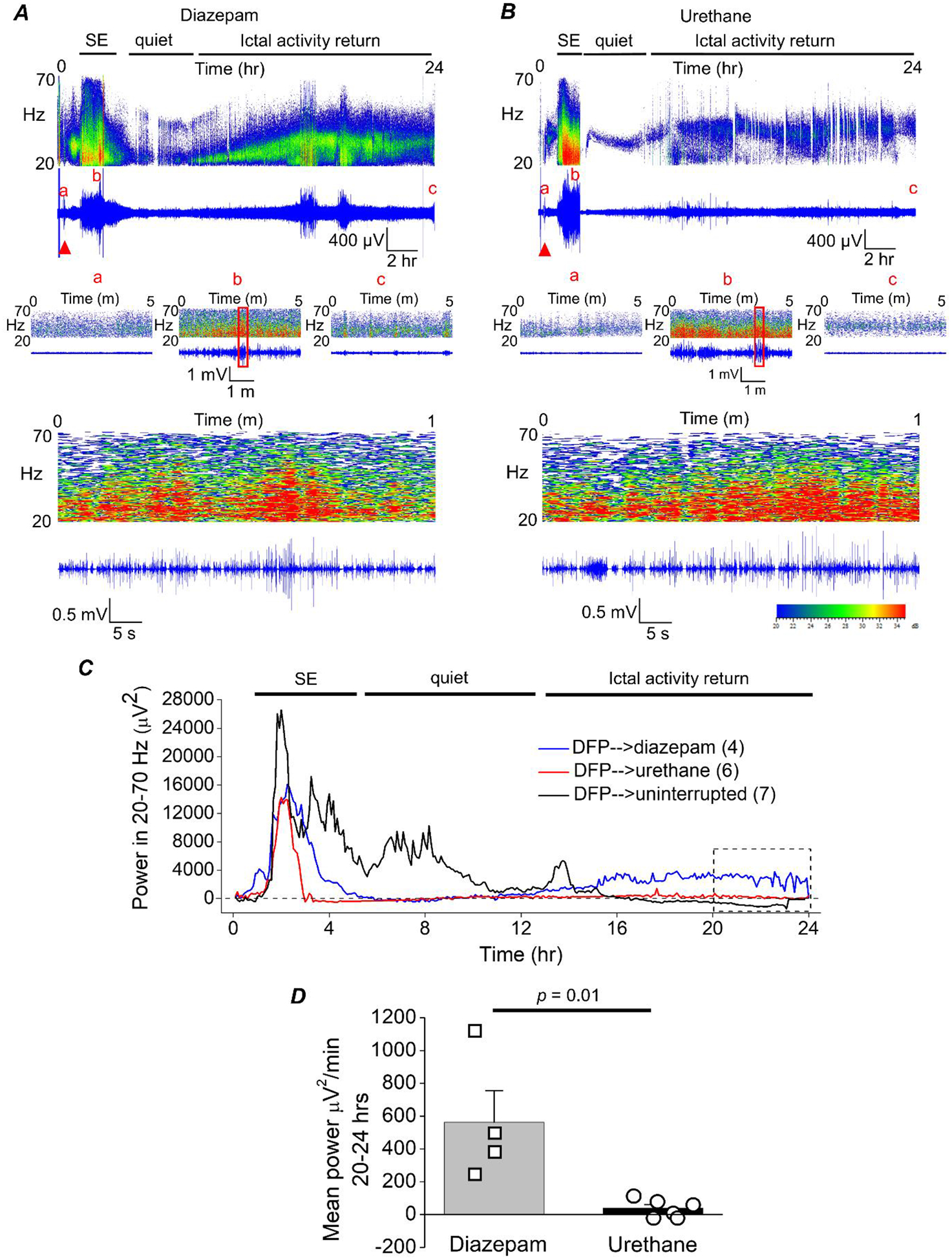
A, cortical electroencephalography (EEG) activity was recorded prior to and during SE induced by exposure to DFP for 24 h. A representative EEG trace and sonogram from the cortical recording of an adult male rat showing increased spike activity just after exposure to DFP (red triangle) that develops into SE (denoted by the bar at the top). Spike activity was quieted by administration of diazepam (10 mg/kg, ip) 1 h after SE onset, but returned within a few hours as denoted by the ictal activity return phase. Above each raw EEG trace is a sonogram of the spike activity obtained in Spike2. The colors of the sonogram indicate the spectral power density in decibels (dB) at the indicated frequency. The middle panels show magnified 5 min intervals of the recording taken from the baseline prior to DFP (a), at the peak intensity of SE (b) and at the end of the recording (c). The area within the red outlined box in panel b was magnified and shown in the bottom panel that represents a 1 min interval of the recording during SE. B, a representative EEG trace from the cortical recording of an adult male rat shows increased spike activity just after exposure to DFP (red triangle) that developed into SE, which was quieted following brief exposure to isoflurane (inhaled) and subsequent injection of urethane (0.8 g/kg, sc) 1 h after SE onset. C, the EEG power in the 20–70 Hz bandwidth averaged over 300 sec epochs during the 24 h period for 4 diazepam-treated, 6 urethane-treated rats and 7 rats that experienced uninterrupted SE. The dashed line indicates baseline power before DFP administration. D, a significant difference was detected between the two treatment groups [diazepam (n = 4) and urethane (n = 6)] in the EEG power in the 20–70 Hz bandwidth using power analysis during the final 4 h of the EEG recording (area within the dashed box in panel C). Data are the mean ± standard error of the mean. p = .01, student’s t-test. The symbols represent each individual rat within the group.
A script written in Python was used to obtain EEG power in the 20–70 Hz band in 300 sec epochs over the 24 h EEG recording. The mean EEG power was measured for the last 4 h of the EEG recording. High frequency seizure activity returned in all rats administered diazepam and the mean power per minute measured in the last 4 h was significantly greater for rats administered diazepam 1 h after SE onset (562 ± 193 μV2/min, n = 4 rats) compared to rats given urethane (37.9 ± 23 μV2/min, n = 6 rats) (p = .01, Mann-Whitney test)d (Figure 3C, D). Urethane effectively blocked the return of large amplitude high frequency spiking (Figure 3C, D). Taken together, these data further strengthen the conclusion that urethane is more effective than diazepam at terminating DFP-induced SE that lasts for at least 1 h, and in contrast to diazepam, urethane blocks the subsequent overnight return of high power seizure activity.
Urethane attenuates early neurodegeneration
Neurodegeneration can be visualized in brain sections using FJB, a dye that enters dead or dying neurons and emits green fluorescence (Schmued et al., 1997). Experiments were performed to determine the extent of neuronal injury using FJB staining performed on coronal hippocampal sections (40 μm) taken from the brains of rats euthanized 24 h after SE onset. Robust neurodegeneration was detected by FJB in the CA1 of the hippocampus of rats that experienced uninterrupted SE (Figure 4A). To determine whether pharmacological interruption of SE after 1 h reduces hippocampal neurodegeneration measured 24 h after SE onset, FJB positive cells were counted in the hilus, CA1 and CA3 in all rats that experienced SE. In the CA1 the total number of FJB positive cells per section was significantly lower in rats administered urethane 1 h after DFP-induced SE compared to rats administered diazepam (40 ± 10 FJB positive cells per hippocampal section for diazepam treated rats, n = 11 rats vs. 8 ± 2 positive cells per hippocampal section for urethane treated rats, n = 8 rats; p = .0002, Mann-Whitney test) (Figure 4B–D)e. There was also reduced neurodegeneration observed in the CA3 of urethane treated rats after 1 h of SE (10 ± 1 FJB positive cells per hippocampal section for 11 diazepam treated rats vs. 5 ± 0.4 positive cells per hippocampal section for 8 urethane treated rats), (p = .002, Mann-Whitney test)f (Figure 4D). Analysis of hilar neurodegeneration revealed no difference in FJB positive cells per section of diazepam treated rats compared to urethane treated rats that experienced 1 h of SE (22 ± 4 FJB positive cells per section for diazepam treated rats, n = 11 vs. 25 ± 4 FJB positive cells per section for urethane treated rats, n = 8) (p > .05, student’s t-test)g (Figure 4D). Overall, the numbers of FJB positive cells in the hippocampus was lower than previously reported on day 4 after SE (Rojas et al., 2016, 2018), which likely reflects a slowly developing injury.
Figure 4. Termination of SE by urethane promotes hippocampal neuroprotection 24 hours after SE onset.
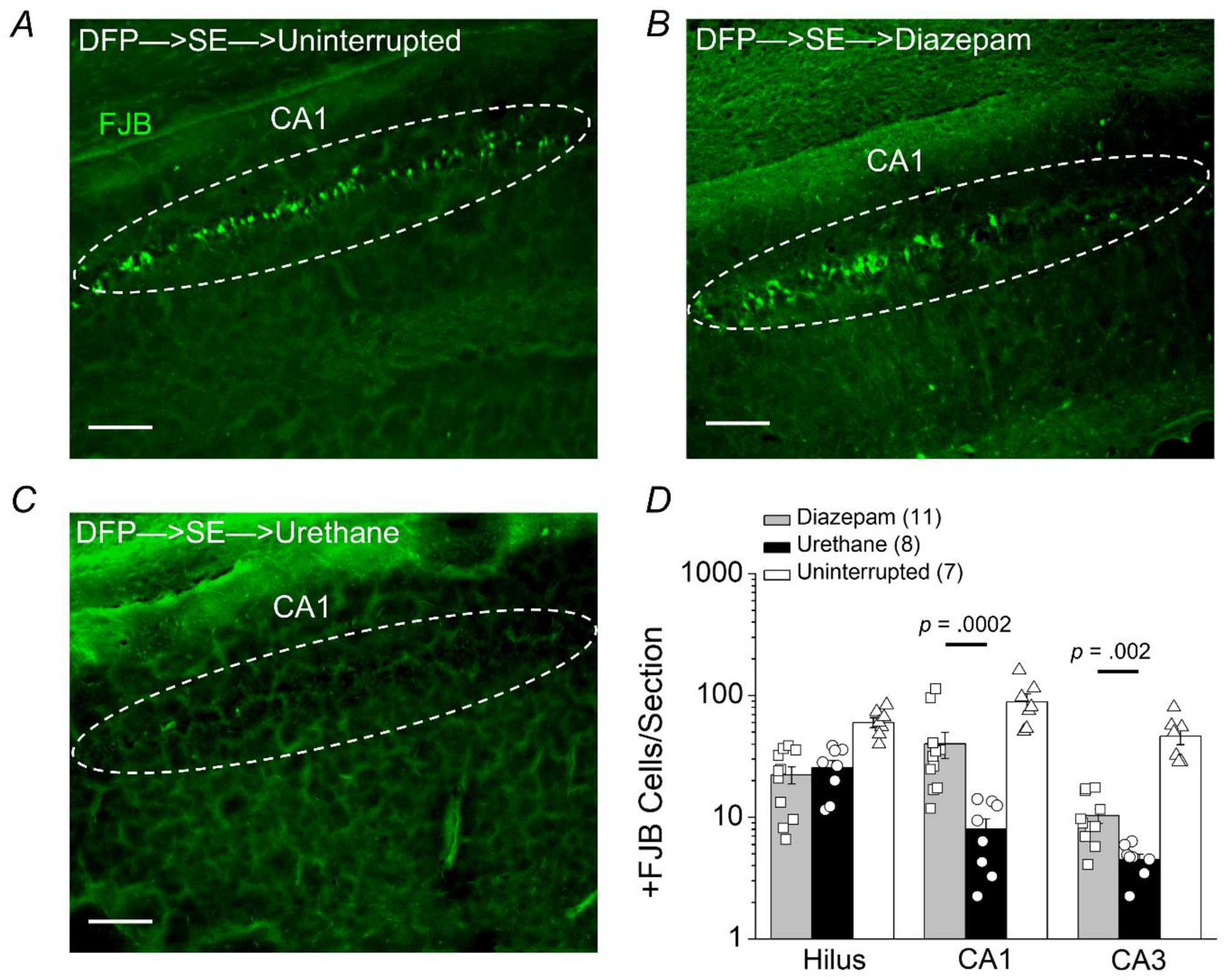
Representative images of FluoroJade B staining in hippocampal sections (40 μm) in the cornu ammonis 1 (CA1) region 24 h after DFP-induced status epilepticus for rats that experienced uninterrupted SE (A), rats treated with diazepam after 1 h of SE (B), and rats injected with urethane after 1 h of SE (C). The images were taken at 100x total magnification. The dashed circles and the arrowheads in the boxed inserts highlight pyramidal cells in CA1. The images are representative of 5 dorsal hippocampal sections per rat. Scale bar = 200 μm. D, the average number of injured neurons per section 24 hours after DFP-induced status epilepticus in three dorsal hippocampal regions (hilus, CA1 and CA3) of rats that experienced uninterrupted SE (white bar, open triangles, n = 7 rats), rats treated with diazepam following 1 h of SE (gray bar, open squares, n = 11 rats) and rats injected with urethane after 1 h of SE (black bar, open circles, n = 8 rats) (p = .0002 in CA1 and p = .002 in CA3, by Mann-Whitney test comparing urethane to diazepam). The bars show the mean and standard error of the mean. The symbols represent each individual rat within the group.
Positive FJB staining was observed and counted in other limbic brain regions including the amygdala, piriform cortex and endopiriform nucleus. Neurodegeneration was present in the medial and lateral amygdala in all rats that experienced SE (Figure 5A–C). However, in the medial amygdala FJB positive cells in rats administered urethane 1 h after DFP-induced SE was significantly lower compared to rats administered diazepam (805 ± 151 FJB positive cells per section for diazepam treated rats, n = 11 rats vs. 303 ± 73 positive cells per section for urethane treated rats, n = 8 rats; p = .01, Mann-Whitney test)h. With the exception of the lateral amygdala all areas investigated displayed significantly lowered FJB positive cells in rats administered urethane 1 h after DFP-induced SE compared to rats administered diazepam (Figure 5D). Neurodegeneration was not observed in rats that were injected with DFP but did not enter SE or non-seizure control rats that were administered sterile water instead of DFP (Figure 6A). Taken together, these data indicate that neurodegeneration detected by FluoroJade B is present in limbic regions 24 h after DFP-induced SE and early termination of SE by urethane significantly reduces neurodegeneration in these seizure sensitive regions.
Figure 5. Neuroprotection by urethane in nuclei of the ventral temporal lobe following DFP-induced status epilepticus.
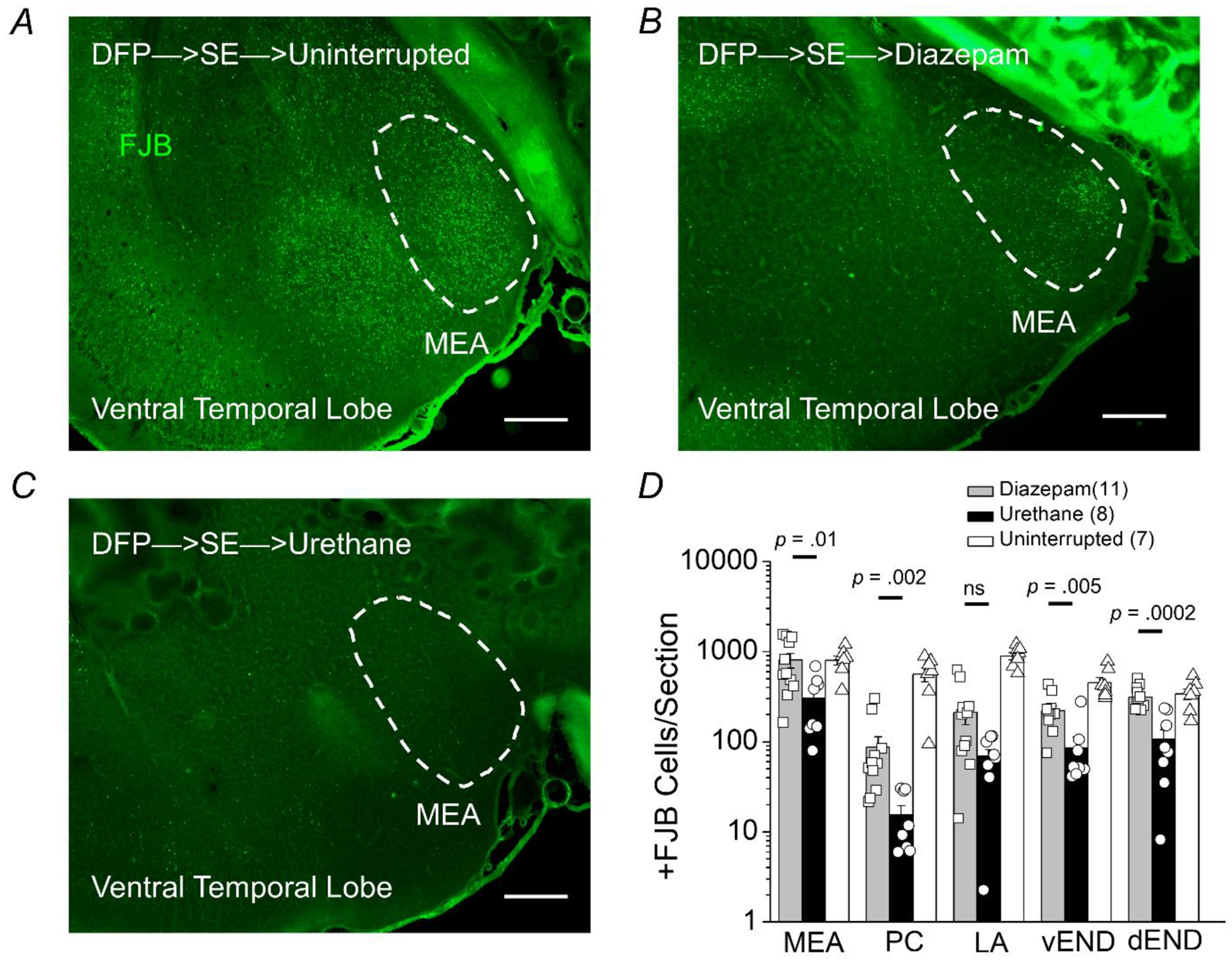
Representative images of FluoroJade B staining in sections (40 μm) of the ventral surface of the temporal lobe 24 hours after DFP-induced status epilepticus for rats that experienced uninterrupted SE (A), rats treated with diazepam after 1 h of SE (B), and rats injected with urethane after 1 h of SE (C). The images were taken at 25x (total magnification). The dashed circles outline the region of the medial amygdala (MEA). The images are representative of 5 sections from the ventral temporal lobe per rat. Scale bar = 500 μm. D, the average number of injured neurons per section 24 h after DFP-induced SE in five nuclei of the ventral temporal lobe (medial amygdala, MEA; piriform cortex, PC; lateral amygdala, LA; ventral endopiriform nucleus, vEND; dorsal endopiriform nucleus, dEND) of rats that experienced uninterrupted SE (white bar, n = 7 rats), rats treated with diazepam following 1 h of SE (gray bar, n = 11 rats) and rats injected with urethane after 1 h of SE (black bar, n = 8 rats). The bars show the mean and standard error of the mean. The symbols represent each individual rat within the group. The Mann-Whitney test was used to compare urethane to diazepam treated rats.
Figure 6. Induction of COX-2 twenty-four hours after DFP-induced SE in rats is attenuated by urethane.
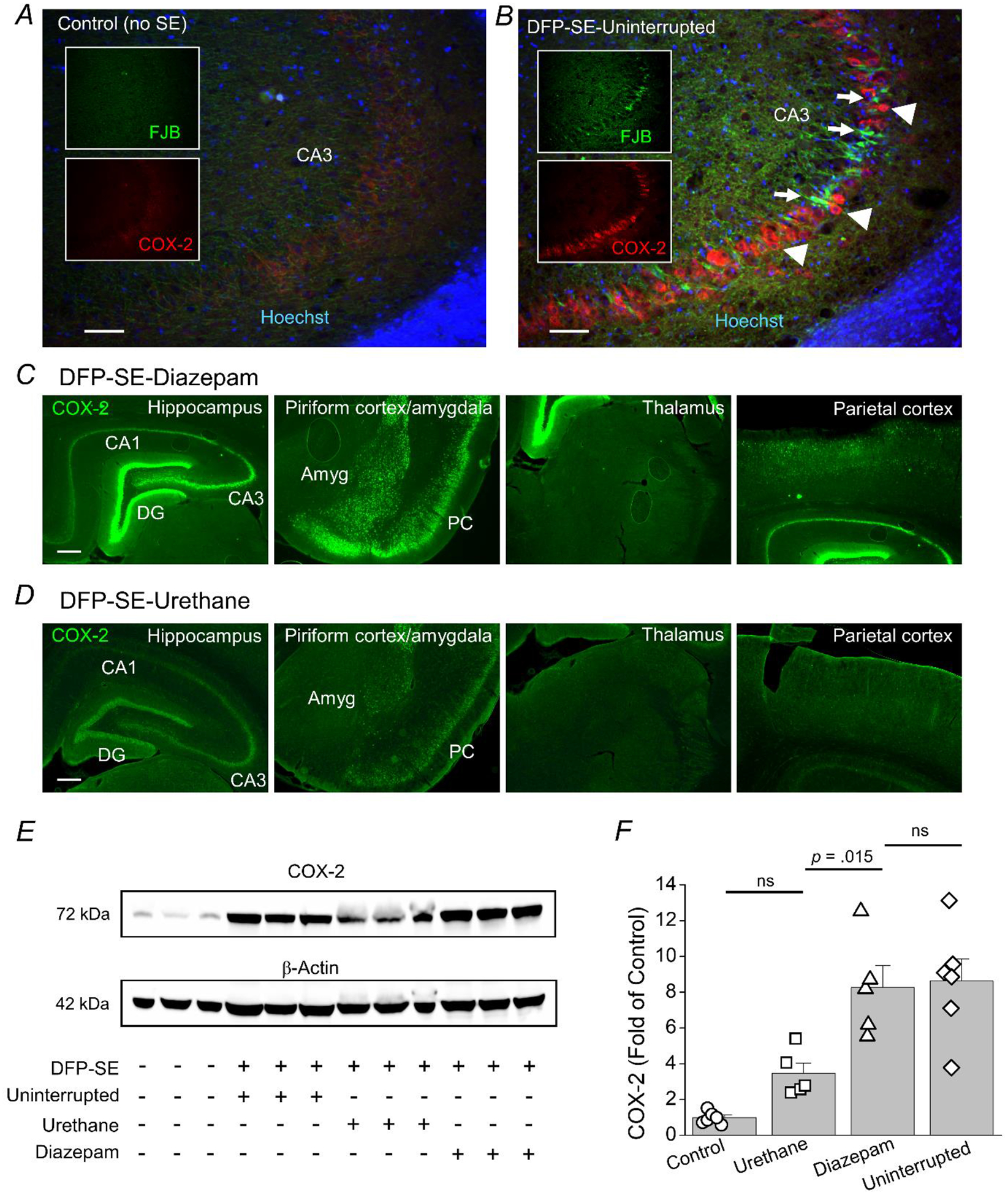
Representative confocal image of cyclooxygenase 2 (COX-2) staining (red), FluoroJade B (FJB) staining (green) and Hoechst staining (blue) in the CA3 region of a hippocampal section (40 μm) obtained from a non-seizure control rat (A) and a rat that experienced uninterrupted SE and was euthanized at 24 h (B). The images were taken at 100x total magnification. The white boxed insert shows COX-2 staining alone in red and FJB staining alone in green. The images shown are representative of five sections each from three rats per treatment. Scale bar = 50 μm. The arrowheads indicate typical COX-2 positive neurons and the arrows indicate FJB positive cells. C, fluorescence images reveals induction of neuronal COX-2 in rats that were administered diazepam 1 h after DFP-induced SE. COX-2 expression (green) is induced in neurons of the hippocampus, amygdala, piriform cortex and parietal cortex. COX-2 was not induced in the thalamus at 24 h. D, rats administered urethane 1 h following DFP-induced SE onset display lower induction of COX-2 in neurons of the hippocampus, amygdala, piriform cortex and parietal cortex compared to diazepam treated rats. The images shown are a single representative of 5 hippocampal sections each from 3 rats in the groups. Scale bar = 100 μm. CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus; Amyg, amygdala; PC, piriform cortex. E, western blot image of COX-2 protein levels (bands in the top box) in the hemibrain (minus cerebellum) of control rats and rats that experienced SE induced by DFP. The bands in the lower box show the level of β-Actin from the same samples used as a loading control. Each lane represents an individual rat and three rats were randomly chosen as representatives of the group. It should be noted that this western blot is for visual interpretation and was not used for quantification. F, the band intensity of COX-2 was normalized to the housekeeping β-Actin for each sample. The bar represents the mean and standard error of the mean. The symbol represents each individual rat within the group. The western blots in Supplemental Figure 1 was used to quantify COX-2 relative to controls. ns = p > 0.05 for control vs. urethane and diazepam vs. uninterrupted, p = 0.015 for diazepam vs. urethane, one-way ANOVA with posthoc Bonferroni.
Seizure-induced upregulation of COX-2 is attenuated by urethane
To determine the extent of COX-2 induction rats administered DFP were euthanized 24 h after SE onset and one brain hemisphere was processed for immunohistochemistry. Fluorescence immunohistochemistry performed on coronal hippocampal sections (40 μm) obtained from non-seizure control rats revealed a low basal level of COX-2 (Figure 6A). Rats that experienced uninterrupted SE induced by DFP displayed robust upregulation of COX-2 in the CA3 region of the hippocampus 24 h following SE onset (Figure 6B). The bright FJB positive cells (green) appear COX-2 negative since these cells are dead or dying (Figure 6B). COX-2 was strongly upregulated 24 h following DFP-induced SE in rats administered diazepam in all cell layers of the hippocampus, the piriform cortex, amygdala and parietal cortex but not the thalamus (Figure 6C). Seizure-induced COX-2 upregulation in limbic nuclei was lower in rats administered urethane 1 h after SE onset compared to rats that received diazepam (Figure 6C, D). The level of COX-2 protein in forebrain homogenates was then quantified using western blot. The western blots in Supplemental Figure 1 shows the band intensity of each individual rat and were used to quantify COX-2 relative to controls. COX-2 was found to be significantly higher in rats administered diazepam compared to rats injected with urethane 1 h after SE onset measured 24 h later (8.3 ± 1.2 fold of control for diazepam treated rats, n = 5 rats vs. 3.5 ± 0.6 fold of control for urethane treated rats, n = 5 rats, p < .015, one-way ANOVA with Bonferroni post hoc test)i (Figure 6E, F; Supplemental Figure 1). Brain COX-2 levels 24 h after SE were similar for rats injected with diazepam compared to rats that experienced uninterrupted SE (8.6 ± 1.3 fold of control) (Figure 6E, F; Supplemental Figure 1). These data suggest that exposure to DFP and subsequent SE induces an inflammatory cascade involving COX-2 in rats, and that urethane but not diazepam administration after SE attenuates this inflammatory response observed 24 h after SE onset.
Urethane attenuates induction of inflammatory mediators
Rats administered DFP were euthanized 24 h after the onset of SE and one hemisphere was processed for RNA extraction and purification. qRT-PCR was carried out to measure the expression of a selected panel of 12 inflammatory mediators (Table 1) in brain tissue (minus cerebellum), a majority of which were previously shown to be up-regulated in rats 4 days following uninterrupted DFP-induced SE (Rojas et al., 2018). An analyte that was not upregulated 4 days following SE is cFos possibly due to the return of normal brain activity four days following SE. Twenty-four hours after SE cFos protein was increased in the hippocampus as determined by immunohistochemistry comparing non-seizure control rats to rats that experienced uninterrupted SE (Figure 7A, B). The level of cFos mRNA was significantly induced in the hippocampus of rats that experienced uninterrupted SE (1.1 ± 0.2 fold change for non-seizure control rats, n = 6 vs. 42 ± 12 fold change for DFP-SE rats, n = 8; p < 0.0001, student’s t-test)j (Figure 7C).
Figure 7. Urethane attenuates induction of brain inflammatory mediators 24 hours after SE.
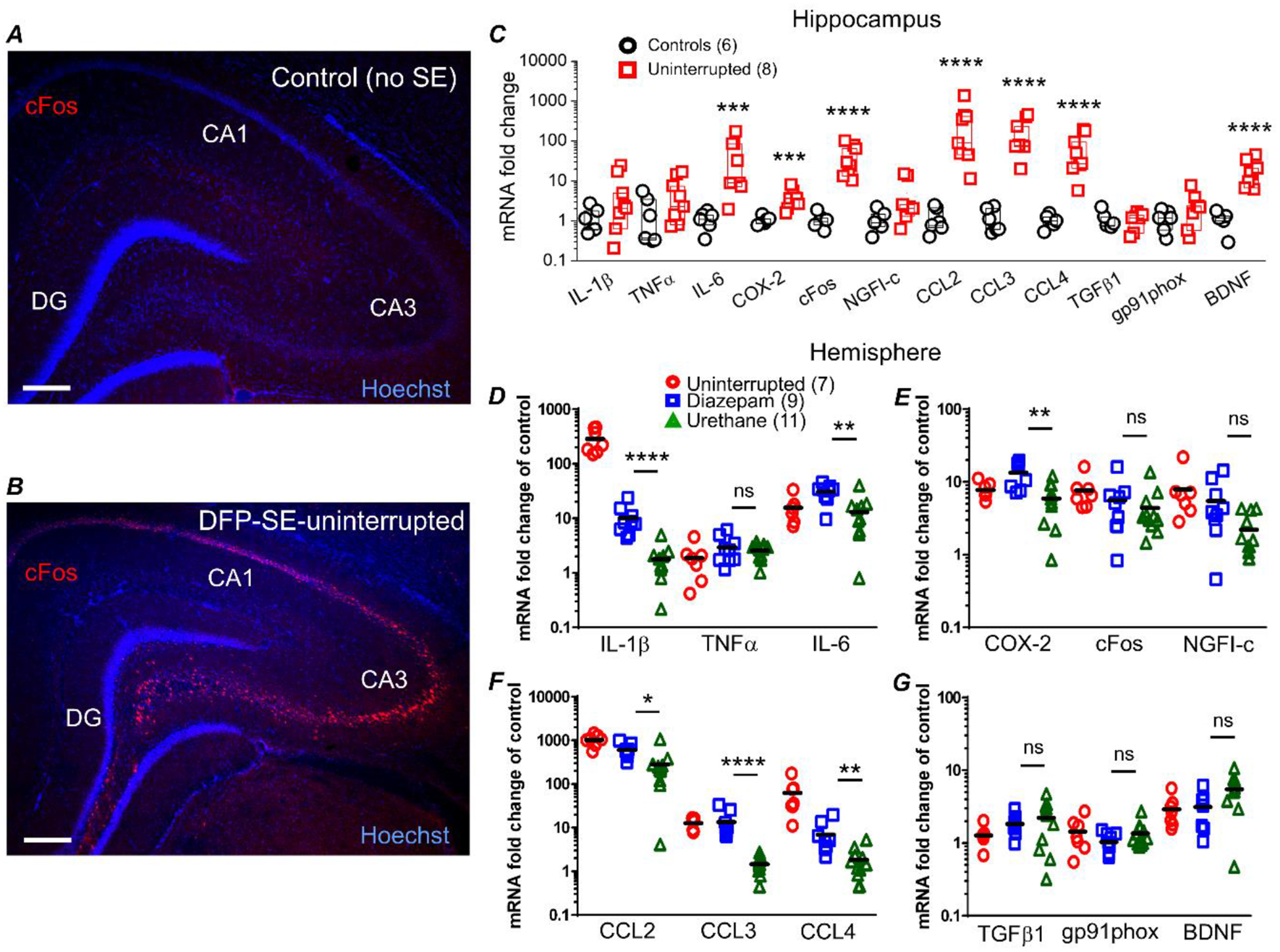
Representative image of cFos staining (red) and Hoechst staining (blue) in a hippocampal section (40 μm) obtained from a non-seizure control rat (A) and a rat that experienced uninterrupted DFP-induced sacrificed at 24 h (B). The images were taken at 25x total magnification. The images shown are representative of five sections each from three rats per treatment. Scale bar = 350 μm. CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus. qRT-PCR was used to quantify the change in abundance of mRNAs of inflammatory mediators, immediate early response factors and growth factors from rats 24 h after injection with water or DFP-induced SE. C, the mRNA fold change of interleukin 6 (IL-6), chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-C motif) ligand 3 (CCL3), chemokine (C-C motif) ligand 4 (CCL4), cyclooxygenase 2 (COX-2), FBJ murine osteosarcoma viral oncogene homolog, (cFos) and brain derived neurotrophic factor (BDNF) were robustly upregulated in the hippocampus of rats that experienced uninterrupted DFP-induced SE. Although the fold changes are shown statistical analysis was carried out using the ΔΔCT values. ns = p >.05, *** = p < .001, **** = p < .0001, student’s t-test. The abundance of mRNA in the brain hemisphere of the 12 genes were separated into 4 panels with three genes shown per panel. IL-1β, interleukin 1 beta; TNFα, tumor necrosis factor alpha and IL-6 are shown in (D). COX-2, cFos and nerve growth factor-induced early-response gene (NGFI-c) are shown in (E). CCL2, CCL3 and CCL4 are shown in (F). Transforming growth factor beta-1 (TGFβ1), NADPH oxidase 2 (GP91phox) and BDNF are shown in (G). The difference in the level of upregulation of the hippocampus compared to the hemi-brain is likely due to contributions of other brain regions in the hemi-brains. The mRNA fold change of IL-1β, IL-6, CCL2, CCL3, CCL4 and COX-2 were significantly reduced in rats that received a single injection of urethane 1 h after SE compared to rats that received diazepam. Although the fold changes are shown statistical analysis was carried out using the ΔΔCT values. ns = p >.05, * = p < .05, ** = p < .01, **** = p < .0001, one-way ANOVA with posthoc Bonferroni (urethane vs. diazepam). The symbols represent each individual rat within the group. The black horizontal bar within the symbols represents the mean.
Rats that experienced uninterrupted SE displayed a robust increase in mRNA levels of most inflammatory mediators investigated, but not the housekeeping genes (β-actin, GAPDH and HPRT1; Table 2), in the hippocampus (Figure 7C) and hemi-brain (Figure 7D–G). For example, mRNA levels were increased 1040-fold for CCL2, 13-fold for CCL3, 63-fold for CCL4, 16-fold for IL-6, and 287-fold for IL-1β above non-seizure controls in hemi-brains. The pattern of upregulation of the inflammatory mediators was similar in the isolated hippocampus compared to hemi-brains. Similarly, most of the inflammatory mediators investigated were upregulated in rats administered diazepam 1 h after SE (Figure 7D–G). On the other hand, rats administered urethane 1 h after DFP-induced SE onset displayed a broadly blunted inflammatory response measured 24 h after SE onset. A significant difference in mRNA upregulation comparing urethane treated rats to diazepam administered rats was observed for the pro-inflammatory cytokine IL-1β (10.1 ± 2 fold induction for diazepam treated rats, n = 9 rats vs. 1.8 ± 0.4 fold induction for urethane treated rats, n = 11 rats; p < .0001, one-way ANOVA with posthoc Bonferroni)k (Figure 7D). Similarly, the SE-induced upregulation of the chemokine CCL3 was strongly blunted by administration of urethane (13.6 ± 3.2 fold induction for diazepam treated rats, n = 9 rats vs. 1.5 ± 0.2 fold induction for urethane treated rats, n = 11 rats; p < .0001, one-way ANOVA with posthoc Bonferroni)l (Figure 7F). Estimation statistics was used to obtain confidence intervals (Table 4) for qRT-PCR data. The confidence intervals provide a more plausible range for the mean of the group and predict the outcome of numerous replications of the experiment.
Early gliosis is unaffected by urethane
Experiments were performed to compare gliosis in rats that experienced SE. Intense immunostaining of Iba1 (a microglia marker) was observed in hippocampal sections of rats that experienced uninterrupted SE but not in non-seizure controls (Figure 8A–C). qRT-PCR was used to quantify changes in mRNA of astrocytic GFAP, microglial CD11b and microglial Iba1 in rat hemi-brains 24 h after SE. Changes in the mRNA levels of these markers can be indicative of the level of gliosis. qRT-PCR revealed that mRNA of GFAP and Iba1 were upregulated above non-seizure control levels in the brains of rats that experienced SE 24 h earlier (Figure 8D). The 24 h induction of Iba1 mRNA was highest in rats that experienced uninterrupted SE (8.8 ± 1 fold above control, n = 7 rats) (Figure 8D). Iba1 mRNA levels were slightly lower in rats administered urethane and similar to controls for rats administered diazepam. The average level of Iba1 mRNA was not different for diazepam treated rats [1.2 ± 0.05 fold above control (n = 5 rats)] compared to rats injected with urethane after SE [4.0 ± 1.2 fold above control (n = 5 rats)] (p > .05, one-way ANOVA with posthoc Bonferroni)m (Figure 8D). Similarly, no difference was detected in GFAP mRNA in rats administered diazepam compared to urethane [5.5 ± 0.8 fold above control (n = 5 rats) for diazepam treated rats vs. 8.2 ± 1.3 fold above control (n = 5 rats) for urethane treated rats] (p > .05, one-way ANOVA with posthoc Bonferroni)n (Figure 8D). Rats administered diazepam displayed similar levels of CD11b mRNA compared to rats injected with urethane (Figure 8D).
Figure 8. DFP-induced gliosis is unaffected by urethane 24 hours after DFP-induced SE.
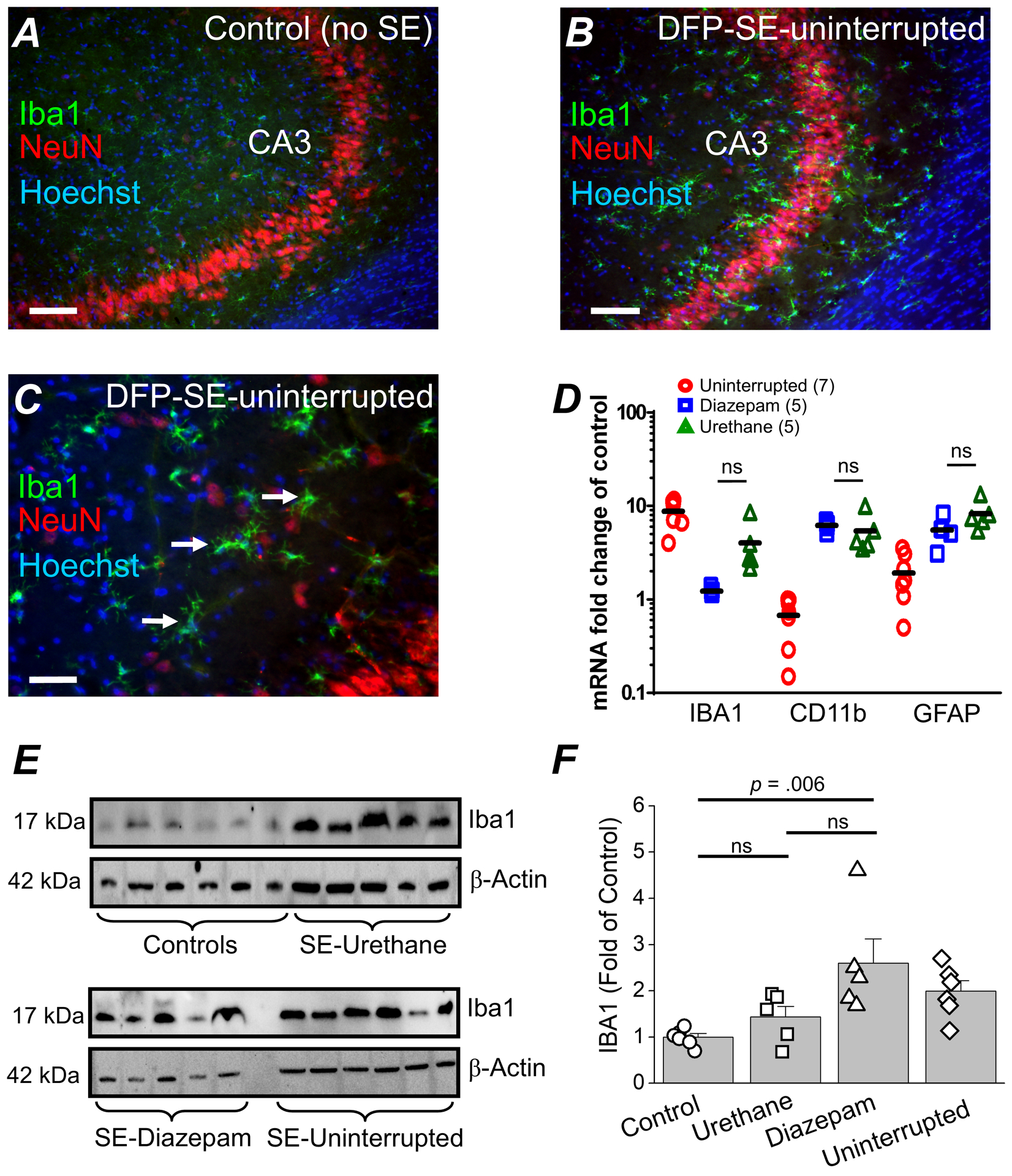
Representative confocal image of ionized calcium-binding adapter molecule 1 (IBA1) staining (green), neuronal nuclei (NeuN) staining (red) and Hoechst staining (blue) in the CA3 region of a hippocampal section (40 μm) obtained from a non-seizure control rat (A) and a rat that experienced uninterrupted DFP-induced SE and was euthanized at 24 h (B). The images were taken at 100x total magnification. C, a magnified image of the CA3 region of a rat that experienced uninterrupted SE. The images shown are representative of five sections each from three rats per treatment. Scale bar = 50 μm (A and B) and 25 μm (C). The arrows indicate typical activated microglia following SE. D, induction of GFAP, CD11B and IBA1 mRNA in the forebrain of rats that experienced uninterrupted DFP-induced SE (n = 7), rats administered diazepam 1 h after SE onset (n = 5 rats) and urethane (1 h SE) treated rats (n = 5 rats) all sacrificed at 24 h. The symbols represent each individual rat within the group. The black horizontal bar within the symbols represents the mean. Although the fold change is shown statistical analysis was performed on the ΔΔCT values. ns = p > .05, one-way ANOVA with posthoc Bonferroni (urethane vs. diazepam). E, western blot image of Iba1 protein (bands in the top box) in the brain of non-seizure control rats and rats that experienced SE induced by DFP and were administered urethane. The bands in the lower box show the level of β-Actin from the same samples used as a loading control. Each bar represents an individual rat. D, the band intensity of Iba1 was normalized to the housekeeping β-Actin for each sample. The data are the mean and standard error of the mean. The symbol represents each individual rat within the group. ns = p > 0.05 for control vs. urethane and diazepam vs. urethane, p = 0.06 for diazepam vs. urethane, one-way ANOVA with posthoc Bonferroni.
The level of Iba1 protein measured by western blot in forebrain homogenates obtained 24 h after SE onset was significantly higher in rats administered diazepam (n = 5 rats) compared to non-seizure controls (n = 6 rats) (2.6 ± 0.5 fold of control for diazepam treated rats vs. 1 ± 0.1 for control rats, p = .006, one-way ANOVA with Bonferroni post hoc test)o (Figure 8E, F). Rats administered urethane (n = 5 rats) 1 h after SE onset displayed a similar level of Iba1 as non-seizure controls (Figure 8E). The level of Iba1 was higher for rats injected with diazepam compared to rats that experienced uninterrupted SE (2.0 ± 0.2 fold of control) or rats administered urethane as measured 24 h after SE onset (Figure 8E, F). Taken together, these data suggest that 24 h after DFP-SE gliosis is apparent, but not reduced by urethane.
Seizure duration correlates with neuropathology after SE
The intensity of status epilepticus experienced by each rat was quantified using EEG power. Power analysis of the entire 24 h EEG recordings revealed that on average the duration of the high power seizure activity in rats that experienced uninterrupted SE was similar to diazepam injected rats when combining the initial duration of SE and the seizure activity return phase (Figure 9A). The longer total duration of seizure activity in rats that experienced uninterrupted SE and rats administered diazepam appears to coincide with the similarities in neuropathology (i.e. neurodegeneration and neuroinflammation) for the two groups of rats. On the other hand, high power seizure activity in rats administered urethane was limited to the 60 min duration of the initial SE experience, and these rats displayed the lowest degree of neuropathology (Figure 9B–D).
Figure. 9. Correlation between seizure duration and neuropathology after SE.
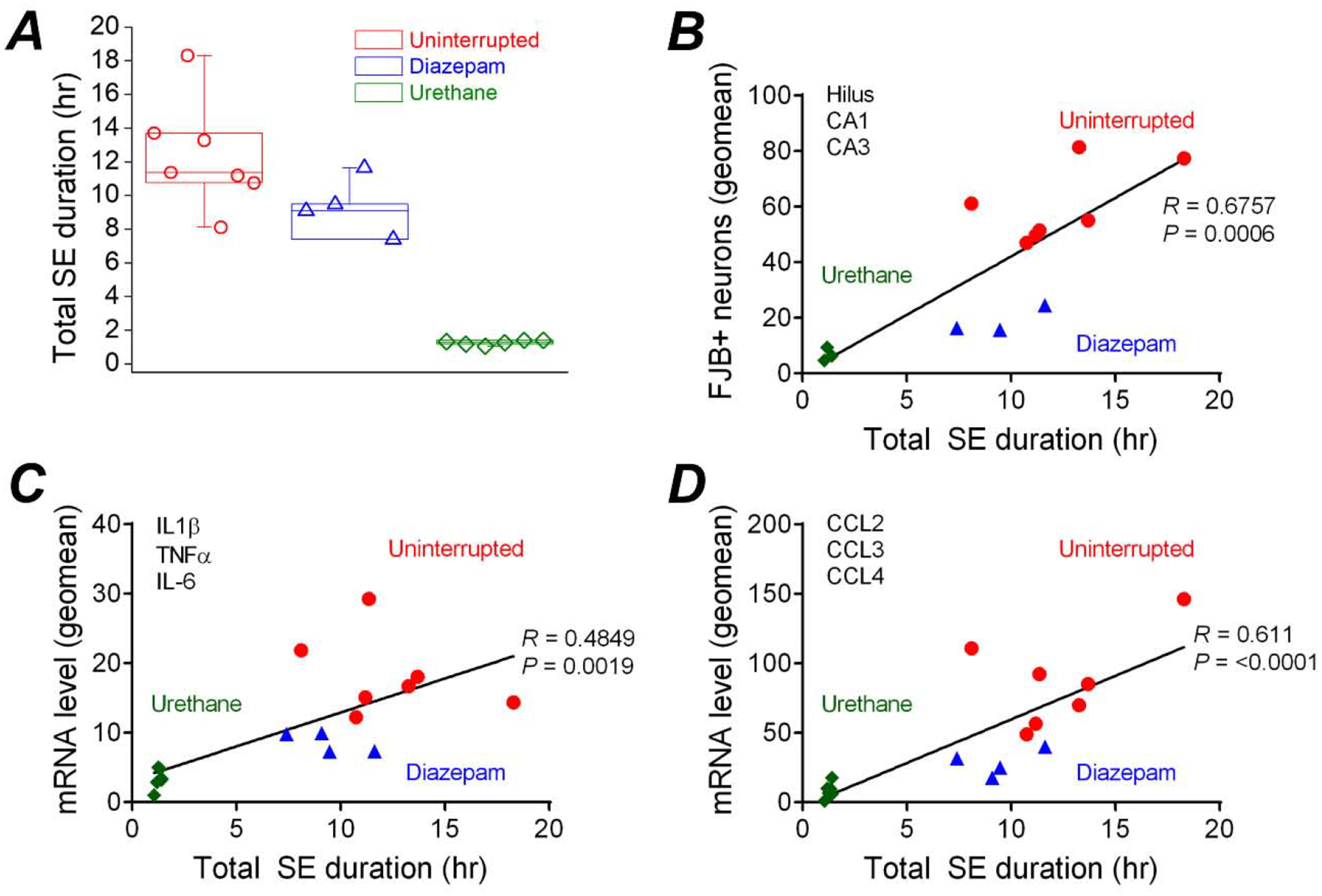
A, total SE duration over 24 hr for rats that experienced uninterrupted DFP-induced SE (n = 7 rats) as well as rats administered diazepam (10 mg/kg, ip) (n = 4 rats) or urethane (0.8 g/kg, sc) (n = 6 rats) one hour after SE onset. The data are the mean and standard error of the mean. The symbol represents each individual rat within the group. Pearson’s correlation coefficient analysis was performed to examine the relationship between total 24 h seizure duration and neuronal damage in the hippocampus shown by FluoroJade B positive (FJB+) cells (B) and neuroinflammation indicated by the mRNA expression levels of three prototypical inflammatory cytokines (IL-1β, IL-6 and TNF-α) (C) and three (CCL2, CCL3 and CCL4) (D) chemokines highly induced in the brain following SE. A geometric mean was obtained from the three hippocampal sub-regions as well as the three inflammatory cytokines and chemokines.
AChE is not a target of urethane
Acetylcholinesterase (AChE) activity was significantly reduced in the brain of rats that experienced DFP-induced SE compared to non-seizure controls measured 2 h after SE onset (100 ± 4% AChE activity for controls, n = 3 rats vs. 67 ± 4% AChE activity for DFP-SE rats, n = 5 rats; p < .001, one-way ANOVA with posthoc Dunnett’s)p (Figure 10A). Brain AChE activity was also inhibited in rats that were administered the same dose of DFP but did not experience (DFP no SE) (70 ± 2% AChE activity for DFP no SE, n = 4 rats; p < .001, one-way ANOVA with posthoc Dunnett’s)q, which demonstrates that failure to experience SE is not due to low overall brain exposure to DFP (Figure 10A). Acetylcholinesterase activity in the brain was unaffected by peripheral administration of pyridostigmine bromide and ethylatropine bromide. However, skeletal muscle obtained from the hind limb of rats administered only pyridostigmine bromide and ethylatropine bromide exhibited reduced AChE activity (85 ± 8%, n = 5 rats), presumably due to pyridostigmine bromide, which is a reversible AChE inhibitor. Acetylcholinesterase activity was also reduced to 82 ± 6% of control in skeletal muscle of rats that received DFP and experienced SE (n = 5 rats). Rats that were administered DFP but did not experience SE also displayed reduced skeletal muscle AChE activity (67 ± 4%, n = 4) which was significantly lower than non-seizure control rats; p < .01, one-way ANOVA with posthoc Dunnett’s)r (Figure 10A). Taken together, these experiments provide evidence that the supporting agents (pyridostigmine bromide and ethylatropine bromide) are unable to cross the blood-brain barrier and protect brain AChE.
Figure 10. Acetylcholinesterase activity is unaltered by urethane or diazepam.
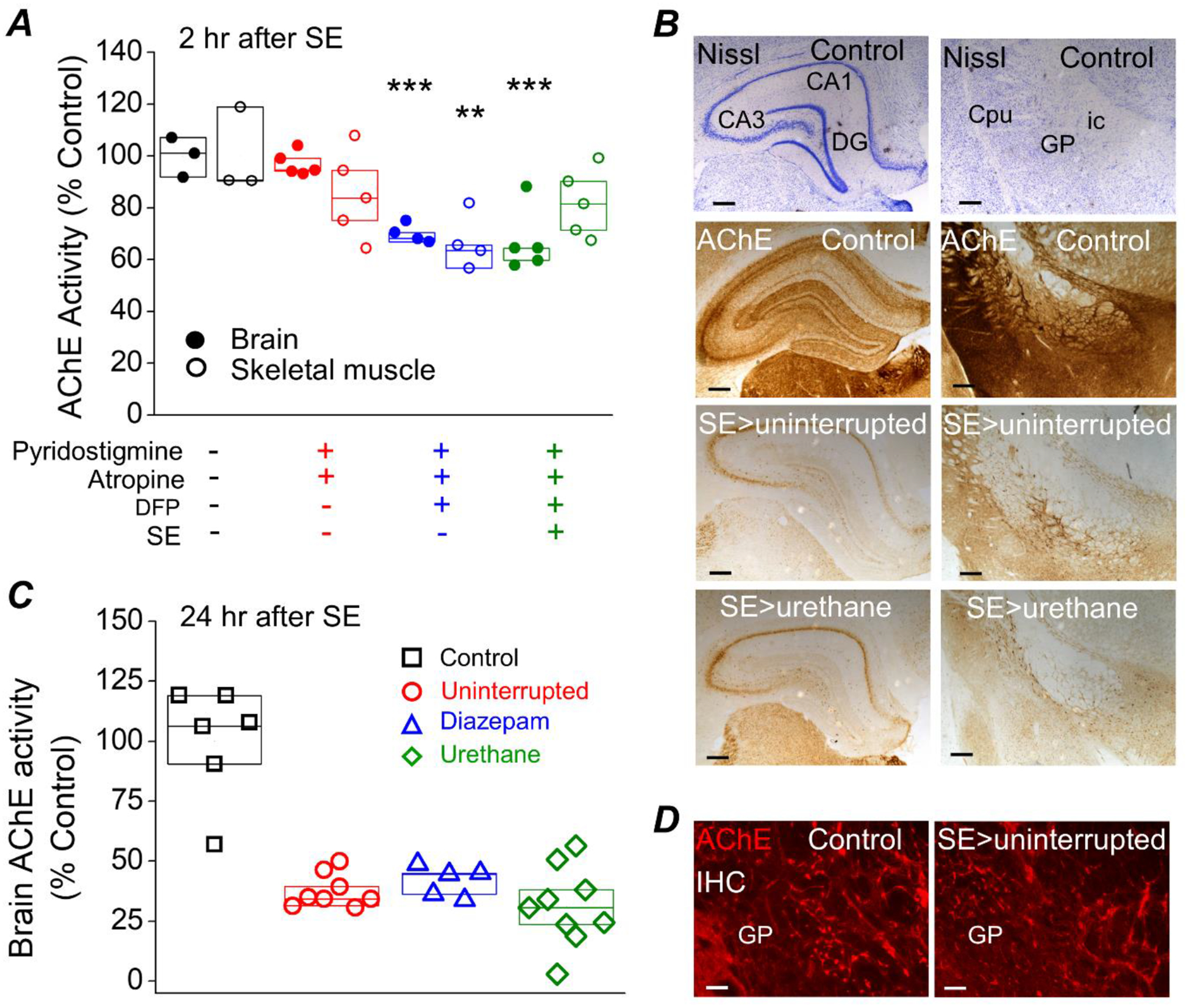
A, AChE inhibition in rat brain and skeletal muscle (from the hindleg, Vastus Lateralis) measured by an acetylcholinesterase assay is significantly reduced measured at 2 hours after SE in rats administered DFP. ** = p < .01, *** = p < .001, one-way ANOVA with posthoc Dunnett’s compared to the “no treatment” controls. Data are box plots with a 25 and 75 range. The symbols represent each individual rat within the group. B, the brain of a non-seizure control rat, a rat that experienced uninterrupted SE and a rat that was administered a single injection of urethane (0.8 mg/kg) 1 hour after SE was rapidly removed and bisected longitudinally at 24 h. One hemisphere was processed for immunohistochemistry. Coronal sections were stained for Nissl and acetylcholinesterase activity using the protocol described by Paxinos et al. (1980). The dark purple indicates the presence of the Nissl bodies and the brown precipitate is indicative of areas of high AChE activity. The top 2 images show the results of Nissl and the bottom 6 images reveal AChE activity in the hippocampus on the left and the globus palladus (GP), internal capsule (ic) and caudate putamen (Cpu) (25x total magnification) on the right. The Nissl and AChE activity stained sections were obtained from the same rats. Scale bar, 100 μm. C, inhibition of acetylcholinesterase in rat forebrain 24 h after DFP-induced SE was determined using an AChE activity assay and protein lysates obtained from half brains. AChE inhibition was similar regardless of whether the rats received diazepam or urethane. The controls were non-seizure rats that were administered pyridostigmine bromide, ethylatropine bromide and water instead of DFP. Data are box plots with a 25 and 75 range. The symbols represent each individual rat within the group. p > .05, one-way ANOVA with posthoc Bonferroni (diazepam vs. urethane). D, immunohistochemistry for AChE was performed on coronal sections revealing positive fluorescent staining of AChE in the globus pallidus (100x total magnification) indicating the presence of acetylcholinesterase (bright red staining). Scale bar, 30 μm. IHC= immunohistochemistry. CA1, cornu ammonis 1; CA3, cornu ammonis 3; DG, dentate gyrus.
To determine whether urethane reactivates acetylcholinesterase, brain AChE activity was measured 24 h after SE onset. Brain AChE activity was strongly inhibited in all rats that experienced DFP-induced SE compared to non-seizure controls as determined by a reduction in the dark brown precipitate in hippocampal coronal sections (Figure 10B). Although AChE activity was lower, the enzyme remained present in the globus pallidus as demonstrated by the bright red fluorescence staining by AChE immunohistochemistry (Figure 10D). No significant difference was detected in brain AChE activity from protein lysates obtained from half-brains lacking the cerebellum in diazepam treated rats versus urethane injected rats (42 ± 3% of non-seizure controls for diazepam, n = 5 rats; 31 ± 5% of non-seizure controls for urethane, n = 9 rats; p > .05, one-way ANOVA with posthoc Bonferroni)s (Figure 10C). These data suggest that the beneficial effects of urethane measured 24 h after SE onset is not a result of lowered brain exposure to DFP or reactivation of AChE.
DISCUSSION
An optimized model of DFP exposure producing status epilepticus in adult male rats was used to investigate the early consequences of DFP-induced SE. Significant inhibition of AChE in the brain and skeletal muscle, as measured 2 h after SE onset, was found in all rats administered DFP including rats that did not experience SE. However, only rats that experienced SE displayed a robust induction of inflammatory mediators (TNFα, IL-1β and COX-2) suggesting that neuroinflammation is the result of increased seizure activity and not a direct consequence of the chemical DFP. The early functional outcomes of terminating SE with urethane were investigated and compared to diazepam, which is still often administered to patients experiencing SE. Since the rats that experienced uninterrupted SE were not administered a pharmacological agent to interrupt or terminate SE we did not consider including them for comparison of efficacy of termination of SE by power analysis. The observation that EEG power in the 20–70 Hz band was lower during the last 4 hours of the EEG recording in these rats might reflect a highly vulnerable metabolic state due to the prolonged SE experience (Fig. 3C). Behaviorally, as determined by the modified Racine scoring, rats that experienced uninterrupted SE appeared very quiescent during the last 4 hours of the EEG recording. The appropriate comparison was diazepam injected rats to urethane administered rats as both groups of rats received a pharmaceutical agent at the same time (1 hr after SE onset) to compare their efficacy to terminate SE. In the rat DFP model of SE a single subanesthetic dose of urethane administered 1 hour after DFP-induced SE resulted in sustained suppression of seizure activity over the 24 hr EEG recording, consistent with a previous study (Rojas et al., 2018). On the other hand, diazepam suppressed SE initially, but high power seizure activity in the 20–70 Hz band returned within 6–10 hours. In our previous study investigating the beneficial effects of urethane in a rat model of DFP-induced SE, the neuropathology was measured 4 days following SE although urethane was administered on day 0 (Rojas et al., 2018). In the 2018 study cohorts of rats were kept alive to investigate the long-term effects of urethane and diazepam on the development of epilepsy (Rojas et al., 2018). It should be noted that none of the urethane treated rats developed any evidence of lung tumors (Rojas et al., 2018) measured seven months after urethane exposure. We believe that none of the rats in the current study developed lung tumors considering the short exposure to a single subanaesthetic dose of urethane. Brief exposure to isoflurane alone (without urethane) was not effective in completely terminating SE as high frequency and large amplitude spiking indicative of SE returned within 5 min after the effect of isoflurane wore off, consistent with the use of isoflurane as a single treatment for paraoxon poisoning (Krishnan et al., 2017). Isoflurane when administered within 30 minutes of paraoxon exposure in rats reduced seizure severity and neurodegeneration (Krishnan et al., 2017). However, when isoflurane exposure was delayed to 60 minutes or longer post-paraoxon the beneficial effect on neurodegeneration and seizure severity was not observed. Therefore, if SE persists for 60 minutes or longer a brief exposure to isoflurane alone may interrupt but not terminate SE. In the current study rats administered isoflurane alone following SE were considered to experience uninterrupted SE since the high intensity seizure activity returned within a few minutes and lasted several hours, however these rats were not included in any comparison analysis.
To determine whether the effective termination of electrographic SE by urethane results in an early beneficial effect, all rats were euthanized 24 h after SE onset. Investigation of neuropathology 24 h post SE allows sufficient time to observe a return or lack of a return of high power seizure activity in EEG experiments, which is integral for demonstration of termination of electrographic SE. In adult rats the benefit of terminating electrographic SE following urethane administration included less neurodegeneration and reduced neuroinflammation measured 24 h after SE.
The return of high power seizure activity was observed in all rats that experienced SE and were administered diazepam 1 h after SE. Although rats that experienced uninterrupted SE endured a longer initial SE they displayed no high power seizure activity towards the end of the EEG recording similar to rats that received urethane to terminate SE (Figures 2 and 3). Brandt et al. (2015) demonstrated that a longer duration of initial SE causes more intense sequelae and this study extends their findings to show that overnight return of high-intensity seizures or a prolonged initial SE experience in rats is associated with the pathological consequences of SE. The prolonged bout of seizures re-occurring during a metabolically vulnerable state is responsible for neuroinflammation and accompanying neurodegeneration.
Rats that experience DFP-induced SE display robust neurodegeneration in the hippocampus (Kadriu et al., 20091; Li et al., 2011; Li et al., 2012; Liu et al., 2012; Rojas et al., 2015, 2018). Neurodegeneration was investigated 24 h after SE onset, at which time substantial neurodegeneration was detected in limbic brain regions. Termination of SE with urethane protected pyramidal cells in the CA1 and CA3 region of the hippocampus as well as neurons in other seizure sensitive regions located on the ventral surface of the temporal lobe. No neuroprotection was detected in the hilus by urethane. We believe that injury to the hilar neurons is caused within the first hour of SE and so was insensitive to urethane. This is consistent with a previous study demonstrating that degeneration of hilar interneurons occurs earlier than pyramidal cells of the CA1 and CA3 regions of the hippocampus after pilocarpine-induced SE in mice (Borges et al., 2003). The variation observed in hippocampal neurodegeneration was large in rats that experienced uninterrupted SE and rats administered diazepam (Figure 9B). Similarly, the variation in the total duration and intensity of the high power seizure activity in the EEG recordings was also large for rats in these two groups (Figure 9A) suggesting that the variable duration of SE experienced by these rats underscores the variation in neuropathology
Neuroinflammation in the brain following DFP-induced SE is characterized by the transient upregulation of a small subset of inflammatory mediators including cytokines (interleukin 1 beta, IL-1β; tumor necrosis factor alpha, TNFα; interleukin 6, IL-6), chemokines (chemokine (C-C motif) ligand 2, CCL2; CCL3; CCL4) and activity dependent genes (cyclooxygenase 2, COX-2; FBJ murine osteosarcoma viral oncogene homolog, cFos; nerve growth factor-induced early-response gene C, NGFI-c). Within hours of DFP exposure, the levels of these inflammatory mediators are significantly upregulated in the brain. A major difference between the previous study that investigated neuropathology on day 4 and the current study is that the inflammatory burst detected in the brain was much higher 24 h after SE compared to 4 days post-SE (Rojas et al., 2018), suggesting that neuroinflammation peaks earlier than neurodegeneration. Changes in the level of inflammatory mediators measured at 2 h after SE onset (Figure 1) preceded neurodegeneration and gliosis, suggesting that neuroinflammation may be a driver of neuropathology that manifests later. Rats that were administered urethane after DFP-induced SE displayed a lower inflammatory burst in the brain twenty-four hours after SE onset as determined by RT-PCR of twelve inflammatory mediators. Urethane strongly antagonized the induction of IL-1β and CCL3. On the other hand, the level of neuroinflammation observed in rats administered diazepam was similar to rats that experienced uninterrupted SE. The changes in brain mRNA of three activity dependent genes (cFos, COX-2 and NGFI-c, encoded by the EGR4 gene) was measured by qRT-PCR. The level of COX-2 protein was also lower in rats administered urethane to terminate SE compared to rats administered diazepam or rats that experienced uninterrupted SE. However, in this rodent OP model the peak induction of COX-2 in rats that experience uninterrupted SE occurs between 24–48 hours (Rojas et al., 2015). The induction of cFos mRNA by DFP-induced SE unaltered by urethane although EEG power analysis revealed that seizure activity was at the basal level at the end of the EEG recording. The rapid induction of cFos mRNA by the initial SE, even in the urethane treated rats, is long lasting and remains elevated 24 h after SE, similar to rats that experience SE following injection of pilocarpine (Dube et al., 1998), kainic acid (Le Gal La Salle, 1988) and amygdaloid stimulation (Hsieh & Watanabe, 2000).
Repetitive exposure to high levels of urethane in rodents is used to induce lung tumors, which are usually associated with a pro-inflammatory local environment similar to that of other tumors. However, in the current study the single subanesthetic dose of urethane does not produce pro-inflammatory effects in the brain as control non-seizure rats administered isoflurane followed by urethane displayed similar levels of all 12 inflammatory mediators as naïve rats. Instead, we believe that the reduction in inflammatory mediators in the brain of urethane treated rats is due mainly to the complete termination of seizure activity by urethane ending the brain injury and allowing for an early resolution of inflammation.
Gliosis is often detected in the brain of rats following exposure to OP agents (Angoa-Perez et al., 2010; Kuruba et al., 2018; Liu et al., 2012; Rojas et al., 2015, 2018; Wu et al., 2018). Astrogliosis (astrocyte activation) and microgliosis (microglia activation) can be revealed by increased immunohistochemical staining of GFAP and IBA1, respectively after DFP-induced SE compared to non-seizure controls (Ferchmin et al., 2014; Kuruba et al., 2018; Li et al., 2011; Li et al., 2015; Liu et al., 2012; Rojas et al., 2015, 2018; Siso et al., 2017; Wu et al., 2018). Alternatively, the mRNA levels of these two glial markers can be quantified by qRT-PCR to determine the relative amount of gliosis that occurs following DFP-induced SE in rats (Rojas et al., 2015, 2018, 2020). Microgliosis and astrogliosis were prominent in the forebrain 24 h after DFP-induced SE consistent with previous studies (Flannery et al., 2016; Liu et al., 2012) and supporting the idea that following DFP-induced SE there is an early gliosis response in the brain that occurs within hours after SE onset and a more robust and delayed response that is observed in the days post SE. There appeared to be no difference in the degree of microgliosis or astrogliosis among rats that experienced uninterrupted SE and rats administered diazepam or urethane 1 h after SE (Figure 8). The delayed astrogliosis following DFP-induced SE was attenuated by urethane 4 days after SE (Rojas et al., 2018), but not 24 hour post-SE suggesting that early astrogliosis is likely a response to the initial SE that is initiated prior to exposure of pharmacological agents used to terminate SE (i.e. urethane and diazepam).
A major finding in the current study is that seizure duration correlates with neuropathology after DFP-induced SE. By investigating pathology 24 hrs after DFP-induced SE we were able to compare the total seizure duration to the degree of neuropathology (hippocampal degeneration and brain inflammation). In the previous study (Rojas et al., 2018) similar comparisons were not made as the delay in investigation of the neuropathology was far out from the EEG recording allowing for partial resolve of neuropathology. For example, in the previous study the interleukins and chemokines investigated were nearing their basal levels by day 4 (Rojas et al., 2018) whereas at 24 hours after SE they were closer to peak induction. The correlation analysis revealed that rats that experienced longer seizure activity displayed a greater neuropathology. Therefore, complete termination of SE by urethane without a return of seizure activity leads to a lesser degree of pathology. The pharmacological agents used did not alter the activity of AChE as it remained inhibited to a similar level in all rats that experienced SE (Figure 10). Therefore, one trivial explanation for the beneficial effects of urethane, that it simply reactivates acetylcholinesterase, has been ruled out.
CONCLUSION
Exposure to high levels of DFP causes SE, which if left untreated results in neuropathology and the inevitable development of epilepsy in surviving rats. In contrast to diazepam, which interrupts but does not terminate SE, a subanesthetic dose of the general anesthetic urethane terminated electrographic SE, and reduced neuroinflammation and neurodegeneration 24 h after DFP-induced SE. Administration of urethane and perhaps other general anesthetics following SE has a number of beneficial consequences that result from termination of electrographic seizures. Preventing the overnight return of the high power seizure activity following SE or shortening the initial SE results in less neuropathology. Together, these studies give insight into therapeutic modalities for the treatment of SE. In the future, it will be important to determine whether neuroprotection and attenuated neuroinflammation afforded by urethane is shared by other general anesthetics that are safer than urethane yet display a dual mechanism of action (inhibition of excitation and facilitation of inhibition) for terminating prolonged OP-induced seizures or SE.
Supplementary Material
Supplemental Figure 1. Induction of COX-2 after DFP-induced SE in rats. A, western blot image of COX-2 (bands in the top box) in the hemibrain (minus cerebellum) of control rats and rats that experienced SE induced by DFP followed by urethane. B, COX-2 (bands in the top box) in the hemibrain of rats that experienced uninterrupted SE and rats that received diazepam following DFP-induced SE. The bands in the lower box in each panel show the level of β-Actin from the same samples used as a loading control. Each lane represents an individual rat. The asterisk are the samples used to rerun the western blot shown in Figure 6E.
Highlights.
Inflammatory mediators are upregulated early during DFP-induced SE
Urethane blocks the return of overnight seizure activity following DFP-induced SE
Urethane attenuates early neurodegeneration in limbic brain regions
Seizure-induced upregulation of inflammatory mediators is attenuated by urethane
Total seizure duration correlates with neuropathology after DFP-induced SE
FUNDING:
This work was supported by the National Institutes of Health (NIH) grants NS097776 (R.D.) and UO1 NS058158 (R.D.).
ABBREVIATIONS:
- SE
status epilepticus
- GFAP
glial fibrillary acidic protein
- IBA1
ionized calcium-binding adapter molecule 1
- CD11B
cluster of differentiation molecule 11B
- DZP
diazepam
- CA1
Cornu Ammonis 1
- CA3
Cornu Ammonis 3
- CT
cycle threshold
- Con
control
- COX-2
cyclooxygenase 2
- NGFI-c
nerve growth factor-induced early-response gene C
- cFos
FBJ murine osteosarcoma viral oncogene homolog
- DFP
diisopropyl fluorophosphate
- OP
organophosphorus compound
- IP
intraperitoneal
- SC
subcutaneous
- FJB
FluoroJade B
- AchE
acetylcholinesterase
- qRT-PCR
quantitative real time polymerase chain reaction
- IL-1β
interleukin −1β
- TNFα
tumor necrosis factor alpha
- CXCL10
C-X-C motif chemokine ligand 10
- CCL2
chemokine (C-C motif) ligand 2
- CCL3
chemokine (C-C motif) ligand 3
- CCL4
chemokine (C-C motif) ligand 4
- IL-6
interleukin 6
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HPRT1
hypoxanthine phosphoribosyltransferase 1
- BDNF
brain derived neurotrophic factor
- TGFβ1
transforming growth factor beta-1
- gp91phox (NOX2)
glycosylated NADPH oxidase
- ELISA
enzyme-linked immunosorbent assay
- RSE
refractory status epilepticus
- IHC
immunohistochemistry
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT:
There are no conflicts of interest in relation to this work. The authors confirm reading the Journal’s position on issues involved in ethical publication and affirm that this manuscript is consistent with the Journal’s guidelines.
REFERENCES
- Angoa-Perez M, Kreipke CW, Thomas DM, Van Shura KE, Lyman M, McDonough JH, & Kuhn DM (2010). Soman increases neuronal COX-2 levels: possible link between seizures and protracted neuronal damage. Neurotoxicology, 31(6), 738–746. doi: 10.1016/j.neuro.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges K, Gearing M, McDermott DL, Smith AB, Almonte AG, Wainer BH, & Dingledine R (2003). Neuronal and glial pathological changes during epileptogenesis in the mouse pilocarpine model. Exp Neurol, 182(1), 21–34. [DOI] [PubMed] [Google Scholar]
- Brandt C, Tollner K, Klee R, Broer S, & Loscher W (2015). Effective termination of status epilepticus by rational polypharmacy in the lithium-pilocarpine model in rats: Window of opportunity to prevent epilepsy and prediction of epilepsy by biomarkers. Neurobiol Dis, 75, 78–90. doi: 10.1016/j.nbd.2014.12.015 [DOI] [PubMed] [Google Scholar]
- Cherian A, & Thomas SV (2009). Status epilepticus. Ann Indian Acad Neurol, 12(3), 140–153. doi: 10.4103/0972-2327.56312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande LS, & DeLorenzo RJ (2019). Novel therapeutics for treating organophosphate-induced status epilepticus co-morbidities, based on changes in calcium homeostasis. Neurobiol Dis. doi: 10.1016/j.nbd.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube C, Andre V, Covolan L, Ferrandon A, Marescaux C, & Nehlig A (1998). C-Fos, Jun D and HSP72 immunoreactivity, and neuronal injury following lithium-pilocarpine induced status epilepticus in immature and adult rats. Brain Res Mol Brain Res, 63(1), 139–154. doi: 10.1016/s0169-328x(98)00282-4 [DOI] [PubMed] [Google Scholar]
- Ferchmin PA, Andino M, Reyes Salaman R, Alves J, Velez-Roman J, Cuadrado B, Carrasco M, Torres-Rivera W, Segarra A, Martins AH, Lee JE, & Eterovic VA (2014). 4R-cembranoid protects against diisopropylfluorophosphate-mediated neurodegeneration. Neurotoxicology, 44, 80–90. doi: 10.1016/j.neuro.2014.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery BM, Bruun DA, Rowland DJ, Banks CN, Austin AT, Kukis DL, Li Y, Ford BD, Tancredi DJ, Silverman JL, Cherry SR, & Lein PJ (2016). Persistent neuroinflammation and cognitive impairment in a rat model of acute diisopropylfluorophosphate intoxication. J Neuroinflammation, 13(1), 267. doi: 10.1186/s12974-016-0744-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guignet M, Dhakal K, Flannery BM, Hobson BA, Zolkowska D, Dhir A, Bruun DA, Li S, Wahab A, Harvey DJ, Silverman JL, Rogawski MA, & Lein PJ (2019). Persistent behavior deficits, neuroinflammation, and oxidative stress in a rat model of acute organophosphate intoxication. Neurobiol Dis. doi: 10.1016/j.nbd.2019.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara K, & Harris RA (2002). The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg, 94(2), 313–318, table of contents. [DOI] [PubMed] [Google Scholar]
- Harvey BD, & Sloviter RS (2005). Hippocampal granule cell activity and c-Fos expression during spontaneous seizures in awake, chronically epileptic, pilocarpine-treated rats: implications for hippocampal epileptogenesis. J Comp Neurol, 488(4), 442–463. doi: 10.1002/cne.20594 [DOI] [PubMed] [Google Scholar]
- Ho J, Tumkaya T, Aryal S, Choi H, & Claridge-Chang A (2019). Moving beyond P values: data analysis with estimation graphics. Nat Methods, 16(7), 565–566. doi: 10.1038/s41592-019-0470-3 [DOI] [PubMed] [Google Scholar]
- Hobson BA, Rowland DJ, Supasai S, Harvey DJ, Lein PJ, & Garbow JR (2018). A magnetic resonance imaging study of early brain injury in a rat model of acute DFP intoxication. Neurotoxicology, 66, 170–178. doi: 10.1016/j.neuro.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honkaniemi J, & Sharp FR (1999). Prolonged expression of zinc finger immediate-early gene mRNAs and decreased protein synthesis following kainic acid induced seizures. Eur J Neurosci, 11(1), 10–17. doi: 10.1046/j.1460-9568.1999.00401.x [DOI] [PubMed] [Google Scholar]
- Hsieh PF, & Watanabe Y (2000). Time course of c-FOS expression in status epilepticus induced by amygdaloid stimulation. Neuroreport, 11(3), 571–574. doi: 10.1097/00001756-200002280-00028 [DOI] [PubMed] [Google Scholar]
- Irwin S (1968). Comprehensive observational assessment: Ia. A systematic, quantitative procedure for assessing the behavioral and physiologic state of the mouse. Psychopharmacologia, 13(3), 222–257. [DOI] [PubMed] [Google Scholar]
- Isojarvi JI, & Tokola RA (1998). Benzodiazepines in the treatment of epilepsy in people with intellectual disability. J Intellect Disabil Res, 42 Suppl 1, 80–92. [PubMed] [Google Scholar]
- Kadriu B, Guidotti A, Costa E, & Auta J (2009). Imidazenil, a non-sedating anticonvulsant benzodiazepine, is more potent than diazepam in protecting against DFP-induced seizures and neuronal damage. Toxicology, 256(3), 164–174. doi: 10.1016/j.tox.2008.11.021 [DOI] [PubMed] [Google Scholar]
- Krishnan JKS, Figueiredo TH, Moffett JR, Arun P, Appu AP, Puthillathu N, Braga MF, Flagg T, & Namboodiri AM (2017). Brief isoflurane administration as a post-exposure treatment for organophosphate poisoning. Neurotoxicology, 63, 84–89. doi: 10.1016/j.neuro.2017.09.009 [DOI] [PubMed] [Google Scholar]
- Kuruba R, Wu X, & Reddy DS (2018). Benzodiazepine-refractory status epilepticus, neuroinflammation, and interneuron neurodegeneration after acute organophosphate intoxication. Biochim Biophys Acta Mol Basis Dis, 1864(9 Pt B), 2845–2858. doi: 10.1016/j.bbadis.2018.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gal La Salle G (1988). Long-lasting and sequential increase of c-fos oncoprotein expression in kainic acid-induced status epilepticus. Neurosci Lett, 88(2), 127–130. doi: 10.1016/0304-3940(88)90112-7 [DOI] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Ford GD, Liu C, Stovall KC, White TE, Bruun DA, Tewolde T, Gates AS, Distel TJ, Surles-Zeigler MC, & Ford BD (2015). Neuregulin-1 inhibits neuroinflammatory responses in a rat model of organophosphate-nerve agent-induced delayed neuronal injury. J Neuroinflammation, 12, 64. doi: 10.1186/s12974-015-0283-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Giulivi C, Ford GD, Tewolde T, Ross-Inta C, & Ford BD (2012). Neuregulin-1 is neuroprotective in a rat model of organophosphate-induced delayed neuronal injury. Toxicol Appl Pharmacol, 262(2), 194–204. doi: 10.1016/j.taap.2012.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lein PJ, Liu C, Bruun DA, Tewolde T, Ford G, & Ford BD (2011). Spatiotemporal pattern of neuronal injury induced by DFP in rats: a model for delayed neuronal cell death following acute OP intoxication. Toxicol Appl Pharmacol, 253(3), 261–269. doi: 10.1016/j.taap.2011.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang LP, Pearson-Smith JN, Huang J, McElroy P, Day BJ, & Patel M (2018). Neuroprotective Effects of AEOL10150 in a Rat Organophosphate Model. Toxicol Sci, 162(2), 611–621. doi: 10.1093/toxsci/kfx283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li Y, Lein PJ, & Ford BD (2012). Spatiotemporal patterns of GFAP upregulation in rat brain following acute intoxication with diisopropylfluorophosphate (DFP). Curr Neurobiol, 3(2), 90–97. [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 25(4), 402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Mack KJ, Yi SD, Chang S, Millan N, & Mack P (1995). NGFI-C expression is affected by physiological stimulation and seizures in the somatosensory cortex. Brain Res Mol Brain Res, 29(1), 140–146. doi: 10.1016/0169-328x(94)00243-8 [DOI] [PubMed] [Google Scholar]
- Paxinos G, & Watson C (1986). The rat brain in stereotaxic coordinates (2nd ed.). Sydney; Orlando: Academic Press. [Google Scholar]
- Paxinos G, Watson CR, & Emson PC (1980). AChE-stained horizontal sections of the rat brain in stereotaxic coordinates. J Neurosci Methods, 3(2), 129–149. [DOI] [PubMed] [Google Scholar]
- Pouliot W, Bealer SL, Roach B, & Dudek FE (2016). A rodent model of human organophosphate exposure producing status epilepticus and neuropathology. Neurotoxicology, 56, 196–203. doi: 10.1016/j.neuro.2016.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ (1972). Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol, 32(3), 281–294. [DOI] [PubMed] [Google Scholar]
- Rojas A, Ganesh T, Lelutiu N, Gueorguieva P, & Dingledine R (2015). Inhibition of the prostaglandin EP2 receptor is neuroprotective and accelerates functional recovery in a rat model of organophosphorus induced status epilepticus. Neuropharmacology, 93, 15–27. doi: 10.1016/j.neuropharm.2015.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Ganesh T, Manji Z, O’Neill T, & Dingledine R (2016). Inhibition of the prostaglandin E2 receptor EP2 prevents status epilepticus-induced deficits in the novel object recognition task in rats. Neuropharmacology, 110(Pt A), 419–430. doi: 10.1016/j.neuropharm.2016.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Ganesh T, Walker A, & Dingledine R (2017). Ethylatropine Bromide as a Peripherally Restricted Muscarinic Antagonist. ACS Chem Neurosci, 8(4), 712–717. doi: 10.1021/acschemneuro.6b00334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Ganesh T, Wang W, Wang J, & Dingledine R (2020). A rat model of organophosphate-induced status epilepticus and the beneficial effects of EP2 receptor inhibition. Neurobiol Dis. doi: 10.1016/j.nbd.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas A, Wang W, Glover A, Manji Z, Fu Y, & Dingledine R (2018). Beneficial Outcome of Urethane Treatment Following Status Epilepticus in a Rat Organophosphorus Toxicity Model. eNeuro, 5(2). doi: 10.1523/ENEURO.0070-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmued LC, Albertson C, & Slikker W Jr. (1997). Fluoro-Jade: a novel fluorochrome for the sensitive and reliable histochemical localization of neuronal degeneration. Brain Res, 751(1), 37–46. [DOI] [PubMed] [Google Scholar]
- Siso S, Hobson BA, Harvey DJ, Bruun DA, Rowland DJ, Garbow JR, & Lein PJ (2017). Editor’s Highlight: Spatiotemporal Progression and Remission of Lesions in the Rat Brain Following Acute Intoxication With Diisopropylfluorophosphate. Toxicol Sci, 157(2), 330–341. doi: 10.1093/toxsci/kfx048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Kuruba R, & Reddy DS (2018). Midazolam-Resistant Seizures and Brain Injury after Acute Intoxication of Diisopropylfluorophosphate, an Organophosphate Pesticide and Surrogate for Nerve Agents. J Pharmacol Exp Ther, 367(2), 302–321. doi: 10.1124/jpet.117.247106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Gupta RC, Aschner M, & Milatovic D (2009). Protection of DFP-induced oxidative damage and neurodegeneration by antioxidants and NMDA receptor antagonist. Toxicol Appl Pharmacol, 240(2), 124–131. doi: 10.1016/j.taap.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Induction of COX-2 after DFP-induced SE in rats. A, western blot image of COX-2 (bands in the top box) in the hemibrain (minus cerebellum) of control rats and rats that experienced SE induced by DFP followed by urethane. B, COX-2 (bands in the top box) in the hemibrain of rats that experienced uninterrupted SE and rats that received diazepam following DFP-induced SE. The bands in the lower box in each panel show the level of β-Actin from the same samples used as a loading control. Each lane represents an individual rat. The asterisk are the samples used to rerun the western blot shown in Figure 6E.