

Abstract
Antibacterial drug resistance is a rapidly growing clinical threat, partially due to expression of β-lactamase enzymes, which confer resistance to bacteria by hydrolyzing and inactivating β-lactam antibiotics. The increasing prevalence of metallo-β-lactamases poses a unique challenge, as currently available β-lactamase inhibitors target the active site of serine β-lactamases but are ineffective against the zinc-containing active sites of metallo-β-lactamases. There is an urgent need for metallo-β-lactamase inhibitors and antibiotics that circumvent resistance mediated by metallo-β-lactamases in order to extend the utility of existing β-lactam antibiotics for treating infection. Here we investigated the antibacterial chelator-releasing prodrug PcephPT (2-((((6R,7R)-2-carboxy-8-oxo-7-(2-phenylacetamido)-5-thia-1-azabicyclo[4.2.0]oct-2-en-3-yl)methyl)thio)pyridine 1-oxide) as an inhibitor of New Delhi metallo-β-lactamase 1 (NDM-1). PcephPT is an experimental compound that we have previously shown inhibits growth of β-lactamase-expressing E. coli using a mechanism that is dependent on both copper availability and β-lactamase expression. Here, we found that PcephPT, in addition to being a copper-dependent antibacterial compound, inhibits hydrolysis activity of purified NDM-1with an IC50 of 7.6 μM without removing zinc from the active site and restores activity of the carbapenem antibiotic meropenem against NDM-1-producing E. coli. This work demonstrates that targeting a metal binding pharmacophore to β-lactamase-producing bacteria is a promising strategy for inhibition of both bacterial growth and metallo-β-lactamases.
Keywords: β-lactamase, resistance, prodrug, antibacterial, zinc, chelation
Graphical Abstract
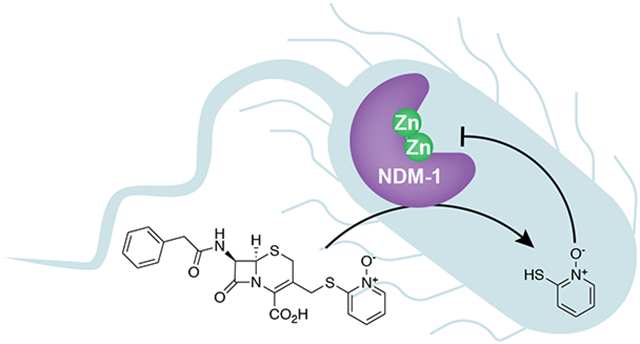
PcephPT, an antibacterial prochelator that releases the metal-binding agent pyrithione (PT) upon cleavage by β-lactamases, inhibits the metallo-β-lactamase NDM-1. PcephPT restores the activity of a carbapenem antibiotic against NDM-1-expressing E. coli and presents a promising strategy for development of targeted metallo-β-lactamase inhibitors.
The spread of antibacterial drug resistance in pathogenic bacteria is a growing clinical threat. A common drug resistance mechanism exploited by bacteria is expression of β-lactamase enzymes that hydrolyze and inactivate a wide range of antibiotics in the β-lactam class, including penicillins, cephalosporins, and carbapenems.1 β-lactamase inhibitors, such as clavulanic acid, sulbactam, and tazobactam, are often used in combination with β-lactam antibiotics to restore bactericidal efficacy. These inhibitors, however, target serine β-lactamases and are not effective against the emerging threat posed by metallo-β-lactamases, which use catalytic zinc ions in their active sites to cleave the β-lactam ring.2–4 There is an urgent need for potent inhibitors of metallo-β-lactamases as well as antibiotics that can bypass metallo-β-lactamase-mediated resistance, if β-lactam antibiotics are to remain effective options for treatment of bacterial infection.
As part of our previous work on developing antibiotics that can overcome β-lactamasemediated resistance, we reported the copper-dependent antibacterial activity of a prochelator we named PcephPT. PcephPT is a prodrug built on a cephalosporin core that is cleaved upon reaction with cephalosporinase and carbapenemase enzymes to release a metal chelating agent (Scheme 1).5 The release of a leaving group at the 3’ position of a cephalosporin after hydrolysis by a β-lactamase has been exploited for use in both antibacterial and diagnostic applications.6–12 The leaving group of PcephPT is pyrithione (PT), a well-studied antimicrobial compound with a bidentate metal-chelating motif. PT interferes with metal homeostasis and can induce hyperaccumulation of metals, including copper.5, 13–16 Copper has antibacterial effects when misregulated or present at high concentrations in bacteria.17–19 We previously reported that PcephPT has antibacterial activity against β-lactamase-expressing E. coli strains and that this activity is dependent on both copper availability and β-lactamase expression.5 PcephPT and related pyrithione-releasing cephalosporins have also been demonstrated to have potent antimycobacterial activity against both replicating and non-replicating Mycobacterium tuberculosis strains that are resistant to other β-lactam antibiotics.20
Scheme 1.
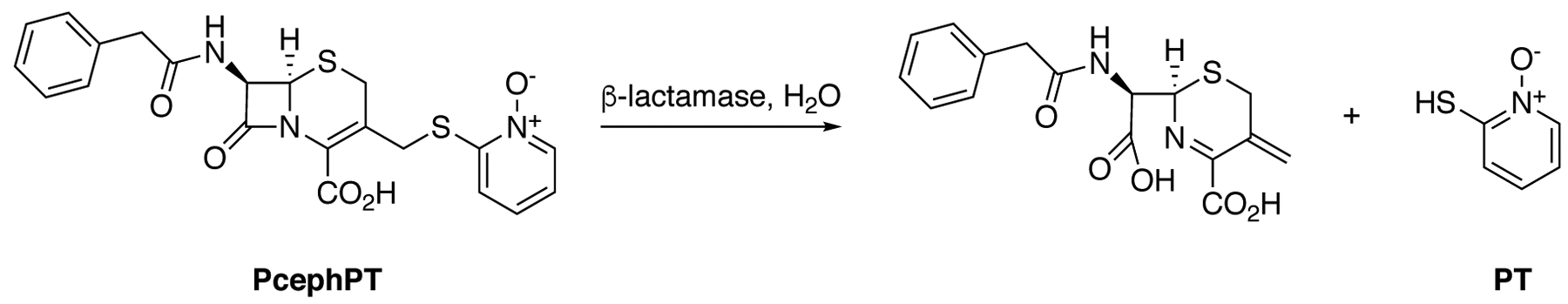
Mechanism of prochelator PcephPT.
We hypothesized that PcephPT, in addition to being a promising antibacterial agent, may also inhibit metallo-β-lactamases by releasing the chelator PT at the zinc-containing active sites of these enzymes. Many reported metallo-β-lactamase inhibitors contain metal-binding pharmacophores that target the active site zinc ions; however, many of these agents may be problematic due to nonselective affinity for metals and metalloproteins, resulting in off-target effects.21 Notably, PT and its copper complex have both been shown to inhibit NDM-1.22, 23 The prochelator design of PcephPT is expected to have a significant mechanistic advantage over other metallo-β-lactamase-inhibiting chelators because it delivers the inhibitor specifically to the resistance-causing enzyme, placing it in close proximity to the active site zinc ions and minimizing its interaction with the rest of the cellular metallome.
Here we explored the ability of PcephPT to inhibit New Delhi metallo-β-lactamase 1 (NDM-1). NDM-1 is a class B1 di-zinc metallo-β-lactamase that hydrolyzes nearly all β-lactam antibiotics, likely due to a flexible binding pocket that accommodates a wide variety of substrates.24, 25 The gene encoding NDM-1 can easily be transferred between bacteria and is often found in pathogenic strains that also have other drug resistance genes.25, 26 Currently there are no NDM-1 inhibitors approved by the US FDA. In the current work, we found that PcephPT has low-μM inhibitory activity of NDM-1 and restores activity of a carbapenem antibiotic in carbapenem-resistant E. coli strains expressing NDM-1. Our mechanistic studies revealed that neither PcephPT nor PT remove the zinc ions from the NDM-1 active site, unlike many known NDM-1-inhibiting chelators. Our results suggest that targeting chelators directly to β-lactamaseexpressing bacteria is a promising strategy for targeted metallo-β-lactamase inhibition in addition to metal-dependent antibacterial activity.
Results and Discussion
PT and PcephPT inhibit NDM-1.
The suitability of PcephPT as a substrate for NDM-1 was validated spectroscopically by monitoring the characteristic absorbance of PT at 340 nm during reaction of PcephPT with purified NDM-1. PcephPT was turned over by 5 nM NDM-1 efficiently at concentrations up to 160 μM (Figure 1A). The final absorbance spectrum of PcephPT treated with NDM-1 at reaction completion matches that of PT, confirming that PT is indeed released during the reaction (Figure 1B).
Figure 1.
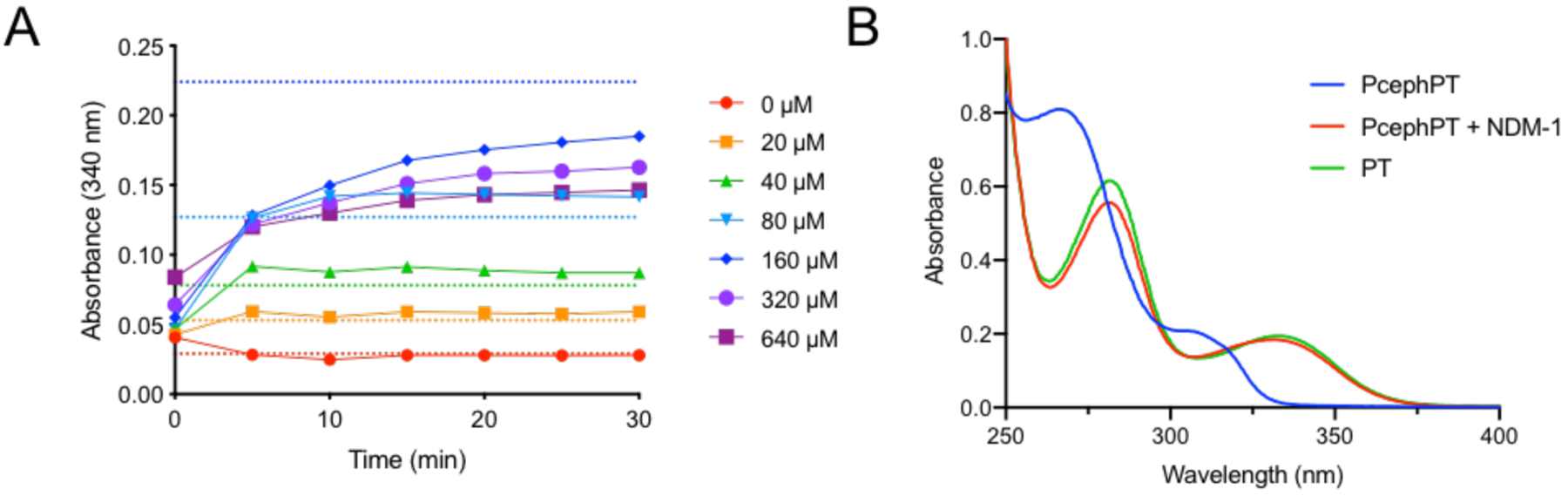
(A) Turnover of PcephPT by NDM-1. PT release from PcephPT was monitored on a plate reader at 340 nm upon treatment with 5 nM NDM-1 in HEPES buffer (50 mM, pH 7.2) at 37 °C. Dashed lines show the expected absorbance of PT (determined from a standard curve) if PcephPT at each concentration is turned over completely. (B) UV-Vis spectrum of 50 μM PcephPT, 50 μM PcephPT treated with 1 nM NDM-1 for 30 minutes at room temperature, and 50 μM PT in HEPES (50 mM, pH 7.2).
The ability of PT and PcephPT to inhibit NDM-1 was tested in an in vitro assay using purified enzyme.27 IC50 values were determined by measuring turnover of the colorimetric β-lactamase substrate nitrocefin by 1 nM NDM-1 in the presence of each compound (Table 1, inhibition curves shown in Figure S1). For comparison, five other β-lactam antibiotics and three conventional chelators known to inhibit NDM-1 were also analyzed (Figure 2). PcephPT was found to inhibit NDM-1 under these conditions with an IC50 of 7.6 μM, the lowest value among the antibiotics tested. Increasing its incubation time with NDM-1 did not affect its IC50 (Figure S2). In comparison, PT was a less effective inhibitor, with an IC50 of 17 μM. Whereas the carbapenems imipenem and meropenem did not inhibit nitrocefin turnover (at the concentrations tested, up to 320 μM), the three cephalosporins tested were active, with IC50 values of 9.1, 16, and 36 μM for cephalothin, ceftriaxone, and cephalexin, respectively. The hydrolysis products of these cephalosporins may be the culprit for this activity, as the isolated hydrolysis product of cephalothin has been previously reported to inhibit NDM-1, although with an IC50 higher than what we found here for cephalothin.28 The discrepancy may be due to treatment with intact cephalothin, which releases the hydrolysis product directly to the active site. None of the β-lactams tested inhibited NDM-1 as well as PcephPT, suggesting that PcephPT has a structural or mechanistic advantage over most other cephalosporins leading to improved inhibition of NDM-1. We hypothesize that this improvement in inhibition could arise from the active inhibitor PT being released from PcephPT near the active site zinc ions, the intact or cleaved cephalosporin core of PcephPT contributing to inhibition, or a combination of these factors. PcephPT was less effective than the known NDM-1 inhibitor dipicolinic acid (DPA) as well as two strong zinc-sequestering chelators, EDTA and TPEN (Figure 2).
Table 1.
IC50 values for inhibition of NDM-1 by PcephPT, zinc chelators, known NDM-1 inhibitor dipicolinic acid (DPA), and β-lactam antibiotics, determined by measuring inhibition of nitrocefin turnover. 95% CI = 95% confidence interval of the IC50.
| Compound | IC50 (μM) | 95% CI (μM) |
|---|---|---|
| PT | 17 | 14–20 |
| PcephPT | 7.6 | 6.5–8.9 |
| Cephalothin | 9.1 | 6.5–13 |
| Ceftriaxone | 16 | 14–17 |
| Cephalexin | 36 | 25–49 |
| Imipenem | >100 | N/A |
| Meropenem | >100 | N/A |
| DPA | 1.8 | 0.58–2.8 |
| EDTA | 0.052 | 0.042–0.063 |
| TPEN | 0.088 | 0–0.10 |
Figure 2:
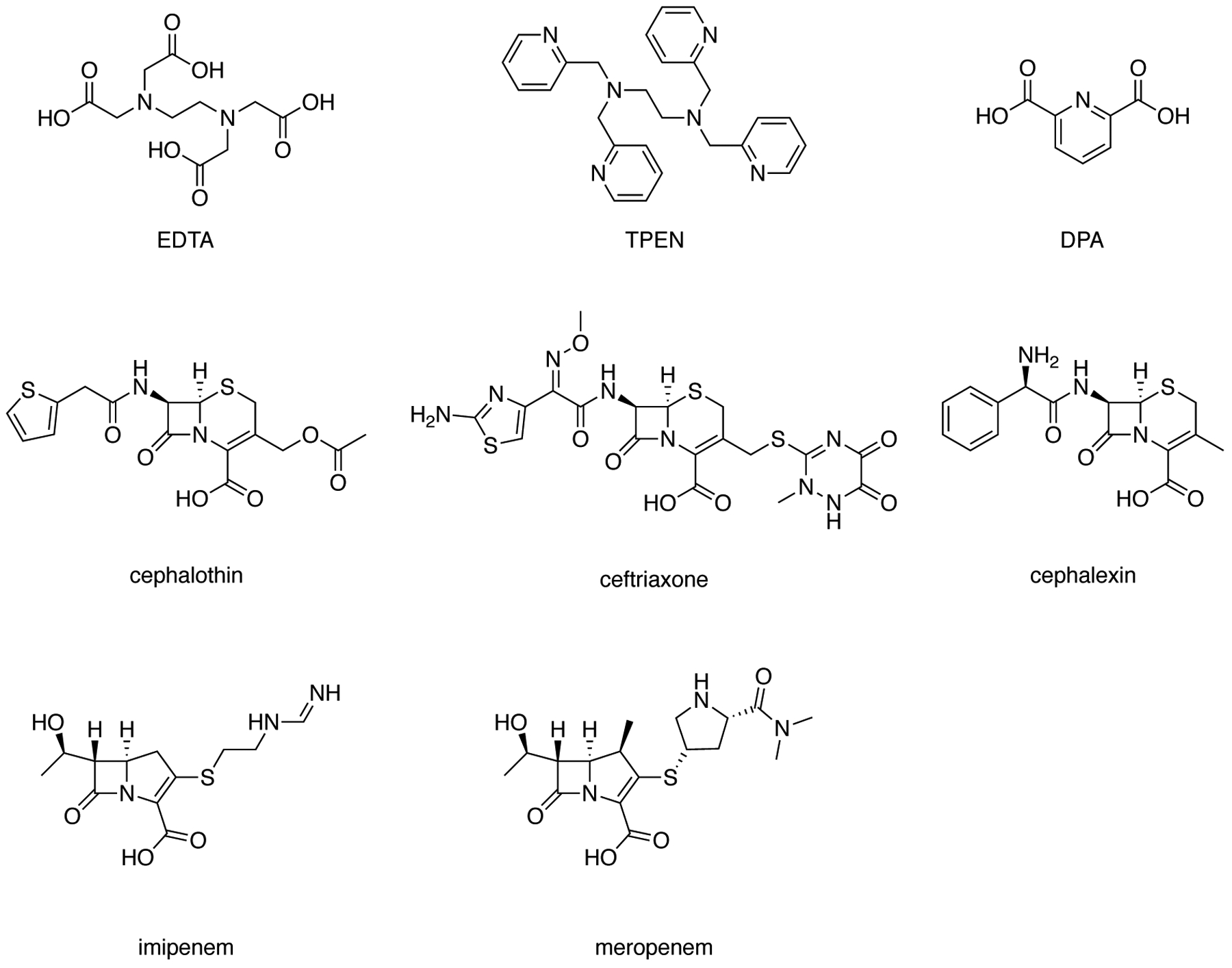
Structures of additional compounds tested for NDM-1 inhibitory activity. EDTA = ethylenediaminetetraacetic acid (used as the disodium salt in assays); TPEN = N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine; DPA = dipicolinic acid.
PT and PcephPT inhibit nitrocefin turnover in E. coli expressing NDM-1.
To determine efficacy of PT and PcephPT to inhibit NDM-1 activity in bacterial cells, nitrocefin turnover was tested in the cell lysate of an E. coli MG1655 strain expressing NDM-1. A culture was grown to an OD600 of ~0.04 and treated with PT or PcephPT at either 50 μM or 100 μM for 30 or 60 minutes at room temperature, lysed by sonication, and immediately treated with nitrocefin. These concentrations and treatment lengths were chosen to minimize killing of the bacterial cells during treatment, while room temperature conditions were chosen to minimize growth. Preliminary experiments revealed that lysis of the bacterial culture was necessary to observe an increase in nitrocefin hydrolysis product, implying that NDM-1 is intracellular and nitrocefin does not penetrate the cell membrane sufficiently during the course of the assay. Even with PT or PcephPT treatment, lysis was necessary to observe nitrocefin turnover, indicating that PT and PcephPT do not cause lysis of the cells on their own. Turnover rates were observed for all treated conditions. At 100 μM, PT and PcephPT reduced nitrocefin turnover by more than 75% when treated for 60 minutes. (Figure 3). When cells were exposed to PcephPT for only 30 minutes, nitrocefin turnover was less inhibited, indicating that inhibition by PcephPT is time-dependent and a result of PcephPT reaching its target during the incubation period rather than inhibiting NDM-1 in the short period of time after sonication. These results indicate that both PT and PcephPT at least partially inhibit NDM-1 in the bacterial cellular environment. PT was able to inhibit nitrocefin turnover to a greater extent than PcephPT, which may be due to a difference in membrane permeability between the two compounds or the relatively slow turnover of PcephPT, or both of these factors.
Figure 3:
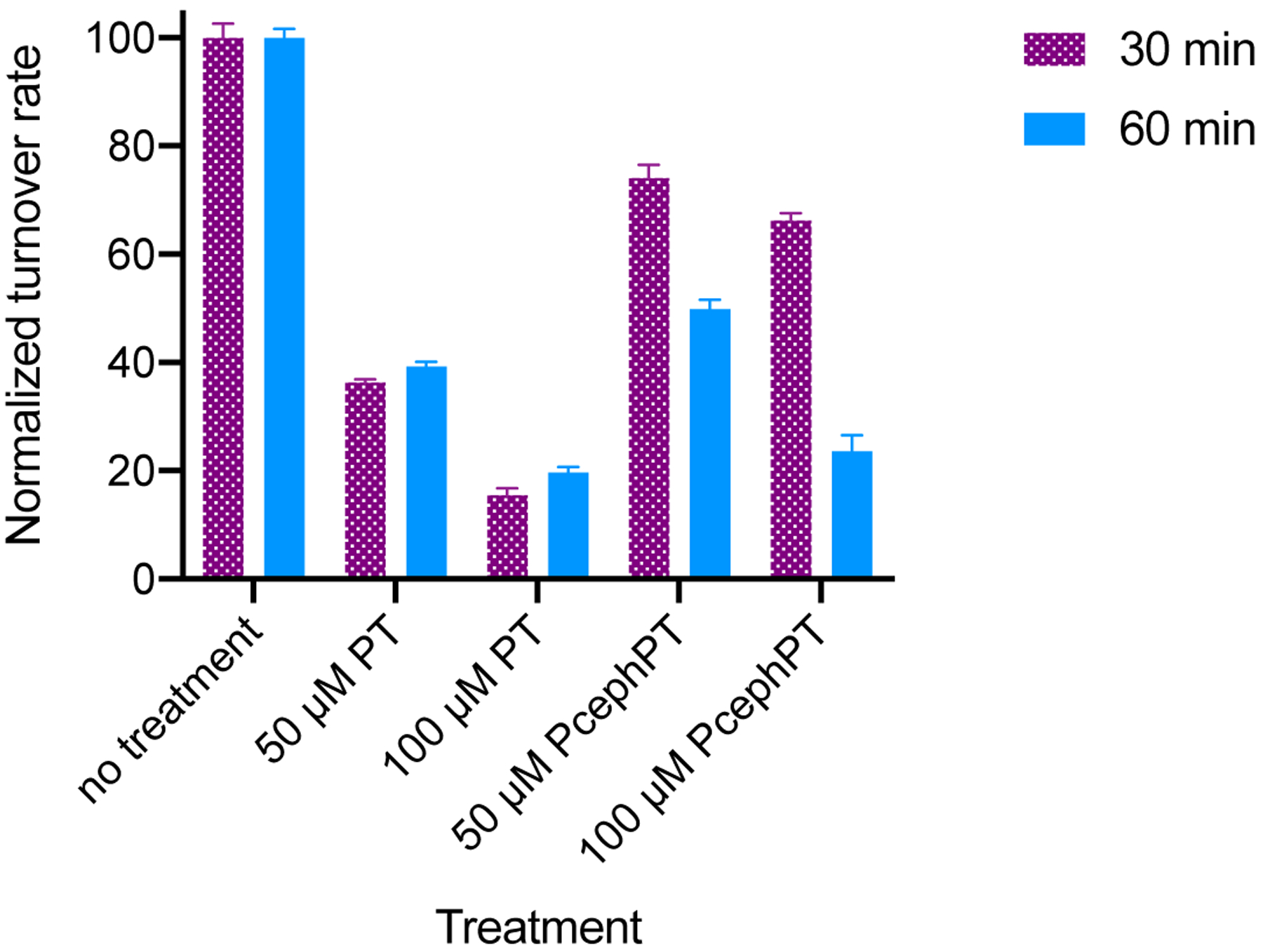
Inhibition of nitrocefin turnover in NDM-1-expressing E. coli MG1655 treated with PT or PcephPT for 30 or 60 minutes.
PcephPT restores activity of meropenem in NDM-1-expressing bacterial strains.
The compounds PT and PcephPT were tested in cultures of E. coli strains expressing NDM-1 to determine whether they can restore antibacterial activity of the carbapenem antibiotic meropenem, which has poor activity in NDM-1-expressing strains (MIC > 160 μM, Table 2). Two strains of E. coli were chosen for our study, the laboratory strain MG1655 and the pathogenic strain UTI89. As shown in our previous work, however, PT and PcephPT have antibacterial activity on their own.5 In our strains expressing NDM-1, PT has an MIC of 20 μM in MG1655 or 40 μM in UTI89, although growth is reduced at 20 μM. PcephPT did not completely inhibit growth in either strain at the highest concentration tested (160 μM), although growth was reduced by half at concentrations above 40 μM. Since NDM-1 inhibition on its own does not inhibit cell growth, these prior results reveal inherent antibacterial activity independent of NDM-1 inhibition. With this activity in mind, we performed broth microdilution assays of meropenem with fixed concentrations of PT (10 μM) or PcephPT (10 or 20 μM). Under such conditions, PT and PcephPT would not inhibit growth, any growth inhibition could be attributed to recovery of meropenem efficacy.
Table 2.
Minimum inhibitory concentrations (MICs) of meropenem, NDM-1 inhibitors, and combinations.
| Compound | MG1655 MIC (μM) | UTI89 MIC (μM) |
|---|---|---|
| Meropenem | >160 | >160 |
| PT | 20 | 40 |
| PcephPT | >160* | >160* |
| Meropenem + 10 μM PT | 160 | 160 |
| Meropenem + 10 μM PcephPT | 160 | 160 |
| Meropenem + 20 μM PcephPT | 10 | 10 |
| Meropenem + 20 μM cephalothin | >160 | >160 |
| Meropenem + 20 μM ceftriaxone | >160 | >160 |
PcephPT caused at least a 50% reduction in growth at 40 μM and higher but did not completely inhibit growth at any concentration tested. 160 μM meropenem = 61 μg/mL, 10 μM meropenem = 3.8 μg/mL, 20 μM PT = 2.5 μg/mL, 40 μM PT = 5.1 μg/mL, 20 μM PcephPT = 9.1 μg/mL, 160 μM PcephPT = 73 μg/mL.
At 10 μM PT or PcephPT, meropenem activity was not restored in either strain. However, 20 μM PcephPT reduced the meropenem MIC from >160 μM to 10 μM in both strains, which is consistent with inhibition of NDM-1 by PcephPT. The higher concentration of PT could not be used to measure meropenem activity recovery due to the antibacterial activity of PT at this concentration. The restoration of meropenem activity by PcephPT at a concentration of PcephPT so close to its IC50 (7.6 μM) with purified enzyme is an impressive result, given that many reported NDM-1 inhibitors must be co-treated at much higher concentrations than their IC50s in order to see a large decrease in carbapenem MIC in biological assays.29–34 This relative potency suggests the delivery of PT directly to the active site of NDM-1 upon PcephPT turnover confers a distinct advantage. In susceptible strains, meropenem has an MIC lower than 10 μM, so it is likely that PcephPT would inhibit NDM-1 more completely and further restore meropenem’s activity if treated at concentrations higher than 20 μM, but growth inhibition caused by meropenem would not be distinguishable from growth inhibition caused by PcephPT itself.
Growth inhibition by PcephPT is delayed in an NDM-1-expressing strain compared to a serine β-lactamase strain.
As noted previously, PcephPT reduces growth significantly at concentrations of 40 μM and higher, but does not completely inhibit growth, even after 20 h at concentrations up to 160 μM. To get a clearer picture of how these compounds influence growth pattern, antibacterial susceptibility assays were repeated by recording OD600 measurements every 30 minutes over the course of the 20-hour incubation (Figure 4) for E. coli MG1655 strains expressing either NDM-1 or the serine β-lactamase CTX-M-1 for comparison. The growth of both strains was fully inhibited by concentrations of PT 20 μM and higher, with delayed growth at 10 μM PT. This growth response is consistent with bacteriostatic activity, where growth is inhibited without killing cells at concentrations near the MIC, allowing regrowth at later timepoints.35 As we found previously, PT is bacteriostatic in the absence of supplemental copper.5
Figure 4.
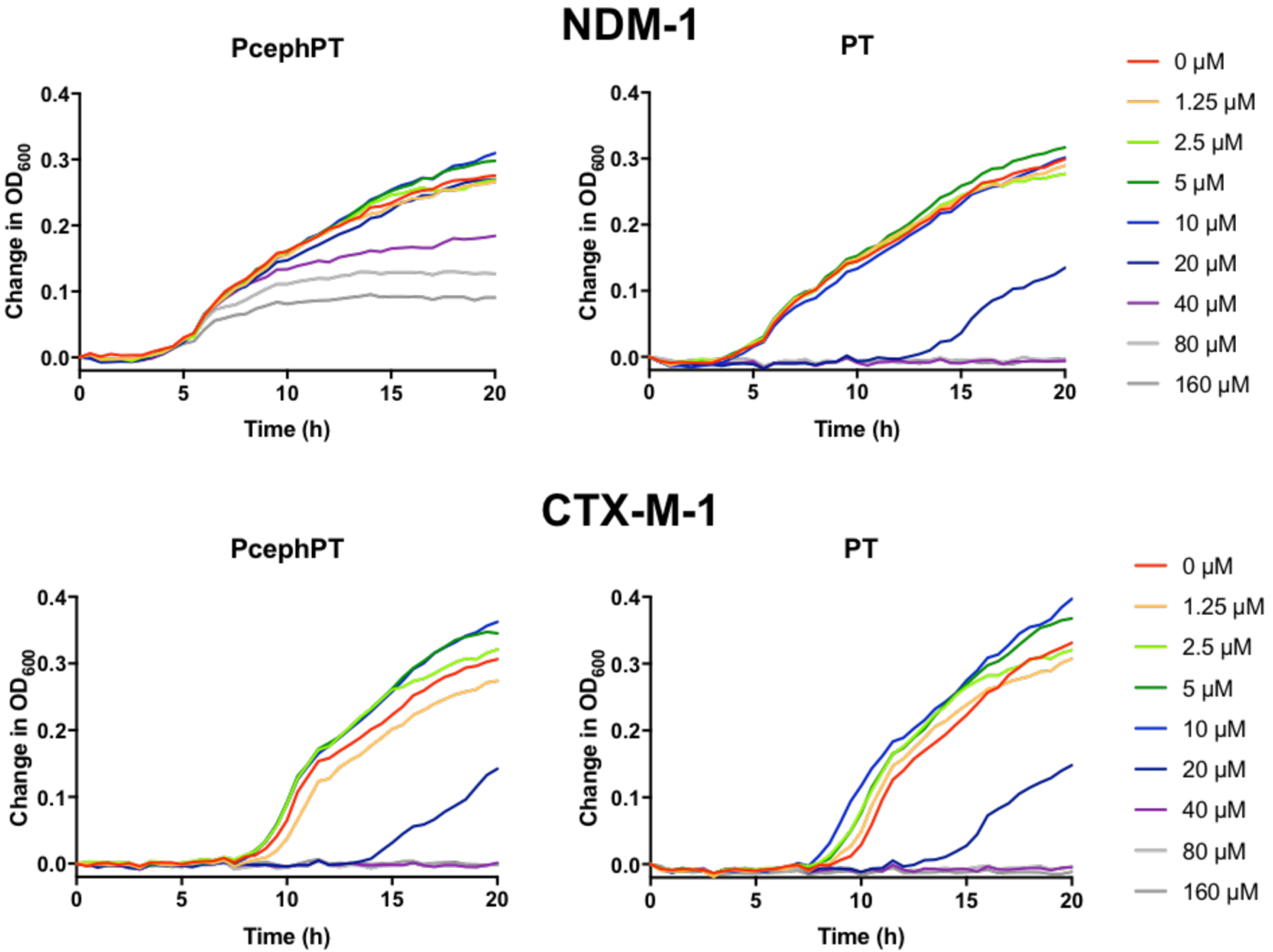
Comparison of time-dependent growth curves of E. coli MG1655 strains expressing the metallo-β-lactamase NDM-1 vs. the serine β-lactamase CTX-M-1 in the presence of varying concentrations of PcephPT and PT, illustrating the delayed antibacterial activity of PcephPT in the NDM-1-expressing strain. Growth in Mueller Hinton media at 37 °C was assessed by measuring OD600 every 30 minutes.
In the CTX-M-1-expressing strain, PcephPT cause d a growth pattern identical to that caused by PT (Figure 4). This result was expected, since PcephPT releases PT quickly in this strain5 and therefore it is expected to behave similarly to PT alone. However, a very different growth pattern was observed for NDM-1-expressing cells treated with PcephPT, with growth in all conditions initially followed by a delayed onset of growth inhibition at concentrations of 40 μM and higher between five and eight hours of growth. This result is consistent with slow NDM-1-activated release of PT from PcephPT in this strain, presumably due to product inhibition. This notion is supported by our previously published data demonstrating that the NDM-1-expressing strain shows slower turnover of PcephPT than strains expressing broad-spectrum serine-β-lactamases.5 Addition of copper made no significant difference in the growth pattern of PcephPT in the NDM-1 strain (Figure S3).
The mode of inhibition is not metal removal from the NDM-1 active site.
The mechanism by which a compound inhibits NDM-1 has implications for its selectivity and is important to determine when developing inhibitors.21 Two common mechanisms of NDM-1 inhibition are removal of zinc from the active site (zinc stripping) and binding of an inhibitor within the active site (ternary complex formation).21 Each strategy has its challenges; for example, highaffinity metal chelators that operate via zinc stripping can interact with metalloproteins nonspecifically and cause other off-target effects, while ternary complex formers may not be effective against the broad spectrum of metallo-β-lactamases.21 The ability of NDM enzymes to evolve, including by developing tighter zinc binding sites under the pressure of zinc deprivation,36 poses additional challenges associated with development of resistance, which may be impacted by the mechanism of inhibition.
To evaluate the mechanism of NDM-1 inhibition by PT and PcephPT, we used an equilibrium dialysis experiment to separate enzyme-bound inhibitors from those free in solution, including inhibitors bound to zinc ions that have been removed from the active site. Briefly, purified holo-NDM-1 enzyme was incubated with 40 μM (four equivalents) or 160 μM (sixteen equivalents) EDTA, PT, or PcephPT and dialyzed against buffer to separate small molecule species. Using methods based on previously published procedures,37 the protein fraction was then denatured and the concentration of protein and zinc quantitated to determine the zinc content per NDM-1 molecule (Figure 5).
Figure 5.
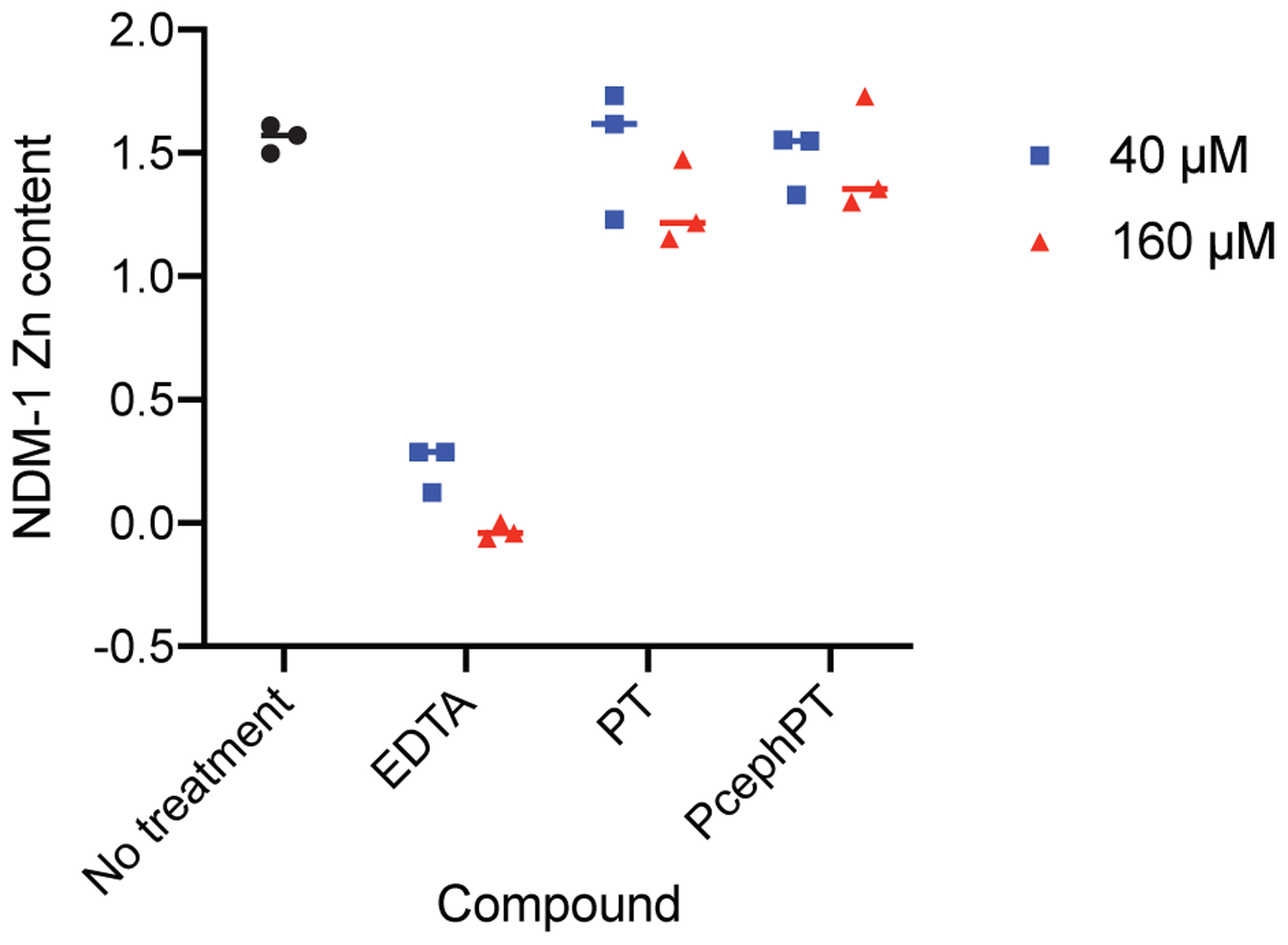
Zinc content of NDM-1 after treatment with compounds followed by equilibrium dialysis. Protein concentration was determined using A280 and zinc concentration was determined using the colorimetric chelator 4-(2-pyridylazo)resorcinol (PAR).
Using this method, untreated NDM-1 was determined to contain 1.6 zinc atoms on average per molecule after dialysis. NDM-1 is capable of binding two zinc atoms, although one binds more strongly than the other,37 which may explain the partial loss of zinc during dialysis. EDTA, a known metal-stripping agent, removed 85% of the zinc from NDM-1 when treated at 40 μM (four equivalents), while 160 μM removed zinc completely. Treatment with PT and PcephPT, however, did not lead to loss of zinc from the active site when treated at either 40 μM or 160 μM. These data suggest that, at these concentrations, these inhibitors do not use the mechanism of zinc removal and are likely to form a ternary complex in the active site of NDM-1. Absorbance peaks corresponding to PT were not observed in the spectra of the dialyzed NDM-1 samples after treatment with PcephPT or PT; however, such peaks are not expected to have a large enough absorbance at the concentrations used to be visible among the other signals present. The potential stable binding of PT to the active site without removal of zinc, in combination with the specific targeting provided by the β-lactamase-targeted core, makes prochelators like PcephPT very promising for use as NDM-1 inhibitors.
UV-Visible spectra of CoCo-NDM-1 treated with PcephPT and PT support ternary complex formation.
Spectroscopy of cobalt(II)-substituted NDM-1 has often been used as a method to study interactions of inhibitors and substrates with the active site of NDM-1.21, 34, 37–40 The two main features of the CoCo-NDM-1 spectrum that have been characterized previously are a strong peak at 330 nm corresponding to a Cys-Co(II) ligand-metal charge transfer band, and several smaller peaks in the 500–650 nm region corresponding to ligand field bands from the pseudotetrahedral Co(II) sites.39 Addition of compounds that remove metal from the NDM-1 active site result in reduction of these absorbance features, while treatment with compounds that form a ternary complex may result in either changes in the shape or intensity of the peaks or an unchanged spectrum. Addition of PT or PcephPT to Co-substituted NDM-1 caused an increase in absorbance in the 500–650 nm region and smoothing of these peaks, as well as a large increase in absorbance in the 300–400 nm region (Figures 6 and S4). The increase in the charge transfer band below 400 nm in our spectrum would be consistent with an additional sulfur ligand bound to an active site cobalt, while the changes in the 500–600 nm region indicate other changes in the coordination environment. These results are consistent with the equilibrium dialysis data in supporting a mechanism involving ternary complex formation for PT binding to the active site metal ions of NDM-1. In comparison, EDTA, a known metal-stripping inhibitor, caused significant decreases in both regions of the spectrum.
Figure 6:
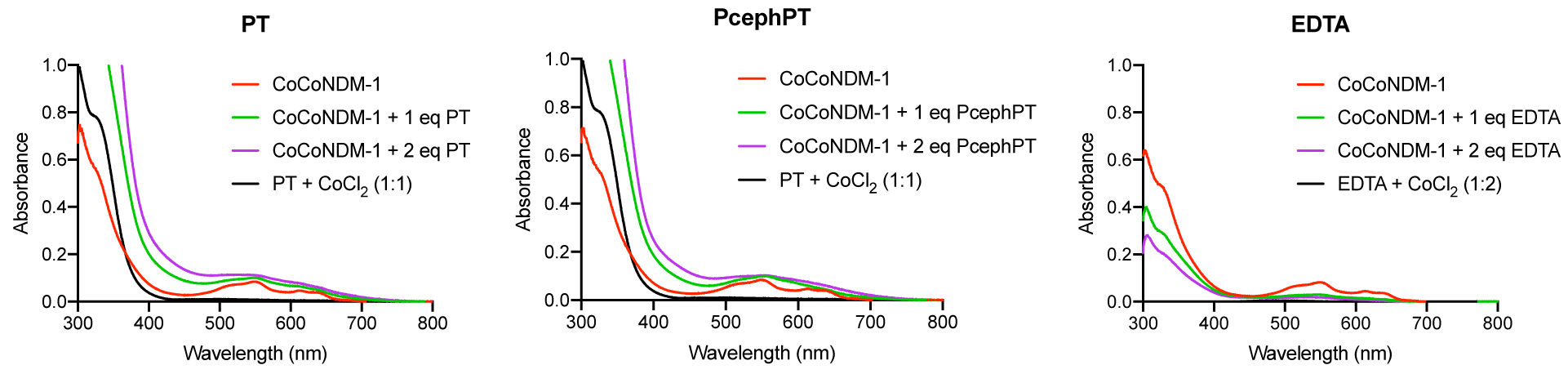
UV-Vis spectra of CoCo-NDM-1 (300 μM) treated with one or two equivalents of PT, PcephPT, or EDTA, compared to the spectra of 300 μM PT or EDTA in the presence of excess CoCl2. Control spectra of apo-NDM-1 (which is subtracted from all NDM-1 spectra here) and expansions of the 500–700 nm peaks are shown in Figure S4.
According to our equilibrium dialysis and UV-Vis experiments, PT and PcephPT do not remove zinc from the NDM-1 active site, suggesting that, if PT is binding to the zinc, it cannot bind strongly enough to compete with active site residues to remove zinc. A competitive inhibition mode may therefore be consistent with this result, since PT may be competing with substrate binding at the active site zinc. We examined the kinetics of nitrocefin turnover in the presence of our inhibitors and found that the kinetics of NDM-1 inhibition by PT or PcephPT do not fit well with any of the classical inhibition modes (competitive, uncompetitive, or noncompetitive). We could not confidently determine the trend in KM values due to poor fitting with the low-concentration part of the Michaelis-Menton curves, potentially resulting from low sensitivity in our assay at these concentrations. However, a clear decrease in Vmax observed with increasing inhibitor concentration (Figure S5) indicates that increasing substrate cannot recover NDM-1 activity, illustrating that the inhibition mode is not competitive.
Conclusions
Here we have demonstrated that an antibacterial prochelator, PcephPT, inhibits the hydrolysis activity of the clinically significant metallo-β-lactamase NDM-1. PT, the molecule released from PcephPT, is a bidentate metal-binding molecule that is known to complex a variety of metal ions, notably in this context the biologically relevant ions of zinc, copper and iron. Its lack of metal selectivity means that it potentially acts on multiple metal-dependent processes to influence cellular metallomes and inhibit microbial growth. Here we have expanded the utility of PT to include targeted NDM-1 inhibition when released from a prodrug. Enzymatic inhibition assays demonstrated an IC50 of 7.6 μM for PcephPT, indicating that it is more potent than the chelator PT alone and comparable in efficacy to other reported NDM-1 inhibitors. PcephPT is able to restore antibacterial activity of meropenem, a carbapenem antibiotic that is readily hydrolyzed by NDM-1, and inhibit turnover of an NDM-1 colorimetric substrate in bacterial culture. Experiments exploring the kinetics of bacterial growth in the presence of PcephPT provide support for the dual mechanism involving both NDM-1 enzyme inhibition and PT-mediated growth inhibition. Equilibrium dialysis and quantification of zinc in PcephPT-treated NDM-1 indicates that zinc removal from the NDM-1 active site is not the mechanism by which PcephPT or PT inhibits NDM-1, suggesting that ternary complex formation is the likely mechanism. Spectroscopic analysis of a cobalt-substituted enzyme treated with PT and PcephPT support this ternary complex hypothesis for the mode of inhibition. Our work demonstrates that the prochelator strategy, in addition to effectively targeting antibacterial agents to drug-resistant pathogens, is a promising method for delivering NDM-1-inhibiting metal-binding agents in a way that improves their efficacy and provides delivery to β-lactamase-expressing strains.
Methods
Materials.
PcephPT was synthesized as described previously.5 Other compounds were purchased from commercial sources and used without further purification. Purified NDM-1 enzyme was generously provided by Professor Bo Li (University of North Carolina – Chapel Hill).
Microbiology.
Preparation of E. coli strains expressing β-lactamases was described previously.5 The gene sequence for NDM-1, which does not include the lipobox sequence necessary for lipidation, is provided in the Supporting Information. Antibacterial susceptibility testing was performed similarly to our previous studies. Prior to all experiments, E. coli carrying the appropriate plasmids was streaked onto LB agar containing 100 μg/mL ampicillin and 50 μg/mL kanamycin. A single colony was used to inoculate 2 mL of Mueller-Hinton broth containing 100 μg/mL ampicillin and 50 μg/mL kanamycin, which was then incubated at 37 °C, 200 rpm, for 16–18 h. This overnight culture was diluted 1:500 in fresh broth and used as the working culture. Compounds to be tested were serially diluted 2-fold in Mueller-Hinton broth to final concentrations ranging from 0 to 160 μM in clear, flat-bottomed 96-well plates. The working culture, spiked with meropenem or no additional treatment was then added to the 96-well plate, giving a final inoculum dilution of 1:1000 (5 × 105 to 1 × 106 CFU/mL) and final volume of 100 μL per well. Wells containing media only were included to verify sterility. Plates were incubated for 20 h at 37 °C, 200 rpm. Plates were covered with AeraSeal film (Sigma-Aldrich) during incubation to minimize evaporation.
Bacterial growth was evaluated by measuring the optical density at 600 nm (OD600) using a PerkinElmer Victor3 V multilabel plate reader at 0 and 20 h. The 0-h time point data were subtracted from the 20 h data to remove background medium signal. The minimum inhibitory concentration (MIC) was defined as the concentration at which no detectable growth occurred after 20 h of incubation (background subtracted OD600 < 0.010). At least two biological replicates were performed with a minimum of at least four total technical replicates for each MIC determination. For timecourse growth assays, plates were incubated in the plate reader at 37 °C for the duration of the assay without shaking or AeraSeal film. The intial OD600 for each condition was subtracted from all later data points.
For turnover experiments in E. coli, an overnight culture prepared as described above, grown for 16–18 h, and diluted 1:100 in fresh Mueller-Hinton broth and incubated at 37 °C while shaking at 200 rpm for 1 h to an OD600 of approximately 0.04. This amount of growth was previously determined as ideal for measuring slow nitrocefin turnover over 30 min (data not shown). The culture was divided and treated with compounds for 1 h at rt. After this treatment period, each sample was lysed by sonication for 1 min and 100 μL was immediately added to a 96-well plate in triplicate wells. Nitrocefin (10 μL) was then added quickly to each well for a final concentration of 50 μM. The absorbance at 492 nm was measured every 5 min in a PerkinElmer Victor3 V multilabel plate reader for 30 min. Turnover rates were calculated and normalized to the untreated control.
Enzyme inhibition assays.
NDM-1 was prepared and reconstituted as described in Chan et al.27 Compounds were serially diluted in 96-well plates in HEPES buffer (50 mM, pH 7.2) and incubated with NDM-1 (1 nM) for 10 min at rt. Nitrocefin (20 μM) was added and a PerkinElmer Victor3 V multilabel plate reader was used to monitor the absorbance at 492 nm over 30 min at 37 °C. The slope of the absorbance increase was normalized to that of the condition containing the least compound to determine relative NDM-1 activity. This activity was plotted against log(concentration) of the compounds and GraphPad Prism software was used to calculate IC50 values and confidence intervals. For analysis of kinetic parameters, PT and PcephPT (0–60 μM) were incubated with 10 nM NDM-1 for 10 min at room temperature before addition of nitrocefin (63–500 μM). The slope of the absorbance increase (492 nm) at rt was converted to normalized velocity; only linear portions of the absorbance plots were used in calculating velocities. GraphPad Prism software was used to fit the activity curves to the Michaelis-Menten model. The average V max for our kinetic experiments was 4.26 μM/min nitrocefin for uninhibited 10 nM NDM-1.
Equilibrium dialysis and zinc quantification.
The following procedure was modified from previous reports.34, 37, 38 NDM-1 (10 μM in a 300-μL sample volume) was incubated in HEPES (50 mM, pH 7.2) with either 4 or 16 equivalents of each compound or no compound for 30 min at rt. Samples were inserted into Thermo Fisher Slyde-A-Lyzer dialysis cassettes with 0.5 mL sample capacity and 10K molecular weight cutoff and dialyzed against fresh HEPES (120 mL) at room temperature for 4–5 h, with buffer replaced after 2 h. Samples were removed from the dialysis cassettes and protein concentration was determined by measuring the absorbance and 280 nm and calculated using ε280 = 29,760 M−1cm−1. The samples were diluted 6-fold in 6-M aqueous guanidinium chloride containing 60 μM 4-(2-pyridylazo)resorcinol (PAR) and the absorbance at 500 nm was measured, for final sample concentrations of 5 M guanidinium chloride and 50 μM PAR. This was used to calculate zinc concentration using a PAR 500-nm standard curve, constructed using samples of zinc sulfate in HEPES, and multiplying by 6 to determine the concentration in the undiluted sample. For the samples treated with 160 μM EDTA, the 500-nm absorbance was just slightly less than that of the 0-μM zinc sample from the standard curve, which we interpret to mean that the dialyzed sample, which is depleted of zinc, contains components that are chelating a small amount of zinc present in the solution of PAR and guanidinium chloride; therefore, these values were not multiplied by the dilution factor of six. Zinc content of NDM-1 was calculated as the ratio of zinc concentration to NDM-1 concentration.
UV-Visible spectroscopy of CoCo-NDM-1 with inhibitors.
Apo NDM-1 was generated by treating purified NDM-1 (300 μM) with excess EDTA in HEPES (50 mM, pH 7.2) containing 2 mM TCEP for 30 min on ice. EDTA and metals were removed by running the sample through a Micro Bio-Spin™ P-6 gel column (Bio-Rad) equilibrated with HEPES (50 mM, pH 7.2). CoCo-NDM-1 was produced by adding two equivalents of CoCl2, producing a pink sample with an absorbance spectrum that is identical to previously published CoCo-NDM-1 spectra.39 Compounds were added from 10 mM stocks in DMSO (PT, PcephPT) or water (EDTA). These volumes of DMSO were confirmed not to have any effect on the absorbance spectrum. The absorbance spectrum was collected immediately after addition of each compound and again after 5 and 10 min, but no changes were observed after the initial scan.
Supplementary Material
Acknowledgments
This project was supported in part with funds from Duke University and the National Institutes of Health (GM084176). A.C.J. acknowledges an NSF Graduate Research Fellowship (DGE 1644868). J.M.Z.-B. acknowledges fellowship support from the Duke Pharmacological Sciences Training Program (T32 GM007105). E.A.P acknowledges a Dean’s Summer Research Fellowship from Duke University. We thank Professor Bo Li and Dr. Andrew N. Chan (UNC-Chapel Hill) for generously providing purified NDM-1 enzyme, Bradford Becken III (Duke University) for transforming E. coli strains, and Vignesh Dhanasekaran for writing scripts to expedite data analysis.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: Enzyme inhibition curves used to generate IC50 values; Enzyme inhibition curves for PT and PcephPT with various incubation times; Growth of E. coli treated with PT and PcephPT with supplemental copper; UV-Visible absorbance spectra of apo NDM-1 and expansions of CoCoNDM-1 spectra; Kinetics of NDM-1 in the presence of PT or PcephPT; NDM-1 gene sequence expressed in E. coli MG1655 and UTI89 strains (PDF)
The authors declare no competing financial interest.
References
- 1.Fisher JF, Meroueh SO, and Mobashery S (2005) Bacterial Resistance to β-Lactam Antibiotics: Compelling Opportunism, Compelling Opportunity, Chem. Rev 105, 395–424, DOI: 10.1021/cr030102i. [DOI] [PubMed] [Google Scholar]
- 2.Crowder MW, Spencer J, and Vila AJ (2006) Metallo-β-lactamases: Novel Weaponry for Antibiotic Resistance in Bacteria, Acc. Chem. Res 39, 721–728, DOI: 10.1021/ar0400241. [DOI] [PubMed] [Google Scholar]
- 3.Palzkill T (2013) Metallo-β-lactamase structure and function, Ann. N.Y. Acad. Sci 1277, 91–104, DOI: 10.1111/j.1749-6632.2012.06796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordmann P, Poirel L, Walsh TR, and Livermore DM (2011) The emerging NDM carbapenemases, Trends Microbiol. 19, 588–595, DOI: 10.1016/j.tim.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 5.Zaengle-Barone JM, Jackson AC, Besse DM, Becken B, Arshad M, Seed PC, and Franz KJ (2018) Copper Influences the Antibacterial Outcomes of a β-Lactamase-Activated Prochelator against Drug-Resistant Bacteria, ACS Infect Dis 4, 1019–1029, DOI: 10.1021/acsinfecdis.8b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Callaghan CH, Sykes RB, and Staniforth SE (1976) A New Cephalosporin with a Dual Mode of Action, Antimicrob. Agents Chemother 10, 245–248, DOI: 10.1128/AAC.10.2.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smyth TP, O’Donnell ME, O’Connor MJ, and St Ledger JO (2000) β-Lactamase-Dependent Prodrugs—Recent Developments, Tetrahedron 56, 5699–5707, DOI: 10.1016/S0040-4020(00)00419-1. [DOI] [Google Scholar]
- 8.Ma Z, and Lynch AS (2016) Development of a Dual-Acting Antibacterial Agent (TNP-2092) for the Treatment of Persistent Bacterial Infections, J. Med. Chem 59, 6645–6657, DOI: 10.1021/acs.jmedchem.6b00485. [DOI] [PubMed] [Google Scholar]
- 9.Majewski MW, Miller PA, Oliver AG, and Miller MJ (2016) Alternate “Drug” Delivery Utilizing β-Lactam Cores: Syntheses and Biological Evaluation of β-Lactams Bearing Isocyanate Precursors, J. Org. Chem 82, 737–744, DOI: 10.1021/acs.joc.6b02272. [DOI] [PubMed] [Google Scholar]
- 10.Phelan RM, Ostermeier M, and Townsend CA (2009) Design and synthesis of a β-lactamase activated 5-fluorouracil prodrug, Bioorg. Med. Chem. Lett 19, 1261–1263, DOI: 10.1016/j.bmcl.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie H, Mire J, Kong Y, Chang M, Hassounah HA, Thornton CN, Sacchettini JC, Cirillo JD, and Rao J (2012) Rapid point-of-care detection of the tuberculosis pathogen using a BlaC-specific fluorogenic probe, Nat. Chem 4, 802–809, DOI: 10.1038/nchem.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu R, Miller PA, Vakulenko SB, Stewart NK, Boggess WC, and Miller MJ (2018) A Synthetic Dual Drug Sideromycin Induces Gram-Negative Bacteria To Commit Suicide with a Gram-Positive Antibiotic, J. Med. Chem 61, 3845–3854, DOI: 10.1021/acs.jmedchem.8b00218. [DOI] [PubMed] [Google Scholar]
- 13.Reeder NL, Kaplan J, Xu J, Youngquist RS, Wallace J, Hu P, Juhlin KD, Schwartz JR, Grant RA, Fieno A, Nemeth S, Reichling T, Tiesman JP, Mills T, Steinke M, Wang SL, and Saunders CW (2011) Zinc Pyrithione Inhibits Yeast Growth through Copper Influx and Inactivation of Iron-Sulfur Proteins, Antimicrob. Agents Chemother 55, 5753–5760, DOI: 10.1128/aac.00724-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reeder NL, Xu J, Youngquist RS, Schwartz JR, Rust RC, and Saunders CW (2011) The antifungal mechanism of action of zinc pyrithione, Br. J. Dermatol 165 Suppl 2, 9–12, DOI: 10.1111/j.1365-2133.2011.10571.x. [DOI] [PubMed] [Google Scholar]
- 15.Yasokawa D, Murata S, Iwahashi Y, Kitagawa E, Kishi K, Okumura Y, and Iwahashi H (2010) DNA microarray analysis suggests that zinc pyrithione causes iron starvation to the yeast Saccharomyces cerevisiae, J. Biosci. Bioeng 109, 479–486, DOI: 10.1016/j.jbiosc.2009.10.025. [DOI] [PubMed] [Google Scholar]
- 16.Park M, Cho Y-J, Lee YW, and Jung WH (2018) Understanding the Mechanism of Action of the Anti-Dandruff Agent Zinc Pyrithione against Malassezia restricta, Sci. Rep 8, 12086, DOI: 10.1038/s41598-018-30588-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Becker KW, and Skaar EP (2014) Metal limitation and toxicity at the interface between host and pathogen, FEMS Microbiol. Rev 38, 1235–1249, DOI: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Djoko KY, Ong C.-l. Y., Walker MJ, and McEwan AG (2015) The Role of Copper and Zinc Toxicity in Innate Immune Defense against Bacterial Pathogens, J. Biol. Chem 290, 18954–18961, DOI: 10.1074/jbc.R115.647099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hodgkinson V, and Petris MJ (2012) Copper Homeostasis at the Host-Pathogen Interface, J. Biol. Chem 287, 13549–13555, DOI: 10.1074/jbc.R111.316406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lopez Quezada L, Li K, McDonald SL, Nguyen Q, Perkowski AJ, Pharr CW, Gold B, Roberts J, McAulay K, Saito K, Somersan Karakaya S, Javidnia PE, Porras de Francisco E, Amieva MM, Dıaz SP, Mendoza Losana A, Zimmerman M, Liang H-PH, Zhang J, Dartois V, Sans S, Lagrange S, Goullieux L, Roubert C, Nathan C, and Aubé J (2019) Dual-Pharmacophore Pyrithione-Containing Cephalosporins Kill Both Replicating and Nonreplicating Mycobacterium tuberculosis, ACS Infectious Diseases 5, 1433–1445, DOI: 10.1021/acsinfecdis.9b00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ju L-C, Cheng Z, Fast W, Bonomo RA, and Crowder MW (2018) The Continuing Challenge of Metallo-β-Lactamase Inhibition: Mechanism Matters, Trends Pharmacol. Sci 39, 635–647, DOI: 10.1016/j.tips.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Adamek RN, Credille CV, Dick BL, and Cohen SM (2018) Isosteres of hydroxypyridinethione as drug-like pharmacophores for metalloenzyme inhibition, JBIC Journal of Biological Inorganic Chemistry 23, 1129–1138, DOI: 10.1007/s00775-018-1593-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Djoko KY, Achard MES, Phan M-D, Lo AW, Miraula M, Prombhul S, Hancock SJ, Peters KM, Sidjabat H, Harris PN, Mitić N, Walsh TR, Anderson GJ, Shafer WM, Paterson DL, Schenk G, McEwan AG, and Schembri MA (2017) Copper ions and coordination complexes as novel carbapenem adjuvants, Antimicrob. Agents Chemother 62, DOI: 10.1128/aac.02280-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Z, Hu L, Sankaran B, Prasad BVV, and Palzkill T (2018) Differential active site requirements for NDM-1 β-lactamase hydrolysis of carbapenem versus penicillin and cephalosporin antibiotics, Nature Communications 9, 4524, DOI: 10.1038/s41467-018-06839-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yong D, Toleman MA, Giske CG, Cho HS, Sundman K, Lee K, and Walsh TR (2009) Characterization of a New Metallo-β-Lactamase Gene, blaNDM-1, and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India, Antimicrob. Agents Chemother 53, 5046–5054, DOI: 10.1128/aac.00774-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walsh TR, Weeks J, Livermore DM, and Toleman MA (2011) Dissemination of NDM-1 positive bacteria in the New Delhi environment and its implications for human health: an environmental point prevalence study, Lancet Infect. Dis 11, 355–362, DOI: 10.1016/S1473-3099(11)70059-7. [DOI] [PubMed] [Google Scholar]
- 27.Chan AN, Shiver AL, Wever WJ, Razvi SZA, Traxler MF, and Li B (2017) Role for dithiolopyrrolones in disrupting bacterial metal homeostasis, Proc. Natl. Acad. Sci. USA 114, 2717–2722, DOI: 10.1073/pnas.1612810114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torelli NJ, Akhtar A, DeFrees K, Jaishankar P, Pemberton OA, Zhang X, Johnson C, Renslo AR, and Chen Y (2019) Active-site druggability of carbapenemases and broad-spectrum inhibitor discovery, ACS Infectious Diseases 5, 1013–1021, DOI: 10.1021/acsinfecdis.9b00052. [DOI] [PubMed] [Google Scholar]
- 29.Krajnc A, Brem J, Hinchliffe P, Calvopiña K, Panduwawala TD, Lang PA, Kamps JJAG, Tyrrell JM, Widlake E, Saward BG, Walsh TR, Spencer J, and Schofield CJ (2019) Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases, J. Med. Chem 62, 8544–8556, DOI: 10.1021/acs.jmedchem.9b00911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meng Z, Tang M-L, Yu L, Liang Y, Han J, Zhang C, Hu F, Yu J-M, and Sun X (2019) Novel Mercapto Propionamide Derivatives with Potent New Delhi Metallo-β-Lactamase-1 (NDM-1) Inhibitory Activity and Low Toxicity, ACS Infectious Diseases 5, 903–916, DOI: 10.1021/acsinfecdis.8b00366. [DOI] [PubMed] [Google Scholar]
- 31.Leiris S, Coelho A, Castandet J, Bayet M, Lozano C, Bougnon J, Bousquet J, Everett MJ, Lemonnier M, Sprynski N, Zalacain M, Pallin TD, Cramp M, Jennings N, Raphy G, Jones MW, Pattipati R, Shankar B, Sivasubrahmanyam R, Soodhagani AK, Juventhala RR, Pottabathini N, Pothukanuri S, Benvenuti M, Pozzi C, Mangani S, De Luca F, Cerboni G, Docquier J-D, and Davies D (2018) SAR studies leading to the identification of a novel series of metallo-β-lactamase inhibitors for the treatment of carbapenem-resistant Enterobacteriaceae infections that display efficacy in an animal infection model, ACS Infectious Diseases 5, 131–140, DOI: 10.1021/acsinfecdis.8b00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schnaars C, Kildahl-Andersen G, Prandina A, Popal R, Radix SL, Le Borgne M, Gjoen T, Andresen AMS, Heikal A, Økstad OA, Fröhlich C, Samuelsen Ø, Lauksund S, Jordheim LP, Rongved P, and Åstrand OAH (2018) Synthesis and preclinical evaluation of TPA-based zinc chelators as metallo-β-lactamase inhibitors, ACS Infectious Diseases 4, 1407–1422, DOI: 10.1021/acsinfecdis.8b00137. [DOI] [PubMed] [Google Scholar]
- 33.Everett M, Sprynski N, Coelho A, Castandet J, Bayet M, Bougnon J, Lozano C, Davies DT, Leiris S, Zalacain M, Morrissey I, Magnet S, Holden K, Warn P, De Luca F, Docquier J-D, and Lemonnier M (2018) Discovery of a Novel Metallo-β-Lactamase Inhibitor That Potentiates Meropenem Activity against Carbapenem-Resistant Enterobacteriaceae, Antimicrob. Agents Chemother 62, DOI: 10.1128/aac.00074-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen AY, Thomas PW, Stewart AC, Bergstrom A, Cheng Z, Miller C, Bethel CR, Marshall SH, Credille CV, Riley CL, Page RC, Bonomo RA, Crowder MW, Tierney DL, Fast W, and Cohen SM (2017) Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-β-lactamase-1, J. Med. Chem 60, 7267–7283, DOI: 10.1021/acs.jmedchem.7b00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pankey GA, and Sabath LD (2004) Clinical Relevance of Bacteriostatic versus Bactericidal Mechanisms of Action in the Treatment of Gram-Positive Bacterial Infections, Clin. Infect. Dis 38, 864–870, DOI: 10.1086/381972. [DOI] [PubMed] [Google Scholar]
- 36.Bahr G, Vitor-Horen L, Bethel CR, Bonomo RA, González LJ, and Vila AJ (2018) Clinical Evolution of New Delhi Metallo-β-Lactamase (NDM) Optimizes Resistance under Zn(II) Deprivation, Antimicrob. Agents Chemother 62, DOI: 10.1128/aac.01849-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas PW, Zheng M, Wu S, Guo H, Liu D, Xu D, and Fast W (2011) Characterization of Purified New Delhi Metallo-β-lactamase-1, Biochemistry 50, 10102–10113, DOI: 10.1021/bi201449r. [DOI] [PubMed] [Google Scholar]
- 38.Chen AY, Thomas PW, Cheng Z, Xu NY, Tierney DL, Crowder MW, Fast W, and Cohen SM (2019) Investigation of Dipicolinic Acid Isosteres for the Inhibition of Metallo-beta-Lactamases, ChemMedChem 14, 1271–1282, DOI: 10.1002/cmdc.201900172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang H, Aitha M, Marts AR, Hetrick A, Bennett B, Crowder MW, and Tierney DL (2014) Spectroscopic and Mechanistic Studies of Heterodimetallic Forms of Metallo-β-lactamase NDM-1, J. Am. Chem. Soc 136, 7273–7285, DOI: 10.1021/ja410376s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bergstrom A, Katko A, Adkins Z, Hill J, Cheng Z, Burnett M, Yang H, Aitha M, Mehaffey MR, Brodbelt JS, Tehrani KHME, Martin NI, Bonomo RA, Page RC, Tierney DL, Fast W, Wright GD, and Crowder MW (2017) Probing the Interaction of Aspergillomarasmine A with Metallo-β-lactamases NDM-1, VIM-2, and IMP-7, ACS Infectious Diseases 4, 135–145, DOI: 10.1021/acsinfecdis.7b00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.