

Abstract
Terpene synthases often catalyze complex carbocation cascade reactions. It has been previously shown that single residue switches involving replacement of a key aliphatic residue with serine or threonine can “short-circuit” such reactions, presumed to act indirectly via dipole stabilization of intermediate carbocations. Here a similar switch was found in the structurally characterized ent-kaurene synthase from Bradyrhizobium japonicum. Application of a recently developed computational approach to terpene synthases, TerDockin, surprisingly indicates direct action of the introduced serine hydroxyl as a catalytic base. Notably, this model suggests alternative interpretation of previous results, and potential routes towards reengineering terpene synthase activity more generally.
Keywords: biosynthesis, terpene synthases, enzymology, natural products, acid-base catalysis
Graphical Abstract:
Terpene synthases produce intricate hydrocarbon backbones that underlie the structural diversity of the extensive family of terpenoid natural products.1 This feat is accomplished by magnesium-assisted lysis of the allylic diphosphate ester in their isoprenyl substrates, which often triggers complex carbocation cascade reactions that are eventually terminated by deprotonation (or, occasionally, carbocation trapping by a nucleophile). To accommodate such reactive intermediates the relevant portion of terpene synthase active sites have been observed to be largely nonpolar, composed of aliphatic and aromatic residues. Indeed, the perceived lack of side chains with suitable basicity has led to the hypothesis that the pyrophosphate anion co-product (−OPP) generally serves as the catalytic (general) base.2
Previous work has demonstrated that single residue changes can switch product outcome in certain plant diterpene synthases.3–11 Arguably the most interesting changes are those involving a key position that controls the complexity of the catalytic reaction. These enzymes are involved in labdane-related diterpenoid biosynthesis. Hence, they react with already bicyclic labdadienyl/copalyl diphosphate (CPP), carrying out initial cyclization to pimarenyl+ intermediates, which can be followed by further cyclization and/or rearrangement (e.g., Scheme 1). Strikingly, the presence of an aliphatic residue, typically alanine or isoleucine, leads to more complex reactions, while serine or threonine at the relevant key position “short-circuits” the carbocation cascade, leading to production of pimaradienes. The key residue is hypothesized to be proximal to the carbocation in the pimarenyl+ intermediate, which continues to react in the presence of the aliphatic residue, but undergoes deprotonation when this is serine or threonine instead. However, based in large part on the perceived difficulty for such a non-activated hydroxyl group to act as a catalytic base, these have been suggested to act via dipole stabilization of the initially formed pimarenyl+ intermediate, enabling deprotonation (presumably by reorientation with respect to −OPP).12
Scheme 1.

Reactions catalyzed by BjKS and A167S mutant.
A number of labdane-related diterpene synthases also have been identified from bacteria. Of particular interest here is the ent-kaurene synthase from Bradyrhizobium japonicum (BjKS),13 which has been shown to be involved in production of gibberellin phytohormones by this rhizobium.14 Notably, high-resolution crystal structures have been determined for BjKS.15 This revealed the expected nonpolar binding pocket for the hydrocarbon portion of its substrate, ent-CPP (1). While other residues were suggested to play particularly important roles in the catalyzed reaction, here alanine-167 was noted to exhibit intriguing parallels to a previously identified single residue switch. In particular, A167 is located at a widely conserved helix-break (G1/2), just as observed for the critical alanine in the only plant diterpene synthase in which both a product switch (alanine to serine) has been identified,4 and that has a crystal structure currently available16 – i.e., the abietadiene synthase from Abies grandis (AgAS).
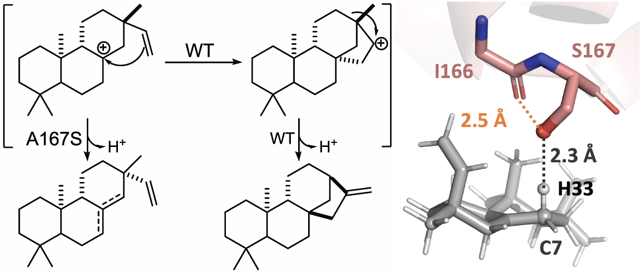
To investigate the hypothesis that A167 might be important in the (bi)cyclization and rearrangement reaction catalyzed by BjKS (Scheme 1), specifically continuation beyond initial cyclization of 1 to an ent-pimara-15-en-8-yl+ intermediate (A) [e.g., to form the ent-beyeranyl+ intermediate (B)], this residue was mutated to serine. The resulting BjKS:A167S mutant was observed to predominantly produce a roughly equal mixture of ent-pimara-8(14),15-diene (2) and ent-pimara-7,15-diene (3), resulting from immediate deprotonation of A (although no ent-pimara-8,15-diene, 4, which also could be formed by deprotonation of A), along with small amounts of ent-kaurene (5), rather than the exclusive production of 5 exhibited by wild-type BjKS (Figure 1).
Figure 1.

Chromatograms from GC-MS analysis of BjKS, either wild-type (WT) or A167S mutant, as indicated. Extracts from E. coli cultures engineered to produce 1 and co-expressing the indicated BjKS. Peaks are numbered as in the text, with 1’ indicating the dephosphorylated derivative of 1 produced by endogenous phosphatases (1’, ent-copalol; 2, ent-pimara-8(14),15-diene; 3, ent-pimara-7,15-diene; 5, ent-kaur-16-ene), as identified by comparison to authentic standards.
Intriguingly, this single residue switch in BjKS differs from that found in plant ent-kaurene synthases (KSs), where the analogous residue is an isoleucine, with threonine substitution leading to predominant production of 2 and only small amounts of 3.3, 6, 8 Moreover, sequence alignment with AgAS suggests that this isoleucine does not fall into the G1/2 helix-break, but rather on the first turn of the G2 helix (i.e., four residues later).4 This difference in location of the critical aliphatic residue between plant KSs and AgAS (which is representative of the family of diterpene synthases involved in conifer resin acid biosynthesis that are distinct from plant KSs17), has been attributed to their use of enantiomeric forms of CPP.12 Regardless, it appears that A may be differentially oriented in BjKS than plant KSs, at least relative to the G1/2 helix, which is perhaps not surprising given that these share <15% sequence identity.13
To gain further insight into the role of the single residue switch in BjKS, computational modeling was undertaken. First, density functional theory (DFT) calculations (PCM(water)-ωB97XD/6–311+G(d,p))18 were carried out to compare the energies of the three possible deprotonation products of carbocation A. No significant difference in energy was found, however (relative energies in kcal/mol: 2, +0.54; 3, +0.73, 4, 0.00), indicating that the observed product distribution is not the result of thermodynamic equilibration, nor its manifestation in transition state structures (TSSs) for deprotonation.
To gain further insight, the recently described TerDockin approach19–20 was employed, using the Rosetta Molecular Modeling Suite.21–22 To perform docking, all available X-ray crystal structures of BjKS were examined.15 The structure with PDB code 4XLX was used because it had the most complete active site density (see Supporting Figure S1 for comparison). Hydrocarbon (carbocation) structures and the diphosphate-magnesium complex were docked into BjKS simultaneously. As no available BjKS structure contains a diphosphate-magnesium complex, the diphosphate conformation was extracted from crystal structure 3P5R, the closest homolog of BjKS with such a complex present. Some conformations of carbocation structures were previously optimized using DFT calculations by Hong and Tantillo.23 Carbocation conformers were identified using Spartan 10 with the MMFF forcefield.24 All conformers generated were then fully optimized using Gaussian0925 with ωB97XD/6–31+g(d,p). TerDockin was applied to both the wild-type BjKS and to the A167S mutant; results for the latter are discussed below, while results for the former can be found in the Supporting Information.
The conformer library of A, along with the diphosphate-magnesium complex, was docked into the BjKS:A167S structure to examine the relative positions of the carbocation center and S167. The first ionization step involves bond breaking between a diphosphate oxygen and the terminal carbon of 1, leading to two possible carbocation-diphosphate ion pair orientations—the terminal carbon near to one or the other oxygen—since only two diphosphate oxygens protrude into the active site; these were examined separately during the docking simulation (Figure 2A; see the Supporting Information for details on the chemically meaningful constraints applied during docking).
Figure 2.

(A) The two diphosphate oxygen atoms to which the terminal carbon of the substrate may have been connected. (B) Model system to identify optimal angles of deprotonation by the S167 hydroxyl group: methanol and 2,3-dimethyl-2-butene. (C) 2D potential energy scan (vertical axis corresponds to relative electronic energies in kcal/mol; other axes correspond to angles from panel (B) in degrees) showing that the optimal angles are ~120° for A and ~180° for B.
As described above, simple alkene stability arguments do not rationalize the distribution of pimaradiene isomers observed. Moreover, the more selective production of 2 by the functionally analogous Ile→Thr mutation in the plant KSs argues against any significant effect from relative stability. Preliminary docking results suggested that S167, rather than diphosphate, may act as the base for the deprotonation step to form the pimaradienes. While an introduced histidine has been suggested to act as the catalytic base for production of cembrene A by the relevant mutant of taxadiene synthase,26 it does not appear to have been previously suggested that a hydroxyl containing residue can act as the catalytic base in terpene synthases. Nevertheless, the pKa of a protonated alcohol is typically around −1 to −4,27 while that of a typical carbocation lacking conjugation is less than −10,28 suggesting that proton transfer from a carbocation to an alcohol is energetically reasonable. In addition, hydrogen atoms at C7, C9 and C14 all appeared to be reasonably close to the S167 oxygen. Consequently, we suspected that the hydroxyl group of S167 acts as a base and that certain Ccarbocation–H–O and H–O–CSer angles in the deprotonation TSS (Figure 2B) were preferred. Optimal angles for proton transfer during deprotonation were identified with DFT calculations on a model system (Figure 2B–C): ~120° for H–O–CSer and ~180° for Ccarbocation–H–O. Constraints favoring these angles were then applied to the docking simulation (see Supporting Information for details and previous papers on terpene docking for the philosophy underpinning this approach and potential limitations19,20).
25,000 Docking simulations for each of the two ion pair orientations and each of five possible deprotonation sites (C7 and C14 bear two hydrogen atoms, while C9 bears one) were then carried out, i.e., 250,000 total docking runs were performed. All docking results were combined and then filtered based on satisfaction of constraints, total protein energy (the lowest 10% were kept) and interface energy (the lowest 5% were kept). Results are summarized in Figure 3 (see Supporting Information for details). In total, deprotonation at C7 (to form 2) is predicted to be most likely, with deprotonation at C14 (to form 3) next most likely and deprotonation at C9 (to form 4) unlikely (Figure 3A). The predicted 59:36:5 ratio for 2:3:4 is consistent with the experimental observation that 2 and 3 are formed in comparable amounts, with slightly more 2 than 3, while 4 is not observed, suggesting that the ability to approach the ideal TSS geometry during deprotonation plays a major role in product selectivity. Note also that the backbone carbonyl oxygen of I166 can hydrogen bond with the S167 hydroxyl group, further increasing the basicity of the Ser side chain (Figure 3B).
Figure 3.

(A) Predicted relative amounts of pimaradiene products. All docking results are combined and filtered based on satisfaction of constraints, total protein energy (the lowest 10% were kept) and interface energy (the lowest 5% were kept; see SI for additional details). (B) A representative pose predicted by docking (C7/Orientation 2/ H33). The distance between the S167 oxygen and H33 is 2.3 Å. The distance between the I166 backbone carbonyl oxygen and the S167 oxygen is 2.5 Å.
In summary, we suggest that the shortening of the BjKS carbocation cascade induced by the A167S substitution is due to direct action of the introduced alcohol as a catalytic base mediating premature deprotonation. Even beyond the implications for BjKS, our results further suggest that the previously identified analogous single residue product switches in plant diterpene synthases may operate in the same fashion – i.e., the introduced serine or threonine may act as a catalytic base to terminate the carbocation cascade reaction. More importantly, appreciation of this ability to directly deprotonate carbocation intermediates immediately indicates that incorporation of hydroxyl containing side chains at appropriate locations provides a means to alter product outcome in enzymatic engineering of terpene synthases more generally, which will be explored in future work.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Terrance E. O’Brien for initial development of TerDockin and feedback on results obtained in this study.
Funding Sources
This work was supported by a grant (GM076324) from the National Institutes of Health (NIH).
ABBREVIATIONS
- CPP
copalyl diphosphate
- KS
ent-kaurene synthase
- BjKS
Bradyrhizobium japonicum KS
- AgAS
Abies grandis abietadiene synthase
- DFT
density functional theory
Footnotes
Financial disclosure
R.J.P. is a member of the scientific advisory board for Manus Bio, Inc.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details (PDF)
Rosetta input files (PDB and text files)
Intermediate A conformers (mol2 files)
Resulting poses (PDB files)
REFERENCES
- 1.Christianson DW, Chem Rev 2017, 117 (17), 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pemberton TA; Christianson DW, The Journal of antibiotics 2016, 69 (7), 486–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu M; Wilderman PR; Peters RJ, Proc. Natl. Acad. Sci. U.S.A 2007, 104, 7397–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilderman PR; Peters RJ, J. Am. Chem. Soc 2007, 129, 15736–15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morrone D; Xu M; Fulton DB; Determan MK; Peters RJ, J. Am. Chem. Soc 2008, 130, 5400–5401. [DOI] [PubMed] [Google Scholar]
- 6.Jia M; Peters RJ, Front. Plant Sci 2016, 7, e1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keeling CI; Weisshaar S; Lin RPC; Bohlmann J, Proc. Natl. Acad. Sci. U.S.A 2008, 105, 1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zerbe P; Chiang A; Bohlmann J, Phytochemistry 2012, 74, 30–39. [DOI] [PubMed] [Google Scholar]
- 9.Kawaide H; Hayashi K; Kawanabe R; Sakigi Y; Matsuo A; Natsume M; Nozaki H, FEBS J 2011, 278 (1), 123–133. [DOI] [PubMed] [Google Scholar]
- 10.Irmisch S; Muller AT; Schmidt L; Gunther J; Gershenzon J; Kollner TG, BMC Plant Biol 2015, 15, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia M; O’Brien TE; Zhang Y; Siegel JB; Tantillo DJ; Peters RJ, ACS Catal 2018, 8 (4), 3133–3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou K; Peters RJ, ChemComm 2011, 47, 4074–4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morrone D; Chambers J; Lowry L; Kim G; Anterola A; Bender K; Peters RJ, FEBS Lett. 2009, 583 (2), 475–480. [DOI] [PubMed] [Google Scholar]
- 14.Nett RS; Montanares M; Marcassa A; Lu X; Nagel R; Charles TC; Hedden P; Rojas MC; Peters RJ, Nat Chem Biol 2017, 13 (1), 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W; Feng X; Zheng Y; Huang CH; Nakano C; Hoshino T; Bogue S; Ko TP; Chen CC; Cui Y; Li J; Wang I; Hsu ST; Oldfield E; Guo RT, Sci Rep 2014, 4, 6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou K; Gao Y; Hoy JA; Mann FM; Honzatko RB; Peters RJ, J. Biol. Chem 2012, 287 (9), 6840–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen F; Tholl D; Bohlmann J; Pichersky E, Plant J 2011, 66 (1), 212–229. [DOI] [PubMed] [Google Scholar]
- 18.Chai JD; Head-Gordon M, Phys Chem Chem Phys 2008, 10 (44), 6615–6620. [DOI] [PubMed] [Google Scholar]
- 19.O’Brien TE; Bertolani SJ; Tantillo DJ; Siegel JB, Chem. Sci 2016, 7, 4009–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Brien TE; Bertolani SJ; Zhang Y; Siegel JB; Tantillo DJ, ACS Catal 2018, 8 (4), 3322–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alford RF; Leaver-Fay A; Jeliazkov JR; O’Meara MJ; DiMaio FP; Park H; Shapovalov MV; Renfrew PD; Mulligan VK; Kappel K; Labonte JW; Pacella MS; Bonneau R; Bradley P; Dunbrack RL Jr.; Das R; Baker D; Kuhlman B; Kortemme T; Gray JJ, J Chem Theory Comput 2017, 13 (6), 3031–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leaver-Fay A; Tyka M; Lewis SM; Lange OF; Thompson J; Jacak R; Kaufman K; Renfrew PD; Smith CA; Sheffler W; Davis IW; Cooper S; Treuille A; Mandell DJ; Richter F; Ban YE; Fleishman SJ; Corn JE; Kim DE; Lyskov S; Berrondo M; Mentzer S; Popovic Z; Havranek JJ; Karanicolas J; Das R; Meiler J; Kortemme T; Gray JJ; Kuhlman B; Baker D; Bradley P, Methods in enzymology 2011, 487, 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong YJ; Tantillo DJ, J Am Chem Soc 2010, 132 (15), 5375–5386. [DOI] [PubMed] [Google Scholar]
- 24.Shao Y; Molnar LF; Jung Y; Kussmann J; Ochsenfeld C; Brown ST; Gilbert AT; Slipchenko LV; Levchenko SV; O’Neill DP; DiStasio RA Jr.; Lochan RC; Wang T; Beran GJ; Besley NA; Herbert JM; Lin CY; Van Voorhis T; Chien SH; Sodt A; Steele RP; Rassolov VA; Maslen PE; Korambath PP; Adamson RD; Austin B; Baker J; Byrd EF; Dachsel H; Doerksen RJ; Dreuw A; Dunietz BD; Dutoi AD; Furlani TR; Gwaltney SR; Heyden A; Hirata S; Hsu CP; Kedziora G; Khalliulin RZ; Klunzinger P; Lee AM; Lee MS; Liang W; Lotan I; Nair N; Peters B; Proynov EI; Pieniazek PA; Rhee YM; Ritchie J; Rosta E; Sherrill CD; Simmonett AC; Subotnik JE; Woodcock HL 3rd; Zhang W; Bell AT; Chakraborty AK; Chipman DM; Keil FJ; Warshel A; Hehre WJ; Schaefer HF 3rd; Kong J; Krylov AI; Gill PM; Head-Gordon M, Phys Chem Chem Phys 2006, 8 (27), 3172–3191. [DOI] [PubMed] [Google Scholar]
- 25.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich A; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark M; Heyd J; Brothers; Kudin; Staroverov; Keith; Kobayashi; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 09, Gaussian Inc., 2016. [Google Scholar]
- 26.Ansbacher T; Freud Y; Major DT, Biochemistry 2018, 57 (26), 3773–3779. [DOI] [PubMed] [Google Scholar]
- 27.Deno NC; Turner JO, J. Org. Chem 1966, 31 (6), 1969–1970. [Google Scholar]
- 28.McCormack AC; McDonnell CM; More O’Ferrall RA; O’Donoghue AC; Rao SN, J Am Chem Soc 2002, 124 (29), 8575–8583. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.