

Abstract
Human T-cell leukemia virus type 1 is an oncovirus that causes aggressive adult T-cell leukemia but is also responsible for severe neurodegenerative and endocrine disorders. Combatting HTLV-1 infections requires a detailed understanding of the viral mechanisms in the host. Therefore, in vitro studies of important virus-encoded proteins would be critical. Our focus herein is on the HTLV-1-encoded regulatory protein p13II, which interacts with the inner mitochondrial membrane, increasing its permeability to cations (predominantly potassium, K+). Thereby, this protein affects mitochondrial homeostasis. We report on our progress in developing specific protocols for heterologous expression of p13II in E. coli, and methods for its purification and characterization. We succeeded in producing large quantities of highly-pure full-length p13II, deemed to be its fully functional form. Importantly, our particular approach based on the fusion of ubiquitin to the p13II C-terminus was instrumental in increasing the persistently low expression of soluble p13II in its native form. We subsequently developed approaches for protein spin labeling and a conformation study using double electron-electron resonance (DEER) spectroscopy and a fluorescence-based cation uptake assay for p13II in liposomes. Our DEER results point to large protein conformation changes occurring upon transition from the soluble to the membrane-bound state. The functional assay on p13II-assisted transport of thallium (Tl+) through the membrane, wherein Tl+ substituted for K+, suggests transmembrane potential involvement in p13II function. Our study lays the foundation for expansion of in vitro functional and structural investigations on p13II and would aid in the development of structure-based protein inhibitors and markers.
Keywords: Human T-cell leukemia virus type 1 encoded protein p13II, high-yield protein production, viral protein-mitochondrial membrane association, DEER-detected p13II self-association, fluorescence-based p13II liposome uptake assay
Graphical Abstract
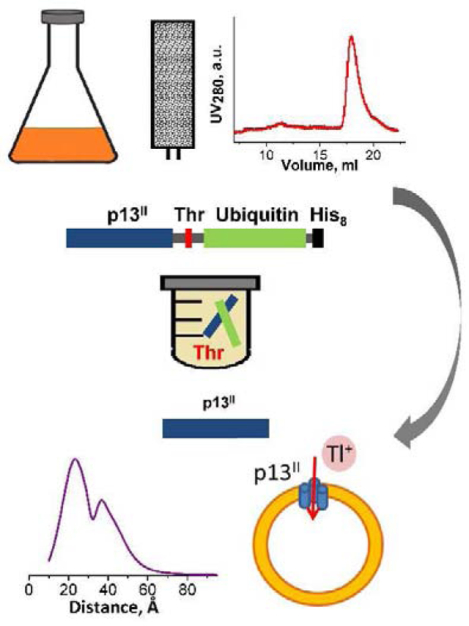
Introduction
Human T-cell leukemia virus type 1 (HTLV-1) was the first human retrovirus discovered (1–5). In infected individuals, it is usually asymptomatic and can linger for decades (6, 7). However, in a substantial number of cases, the HTLV-1 infection progresses to severe illnesses, such as adult T-cell leukemia/lymphoma (ATLL) or myelopathy/tropical spastic paraparesis (HAM/TSP) (6, 8–11). ATLL is an aggressive cancer, which in most cases advances rapidly to a fatal outcome within a year (1, 5, 6, 12). HAM/TSP is an irreversibly progressive neurological disease manifested in motor disorders, leading to a low quality of life (10, 11, 13). An estimated 10–20 million people worldwide (14, 15), or at least 5–10 million according to a more recent report (16), are infected with HTLV-1. Of these individuals, 5–10% will develop either ATLL or HAM/TSP (17, 18). HTLV-1 is also linked to infective dermatitis, Sjögren’s syndrome—an immune and endocrine-metabolic condition—and several other serious illnesses (5, 19). Unfortunately, the current knowledge about HTLV-1 mechanisms in the host and the factors triggering the virus-associated diseases is very insufficient (6, 15, 20). Owing to this, no effective treatment exists for these HTLV-1-associated diseases (5, 12, 15). Multiple studies suggest that proteins encoded by HTLV-1 play critical roles in the adaptation and proliferation of this virus through beneficial interactions with the components of infected cells (21–23). Elucidating the molecular mechanisms of proteins key to the lifecycle of this virus would help with understanding and possibly intervening in HTLV-1 infectivity and pathogenesis.
Of special interest to us is the nonstructural regulatory protein p13II encoded by the open reading frame II of the HTLV-1 genome (24). The protein has 87 amino acids and is positively charged, and its secondary structure was predicted to contain the following domains (Fig. 1 A): (i) a hydrophobic N-terminus; (ii) an amphipathic alpha helix with residues 20–30, including the mitochondrial targeting sequence LRVWRLCTRR; (iii) a transmembrane region of residues 31–41; (iv) a highly flexible region with residues 42–49; (v) a predicted β-sheet hairpin region with residues 60–75; and (vi) C-terminal PXXP motifs that could be binding SH3 domains (Fig. 1 A) (25–27). Although the protein was found in the nuclei of infected cells (28, 29), its function there was not understood. But it was established to a high degree of confidence that p13II associates with the inner mitochondrial membrane (IMM), where it forms a cation-conducting oligomeric channel, which most likely is selective to potassium ions (K+) (24, 25, 30, 31). Through its channel activity, the protein alters the IMM permeability, leading to dissipation of the mitochondrial membrane potential (25, 30). As a result of the IMM depolarization, the levels of reactive oxygen species (ROS) in mitochondria increase due to the elevation of proton (H+) transport through the H+ pumps of the electron transport chain, which, depending on the concentration of ROS, leads to either apoptosis in the transformed cancerous cells or cell activation and division in normal cells (24, 30, 32, 33). Through these mechanisms, p13II aids virus proliferation (24–26, 32, 34, 35).
Figure 1. p13II protein and the current view of its mitochondrial function:
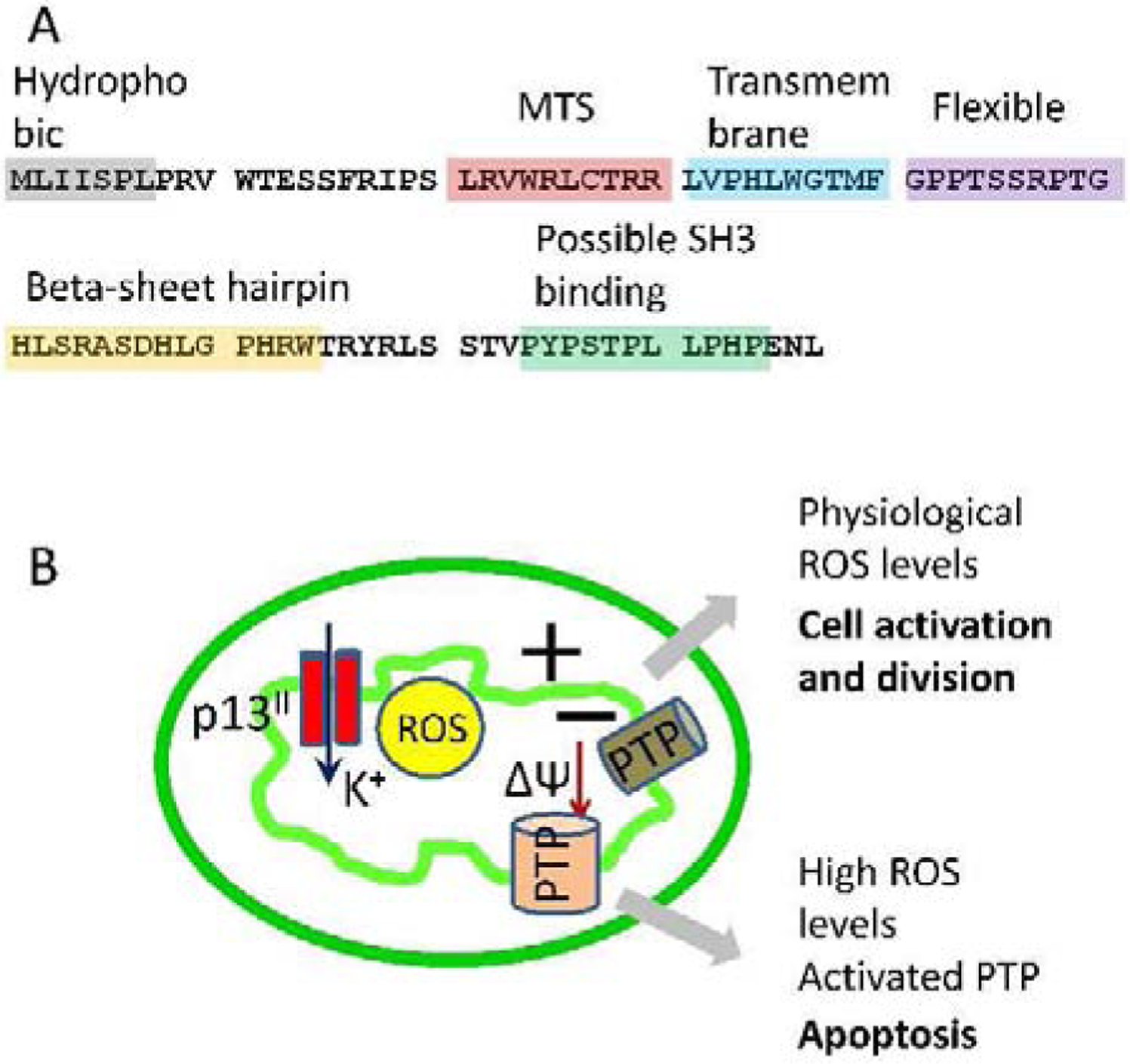
(A) Amino acid sequence and proposed domain architecture of p13II - The protein contains mitochondrion targeting sequence (MTS), transmembrane segment, and possible protein-protein interaction (presumably with proteins containing SH3 domain) region; (B) The current view on how mitochondrion may be affected by p13II functions. In the IMM the protein forms cation-conducting (predominantly K+) oligomers, leading to dissipation of IMM potential, ΔΨm, thereby increasing the level of reactive oxygen species (ROS) and, depending on the levels of ROS, leading to either cell activation or opening of the mitochondrial permeability transition pore (PTP) and apoptosis.
Because of the critical role of p13II in the viral physiology, a detailed understanding of this protein structure and function is needed. To gain such knowledge, in vitro investigations on sufficient amounts of heterologously produced and purified p13II protein are needed to acquire the molecular details of its function and structure. To the best of our knowledge, this work provides the first detailed description on the production in E. coli of milligram quantities of high-purity full-length (FL) functional p13II. Having enough of pure protein, we were able to provide initial characterization of its conformation in a solution and upon interaction with lipid membrane mimetics carried out with size exclusion chromatography (SEC), pulse electron paramagnetic resonance (EPR), and circular dichroism (CD). Importantly, the fluorescence-based activity assay, conducted herein on liposome-bound p13II, suggests that membrane potential may play a role in p13II function in IMM.
Materials and methods
Design, cloning, and mutagenesis of p13II-Ub and p13II-StrII fusion constructs
We designed two fusion constructs of p13II (GenBank: BAH85792.1) with either ubiquitin (Ub) plus an octohistidine (His8) tag or streptavidin-binding peptide II (StrII) plus a His8 tag at the C-terminus; a thrombin cleavage site was introduced between p13II and the Ub or StrII (Fig. 2 A and B). The genes encoding these protein constructs were commercially synthesized (GenScript [GenScript Biotech Corp.]) and cloned into the pET15b vector (Novagen) at the Nde I and BamH I cloning sites, which produced two plasmids, pET15b/p13II-Ub and pET15b/p13II-StrII, respectively. The plasmids were transformed in E. coli BL21(DE3) cells for protein expression under the control of a T7 promoter.
Figure 2. Schematic representation (upper) and aa sequence (lower) of p13II fusion constructs used in this study:

(A) p13II-Ub construct - p13II (green) was fused to Ub (blue) and His8 tag (black) at the C-terminus; (B) p13II-StrII construct - p13II (green) was fused to StrII (magenta) and His8 tag (black) at the C-terminus. The thrombin (Thr) cleavage site between the p13II and Ub/StrII aa sequences is shown in red. The additional Gly2 residue introduced in p13II aa sequence due to cloning strategy is in orange.
Two single cysteine (Cys) mutants of p13II—S47C and H63C—were created by site-directed mutagenesis in the p13II-Ub construct. In these mutants, the native C28 residue in wild-type (WT) p13II was replaced with serine. For mutagenesis, a QuikChange lightning multi-site-directed mutagenesis kit (Agilent Technologies) was used according to the manufacturer protocol.
Expression and purification of p13II-Ub and p13II-StrII constructs
We expressed p13II-Ub (both WT and mutants) and p13II-StrII in E. coli BL21(DE3) cells. Each of the pET15b/p13II-Ub and pET15b/p13II-StrII plasmids were transfected separately in BL21(DE3) competent cells (New England BioLabs), and colonies were grown overnight on LB/agar/ampicillin-100 plates (Teknova) at 37 °C. In each case, a flask containing 200 mL LB broth (Millipore-Sigma) and 100 μg/mL ampicillin (Amp) (Gold Biotechnology) was inoculated with a single colony, and a bacterial stock solution was grown at 37 °C overnight for 17–18 h in a shaking incubator. The next day, 4 L flasks containing 2 L LB broth with 100 μg/mL Amp were inoculated with 30 ml of the overnight cell culture and grown at 37 °C until the absorbance at 600 nm (OD600) reached 0.75–0.8. Thereafter, the temperature was reduced to 28 °C, and protein expression was induced by the addition of IPTG (Isopropyl β-D-1-thiogalactopyranoside) (Gold Biotechnology) to a final concertation of 1 mM. The protein expression proceeded for 7 h. These protein expression conditions were found to be optimal after testing the expression at 37 °C for 4 h at 18 °C overnight. Then the cells were spun down in an Allegra X-14R centrifuge (Beckman Coulter) at 4,100 RPM (3,912×g), and the cell pellets were collected and resuspended in a buffer of 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (Sigma) pH 7.5, 300 mM NaCl (Sigma), 5% (w/v) glycerol (Fisher Scientific) and 200 μM TCEP (tris(2-carboxyethyl)phosphine) (Aldrich). Then PMSF (phenylmethylsulfonyl fluoride) (Gold Biotechnology) and a chicken egg lysozyme (Sigma) were added to the final concentrations of 1 mM and 0.5 mg/mL, respectively. The cells were broken with sonication, and the cell debris was separated with centrifugation at 9,000 RPM (8,891×g). Both the p13II-Ub and p13II-StrII were purified from the supernatants containing the soluble fraction of cell lysates.
We used nickel (Ni)-affinity chromatography and size exclusion chromatography (SEC) to purify WT and mutant p13II-Ub. The soluble cell lysate in the resuspension buffer was incubated with Ni-NTA agarose resin (Qiagen) at 1.5 ml resin per 1 L cell culture for 1 h at 4 °C under constant agitation. Thereafter, the Ni-NTA agarose resin with the bound protein was transferred to a gravity column and washed with 10 resin volumes of the resuspension buffer supplemented with 40 mM imidazole to remove most of the weakly bound impurities. Then the p13II-Ub was eluted with 300 mM imidazole in the resuspension buffer. The protein was concentrated in Amicon® ultra centrifugal filters with 10 kDa MWCO and then loaded on a Superdex™ 200 increase 10/300 GL size-exclusion column (GE Healthcare) connected to a BioRad Biologic DuoFlow protein purifier for further SEC purification in a buffer containing 20 mM HEPES pH 7.5, 220 mM NaCl, 20 mM KCl, and 2.5 % (w/v) glycerol.
After the p13II-Ub was purified by SEC, the buffer was exchanged with 50 mM Tris pH 8, 100 mM NaCl, 5 mM KCl, 200 μM TCEP, and 2.5% glycerol, and the protein was concentrated again to several mg/mL. Thrombin (Sigma) and CaCl2 were added to 16–20 units per mg of protein and 4 mM, respectively. The thrombin digestion reaction was allowed to proceed for 16–17 h at room temperature (RT). Thereafter, the protein was diluted 3–4 times with a buffer of 50 mM Tris/HCl pH 7.5, 200 mM NaCl, 10 mM KCl, and 200 μM TCEP; imidazole was added to 10 mM in the final concentration. Then the protein solution was mixed with the Ni-NTA agarose resin at 1 mL resin per 1 mg protein to separate the tag-free p13II from the non-digested p13II-Ub and Ub-His8 tag with Ni-affinity binding. This mixture was incubated for 1 h at 4 °C and then transferred to a gravity column, and the flow-through fraction containing the tag-free p13II was collected. The column was washed with one bed volume of binding buffer, this fraction was also collected and combined with the solution containing tag-free p13II, and the protein was concentrated in centrifuge concentrators with 3 kDa MWCO. Thereafter, the tag-free p13II was purified by SEC in phosphate-buffered saline (PBS) pH 7.4 (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4) to remove the leftover thrombin and concentrated again.
To purify the WT p13II-StrII, we developed a protocol similar to the one used for p13II-Ub. However, the quantity and purity of the p13II-StrII after the Ni-affinity purification was insufficient for proceeding with SEC. Therefore, we subjected it to an additional purification step based on cation-exchange chromatography. To do so, we exchanged the protein buffer with 50 mM sodium phosphate (NaPi) pH 6.1 and 1 mM EDTA and then bound the protein to CM sepharose cation exchange resin (Sigma) for 1 h at 4 °C. After removing the flow-through fraction, we washed the resin with 10 bed volumes of NaPi pH 6.1 supplemented with 40 mM NaCl and eluted the protein with 2 bed volumes of NaPi pH 6.1 supplemented with 500 mM NaCl. The protein was concentrated in centrifuge concentrators with 3 kDa MWCO and subjected to SEC on a Superdex™ 200 increase 10/300 GL column with the BioRad protein purifier.
To access the oligomeric state of the purified p13II versions, we conducted SEC on the proteins used as SEC standards (GE Healthcare), ribonuclease A (13.7 kDa) and carbonic anhydrase (29 kDa); 0.5 mg of ribonuclease A and 0.5 mg of carbonic anhydrase were mixed, solubilized in 0.9 ml of either 20 mM HEPES pH 7.5, 220 mM NaCl, 20 mM KCl, and 2.5 % (w/v) glycerol or PBS and loaded on the SEC column. We found that the elution of these standard proteins was virtually insensitive to the buffer composition. Then the positions of the p13II-Ub, tag-free p13II, and p13II-StrII elution peaks were referenced with those of the standard proteins.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting
All the samples of p13II variants for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting (WB) contained 10 mM DTT (Dithiothreitol) (Sigma). After these samples were heated for 10 min at 95 °C, they were loaded on 4–20 % Criterion™ TGX™ precast midi protein gels (BioRad), which were immersed in Tris/glycine/SDS buffer (BioRad), and the electrophoresis was conducted at 200 V in a Criterion™ vertical electrophoresis cell (BioRad). The purified protein quantities used for the gel visualization and WB transfer were 15–50 μg and 6–12 μg, respectively. For WB, the protein was transferred from the gel to a 0.2 μm nitrocellulose membrane (BioRad) at 10 V overnight using a Criterion™ blotter (BioRad). The protein gels and the WB membrane with transferred p13II proteins were visualized via Coomassie Blue staining and colorimetric detection using an anti-His tag antibody, respectively.
Circular dichroism spectroscopy and data analysis
The following four types of samples were studied using Circular dichroism (CD) spectroscopy: (1) 10 μM tag-free WT p13II in a buffer solution, (2) 10 μM tag-free WT p13II in 3.5 mM LPPG (1-palmitoyl-2-hydroxy-sn-glycero-3-phospho-(1’-rac-glycerol) (sodium salt), 16:0 Lyso PG), (3) 10 μM tag-free WT p13II in liposomes made of 2.2 mM total DOPC(1,2-dioleoyl-sn-glycero-3-phosphocholine) and POPG (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1’-rac-glycerol) (sodium salt)) lipids, and (4) 10 μM tag-free WT p13II in liposomes made of 2.2 mM DOPC/POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (sodium salt)). In all cases, the buffer used was PBS supplemented with 100 μM TCEP. A stock solution of 200 mM LPPG in PBS was used to prepare the sample of 10 μM p13II in 3.5 mM LPPG. The liposomes stock solutions with a total lipid concentration of 20 mM and a molar ratio of non-charged to charged lipids of 60%-to-40% (60/40) were prepared as follows: Lipids from chloroform or chloroformmethanol-H2O solutions were mixed; the organic solvent was removed using evaporation with a flow of argon gas followed by desiccation in a vacuum chamber for 3–4 h at RT. The dried lipid mixtures were then rehydrated with PBS/TCEP and stored for 30–60 min at 4 °C. The liposomes were extruded using an Avanti Polar Lipids mini-extruder and Whatman polycarbonate membranes with a 100 nm pore size. Thereafter, aliquots of p13II, liposome stock solution, and PBS/TCEP were mixed to obtain the final concentrations. All proteo-LPPG and proteoliposome samples were incubated for 30–40 min at RT before recording their CD spectra. Additionally, protein-free samples of just PBS/TCEP, 3.5 mM LPPG in PBS/TCEP, 2.2 mM DOPC/POPG liposomes in PBS/TCEP, and 2.2 mM DOPC/POPS liposomes in PBS/TCEP were also prepared and used as background material. All lipids and LPPG were purchased from Avanti Polar Lipids, Inc., and used as received.
The CD spectra of the p13II in a solution and in the presence of lipid membrane mimetics as well as the respective background referencing samples were recorded at 25 °C in the range between 200 nm and 260 nm using the model No. 202–01 CD spectropolarimeter (Aviv Biomedical, Lakewood, NJ). For each protein sample, three scans were recorded, with the background recorded in a single scan. For each condition, the backgrounds were subtracted from the averaged protein scans. The parameters set to record the CD spectra were as follows: The wavelength step was 1 nm, the averaging time was 5 s, and the time constant was 100 ms.
The CD data deconvolution was performed using the available online BeStSel program (36, 37).
Spin labeling and EPR spectroscopy
For labeling the two single cysteine mutants S47C and H63C of FL p13II-Ub, we used two types of spin labels (SLs): MTSL (S-(1-oxyl-2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate) and ISL (3-(2-Iodoacetamido)-PROXYL). When labeling in a buffer solution was attempted, the protein precipitated out upon the addition of MTSL within a broad range of MTSL concentrations and protein-to-SL molar ratios (1:5 to 1:30). Consequently, in buffer-only solutions, we utilized the labeling with ISL, which worked well for the H63C substitution, but not for S47C. For this reason, we exercised specific spin-labeling protocols for both mutant constructs.
The mutant H63C of p13II-Ub was labeled with ISL in a buffer solution containing 50 mM Tris/HCl pH 8, 300 mM NaCl, 2.5 % (w/v) glycerol, 1 mM EDTA, and 40 μM TCEP for 12–14 h at RT. The protein-to-ISL molar ratio was 1:30. The unreacted ISL was removed using several washes in centrifuge concentrators with 10 kDa MWCO. Part of the spin-labeled protein construct was used for pulse EPR (double electron-electron resonance, DEER) measurements as labeled. The rest had the tag cleaved off by thrombin, and the spin-labeled tag-less p13II was separated from the Ub-His8 tag and thrombin by using the procedure described above. In both cases, the final spin-labeled protein was in PBS.
The mutant S47C was spin-labeled with MTSL in PBS supplemented with 4 mM LPPG for 12–14 h at RT. The protein-to-MTSL molar ratio used in this case was 1:30 as well. The unreacted SL was removed by the dialysis against PBS supplemented with 2 mM LPPG using a membrane with10 kDa MWCO.
Several samples for DEER measurements were prepared based on the spin-labeled S47C and H63C mutants. For the p13II-Ub S47C, these samples were as follows: (1) 50 μM protein in LPPG from 6 mM to 18 mM and (2) 50 μM protein in liposomes of DOPC/POPG in 60/40 molar ratio, yielding protein to total lipid molar ratio of 1:200. The liposome samples also contained 1.48 mM of residual LPPG, due to its 2 mM content in the protein stock. For H63C, these samples were, respectively: (1) 100 μM of either the tag-free p13II or p13II-Ub protein in PBS or (2) 60–110 μM of either the tag-free p13II or p13II-Ub protein prepared in 4–12 mM LPPG, (3) 97 μM protein in 14 mM LPPC (1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine, 16:0 Lyso PC), and (4) in DOPC/POPG lipids in 60/40 molar ratio and protein to lipid molar ratio of 1:200. In all cases, the protein was incubated with the membrane mimetics for 1 h at RT. Glycerol was added to the final 25% (w/v) to all solution and LPPG samples before freezing. No glycerol was added to the proteoliposome samples. The samples were transferred into 1.8 mm i.d. Pyrex™ capillary sample tubes and plunge-frozen in liquid nitrogen for the DEER measurements.
DEER spectroscopy was conducted at 17.3 GHz and 60 K using a home-built Ku-band pulse EPR spectrometer (38). The standard four-pulse DEER setup (39) was employed with the detection π/2-π -π pulse sequence having π/2- and π-pulse widths of 16 and 32 ns respectively, and applied at the low-field edge of the EPR spectrum. The 16 ns pump π-pulse was applied at a 70 MHz lower frequency corresponding to the central maximum of the EPR spectrum. Usually, 1–2.5 μs evolution time, tm, was used for recording, with the data averaging time taking from 4 to 24 hours depending on phase relaxation time, tm and distance range. The as-recorded DEER data were subjected to the standard procedure of background subtraction. The latter points (about half of the record) in the logarithm of the data were fit to the first- or second-degree polynomial and the fits were subtracted out. The resulting linear-scale data V(t) were modified as V′(t) = (V(t) − 1)/V(0) to give the amplitude at zero equal to DEER signal “modulation depth” and the asymptotic value of zero (40). (A frequently-used alternative presentation is to plot the signal as V(t)/V(0), i.e. unity at t = 0) The background-free data were then processed into the distance distributions using either L-curve Tikhonov regularization (41) or denoising/SVD method (42, 43) with the outcomes being close enough, so just the Tiknonov distributions are shown in Figure 7.
Figure 7. Spin labeling and DEER spectroscopy on p13II single Cys mutants.
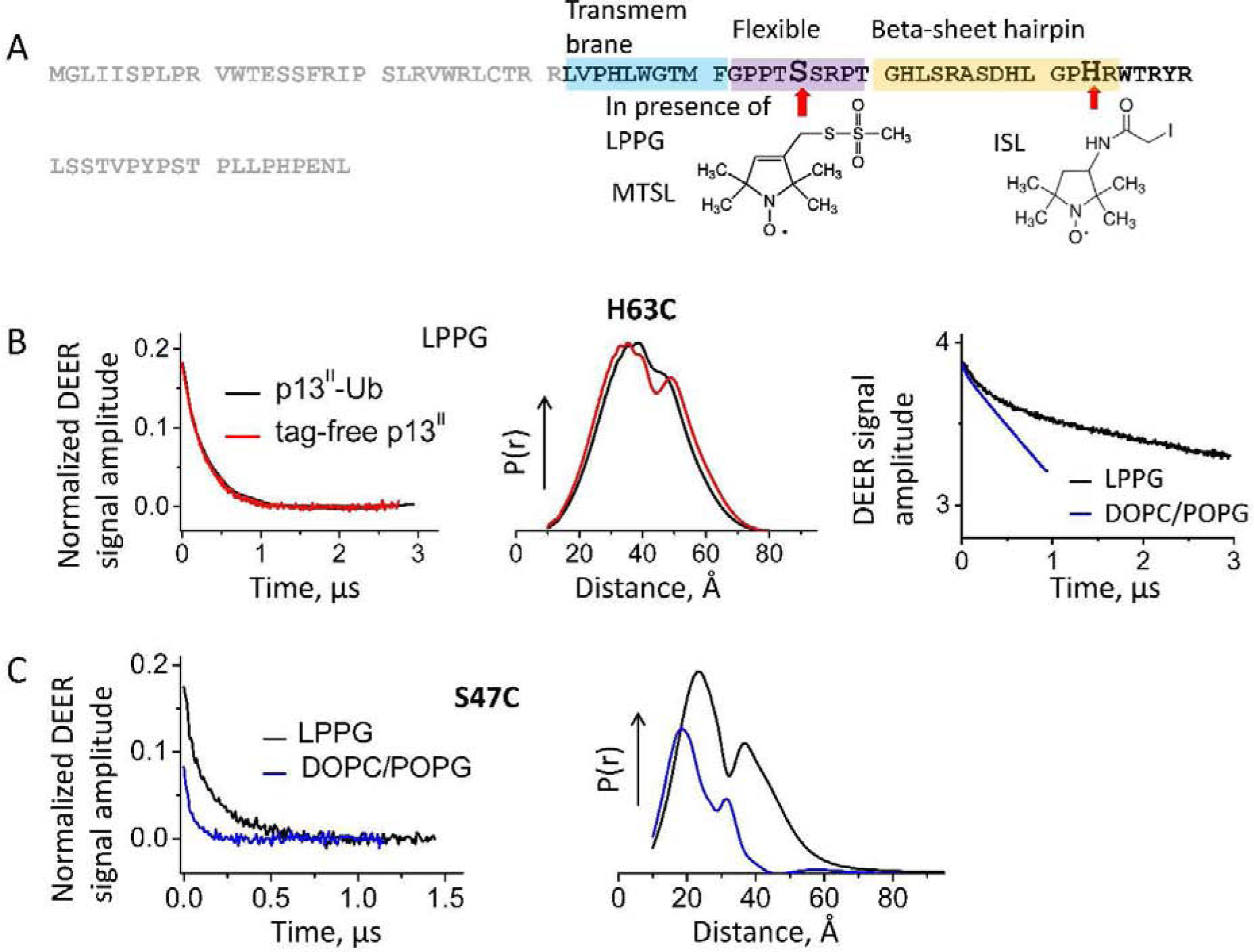
(A) The positions of single substitutions to Cys in the putative secondary structure of p13II for S47C and H63C constructs. S47C was spin-labeled with MTSL in the presence of LPPG, but H63C was labeled with ISL in solution. (B) Background-subtracted normalized DEER data for H63C mutant in LPPG (left) and corresponding distances (middle) for proteins expressed with and without Ub+His8 tag. The raw DEER data for H63C in LPPG and lipid are shown on the right. (C) Background-subtracted and normalized DEER signals (left) and reconstructed distances (right) for residue S47C in LPPG and lipid.
Continuous wave (cw) EPR experiments were conducted on samples of p13II-Ub spin-labeled at position H63C. For cw EPR measurements the solutions were placed into precision 50 μL closed bottom capillaries (Kimble Glass, Vineland, NJ), filling ~10 mm tube length yielding ~6.7 μl sample volume. All measurements were performed at RT with the Bruker ELEXIS E500 spectrometer(Bruker Spectrosopin, Billerica, MA) equipped with ER 4122-SHQE super-high Q resonator. The full extent of nitroxide cw EPR spectra were recorded under non-saturating conditions and field modulation amplitude of 1.5 G. Three different samples were measured: 75 μM protein in buffer; 75 μM protein in 8 mM LPPG; ca. 50 μM protein in 10 mM DOPC/POPG lipids using 1:200 protein-to-lipid molar ratio. In all cases PBS was used for buffer.
Additionally, for a control experiment, 50 μM MTSL in 6 mM LPPG was incubated for 1.5 h at RT. The 50 μM MTSL concentration used was certainly much higher than that, which could remain after the dialysis following protein spin labeling. For this control sample, in addition to RT cw EPR spectrum, the DEER data were recorded to establish whether free MTSL could contribute to distances observed in LPPG. This was needed to test if significant amount of MTSL was trapped in LPPG micelles during the process of residue S47C spin labeling.
Fluorescence-based activity assay in liposomes
We conducted a bulk fluorescence assay to evaluate the increase of liposome permeability to Tl+ upon WT p13II-liposome association. The assay was based on the quenching of ANTS fluorescence dye encapsulated in liposomes by Tl+ added to the solution surrounding the liposomes. Two types of liposomal formulations were made using the same 60/40 molar ratio of DOPC to POPS or POPG. Firstly, the proper aliquots of lipids from the stock solutions in chloroform or chloroform-methanol-H2O were mixed together and dried as described above (cf. CD spectroscopy). Then the buffer containing either 20 mM HEPES pH 7, 90 mM K2SO4, 2 mM K3PO4, and 25 mM fluorescence dye ANTS (8-aminonaphthalene-1,3,6-trisulfonic Acid) or 20 mM Tris/HCl pH 8, 90 mM K2SO4, 2 mM K3PO4, and 25 mM ANTS was added to the dried lipids to obtain a 20 mM total lipid concentration and allowed to hydrate for 30–60 min at 4 °C. Then liposomes were extruded 15 times through Whatman polycarbonate membranes with a pore size of 100 nm using an Avanti Polar Lipids mini-extruder. Thereafter, the liposome solutions were passed in succession through NAP5 and NAP10 columns using the same buffer compositions but no ANTS. This produced liposomes with encapsulated fluorescence dye.
Next, the WT tag-free p13II in the buffer containing 20 mM HEPES pH 7, 100 mM K2SO4, and 200 μM TCEP was mixed with the liposomes to give 1:605 protein-to-lipid molar ratio and incubated for 1 h at RT. These proteoliposome solutions were mixed in fluorescent cuvettes using the ratio of 1:5.7, with the bath buffer containing either 20 mM HEPES pH 7, 70 mM K2SO4, 2 mM K3PO4, and 60 mM TlNO3 or 20 mM MES pH 5.5, 70 mM K2SO4, 2 mM K3PO4, and 60 mM TlNO3. The second buffer was used to create a pH gradient across the liposome wall. In this case, the final outside pH was 6.2 (vs. the liposome’s internal pH of 8) after the proteoliposome solution was mixed with the bath buffer.
The fluorescence of ANTS was recorded immediately following the mixing step (0 min) and after an 18-min delay. Similarly prepared reference samples not loaded with the protein were also made and measured to serve as controls. The quenching of fluorescence intensity (via fluorescence intensity reduction) from 0 min to 18 min after the proteoliposomes or reference liposomes were placed in the bath buffer was used to estimate the liposome permeability to Tl+. The effect of the p13II on the liposomes permeability was estimated by subtracting the fluorescence intensity reduction data in protein-free liposomes from that in the proteoliposomes. Three separate experiments of each type were performed. For each of these experiments, three or four repetitive samples with and without protein were prepared and measured. The final result for each experiment is the average of the results for these samples.
In all cases, each experiment that includes measurements on samples with and without protein were conducted using the same experimental conditions and within 2 h after samples preparation. However, we observed variations in the fluorescence intensity for background samples (liposomes without p13II) with and without pH gradient when each of these sample batches were measured on different days. To establish whether this is due to experimental setup, we conducted control measurements for all the protein-free liposome samples on the same day within 2 h interval and using the same instrument setup: 3 samples of DOPC/POPS liposomes at pH 7 with incapsulated ANTS in bath buffer at pH 7 and 3 samples of DOPC/POPS liposomes at pH 8 with incapsulated ANTS in bath buffer at pH 5.5 were measured. The result established that the data with and without pH gradient are virtually the same.
All fluorescence measurements were conducted using a Photon Technology International fluorimeter with the following settings: The excitation wavelength was 325 nm, the emission was recorded between 455 nm and 575 nm, slit width for both excitation and emission was 4 nm, the step size was 1 nm; and the integration time was 0.3 sec.
Results
Design and cloning of p13II-Ub and p13II-StrII fusion constructs
In this work, we aimed to establish a cloning strategy and develop protocols for the expression in E. coli and purification of the HTLV-1 encoded p13II protein on milligram scale. Designing a p13II construct containing a polyhistidine tag (His-tag) for Ni-affinity purification was our first choice because this is a well-established and relatively inexpensive purification technique (44). We decided to fuse the His-tag to the soluble C-terminus rather than the N-terminus of p13II; this choice was essentially based on the prediction that the N-terminus should be hydrophobic (24) and on the lack of structural information (Fig. 1 A). Because of the high hydrophobicity of p13II N-terminus, this region could be important for p13II association with lipid membranes. Thus, the addition of His-tag and protease at the N-terminus could disturb this possible protein-lipid interaction. In our preliminary experiments, we found that p13II with a hexahistidine (His6) tag at the C-terminus does not bind to Ni-NTA resin; therefore, it could not be purified using Ni-affinity chromatography. A possible reason may be an occluded C-terminal region and consequently inaccessible His6 tag. To overcome this hurdle, we designed two fusion constructs of full-length p13II, namely p13II-Ub and p13II-StrepII (Fig. 2 A and B). These constructs include either Ub (45) or StrepII peptide (46, 47) plus His8 at the C-terminus of the p13II construct (Fig. 2). Thus, the His8 tag was located far enough from the presumably occluded C-terminus of p13II. A thrombin protease cleavage site located between p13II and Ub or StrII was also introduced so the tag can be removed after the protein is purified. Short SGSG and SGS aa sequences flanking the thrombin recognition site serve as flexible linkers (Fig. 2). The gene insertion between the Nco I and BamH I restriction sites in the pET15b vector allowed a bypass of the vector-encoded His6 tag at the protein N-terminus. Thus, we engineered two p13II expression vectors, namely pET15b/p13II-Ub and pET15b/p13II-StrepII. Due to the cloning at the Nco I site, an additional glycine residue (Gly2) was introduced after the methionine at the p13II N-terminus (Fig. 2). Therefore, our final p13II construct had 88 aa residues instead of the 87 in the original protein sequence. Also, eight extra aa residues remained fused to its C-terminus after the thrombin digestion (Fig. 2). Nevertheless, we see it as appropriate to refer to the resulting tag-free version of p13II as “wild-type” (WT) and use it throughout the text of this paper.
Expression and purification of WT and mutant p13II
We expressed both WT p13II-Ub and WT p13II-StrII in E. coli, and the proteins were purified from the soluble fractions of cell lysates. In the case of p13II-Ub, we used two-step purification employing Ni-affinity chromatography followed by SEC. The presence of eight histidine residues in the affinity purification tag instead of the standard His6 provided stronger binding to the Ni-NTA agarose resin, allowing for extensive washing with 40 mM imidazole to efficiently remove weakly bound protein impurities. In the size-exclusion column, p13II-Ub was eluted as a well-defined peak showing a maximum at ca. 17 ml (Fig. 3 A). The Ub-His8 tag sequence was removed from the SEC-purified p13II-Ub using thrombin digestion and then binding it to the Ni-NTA agarose column. The tag-free p13II was collected in flow-through fraction that was not bound to the Ni-NTA agarose resin. We found that the most efficient digestion could be achieved with 16–20 units of thrombin per mg p13II-Ub. Finally, extra SEC purification was applied to separate the tag-free p13II from the thrombin (Fig. 3 B). Thus, we produced two variants of WT p13II: p13II-Ub and tag-free p13II, with respective molecular weights of 20,980 Da and 10,873.62 Da. The calculated molar extinction coefficients (ε) at 280 nm of p13II-Ub and tag-free p13II are 26,470 M−1cm−1 and 24,980 M−1cm−1. These values were used to estimate protein concentrations from their absorbance at 280 nm.
Figure 3. Purification and detection of p13II-Ub and tag-free p13II - SEC, SDS-PAGE and WB:
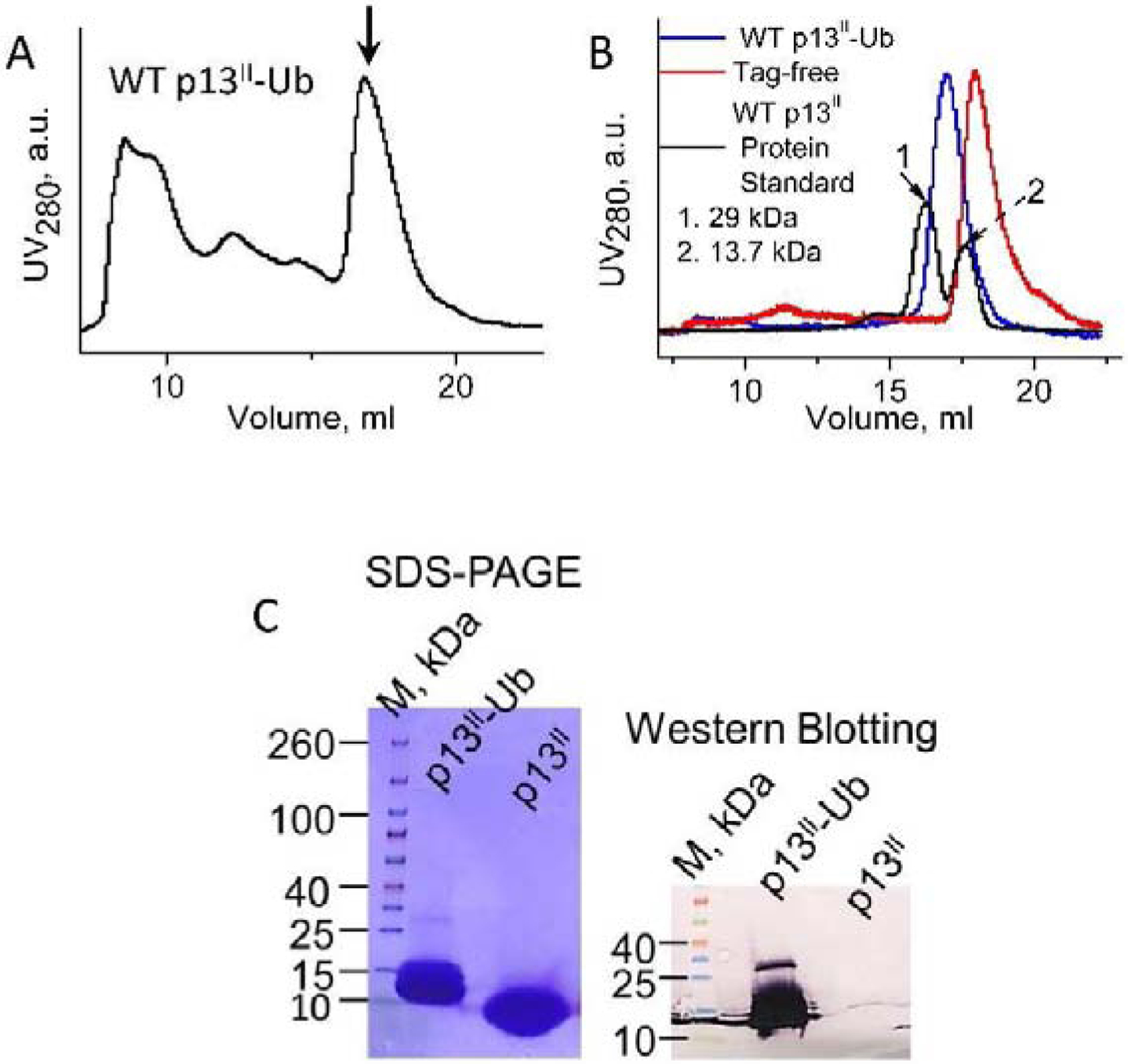
(A) Size-exclusion chromatogram of Ni-affinity purified p13II-Ub. The arrow indicates the elution peak of p13II-Ub. (B) Size exclusion chromatograms of fully purified p13II-Ub (blue), tag-free p13II (red) (the low-intensities peaks on the left are from thrombin), and a mixture of protein standards (black) - Carbonic anhydrase (29 kDa) and Ribonuclease A (13.7 kDa). (C) SDS-PAGE (left) and WB (right) of fully-purified p13II-Ub and tag-free p13II - ca. 25 μg and ca. 8.3 μg from each protein were used for SDS-PAGE and WB, respectively. As expected, only p13II-Ub was WB-positive.
The final p13II-Ub and tag-free p13II products had very high purity, as reported by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie Blue staining (Fig. 3 C, left). As expected, the p13II-Ub carrying the His8 tag was WB-positive, whereas WB showed no protein band for p13II without the Ub-His8 tag (Fig. 3 C, right), as we used an anti-His tag antibody and colorimetric detection.
The yield of the SEC-purified WT p13II-Ub was ca. 0.9±0.1 mg protein per liter of bacterial culture, based on more than three consecutive protein expressions and purifications using the same protocol. In comparison, the yield of the tag-free WT p13II was 20 times less than the p13II-Ub in mg/ml. As a better measure, this corresponds to the p13II molar concentration of nearly 10 times smaller than that of p13II-Ub, both produced using equal volumes of bacterial culture. Thus, on average, ca. 9 mg (4.3 mL of 100 μM) of highly pure p13II-Ub and ca. 0.450 mg (only 0.414 mL of 100 μM) of highly pure tag-free p13II can be produced from reasonable 10 L of bacterial culture. These quantities are adequate for conducting detailed functional and structural studies, e.g. conducting crystallization screening or NMR measurements.
The analysis of SEC chromatogram of purified p13II-Ub shows that the protein is monomeric in solution, as its elution peak appears between those of carbonic anhydrase (29 kDa) and ribonuclease A (13.7 kDa), which are used as standard monomeric protein markers (Fig. 3 B). The tag-free p13II is also monomeric as its SEC elution peak shifts toward a lower molecular weight than that of p13II-Ub (Fig. 3 B) and is closer to that of ribonuclease A.
Using site-directed mutagenesis on the background of a cysteine (Cys)-free construct, we generated S47C and H63C single Cys mutants of p13II-Ub and the corresponding tag-free p13II. WT p13II has a single native Cys residue at position 28 (position 27 in the native aa sequence lacking the extra Gly2 at the N-terminus), which was substituted with serine to give the Cys-free construct. Notably, the fused Ub has no native cysteines (Fig. 2 A); thereby, no additional mutations were required to produce a Cys-less construct for engineering single Cys mutants of p13II-Ub. These mutants were expressed and purified following the protocols developed for WT proteins (Supplementary Fig. 1) with WT-comparable yields.
We found that the expression level of soluble WT p13II-StrII construct was considerably less than that of p13II-Ub, but the use of Ni-affinity purification was possible since protein His8-tag is accessible and binds to the Ni-NTA agarose resin. Therefore, we studied the expression and purification of the potentially useful p13II-StrII construct. But due to a relatively low concentration of the targeted protein, Ni-affinity purification followed by SEC was insufficient for its complete separation from the impurities. Mitigating this issue, we subjected the Ni-affinity-purified p13II-StrII to cation-exchange purification step at pH 6.1, since at this pH p13II is highly positively charged. The theoretical pI of p13II-StrII is 11.71 as reported by the ExPASy ProtParam program. This step was followed by SEC, yielding highly pure p13II-StrII (Fig. 4 A) as confirmed by SDS-PAGE and WB (Fig. 4 B). The calculated molecular weight of p13II-StrII is 13,674.50 Da, and its molar extinction coefficient at 280 nm is 30,480 M−1 cm−1. The final yield of p13II-StrII was close to 20 μg per liter of culture. Thus, conservatively, about 200 μg of p13II-StrII can be produced from 10 L of E. coli culture, yielding ca. 0.146 mL of 100 μM protein concentration, which is quite a workable amount for both functional and structural studies since more sensitive experimental techniques, such as immunoblotting (27) or EPR spectroscopy (40, 48), are satisfied with smaller (ng to μg) amounts or concentrations in the low micromolar range.
Figure 4: Purification and detection of p13II-StrII by SEC, SDS-PAGE, and WB:
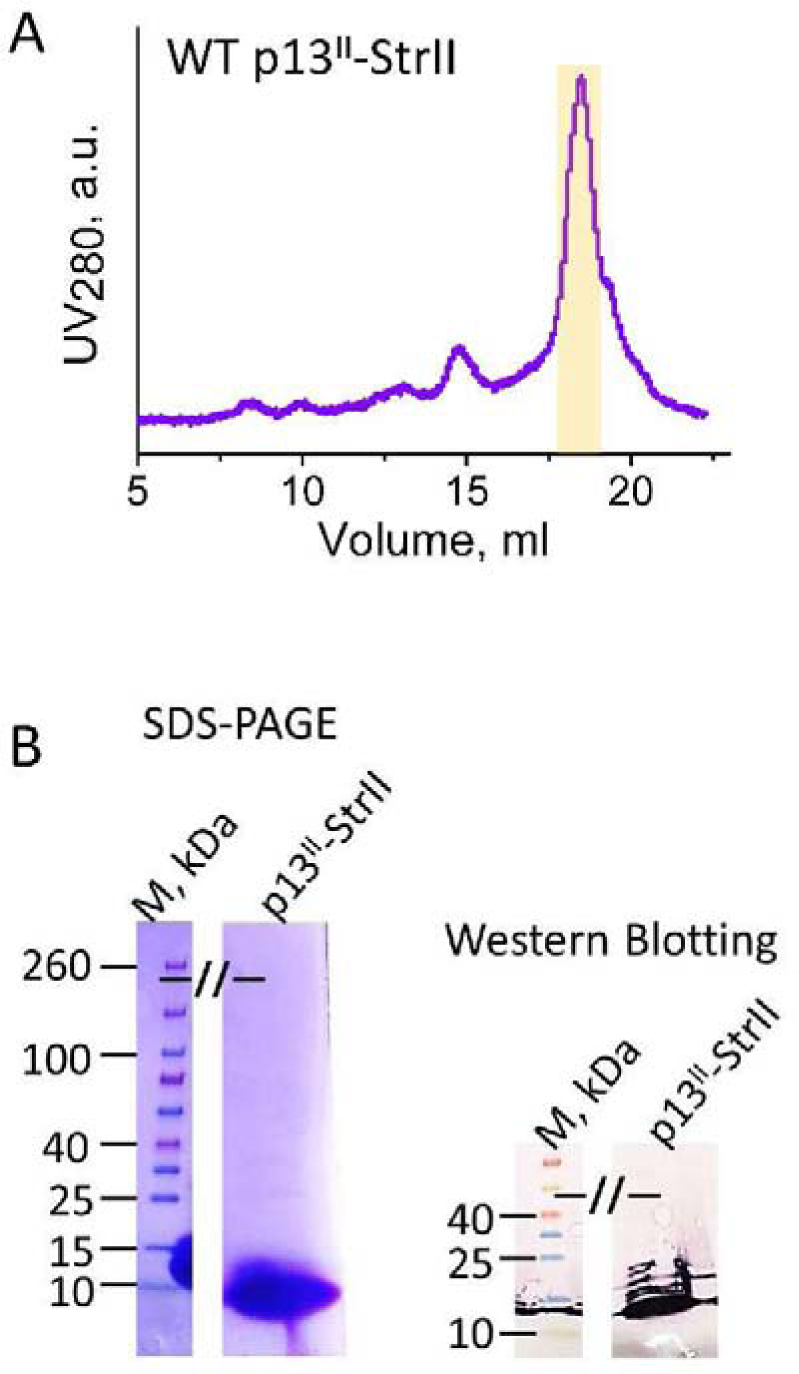
(A) The size exclusion chromatogram of p13II-SrtII, purified by Ni-affinity and cation exchange. The fractions corresponding to p13II-StrII main peak (highlighted in yellow) were collected. (B) SDS-PAGE (left) and WB (right) of fully-purified p13II-StrII. In these assays ~19.5 μg and ~6 μg protein was used for SDS-PAGE and WB, respectively.
The procedures for p13II-Ub, tag-free p13II, and p13II-SrtII expression and purification are illustrated in Figure 5.
Figure 5. Expression and purification scheme p13II variants:
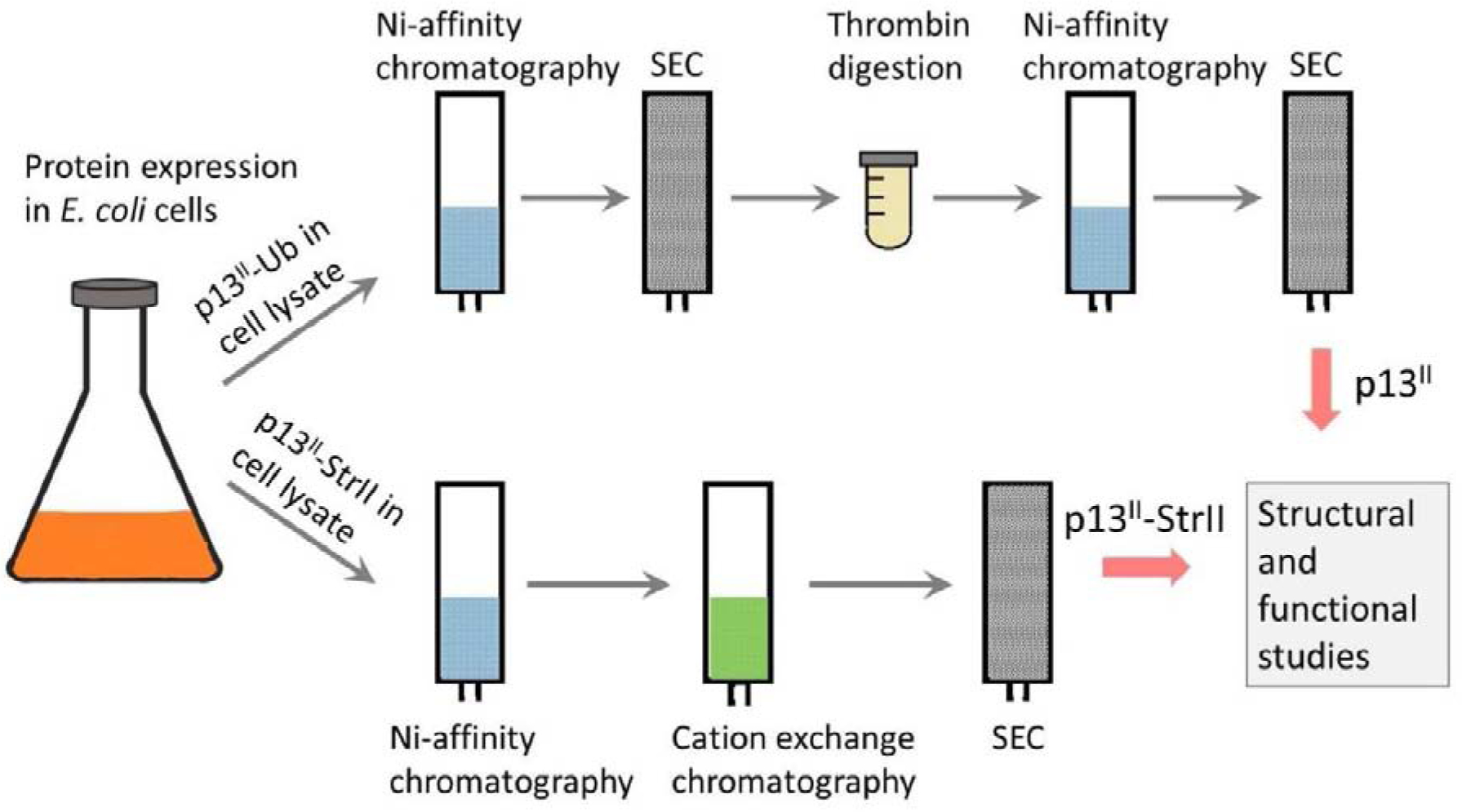
Both p13II-Ub and p13II-StrII were expressed in E. coli. p13II-Ub was purified by using Ni-affinity chromatography followed by SEC (upper route). The separation of p13II from Ub-His8 tag was achieved by thrombin digestion of p13II-Ub, binding the tag to Ni-NTA agarose, and SEC separation of the tag-free p13II from thrombin. p13II-StrII was purified using Ni-affinity chromatography, followed by cation-exchange chromatography and SEC (lower route).
CD spectroscopy of tag-free p13II in solution, LPPG, and liposomes
We recorded the far-UV CD spectra of the tag-free p13II in a buffer solution as well as in the presence of membrane mimetics that are LPPG, DOPC /POPG, and DOPC/POPS liposomes. The visual observation showed that a substantial part of the protein structure is helical, according to a well-defined minimum observed at 222 nm (Fig. 6). Interestingly, the protein association with LPPG led to a mild increase of protein helical content. We further subjected the experimental data to CD spectra deconvolution using the BeStSel software (36, 37). The results indicated that LPPG-bound p13II has the highest ~38% helical content (both regular and distorted α-helix), whereas in buffer solution it was ~30%, with the lowest value of 25–26% found for liposomes made of either DOPC/POPS or DOPC/POPG. A slight increase, from ca. 10% to ca. 16%, in the antiparallel β-sheet structure was observed upon transition from buffer to lipid environment. The turn content is almost equal in all p13II states, about 10%–13%. However, the large part of the protein (between 39% in LPPG to 49% in buffer) corresponds to so-called ‘other’ structure content, which includes 3,10-helix, π-helix, β-bridge, bend, loop etc. (36). Thus, the accuracy in assessing the p13II secondary structure based just on its CD spectra is expectedly limited.
Fig 6. CD spectra of tag-free FL p13II:
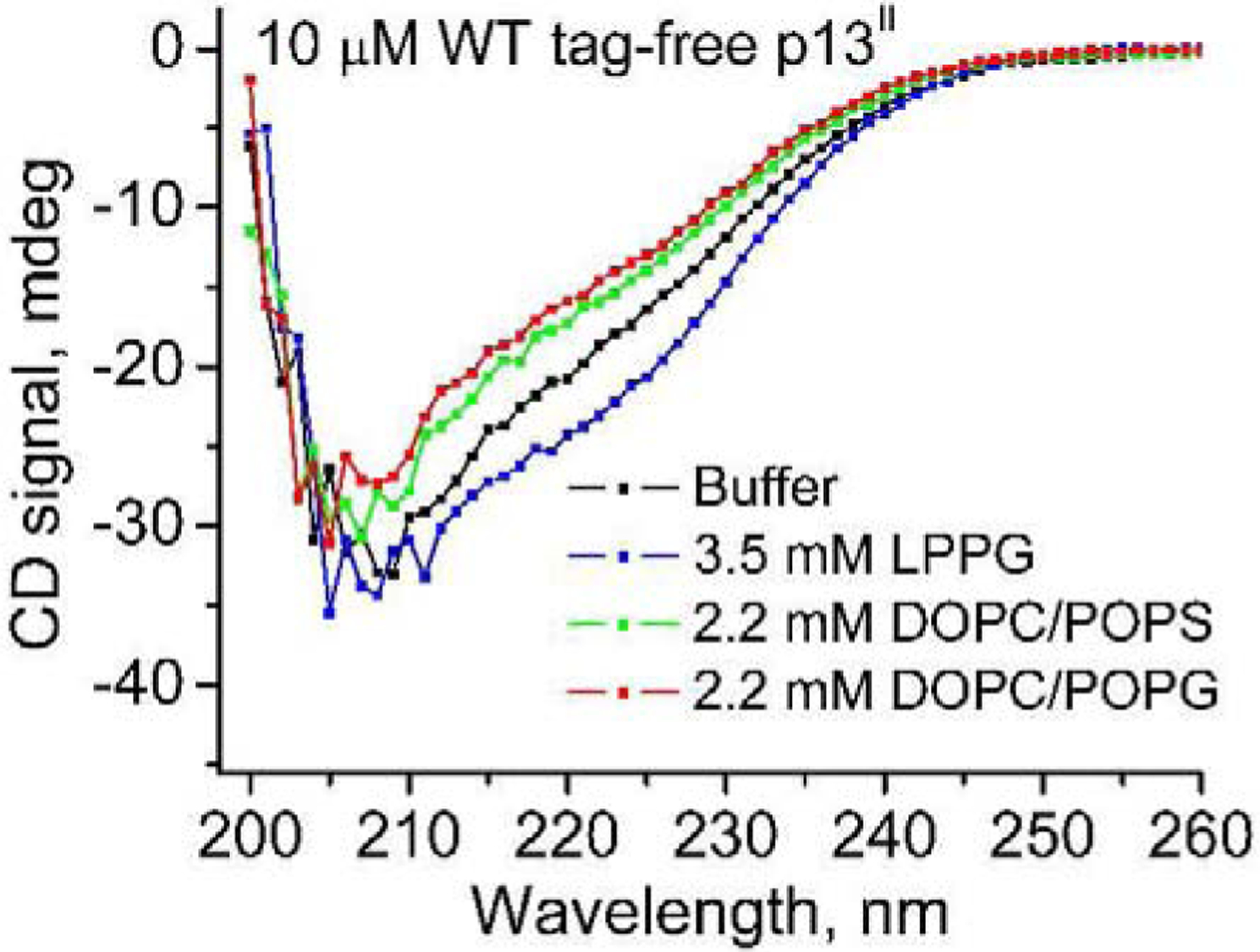
The data are shown for tag-free p13II in PBS buffer solution (black), in LPPG (blue), in DOPC/POPS membranes (green), and in DOPC/POPG membranes (red). Protein concentration was 10 μM in all cases. The concentration of LPPG was 3.5 mM and of total lipid 2.2 mM.
Spin labeling of p13II single Cys mutants and pulse EPR spectroscopy in LPPG- and lipid-bound states as well as in solution
We used Cys-specific spin labeling to introduce covalently bound paramagnetic reporting groups for two single Cys mutants, S47C and H63C, of p13II-Ub. The Cys residues of these mutants are located in the p13II C-terminal half of the protein, with S47C positioned close to the proposed TM domain and H63C located in the predicted soluble β-sheet hairpin region (Fig. 7 A). We selected these residues expecting them to be more amenable to spin labeling than the native C28 residue located within the hydrophobic part of the protein. Our initial attempts to label S47C and H63C mutants of p13II-Ub with a MTSL reagent in a buffer solution led to protein precipitation. This issue prompted us to use a more hydrophilic ISL spin label, with which we successfully labeled H63C, but the S47C residue might have been occluded and labeled poorly. However, we noticed that S47C residue can be spin-labeled with MTSL in the presence of LPPG. It is possible that the interaction of the p13II with LPPG stabilizes a protein conformation that is less sensitive to the presence of MTSL but also makes the labeling site of S47C more accessible to the reagent.
DEER spectroscopy (49, 50) was used to study the samples of the spin-labeled H63C mutant at three conditions: in solution, in LPPG micelles, and in lipid bilayers made of DOPC/DOPG. Also, the samples of the spin-labeled S47C mutant in the LPPG and lipid bilayers of DOPC/POPG were measured. For the singly spin-labeled mutant H63C in a buffer solution, we observed no DEER signal, telling that no spin pairs were present. Thus the protein is monomeric in solution. On the contrary, DEER signals indicative of spin coupling were recorded for both single-spin labeled H63C and S47C mutants in the LPPG-bound case, pointing to protein oligomerization taking place upon interaction with this membrane mimetic (Fig. 7 B and C). These types of DEER signals were observed in a 1:120–1:360 range of protein-to-LPPG molar ratios (P/LPPG) (Suppl. Fig. S2). These ratios were used to test whether the observed DEER signals might be contributed by unspecific protein pairing caused by statistical distribution of p13II in LPPG micelles if this scenario is possible. In such a case, the p13II aggregation number would likely be determined by the Poisson distribution of protein in LPPG micelles, thereby increasing the modulation depth (amplitude) of the DEER signal with the increase of the protein fraction. The micellar aggregation number of LPPG is ca. 125 (51), so the unspecific pairing at the highest P/LPPG could not be ruled out. However, the order of such unspecific protein aggregates would be substantially reduced with the decrease of P/LPPG and therefore, we would observe DEER signals with much reduced amplitude. However, the effect of P/LPPG on DEER signal was negligible, which renders the unspecific effects insignificant and is supportive of protein-specific oligomerization in a lipid-like environment. Also, in the case of residue S47C, a possible contribution to the observed distances due to a spin label trapping in the LPPG micelles was ruled out, since no DEER signal for the control sample of MTSL/LPPG without protein was observed (Suppl. Fig. S3).
From the DEER signals recorded for the p13II-LPPG samples, the inter-spin distances were reconstructed. The distance distributions showed substantial spread in all cases: For residue H63C, they ranged from ca. 20 Å to 60 Å (Fig. 7 B, middle panel, FWHM 30Å), whereas for S47C, they ranged from ca. 20 Å to 50 Å, exhibiting two maxima at ca. 25 Å and 38 Å (Fig. 7 C, right panel). This bimodal character could originate from two protein conformations, but could be also a result of spin label flexibility, although less likely. A contribution from oligomers with oligomeric orders higher than a dimer is also possible (40, 48). The recorded cw EPR spectrum of H63C in LPPG (Suppl. Fig. S4) also supports a bimodal conformation as two spectral components different in motional range (52) are clearly visible. However, further effort would be necessary to characterize the p13II oligomer in LPPG in sufficient detail.
Interestingly, based on the observed DEER signals for residue H63C, the p13II self-association was significantly reduced in non-charged lyso phospholipid LPPC (1-palmitoyl-2-hydroxy-sn-glycero-3-phosphocholine, 16:0 Lyso PC) compared to that in LPPG (Suppl. Fig. S3) emphasizing the role of lipid charge in binding and stabilizing the protein.
It should be noted that for p13II/H63C in LPPG, we obtained similar DEER data regardless of the presence of the Ub-His8 tag (Fig. 7 B). This indicates that the soluble fusion tag has no perceivable effect on p13II conformation in context of lipids.
In a similar measurement of the spin-labeled H63C mutant in DOPC/POPG lipids the DEER signal showed reduced amplitude and steeper slope than the DEER signal in LPPG (Fig. 7 B, right panel). The data indicated a change of p13II conformation to one that is more confined in the lipid-bound state compared to the LPPG-bound state. The steeper slope of a DEER signal is typical for membrane proteins due to increased local concentration of the solute (48). In addition, the spin-label phase memory relaxation time was shortened, apparently caused by methyl group rotations and acyl chain collective fluctuations, as expected for a lipid environment where spin-label moiety is near or in contact with the bilayer. (49, 53–57) For the residue S47C in DOPC/POPG lipid membranes, we observed a DEER signal with reduced amplitude and a less prominent distance distribution (Fig. 7 C, right panel), which confirms that the protein changes its conformation (or possibly oligomerization number) depending on the lipid environment.
Fluorescence-detected permeability of p13II-bound liposomes to Tl+
The current view is that p13II increases lipid membrane permeability to cations, particularly K+, and this effect is related to its function in IMM (24, 26, 30, 31). To study this idea, we developed an in vitro assay for detecting the effect of p13II on liposome permeability to thallium ions (Tl+) based on fluorescence quenching of ANTS dye encapsulated in liposomes (Fig. 8) by the ions. This type of activity assay has been used previously in studies of potassium channels (58). We tested liposomes prepared using two lipid compositions: DOPC/POPS and DOPC/POPG. However, the DOPC/POPG liposomes produced large background fluorescence, as they turned out to be significantly permeable to Tl+ even before the addition of p13II; therefore, they were not used any further to elucidate p13II activity. Contrariwise, protein-free DOPC/POPS liposomes afforded much lower Tl+ leakage and were selected to study p13II activity.
Figure 8. p13II fluorescence-based Tl+ uptake assay in liposomes.
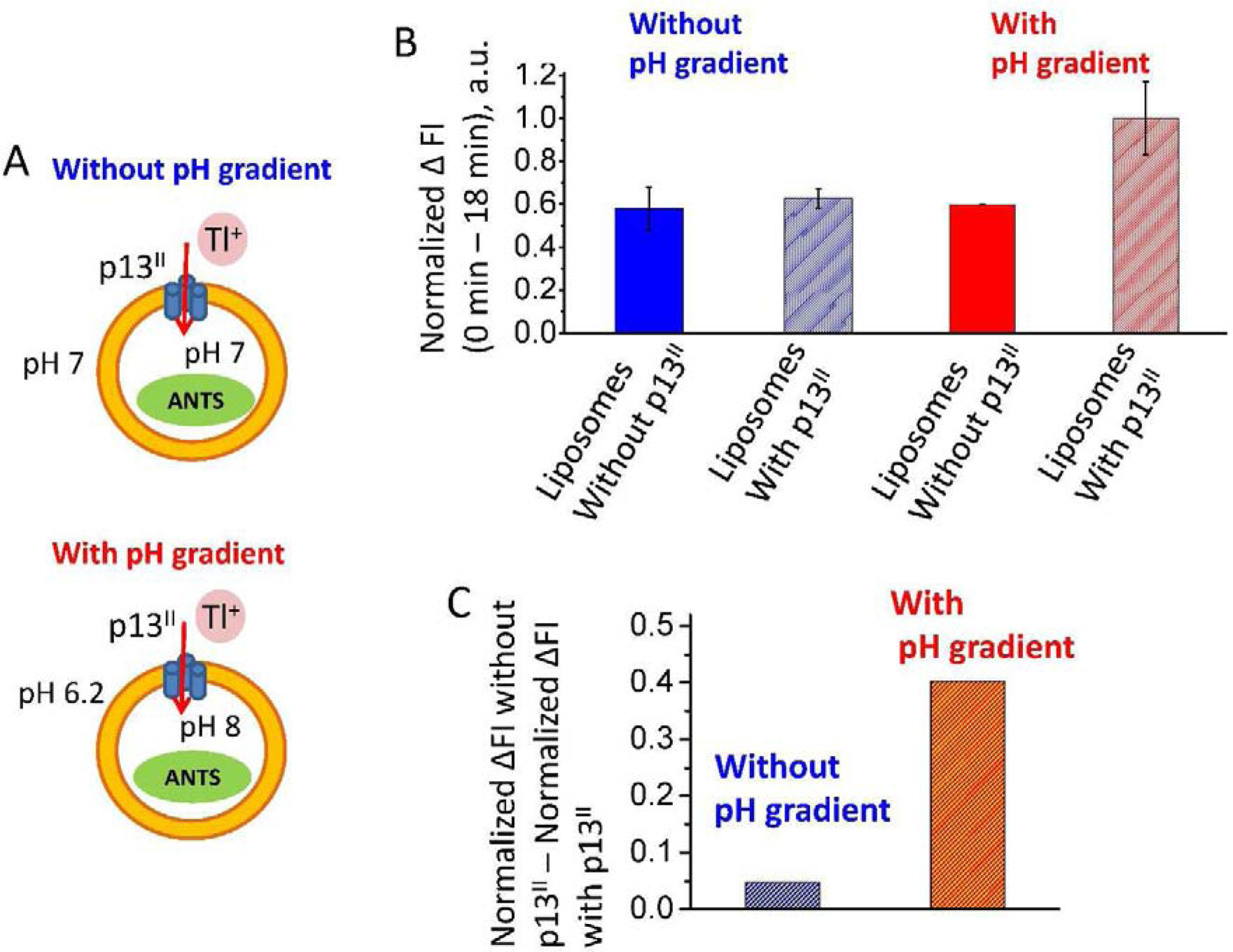
(A) Two types of Tl+ uptake experiments were carried out, namely with or without transmembrane pH gradient. After ANTS fluorescent dye encapsulation in liposomes, WT p13II was added to the liposome solution to give protein-to-lipid molar ratio of 1:605. The mixture was incubated for 1 h at RT to form proteoliposomes which were transferred into Tl+-containing bath buffer. Starting from this post-mixing state, the ANTS fluorescence quenching was measured to estimate the contribution caused by Tl+ influx. The same type of experiments was carried out on liposomes without protein to serve as a reference. (B) The plot shows the normalized drop in the fluorescence intensity (ΔFI) recorded at 18 min waiting time and right after (0 min) the transfer of liposomes to the bath buffer for the cases of protein-bound and free liposomes. The data with and without pH gradient are in red and blue, respectively. The bars corresponding to the presence of protein are rendered in pattern. (C) The difference between normalized ΔFI obtained with and without p13II is plotted in B for samples with and without pH gradient. Tl+ influx for protein-containing samples becomes prominent in the presence of transmembrane pH gradient.
We first studied ANTS fluorescence quenching experiments at pH7 without transmembrane pH gradient (Fig. 8 A, upper panel). In this case, p13II increased the Tl+ influx just marginally, as the quenching of ANTS fluorescence was only slightly greater than in protein-free liposomes (Fig. 8 B and C, and Suppl. Fig. S4). This necessitated a test of whether other factors, such as membrane potential, would increase the Tl+ influx, as suggested by studies of p13II transport of K+ in the IMM (26, 32). For this purpose, we introduced a transmembrane pH gradient with a ΔpH of −1.8 (Fig. 8 A, lower panel). This value of the ΔpH produces a membrane potential of ca. 108 mV since a ΔpH of 1.0 corresponds to −60 mV (59). Remarkably, under these conditions, the influx of Tl+ over the same waiting time of 18 min has increased by a factor of 4 to 6.5 for p13II-bound liposomes, as compared to free liposomes (Fig. 8 B and C, and Supplemental Fig. S6 and Supplemental Fig. S7), which suggests that the membrane potential is essential for p13II function.
Discussion
The p13II protein is essential for HTLV-1 survival and proliferation in the host (20, 26, 32, 60–62), which emphasizes this protein potential as a therapeutic target. Moreover, the methods of HTLV-1 detection in infected individuals are very limited, and even though HTLV-1 is an enveloped virus, it is transmitted predominantly through cell-to-cell contact rather than by the spread of mature virions (63). Such an intimate integration with the host cell makes it an elusive target, requiring identification of any weak spots that could be attacked. For instance, the HTLV-1 regulatory proteins, such as p13II, which are expressed and function in the host cell, could potentially serve as markers for HTLV-1 detection in infected individuals. Acquiring comprehensive knowledge about p13II structure and function at the molecular level would be of great value in this endeavor. However, to date, the strategies to produce highly pure p13II have been limited and employed the more expensive protein expression in mammalian cells or in E. coli, but as an N-terminal GST-tagged construct followed by GST affinity purification; the yields and purity of obtained protein were not described as well (35). The availability of efficient and highly robust protocols for the expression and purification of large quantities of p13II would significantly advance the detailed in vitro molecular studies.
Here, we developed such protocols and produced milligram-scale quantities of highly-pure soluble p13II proteins in E. coli, (Figs. 2–5). To do so, we engineered a fusion construct of p13II-Ub containing the aa sequence of a p13II protein and Ub+His8 tag at the p13II C-terminus. Careful consideration was given to making the fusion at the C-terminus, since the N-terminus of p13II is hydrophobic and could possibly be involved in an interaction with the lipid membrane. In previous studies, Ub has been employed for fusion—although at the N-terminus of the targeted protein—and the solubility of the fusion construct has increased significantly (45, 64). Utilizing other than Ub tag, such as GST (35) could potentially also improve protein solubility. However, the prospective usage of p13II fusion construct for cys labeling makes Ub more advantageous, as no additional cys residues need to be mutated out. Moreover, we improved the specificity of the Ni-affinity purification by introducing a His8 tag instead of the typical His6 tag, thereby enhancing the protein binding to the Ni-NTA agarose resin. Increasing the number of His residues in the affinity tag proved to be successful in other studies as well (65). Additionally, using the thrombin digestion site introduced into the p13II-Ub construct yielded the highly pure tag-free p13II in quantities sufficient for conducting extensive functional and structural studies.
As a matter of experimenting with fusion constructs, we also produced highly pure p13II-StrII (Fig. 2, 4, 5), although with a moderate yield. StrII is a short non-perturbing peptide selected from a random peptide library that tightly binds streptavidin and, generally, can be used as a purification tag (66). In this work, we did not need to employ StrII for affinity purification; however, this has potential for use in future studies. Our motivation, however, is in the possibility of utilizing StrII as a functional tag for interacting with streptavidin, which could be advantageous in studies based on the immobilization of p13II (67), for example, the immobilization of p13II-liposome systems for activity assays using high-resolution fluorescence microscopy (68). Such a possibility is a matter for future investigations.
We cumulatively applied several biochemical and biophysical techniques to characterize the p13II protein. We found that, under our experimental conditions, all three constructs, p13II-Ub, p13II-StrII, and tag-free p13II, are monomeric in solution, based on SEC (Fig. 3 and 4) and EPR spectroscopy. CD spectroscopy suggests that the tag-free p13II is highly folded in solution, showing substantial content of alpha-helical and β-sheet structures (Fig. 6). Binding of the protein to lipid membrane mimetics-such as LPPG and liposomes–affected its secondary structure to some extent (Fig. 6): LPPG increased helical content, but in liposomes the effect was rather opposite; Both LPPG and liposomes increased the antiparallel β-sheet structure. It has been shown previously, that a peptide containing the proposed transmembrane segment of p13II folds into a helical structure when dissolved in lipid membranes (26). Therefore, it could be expected that this protein region contributes to p13II helical content. However, mapping the secondary structure changes throughout the entire p13II polypeptide based just on the available CD spectra is not feasible. Higher resolution structures for protein in solution and LPPG/lipid are needed. Our CD data indicate no major qualitative alterations in p13II secondary structure upon interaction with lipid membranes, since all structural elements are preserved. This might suggest that the structural transitions from solution to the lipid-bound state affect mostly the protein tertiary and quaternary structure, as detected by DEER spectroscopy and sensed indirectly through the spin-labeling procedures.
Since it was proposed that p13II functions in IMM as an oligomer, we tested this hypothesis with DEER measurements of spin-labeled proteins utilizing two selected single Cys substitutions, S47C and H63C. DEER was carried out on proteins reconstituted into micelles of lyso-lipid with charged headgroups (LPPG) and in lipid bilayers made of mixture of lipids with and without charged headgroups (Fig. 7). DEER is a well-established, robust technique for studying protein oligomerization, particularly in lipid membranes (40, 48, 53, 69, 70). The acquired DEER data evidenced that p13II does associate with the lipid membrane mimetics and the protein oligomerizes in a lipid environment when charged lipids are present. However, the results suggest that the p13II conformation is affected by the type of lipid environment, as the DEER signals for both labeled sites, S47C and H63C, are distinct between the cases of LPPG and DOPC/POPG liposomes (Fig. 7). Moreover, in LPPG, we recorded a more pronounced DEER signal and better resolved inter-spin distance for the H63C residue, which is located in the soluble C-terminal domain, indicating increased conformational stability in this region when a protein is bound to this membrane mimetic. This result is also buttressed by the increased folding of WT p13II as well as slower p13II and nitroxide label mobility in LPPG, based respectively on CD (Fig. 6) and cw EPR (Suppl. Fig. S4). The reason for this observation could be in strong electrostatic interactions between the positively charged residues in p13II (including those in close proximity to H63C) with LPPG acidic headgroups, as previously hypothesized for negatively charged lipids and found important for the proper function of p13II in K+ transport across IMM (26, 30). This assumption, along with the DEER results for p13II in LPPC environment (Suppl. Fig. S3), was also the reason for us to use highly charged membrane mimetics in our study. The difficulty of recording a well-defined DEER signal for the spin-labeled H63C mutant in the lipid bilayer could be because the residue 63 is in the protein C-terminal soluble region; when p13II’s oligomer resides in the membrane bilayer, its C-terminus possibly is dynamic and conformationally heterogeneous, resulting in a multicomponent smeared DEER signal for the H63C residue. This is also supported by CD and cw EPR, which suggest that in general p13II is slightly less folded and the region of residue 63 remains highly dynamic in lipid environment. However, more investigation would be necessary to shed light on how p13II associates with the membrane and to elucidate the probable multiple protein conformations.
Furthermore, we conducted an in vitro fluorescence-based assay for testing the functionality of liposome-bound p13II. We found that p13II increases the permeability to Tl+ (K+) of membranes made of DOPC/POPS lipids. However, to amplify this effect, a pH gradient was necessary to produce the transmembrane potential, which is believed to activate the channel. This result is in line with previous studies on synthetic FL p13II constructs in isolated mitochondria (26, 32). Certainly, in IMM, p13II functions under the condition of a high electrochemical gradient, which depends on both the transmembrane potential and ΔpH (59, 71). Thus, increasing the concentration of positively charged ions outside of liposomes mimics, to a certain extent, the native mitochondrial conditions and expectedly facilitates the translocation of Tl+ across the bilayer due to p13II function. Importantly, our observations suggest that p13II is capable of increasing the membrane leakage. However, one would reasonably expect that in vivo the complex IMM environment could bring into play several other factors affecting protein function, such as protein-protein interactions, unidentified effectors, or association with specific mitochondrial lipids, such as cardiolipin. Further investigations of membrane compositions mimicking those of IMM and of membrane permeability to other cations, such as Ca2+ and Na+, would be necessary to determine the lipid and cation specificity of p13II.
In conclusion, we successfully expressed in E. coli and isolated highly-pure soluble FL p13II in milligram quantities. The approaches, which we developed, present a significant advancement in obtaining a difficult to produce protein and can be applied to other proteins as well. The p13II, which we produced, is functional when reconstituted into lipid membranes, according to DEER spectroscopy measurements of conformational changes taking place in the transition from a solution to a membrane-bound state and in a liposome-based assay of Tl+ uptake. The significant progress made by this work provides a solid base for us to further advance to more specific in vitro functional and structural investigations.
Supplementary Material
HIGHLIGHTS.
The p13II regulatory protein from human T-cell leukemia virus type 1 was heterologously expressed in E. coli in large quantities on a milligram scale.
Custom p13II fusion constructs and protocols to extract highly-pure protein were developed.
Purified p13II is monomeric in solution, but oligomerizes upon interaction with membrane mimetics containing charged lipids.
The developed in vitro fluorescence-based activity assay suggests that p13II increases liposome permeability to Tl+ in the presence of proton gradient (transmembrane potential).
Our advances will facilitate further studies of p13II, which is a potential druggable target.
Acknowledgements
This work was supported by grants from the National Institutes of Health R01GM123779 and P41GM103521. CF salary was supported by grant R03 AI137735. Fluorescence spectroscopy was conducted at the Imaging Facility of Cornell University Institute of Biotechnology. We thank Carol Bayles for helping us with setting up the fluorescence experiment and fluorimeter. We thank Michael Lynch for assistance with CD spectrometer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare no competing financial interest.
REFERENCES
- 1.Yoshida M (2005) Discovery of HTLV-1, the first human retrovirus, its unique regulatory mechanisms, and insights into pathogenesis, Oncogene 24, 5931–5937. [DOI] [PubMed] [Google Scholar]
- 2.Gallo RC (2005) The discovery of the first human retrovirus: HTLV-1 and HTLV-2, Retrovirology 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miyoshi I, Kubonishi I, Yoshimoto S, Akagi T, Ohtsuki Y, Shiraishi Y, Nagata K, and Hinuma Y (1981) Type C virus particles in a cord T-cell line derived by co-cultivating normal human cord leukocytes and human leukaemic T cells, Nature 294, 770–771. [DOI] [PubMed] [Google Scholar]
- 4.Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, and Gallo RC (1980) Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma, Proc Natl Acad Sci U S A 77, 7415–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verdonck K, Gonzalez E, Van Dooren S, Vandamme AM, Vanham G, and Gotuzzo E (2007) Human T-lymphotropic virus 1: recent knowledge about an ancient infection, Lancet Infect Dis 7, 266–281. [DOI] [PubMed] [Google Scholar]
- 6.Barmak K, Harhaj E, Grant C, Alefantis T, and Wigdahl B (2003) Human T cell leukemia virus type I-induced disease: pathways to cancer and neurodegeneration, Virology 308, 1–12. [DOI] [PubMed] [Google Scholar]
- 7.Mortreux F, Gabet AS, and Wattel E (2003) Molecular and cellular aspects of HTLV-1 associated leukemogenesis in vivo, Leukemia 17, 26–38. [DOI] [PubMed] [Google Scholar]
- 8.Troisgros O, Barnay JL, Darbon-Naghibzadeh F, Olive P, and Rene-Corail P (2016) Retrospective clinic and urodynamic study in the neurogenic bladder dysfunction caused by human T cell lymphotrophic virus type 1 associated myelopathy/tropical spastic paraparesis (HAM/TSP), Neurourol Urodyn. [DOI] [PubMed] [Google Scholar]
- 9.Ratner L (2004) Adult T cell leukemia lymphoma, Front Biosci 9, 2852–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Godoy AJ, Kira J, Hasuo K, and Goto I (1995) Characterization of cerebral white matter lesions of HTLV-I-associated myelopathy/tropical spastic paraparesis in comparison with multiple sclerosis and collagen-vasculitis: a semiquantitative MRI study, J Neurol Sci 133, 102–111. [DOI] [PubMed] [Google Scholar]
- 11.Levin MC, and Jacobson S (1997) HTLV-I associated myelopathy/tropical spastic paraparesis (HAM/TSP): a chronic progressive neurologic disease associated with immunologically mediated damage to the central nervous system, J Neurovirol 3, 126–140. [DOI] [PubMed] [Google Scholar]
- 12.Taylor GP, and Matsuoka M (2005) Natural history of adult T-cell leukemia/lymphoma and approaches to therapy, Oncogene 24, 6047–6057. [DOI] [PubMed] [Google Scholar]
- 13.Cruickshank JK, Rudge P, Dalgleish AG, Newton M, McLean BN, Barnard RO, Kendall BE, and Miller DH (1989) Tropical spastic paraparesis and human T cell lymphotropic virus type 1 in the United Kingdom, Brain 112 (Pt 4), 1057–1090. [DOI] [PubMed] [Google Scholar]
- 14.Edlich RF, Arnette JA, and Williams FM (2000) Global epidemic of human T-cell lymphotropic virus type-I (HTLV-I), J Emerg Med 18, 109–119. [DOI] [PubMed] [Google Scholar]
- 15.Willems L, Hasegawa H, Accolla R, Bangham C, Bazarbachi A, Bertazzoni U, Carneiro-Proietti AB, Cheng H, Chieco-Bianchi L, Ciminale V, Coelho-Dos-Reis J, Esparza J, Gallo RC, Gessain A, Gotuzzo E, Hall W, Harford J, Hermine O, Jacobson S, Macchi B, Macpherson C, Mahieux R, Matsuoka M, Murphy E, Peloponese JM, Simon V, Tagaya Y, Taylor GP, Watanabe T, and Yamano Y (2017) Reducing the global burden of HTLV-1 infection: An agenda for research and action, Antiviral Res 137, 41–48. [DOI] [PubMed] [Google Scholar]
- 16.Gessain A, and Cassar O (2012) Epidemiological Aspects and World Distribution of HTLV-1 Infection, Front Microbiol 3, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Proietti FA, Carneiro-Proietti AB, Catalan-Soares BC, and Murphy EL (2005) Global epidemiology of HTLV-I infection and associated diseases, Oncogene 24, 6058–6068. [DOI] [PubMed] [Google Scholar]
- 18.Uchiyama T (1997) Human T cell leukemia virus type I (HTLV-I) and human diseases, Annu Rev Immunol 15, 15–37. [DOI] [PubMed] [Google Scholar]
- 19.Alves C, and Dourado L (2010) Endocrine and metabolic disorders in HTLV-1 infected patients, Braz J Infect Dis 14, 613–620. [PubMed] [Google Scholar]
- 20.Georgieva ER (2018) Non-Structural Proteins from Human T-cell Leukemia Virus Type 1 in Cellular Membranes-Mechanisms for Viral Survivability and Proliferation, Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bai XT, and Nicot C (2012) Overview on HTLV-1 p12, p8, p30, p13: accomplices in persistent infection and viral pathogenesis, Front Microbiol 3, 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo YL, and Giam CZ (2006) Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence, EMBO J 25, 1741–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sinha-Datta U, Datta A, Ghorbel S, Dodon MD, and Nicot C (2007) Human T-cell lymphotrophic virus type I rex and p30 interactions govern the switch between virus latency and replication, J Biol Chem 282, 14608–14615. [DOI] [PubMed] [Google Scholar]
- 24.Silic-Benussi M, Biasiotto R, Andresen V, Franchini G, D’Agostino DM, and Ciminale V (2010) HTLV-1 p13, a small protein with a busy agenda, Mol Aspects Med 31, 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Biasiotto R, Aguiari P, Rizzuto R, Pinton P, D’Agostino DM, and Ciminale V (2010) The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake, Biochim Biophys Acta 1797, 945–951. [DOI] [PubMed] [Google Scholar]
- 26.D’Agostino DM, Ranzato L, Arrigoni G, Cavallari I, Belleudi F, Torrisi MR, Silic-Benussi M, Ferro T, Petronilli V, Marin O, Chieco-Bianchi L, Bernardi P, and Ciminale V (2002) Mitochondrial alterations induced by the p13II protein of human T-cell leukemia virus type 1. Critical role of arginine residues, J Biol Chem 277, 34424–34433. [DOI] [PubMed] [Google Scholar]
- 27.Tibaldi E, Venerando A, Zonta F, Bidoia C, Magrin E, Marin O, Toninello A, Bordin L, Martini V, Pagano MA, and Brunati AM (2011) Interaction between the SH3 domain of Src family kinases and the proline-rich motif of HTLV-1 p13: a novel mechanism underlying delivery of Src family kinases to mitochondria, Biochem J 439, 505–516. [DOI] [PubMed] [Google Scholar]
- 28.Koralnik IJ, Fullen J, and Franchini G (1993) The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments, J Virol 67, 2360–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andresen V, Pise-Masison CA, Sinha-Datta U, Bellon M, Valeri V, Washington Parks R, Cecchinato V, Fukumoto R, Nicot C, and Franchini G (2011) Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles, Blood 118, 1549–1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Silic-Benussi M, Cannizzaro E, Venerando A, Cavallari I, Petronilli V, La Rocca N, Marin O, Chieco-Bianchi L, Di Lisa F, D’Agostino DM, Bernardi P, and Ciminale V (2009) Modulation of mitochondrial K(+) permeability and reactive oxygen species production by the p13 protein of human T-cell leukemia virus type 1, Biochim Biophys Acta 1787, 947–954. [DOI] [PubMed] [Google Scholar]
- 31.Silic-Benussi M, Marin O, Biasiotto R, D’Agostino DM, and Ciminale V (2010) Effects of human T-cell leukemia virus type 1 (HTLV-1) p13 on mitochondrial K+ permeability: A new member of the viroporin family?, FEBS Lett 584, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.D’Agostino DM, Silic-Benussi M, Hiraragi H, Lairmore MD, and Ciminale V (2005) The human T-cell leukemia virus type 1 p13II protein: effects on mitochondrial function and cell growth, Cell Death Differ 12 Suppl 1, 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Silic-Benussi M, Cavallari I, Vajente N, Vidali S, Chieco-Bianchi L, Di Lisa F, Saggioro D, D’Agostino DM, and Ciminale V (2010) Redox regulation of T-cell turnover by the p13 protein of human T-cell leukemia virus type 1: distinct effects in primary versus transformed cells, Blood 116, 54–62. [DOI] [PubMed] [Google Scholar]
- 34.Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, and Kroemer G (2004) Viral proteins targeting mitochondria: controlling cell death, Biochim Biophys Acta 1659, 178–189. [DOI] [PubMed] [Google Scholar]
- 35.Ciminale V, Zotti L, D’Agostino DM, Ferro T, Casareto L, Franchini G, Bernardi P, and Chieco-Bianchi L (1999) Mitochondrial targeting of the p13II protein coded by the x-II ORF of human T-cell leukemia/lymphotropic virus type I (HTLV-I), Oncogene 18, 4505–4514. [DOI] [PubMed] [Google Scholar]
- 36.Micsonai A, Wien F, Kernya L, Lee YH, Goto Y, Refregiers M, and Kardos J (2015) Accurate secondary structure prediction and fold recognition for circular dichroism spectroscopy, Proc Natl Acad Sci U S A 112, E3095–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Micsonai A, Wien F, Bulyaki E, Kun J, Moussong E, Lee YH, Goto Y, Refregiers M, and Kardos J (2018) BeStSel: a web server for accurate protein secondary structure prediction and fold recognition from the circular dichroism spectra, Nucleic Acids Res 46, W315–W322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Borbat PP, Crepeau RH, and Freed JH (1997) Multifrequency two-dimensional Fourier transform ESR: an X/Ku-band spectrometer, J Magn Reson 127, 155–167. [DOI] [PubMed] [Google Scholar]
- 39.Pannier M, Veit S, Godt A, Jeschke G, and Spiess HW (2000) Dead-time free measurement of dipole-dipole interactions between electron spins, J Magn Reson 142, 331–340. [DOI] [PubMed] [Google Scholar]
- 40.Georgieva ER, Borbat PP, Norman HD, and Freed JH (2015) Mechanism of influenza A M2 transmembrane domain assembly in lipid membranes, Sci Rep 5, 11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chiang YW, Borbat PP, and Freed JH (2005) The determination of pair distance distributions by pulsed ESR using Tikhonov regularization, J Magn Reson 172, 279–295. [DOI] [PubMed] [Google Scholar]
- 42.Srivastava M, and Freed JH (2017) Singular Value Decomposition Method to Determine Distance Distributions in Pulsed Dipolar Electron Spin Resonance, Journal of Physical Chemistry Letters 8, 5648–5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Srivastava M, Georgieva ER, and Freed JH (2017) A New Wavelet Denoising Method for Experimental Time-Domain Signals: Pulsed Dipolar Electron Spin Resonance, J Phys Chem A 121, 2452–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bornhorst JA, and Falke JJ (2000) Purification of proteins using polyhistidine affinity tags, Methods Enzymol 326, 245–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baker RT, Smith SA, Marano R, McKee J, and Board PG (1994) Protein expression using cotranslational fusion and cleavage of ubiquitin. Mutagenesis of the glutathione-binding site of human Pi class glutathione S-transferase, J Biol Chem 269, 25381–25386. [PubMed] [Google Scholar]
- 46.Schmidt TG, and Skerra A (1994) One-step affinity purification of bacterially produced proteins by means of the “Strep tag” and immobilized recombinant core streptavidin, J Chromatogr A 676, 337–345. [DOI] [PubMed] [Google Scholar]
- 47.Vassylyeva MN, Klyuyev S, Vassylyev AD, Wesson H, Zhang Z, Renfrow MB, Wang H, Higgins NP, Chow LT, and Vassylyev DG (2017) Efficient, ultra-high-affinity chromatography in a one-step purification of complex proteins, Proc Natl Acad Sci U S A 114, E5138–E5147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Georgieva ER, Borbat PP, Grushin K, Stoilova-McPhie S, Kulkarni NJ, Liang Z, and Freed JH (2016) Conformational Response of Influenza A M2 Transmembrane Domain to Amantadine Drug Binding at Low pH (pH 5.5), Front Physiol 7, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Borbat PP, and Freed JH (2007) Measuring distances by pulsed dipolar ESR spectroscopy: spin-labeled histidine kinases, Methods Enzymol 423, 52–116. [DOI] [PubMed] [Google Scholar]
- 50.Jeschke G (2012) DEER distance measurements on proteins, Annu Rev Phys Chem 63, 419–446. [DOI] [PubMed] [Google Scholar]
- 51.Chou JJ, Baber JL, and Bax A (2004) Characterization of phospholipid mixed micelles by translational diffusion, J Biomol NMR 29, 299–308. [DOI] [PubMed] [Google Scholar]
- 52.Hubbell WL, Cafiso DS, and Altenbach C (2000) Identifying conformational changes with site-directed spin labeling, Nat Struct Biol 7, 735–739. [DOI] [PubMed] [Google Scholar]
- 53.Georgieva ER, Borbat PP, Ginter C, Freed JH, and Boudker O (2013) Conformational ensemble of the sodium-coupled aspartate transporter, Nat Struct Mol Biol 20, 215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lindgren M, Eaton GR, Eaton SS, Jonsson BH, Hammarstrom P, Svensson M, and Carlsson U (1997) Electron spin echo decay as a probe of aminoxyl environment in spin-labeled mutants of human carbonic anhydrase II, J. Chem. Soc.-Perkin Trans 2, 2549–2554. [Google Scholar]
- 55.Zecevic A, Eaton GR, Eaton SS, and Lindgren M (1998) Dephasing of electron spin echoes for nitroxyl radicals in glassy solvents by non-methyl and methyl protons, Mol. Phys 95, 1255–1263. [Google Scholar]
- 56.Fresch B, Frezzato D, Moro GJ, Kothe G, and Freed JH (2006) Collective fluctuations in ordered fluids investigated by two-dimensional electron-electron double resonance spectroscopy, J. Phys. Chem. B 110, 24238–24254. [DOI] [PubMed] [Google Scholar]
- 57.Freed JH (1992) CRITICAL FLUCTUATIONS AND MOLECULAR-DYNAMICS AT LIQUID-CRYSTALLINE PHASE-TRANSITIONS .1. THEORETICAL ASPECTS OF THE NEMATIC-SMECTIC-A TRANSITION, J. Chem. Phys 96, 3901–3911. [Google Scholar]
- 58.Posson DJ, Rusinova R, Andersen OS, and Nimigean CM (2018) Stopped-Flow Fluorometric Ion Flux Assay for Ligand-Gated Ion Channel Studies, Methods Mol Biol 1684, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, and Walter P (2008) Molecular Biology of the Cell, https://www.ncbi.nlm.nih.gov/books/NBK26894/ ed., New York. [Google Scholar]
- 60.Hiraragi H, Kim SJ, Phipps AJ, Silic-Benussi M, Ciminale V, Ratner L, Green PL, and Lairmore MD (2006) Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13(II) is required for viral infectivity in vivo, J Virol 80, 3469–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiraragi H, Michael B, Nair A, Silic-Benussi M, Ciminale V, and Lairmore M (2005) Human T-lymphotropic virus type 1 mitochondrion-localizing protein p13II sensitizes Jurkat T cells to Ras-mediated apoptosis, J Virol 79, 9449–9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Silic-Benussi M, Cavallari I, Zorzan T, Rossi E, Hiraragi H, Rosato A, Horie K, Saggioro D, Lairmore MD, Willems L, Chieco-Bianchi L, D’Agostino DM, and Ciminale V (2004) Suppression of tumor growth and cell proliferation by p13II, a mitochondrial protein of human T cell leukemia virus type 1, Proc Natl Acad Sci U S A 101, 6629–6634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, and Bangham CR (2003) Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton, Science 299, 1713–1716. [DOI] [PubMed] [Google Scholar]
- 64.Kasi D, Nah HJ, Catherine C, Kim ES, Han K, Ha JC, and Kim DM (2017) Enhanced Production of Soluble Recombinant Proteins With an In Situ-Removable Fusion Partner in a Cell-Free Synthesis System, Biotechnol J 12. [DOI] [PubMed] [Google Scholar]
- 65.Molbaek K, Scharff-Poulsen P, Helix-Nielsen C, Klaerke DA, and Pedersen PA (2015) High yield purification of full-length functional hERG K+ channels produced in Saccharomyces cerevisiae, Microb Cell Fact 14, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt TG, and Skerra A (1993) The random peptide library-assisted engineering of a C-terminal affinity peptide, useful for the detection and purification of a functional Ig Fv fragment, Protein Eng 6, 109–122. [DOI] [PubMed] [Google Scholar]
- 67.Johar SS, and Talbert JN (2017) Strep-tag II fusion technology for the modification and immobilization of lipase B from Candida antarctica (CALB), J Genet Eng Biotechnol 15, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wazawa T, and Ueda M (2005) Total internal reflection fluorescence microscopy in single molecule nanobioscience, Adv Biochem Eng Biotechnol 95, 77–106. [DOI] [PubMed] [Google Scholar]
- 69.Georgieva ER (2017) Nanoscale lipid membrane mimetics in spin-labeling and electron paramagnetic resonance spectroscopy studies of protein structure and function, Nanotechnol Rev 6, 75–92. [Google Scholar]
- 70.Milikisiyants S, Wang S, Munro RA, Donohue M, Ward ME, Bolton D, Brown LS, Smirnova TI, Ladizhansky V, and Smirnov AI (2017) Oligomeric Structure of Anabaena Sensory Rhodopsin in a Lipid Bilayer Environment by Combining Solid-State NMR and Long-range DEER Constraints, J Mol Biol. [DOI] [PubMed] [Google Scholar]
- 71.Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, and Zorov DB (2018) Mitochondrial membrane potential, Anal Biochem 552, 50–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.