

Abstract
Ubiquitin/ISG15‐conjugating enzyme E2L6 (UBE2L6) is a critical enzyme in ISGylation, a post‐translational protein modification that conjugates the ubiquitin‐like modifier, interferon‐stimulated gene 15 (ISG15), to target substrates. Previous gene expression studies in acute promyelocytic leukemia (APL) cells showed that all‐trans‐retinoic acid (ATRA) altered the expression of many genes, including UBE2L6 (200‐fold) and other members of the ISGylation pathway. Through gene expression analyses in a cohort of 98 acute myeloid leukemia (AML) patient samples and in primary neutrophils from healthy donors, we found that UBE2L6 gene expression is reduced in primary AML cells compared with normal mature granulocytes. To assess whether UBE2L6 expression is important for leukemic cell differentiation—two cell line models were employed: the human APL cell line NB4 and its ATRA‐resistant NB4R counterpart, as well as the ATRA‐sensitive human AML HL60 cells along with their ATRA‐resistant subclone—HL60R. ATRA strongly induced UBE2L6 in NB4 APL cells and in ATRA‐sensitive HL60 AML cells, but not in the ATRA‐resistant NB4R and HL60R cells. Furthermore, short hairpin (sh)RNA‐mediated UBE2L6 depletion in NB4 cells impeded ATRA‐mediated differentiation, suggesting a functional role for UBE2L6 in leukemic cell differentiation. In addition, ATRA induced ISG15 gene expression in NB4 APL cells, leading to increased levels of both free ISG15 protein and ISG15 conjugates. UBE2L6 depletion attenuated ATRA‐induced ISG15 conjugation. Knockdown of ISG15 in NB4 APL cells inhibited ISGylation and also attenuated ATRA‐induced differentiation. In summary, we demonstrate the functional importance of UBE2L6 in ATRA‐induced neutrophil differentiation of APL cells and propose that this may be mediated by its catalytic role in ISGylation.
Keywords: AML, APL, ATRA, differentiation, ISG15, UBE2L6
Knockdown of UBE2L6 or ISG15 in NB4 APL cells inhibits ISGylation of protein targets and attenuates ATRA‐induced differentiation.
Abbreviations
- AML
acute myeloid leukemia
- APL
acute promyelocytic leukemia
- ATRA
all‐trans‐retinoic acid
- CML
chronic myeloid leukemia
- HERC5
HECT and RLD domain containing E3 ubiquitin protein ligase 5
- ISG15
interferon‐stimulated gene 15
- NBT
nitro blue tetrazolium
- RARα
retinoic acid receptor alpha
- UBE1L
ubiquitin‐like modifier‐activating enzyme 7
- UBE2L6
ubiquitin/ISG15-conjugating enzyme E2L6
- UBLs
ubiquitin‐like modifiers
- USP18
ubiquitin‐specific peptidase 18
1. Introduction
Acute myeloid leukemia (AML) is a clonal disorder characterized by the accumulation of immature hematopoietic precursors in the bone marrow and peripheral circulation (Caceres‐Cortes, 2013). Overall survival is poor, particularly for patients over 60 years of age, who have an overall 5‐year survival of ~ 10% (Kantarjian and O'Brien, 2010; Thein et al., 2013). Improved therapeutic strategies with tolerable toxicity profiles are needed.
Acute promyelocytic leukemia (APL) is a clinically, pathologically, and molecularly distinct subtype of AML (Tallman and Altman, 2009). It is distinguished in 95% of cases by a translocation of chromosomes 15 and 17, which leads to the expression of a fusion oncoprotein PML‐retinoic acid receptor alpha (RARα) (Tallman and Altman, 2009). This protein disrupts functional retinoid signaling in APL cells, repressing gene transcription and halting myeloid maturation at the promyelocyte stage (Tang and Gudas, 2011). Therapeutic doses of all‐trans‐retinoic acid (ATRA) reactivate gene transcription and overcome this differentiation block allowing clinical remission (Tang and Gudas, 2011). As we have previously reviewed, ATRA also encourages the degradation of the PML‐RARα protein through cooperating pathways including proteasomal degradation and autophagy (Orfali et al., 2014).
Cellular protein activity and stability is regulated by post‐translational modification (Krishna and Wold, 1993). One such modification is ‘ubiquitination’, the reversible addition of ubiquitin protein (8.5kDa) to the lysine residues of target substrates (Komander and Rape, 2012). This is catalyzed by a series of enzymes: (a) Ubiquitin‐activating enzymes (E1) use ATP to convert ubiquitin to a high‐energy thioester; (b) ubiquitin‐conjugating enzymes (E2) bind active ubiquitin on their cysteine residues; and (c) ubiquitin‐ligase enzymes (E3) interact with E2 enzymes and catalyze the formation of a covalent bond between ubiquitin and its target substrate. E3 ligases regulate substrate specificity. Ubiquitin can also be removed from target proteins through the action of deubiquitinases (Friend et al., 2014). Ubiquitin‐like modifiers (UBLs) are proteins that share significant structural and some sequence homology with ubiquitin and modify substrates in a similar enzyme‐controlled fashion (Hochstrasser, 2009). While protein ubiquitination is known to alter protein activity or target substrates for degradation by the 26S proteasome, the functional roles of UBL modifications are less well‐defined and remain under investigation (Hochstrasser, 2009). We now recognize at least ten UBLs, including small ubiquitin‐like modifier, autophagy‐related proteins 8 and 12 (autophagy‐related protein 8, autophagy‐related protein 12), and interferon‐stimulated gene 15 (ISG15)—which is an important modification in the present study (Hochstrasser, 2009).
ISG15 is a 15 kDa protein, which contains 2 UBL domains that share 33% and 32% homology with ubiquitin (Sgorbissa and Brancolini, 2012). The conjugation of ISG15 to substrate lysine residues, known as ISGylation, relies on a narrow range of E1, E2, and E3 enzymes (Fig. S1). Ubiquitin‐like modifier‐activating enzyme 7 (UBA7/UBE1L) is the E1 enzyme of ISGylation (Jeon et al., 2010). Ubiquitin/ISG15‐conjugating enzyme E2 L6 (UBE2L6) operates as an E2 enzyme for both ISGylation and ubiquitination (Jeon et al., 2010). Three known E3 ligases determine ISGylation targets, over 300 of which have been proposed (Jeon et al., 2010; Sgorbissa and Brancolini, 2012). HECT and RLD domain containing E3 ubiquitin protein ligase 5 (HERC5) is the dominant E3 for ISGylation and associates with ribosomes to target newly synthesized proteins in a nonspecific manner (Durfee et al., 2010). Tripartite motif containing 25 and ariadne RBR E3 ubiquitin protein ligase 1 show specificity for 14‐3‐3 and 4EHP, respectively (Sgorbissa and Brancolini, 2012). Ubiquitin‐specific peptidase 18 (USP18) removes ISG15 from its substrates and thus negatively regulates the ISGylation pathway (Malakhov et al., 2002). The precise functions of ISGylation remain under investigation and may be contextual. All components of the system are induced on type I interferon stimulation suggesting a functional role in antiviral responses (Sgorbissa and Brancolini, 2012). Free ISG15 has interferon‐stimulating cytokine activity when secreted outside of the cell (Bogunovic et al., 2013).
ISG15 expression and ISGylation are induced during erythropoiesis, and primary erythroblasts harvested from ISG15−/− knockout mice show impaired differentiation in ex vivo culture (Maragno et al., 2011). Transcriptional profiling of human granulopoiesis has shown that ISG15 expression is similarly induced during terminal neutrophil differentiation and a PU.1 binding site has been identified within the ISG15 promoter region (Meraro et al., 2002; Theilgaard‐Monch et al., 2005). To date, however, a functional role for ISGylation in granulopoiesis has not been proven.
Our work has found that UBE2L6, the gene encoding the E2 enzyme of ISGylation, is strongly upregulated following ATRA treatment of APL cells. Through a series of short hairpin (sh)RNA knockdown experiments, we have investigated for the first time the functional importance of this enzyme in the ATRA‐mediated granulocytic differentiation of APL cells. We report that inhibiting UBE2L6 expression results in reduced ISGylation and impaired APL cell differentiation. Interference with ISG15 expression similarly impedes differentiation. Through improving our understanding of ISGylation and protein PTMs involved in ATRA‐mediated differentiation of APL cells, we hope to identify ways of promoting differentiation therapy in other AML subtypes.
2. Materials and methods
2.1. Cell lines and culture conditions
The human APL cell line NB4 and its ATRA‐resistant NB4R counterpart were kindly gifted by B.E. Torbett and P. Paolo‐Pandolfi, respectively. ATRA‐sensitive human M2 AML HL60 cells were obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany). Their ATRA‐resistant subclone, HL60R cells were kindly gifted by M. Tschan. All cell lines were maintained in RPMI 1640 (Sigma R8758, Sigma‐Aldrich, Merck, NJ, USA) medium supplemented with 10% fetal calf serum (Sigma F7524) and 1% penicillin/streptomycin (Gibco 15070‐063, ThermoFisher Scientific, Waltham, MA, USA) in a humidified atmosphere containing 5% CO2 at 37 °C. For differentiation experiments, cells were seeded at 0.2 × 105 cells per mL and treated for 4 days with 1 μm ATRA (Sigma R2625) diluted from a 1 mm stock in 100% EtOH.
2.2. Patient study
A cohort of 98 AML patient samples, collected through the HOVON/SAKK (Dutch‐Belgian Hematology‐Oncology/Swiss Group for Clinical Cancer Research Cooperative Group) protocols 04, 04A, 29, and 42 between 1987 and 2006, were provided by P. Valk and B. Lowenberg. Patient characteristics have been previously outlined (Schlafli et al., 2012). Primary neutrophils from healthy donors were isolated using Polymorphprep (Axis‐Shield, Dundee, Scotland). All patients provided written informed consent in accordance with the Declaration of Helsinki.
2.3. RNA extraction, quantitative real‐time PCR (qPCR), TaqMan low‐density array
Total cellular RNA was harvested using TRIzol (Invitrogen 15596‐018, ThermoFisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. 1μg of RNA was reverse‐transcribed using qScript (Quanta Biosciences #95047, Beverly, MA, USA) as per product protocol at a final reaction volume of 20 μL, and the resulting cDNA was diluted 1 : 10 in H20. Subsequent qRT–PCRs were carried out using 2 μL of template together with 1× SYBR Green Supermix (Quanta Biosciences #84091), forward and reverse primers at 0.25 µm and 2.5 μL H20 in a final reaction volume of 15 μL. Reactions were run on a Bio‐Rad MyiQ™ (Irvine, CA, USA) Single Color Real‐time PCR detection system with each cycle including a 94 °C × 20 s denaturation step, 60 °C × 20 s annealing step, and a 72 °C x 30 s extension step. Primer pairs were designed to span distinct exon software to avoid genomic DNA signaling, and gene expression amplicons were validated with sequencing at the Genomics Resources Core Facility, WCMC. Sequences of specific primers were as follows: UBE2L6_F CTGGAAGCCTTGCACCAAGA, UBE2L6_R GAACATGAGTTAGGAGGGCCG, ISG15_F GGTGGACAAATGCGACGAAC, and ISG15_R TCGAAGGTCAGCCAGAACAG. The transcript levels in biological replicates (n = 3) were normalized to hPRT transcript levels, and relative differences were calculated using the Pfaffl method. Graphical displays and measurements of statistical significance were performed on graphpad prism software (San Diego, CA, USA).
2.4. Lentiviral shRNA transduction
pLKO.1 lentiviral vectors expressing small hairpin shRNAs targeting both UBE2L6 and ISG15 were purchased from Sigma‐Aldrich along with a nontargeting shRNA control (SCH002) in bacterial glycerol stocks. For each gene, five shRNAs were initially tested for efficiency by measuring mRNA levels by qPCR and two shRNAs were then selected for use in further experiments. (shUBE2L6_499 = NM_004223.3‐499s1c1/ TRCN0000007284, shUBE2L6_1082 = NM_004223.3‐1082s1c1/ TRCN0000007281, shISG15_319 = NM_005101.3‐319s21c1/ TRCN0000237825, and shISG15_352 = NM_005101.3‐352s21c1/ TRCN0000237824). Lentiviral production and transduction was performed as previously described (Tschan et al., 2003). All vectors contain a puromycin resistance gene, and transduced cell clones were selected for 4 days using 1.5 µg·mL−1 puromycin.
2.5. Morphology examination
Cells were cytospun onto glass slides and stained with Rapi‐Diff (Braidwood Laboratories 22007, 22008, 22009, London, UK) according to product guidelines. Morphology was examined using an Olympus DP70 digital microscope at 400X magnification (Mason Technology, Cork, Ireland).
2.6. Nitro Blue Tetrazolium (NBT) assay
Cells were incubated with 0.2% nitro blue tetrazolium (Sigma N5514) and 40 ng·mL−1 phorbol 12‐myristate 13‐acetate (PMA) (Sigma P8139) in RPMI for 20 min at 37 °C, washed, and cytospun onto glass slides on a base of PBS/1% FBS. Slides were then counterstained with 0.5% safranin O (Sigma 84120) in 20% EtOH and coverslipped with Entellan (Merck 1076910100, Branchburg, NJ, USA). Nitro blue tetrazolium‐positive cells were then examined, counted in triplicate, and presented accordingly.
2.7. Western blotting
Cellular protein extracts were lysed in modified RIPA buffer (50 mm Tris/HCl—pH 7.4, 150 mm NaCl, 0.25% sodium deoxycholate, 1% Igepal, 1 mm EDTA, 1× Pefabloc, 1× protease inhibitor cocktail, 1 mm Na3VO4, 1 mm NaF). Protein samples were separated on NuPAGE 4–12%, Bis/Tris gels (Invitrogen NP0322), and electrophoretically transferred onto PVDF membranes (Invitrogen IB401001). Primary antibodies were as follows: anti‐UBE2L6 (Abgent AP2118A, San Diego, CA, USA), anti‐ISG15 (Proteintech 15981‐1‐AP, Manchester, UK), and anti‐β‐actin (Sigma A5441). Proteins were visualized using relevant IR‐DYE secondary antibodies and quantified on the Odyssey IR imaging system (Li‐Cor, Cambridge, UK).
2.8. Flow cytometry
Live cells were incubated for 30 min with PE‐conjugated anti‐CD11b antibody (eBioscience 12‐0118 or Immunotools #21279114, San Diego CA, USA) in 1% albumin/ PBS, and washed with PBS prior to analysis. Fluorescence was detected using a BD‐LSRII flow cytometer (BD Biosciences, Oxford, UK). Data analysis and histogram overlays were performed on flowjo software (FlowJo, Becton, Dickinson & Company, Franklin Lakes, NJ, USA).
3. Results
3.1. UBE2L6 is induced during the neutrophil differentiation of leukemic cells
We have previously examined the gene expression changes induced by ATRA in APL cells by sequencing RNA extracted from NB4 cells treated with 1 μm ATRA for 72 h alongside untreated controls (Orfali et al., 2019). These data showed that ATRA induced a 200‐fold increase in UBE2L6 expression. Other members of the ISGylation pathway were also found to be coregulated (RNAseq data reproduced in Table 1). As NB4 cells respond to ATRA by differentiating toward mature neutrophils, this prompted us to question whether UBE2L6 expression is important for leukemic cell differentiation.
Table 1.
ATRA‐induced expression changes in ISGylation genes.
| Gene | Name | Fold change in expression |
|---|---|---|
| UBE2L6 | Ubiquitin/ISG15‐conjugating enzyme E2L6 | 200.93 |
| ISG15 | Interferon‐stimulated gene 15 | 17.54 |
| USP18 | Ubiquitin‐specific peptidase 18 | 12.53 |
| UBE1L | Ubiquitin‐like modifier‐activating enzyme 7 | 7.77 |
| HERC5 | HECT and RLD domain containing E3 ubiquitin protein ligase 5 | 3.62 |
| TRIM25 | Tripartite motif containing 25 | 2.16 |
We first examined UBE2L6 mRNA expression in 98 primary AML patient samples (M0–M4), six samples of normal CD34+ (HSC) cells, and 24 donated mature granulocyte samples using a TaqMan low‐density array. Relative UBE2L6 mRNA levels are shown as differences in Ct values as compared to mRNA levels for the housekeeping genes HMBS and ABL1. Expression was significantly lower in AML patient samples and HSC cells than in granulocytes, suggesting that increased expression may be important for the mature granulocyte phenotype (Mann–Whitney U‐test, ****P ≤ 0.0001) (Fig. 1A).
Figure 1.
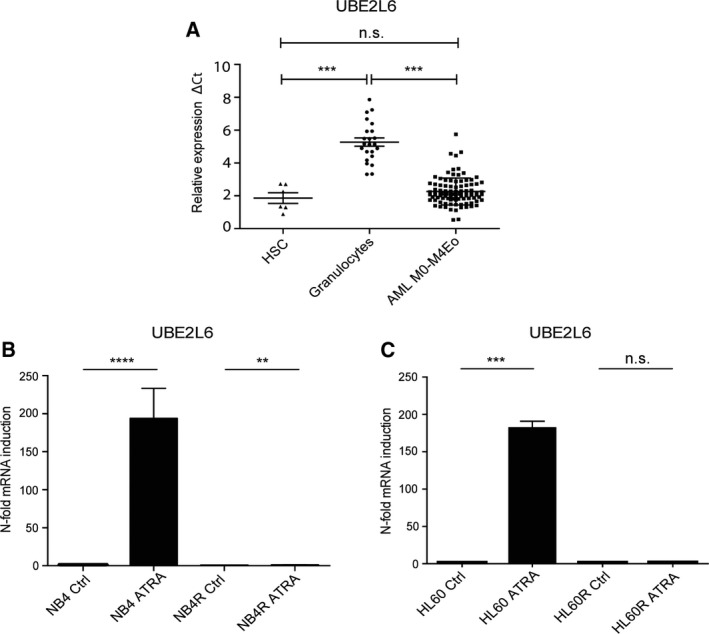
UBE2L6 expression is increased during leukemic cell differentiation. (A) UBE2L6 mRNA levels of primary AML patient samples, normal CD34+(HSC) cells, and mature granulocytes from healthy donors were quantified using qPCR. The relative ΔCt expression was calculated by the difference in UBE2L6 expression to the housekeeping genes HMBS and ABL (Mann–Whitney U‐test ****P ≤ 0.0001). (B) NB4 and NB4R cells were seeded at 0.2 × 105 cells per mL and treated with 1 μm ATRA for 72 h. Successful differentiation was confirmed in NB4 cells by flow cytometric analysis of CD11b expression. ATRA‐resistant NB4R cells did not differentiate (data not shown). Total RNA was extracted, and UBE2L6 mRNA expression was quantified by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene hPRT (n = 3) (t‐test ****P ≤ 0.0001, **P ≤ 0.01). (C) HL60 and HL60R cells were treated with 1 μm ATRA for 96 h. Successful HL60 differentiation was confirmed by qPCR measurement of GCSFR expression. ATRA‐resistant HL60R cells failed to differentiate (data not shown). Total RNA was extracted, and UBE2L6 expression was quantified by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene HMBS (n = 3) (t‐test ***P ≤ 0.001).
To test this hypothesis further, we treated NB4 cells with ATRA along with their ATRA‐resistant counterparts NB4R cells. We measured UBE2L6 expression by quantitative real‐time (q)PCR at 72 h, assessing Ct values relative to the housekeeping gene hPRT. Validating our earlier RNA sequencing observations, we detected a 180‐fold increase in UBE2L6 expression in differentiating NB4 cells (****P ≤ 0.0001), but only a 0.23‐fold difference in NB4R cells (**P = 0.0021) (Fig. 1B). The HL60 cell line (human M2 AML), although it does not carry the PML‐RARα oncoprotein, also differentiates down a granulocytic lineage in response to ATRA therapy and can be used as a second model of leukemic cell differentiation. Following 96 h of ATRA treatment, we found a 189‐fold increase in UBE2L6 expression in HL60 cells (***P = 0.0003). ATRA‐resistant HL60R cells, however, failed to induce UBE2L6 (Fig. 1C).
These results indicate that UBE2L6 is prominently upregulated during leukemic cell differentiation rather than solely on ATRA treatment and that this effect is not restricted to APL cells carrying the PML‐RARα fusion oncoprotein.
3.2. Knockdown of UBE2L6 inhibits ATRA‐induced neutrophil differentiation of NB4 APL cells
In order to investigate whether UBE2L6 has a functional role in leukemic cell differentiation, we generated UBE2L6 knockdown NB4 cells using a lentiviral delivery system to deliver target‐specific shRNA. NB4 cells transduced with a nontargeting shRNA were used as a control (SHC). Functional knockdown was confirmed by detecting reduced UBE2L6 protein levels following ATRA treatment in two knockdown clones: shUBE2L6_499 and shUBE2L6_1082. Superior knockdown efficiency is evident in shUBE2L6_1082 (Fig. 2A).
Figure 2.
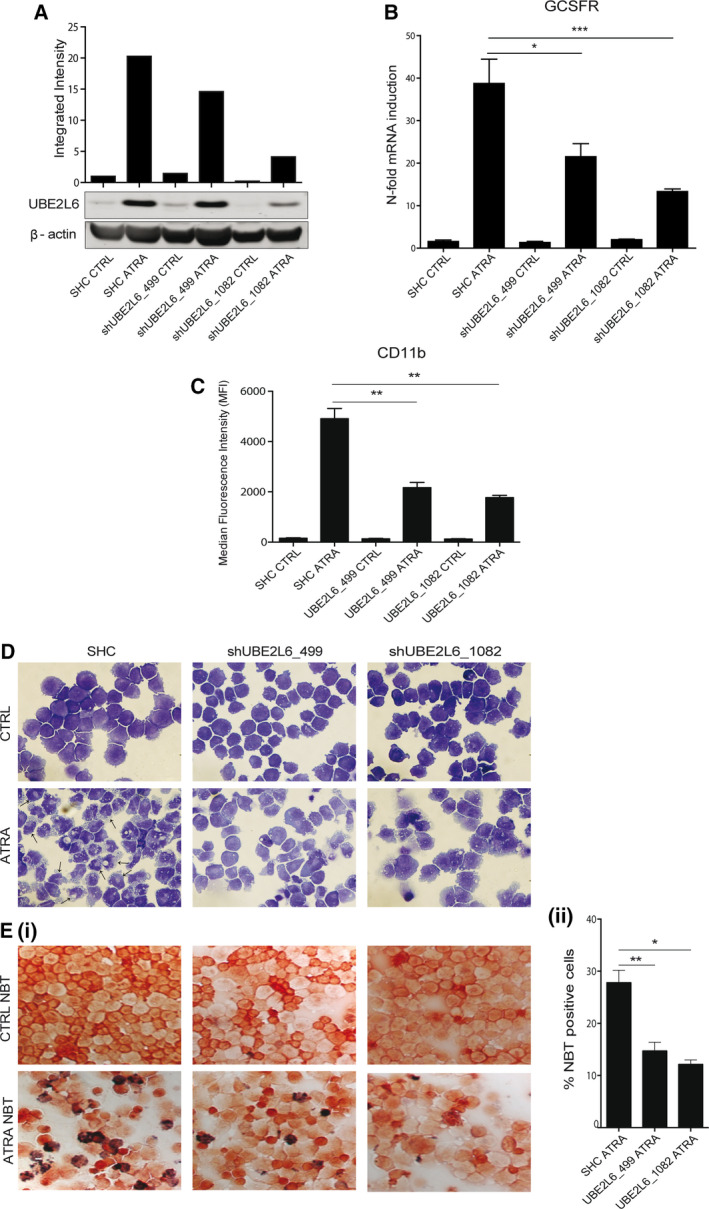
UBE2L6 inhibition attenuates APL cell differentiation. NB4 cells expressing nontargeting shRNA (SHC) or shRNA targeting UBE2L6 (shUBE2L6_499 and shUBE2L6_1082) were seeded at 0.2 × 105 cells per mL and treated for 72 h with 1 μm ATRA. (A) Functional knockdown efficiency was tested by measuring UBE2L6 protein levels in whole‐cell lysates by immunoblot at 72 h. β‐actin was used as a loading control. (B) Total RNA was extracted, and differentiation was assessed by measuring GCSFR mRNA expression by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene HMBS (n = 3) (t‐test ***P ≤ 0.001, *P ≤ 0.05). (C) Surface CD11b protein expression on live cells was measured by flow cytometry as a second assay of differentiation. Median fluorescence intensities (MFIs) are shown at 72 h (n = 3) (t‐test **P ≤ 0.01). (D) Morphologic appearance of treated cells at 72 h. Neutrophil differentiation evidenced by increased cytoplasmic volume and nuclear lobulation, indicated with arrows. (E) Neutrophil function was tested using nitro blue tetrazolium at 72 h (i) Differentiated cells reduce nitro blue tetrazolium to a blue color. (ii) nitro blue tetrazolium ‐positive cells were counted in triplicate and presented as mean ± SEM (t‐test **P ≤ 0.01, *P ≤ 0.05) (magnification 400×).
ATRA‐induced neutrophil differentiation was reduced in UBE2L6 knockdown NB4 clones compared with control cells. At a transcript level, we detected reduced expression of granulocyte colony‐stimulating factor receptor (GCSFR), a marker of neutrophil differentiation, by qPCR at 72 h (*P = 0.0123, ***P = 0.0004) (Fig. 2B). At a protein level, surface CD11b expression was reduced in ATRA‐treated knockdown cells when analyzed by flow cytometry at 72 h. A direct correlation was observed between UBE2L6 knockdown efficiency and detectable CD11b levels, with both shUBE2L6_499 and shUBE2L6_1082 showing a significant reduction in CD11b (**P = 0.0017 and P = 0.0067, respectively) (Fig. 2C). Morphologically, ATRA‐treated control cells displayed characteristic features of granulocytic differentiation with increased cytoplasmic volume and visible nuclear indentation (Fig. 2D lower left panel, arrows). This phenotype was stunted in knockdown clones (Fig. 2D lower middle and right panels). Finally, we assessed functional differentiation using the nitro blue tetrazolium assay, which tests the reducing power of the neutrophil enzyme alkaline phosphatase. We observed and quantified a decreased nitro blue tetrazolium reduction in UBE2L6 knockdown clones (**P = 0.0069, *P = 0.0354), with a direct correlation again seen between knockdown efficiency and functional differentiation (Fig. 2E (i) lower panels and Fig. 2E (ii)).
Together, these results demonstrate that UBE2L6 depletion impedes ATRA‐mediated granulocytic differentiation of APL cells and prompted us to question the potential mechanism involved in this differentiation block.
3.3. UBE2L6 mediates protein ISGylation in ATRA‐treated APL cells
As described earlier, UBE2L6 is an E2 ligase critical in the conjugation of ISG15 to target proteins during ISGylation, a process with an unknown role in leukemic cell differentiation. A search of UBE2L6 human protein interactions on the publicly available STRING database (Search Tool for the Retrieval of Interacting Genes/Proteins) (http://www.string-db.org) (Franceschini et al., 2013) depicts a high confidence of interaction between UBE2L6 and ISG15, as well as interactions with the E1 and all E3 ligases of the ISGylation pathway (Fig. 3A).
Figure 3.
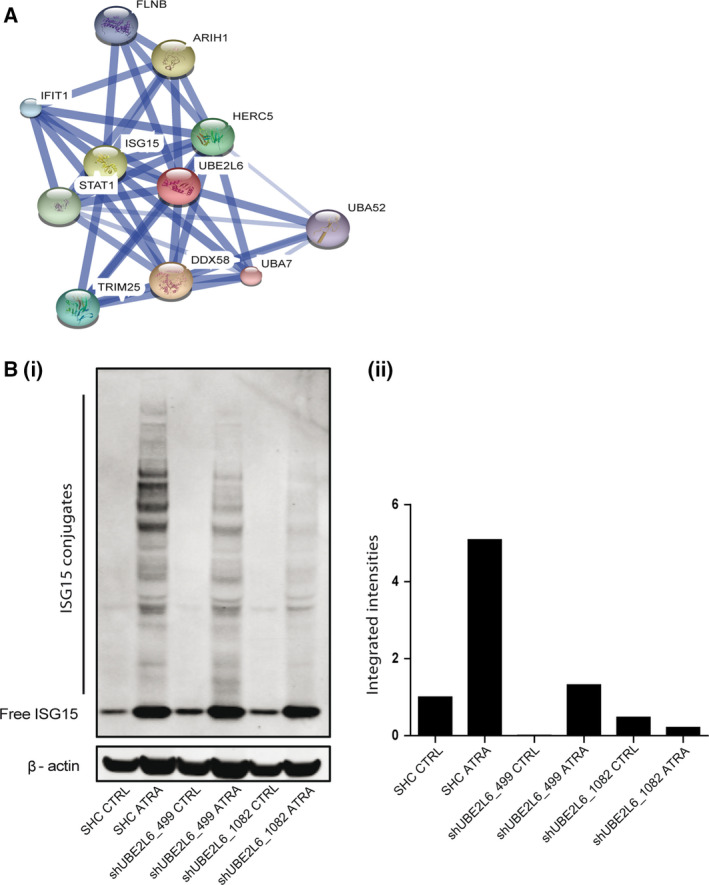
UBE2L6 regulates ATRA‐induced ISGylation. (A) Proteins known with a high confidence to interact with UBE2L6 are shown. Image created using the STRING proteomics database (http://www.string-db.org). (B) (i) Levels of free and conjugated ISG15 were measured by immunoblot in whole‐cell lysates extracted from NB4 cells expressing either nontargeting shRNA (SHC) or shRNA targeting UBE2L6 (shUBE2L6_499 and shUBE2L6_1082) following a 72 h of treatment with 1 μm ATRA. β‐actin was used as a loading control. (ii) Conjugated ISG15 levels were normalised to β‐actin and presented as integrated intensities.
We tested levels of both free and conjugated ISG15 proteins in UBE2L6 knockdown NB4 cells using western blot analysis. We found a prominent induction of free ISG15 protein following ATRA treatment in control cells and in knockdown clones. Notably, conjugated ISG15 was markedly increased after 72 h of ATRA treatment in control cells but was impaired in UBE2L6 knockdown cells, with the most efficient knockdown clone shUBE2L6_1082 having the least amount of visible conjugates (Fig. 3B (i,ii)).
3.4. ISG15 is induced during the neutrophil differentiation of leukemic cells
Our RNA sequencing data showed a 17.54‐fold increase in ISG15 gene expression in NB4 cells with ATRA treatment (Orfali et al., 2019) (Table 1). We validated this finding by qPCR measurement of ISG15 expression in ATRA‐treated NB4 cells at 72 h, which showed a 23‐fold increase in expression in differentiating cells (****P ≤ 0.0001). No significant change in expression was found in ATRA‐treated NB4R cells (Fig. 4A). HL60 cells undergoing differentiation with ATRA treatment showed a fourfold induction of ISG15 at 96 h (**P = 0.0011), whereas no induction was seen in HL60R cells (Fig. 4B). These findings associate ISG15 induction with leukemic cell differentiation rather than solely with ATRA treatment.
Figure 4.
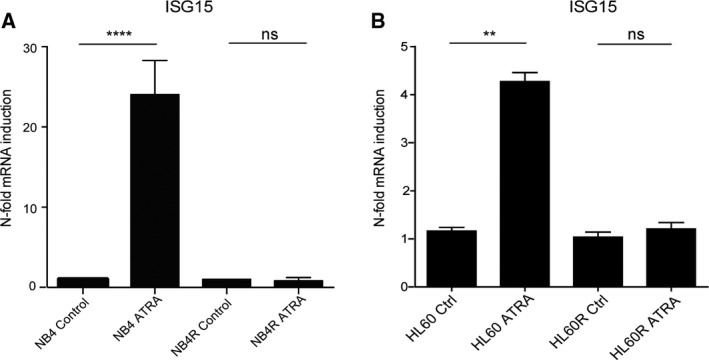
ISG15 expression is induced during leukemic cell differentiation. (A) NB4 and NB4R cells were seeded at 0.2 × 105 cells per mL and treated with 1 μm ATRA for 72 h. Successful differentiation was confirmed in NB4 cells by flow cytometric analysis of CD11b expression. ATRA‐resistant NB4R cells did not differentiate (data not shown). Total RNA was extracted, and ISG15 mRNA expression was quantified by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene hPRT (n = 3) (t‐test ****P ≤ 0.0001). (B) HL60 and HL60R cells were treated with 1 μm ATRA for 96 h. Successful HL60 differentiation was confirmed by qPCR measurement of GCSFR expression. ATRA‐resistant HL60R cells failed to differentiate (data not shown). Total RNA was extracted, and ISG15 expression was quantified by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene HMBS (n = 3) (t‐test **P ≤ 0.01).
3.5. Knockdown of ISG15 inhibits ATRA‐induced neutrophil differentiation of NB4 APL cells
To investigate whether the induction of ISG15 during ATRA‐mediated leukemic cell differentiation had functional significance, we knocked down ISG15 in NB4 APL cells. We confirmed efficient ISG15 protein knockdown in two clones, shISG15_319 and shISG15_352, detecting reduced basal levels of free ISG15 in knockdown cells compared with controls and by detecting reduced induction of free ISG15 by 72 h of ATRA treatment. ISG15 conjugates were not seen before or after ATRA treatment in knockdown cells consistent with the knockdown blocking ATRA‐induced ISGylation (Fig. 5A).
Figure 5.
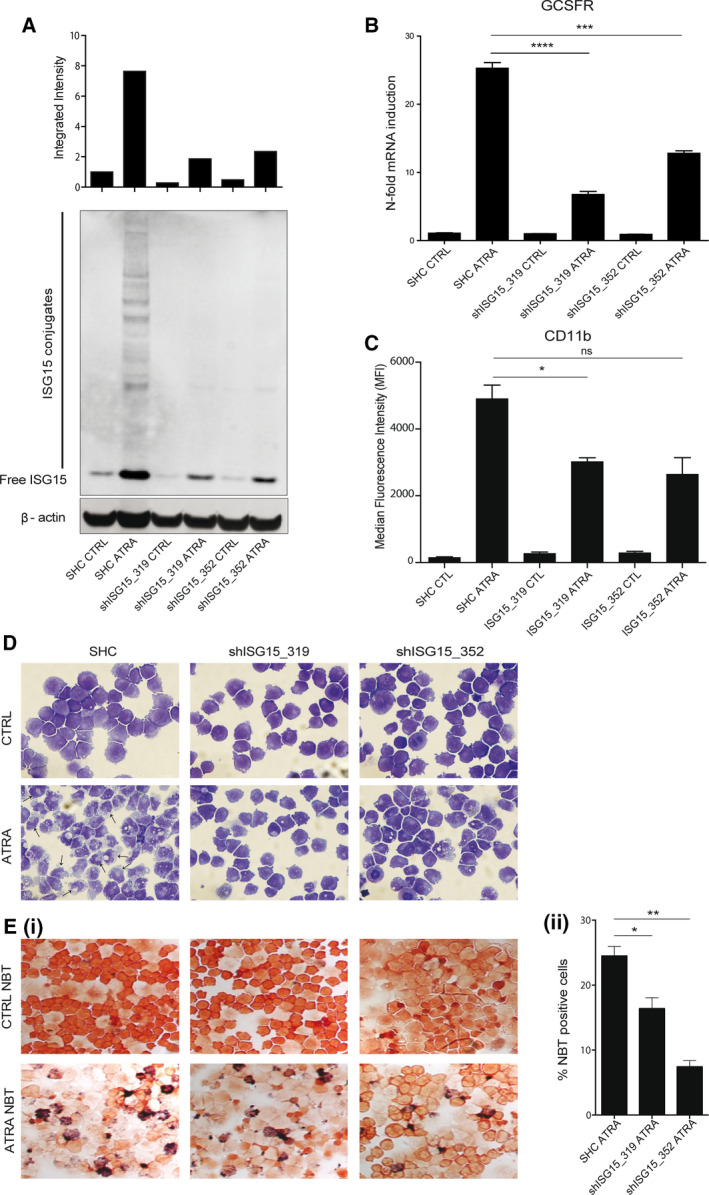
Inhibition of ISG15 impedes APL cell differentiation. NB4 cells expressing nontargeting shRNA (SHC) or shRNA targeting ISG15 (shISG15_319 and shISG15_352) were seeded at 0.2 × 105 cells per mL and treated for 72 h with 1 μm ATRA. (A) Functional knockdown efficiency was tested by measuring protein levels of both free and conjugated ISG15 in whole‐cell lysates by immunoblot at 72 h. β‐actin was used as a loading control. (B) Total RNA was extracted, and differentiation was assessed by measuring GCSFR mRNA expression by qPCR. Values are given as n‐fold induction compared with untreated cells and normalized to housekeeping gene HMBS (n = 3) (t‐test ****P ≤ 0.0001, ***P ≤ 0.001). (C) Surface CD11b protein expression on live cells was measured by flow cytometry as a second assay of differentiation. MFIs are shown at 72 h (n = 3) (t‐test *P ≤ 0.05). (D) Morphologic appearance of treated cells at 72 h. Neutrophil differentiation evidenced by increased cytoplasmic volume and nuclear lobulation indicated with arrows. (E) Neutrophil function was tested using nitro blue tetrazolium at 72 h. (i) Differentiated cells reduce nitro blue tetrazolium to blue color. (ii) nitro blue tetrazolium‐positive cells were counted in triplicate and presented as mean ± S.E.M (t‐test **P ≤ 0.01, *P ≤ 0.05) (magnification 400×).
ATRA‐induced neutrophil differentiation was reduced in both ISG15 knockdown clones compared with control cells, analogous to UBE2L6‐depleted cells. At the transcript level, we found reduced GCSFR mRNA expression in ATRA‐treated knockdown clones compared with ATRA‐treated control cells at 72 h (****P ≤ 0.0001, **P = 0.0002, respectively) (Fig. 5B). At the protein level, surface CD11b expression was reduced in both ATRA‐treated knockdown clones (*P = 0.0434 in shISG15_319 clone), but did not achieve significance in the shISG15_352 knockdown clone (Fig. 5C). ISG15 knockdown cells failed to morphologically differentiate into mature myeloid forms after 72 h of ATRA treatment (Fig. 5D lower middle and right panels), and their ability to reduce nitro blue tetrazolium was also diminished as is shown in Fig. 5E (i) lower middle and right panels. Decreased nitro blue tetrazolium reduction was quantified and is presented in Fig. 5E (ii) (*P = 0.0312, **P = 0031).
Our findings suggest a functional role for ISGylation in the ATRA‐mediated neutrophil differentiation of APL cells. In the context of our earlier results, ISGylation may be the prominent mechanism by which UBE2L6 regulates differentiation.
4. Discussion
Our results demonstrate that UBE2L6 is underexpressed in AML cells compared with their mature myeloid counterparts. Using two cell line models of AML cell differentiation, we show that cells undergoing ATRA‐mediated neutrophil differentiation strongly induce UBE2L6. We show that shRNA depletion of UBE2L6 in leukemic cells impedes their ability to differentiate, reporting for the first time a functional importance for this enzyme in ATRA‐mediated leukemic cell differentiation.
While UBE2L6 was first identified as an E2‐conjugating enzyme in ubiquitination, it is now thought to preferentially function as an E2 enzyme for ISG15 conjugation (Jeon et al., 2010; Kim et al., 2004; Zhao et al., 2004). We demonstrate that UBE2L6 modulates ISGylation in leukemic cells and further show that genetic inhibition of ISG15 strongly interferes with the neutrophil differentiation of ATRA‐treated APL cells. We hence propose that the effects of UBE2L6 on leukemic cell differentiation are likely to involve its activity in ISGylation.
In addition to ISG15 and other elements of the cellular ISGylation machinery, UBE2L6 is induced by type 1 interferon signaling and contains an interferon‐stimulated response element in its promoter region (Kim et al., 2004). ATRA has previously been shown to upregulate ISGylation machinery in NB4 cells and to stimulate ISGylation (Dao et al., 2006). This effect is thought to be due to ATRA stimulating the secretion of type I interferon as antibody‐mediated blockade of the interferon relative receptor complex suppresses this ISGylation (Dao et al., 2006). Experiments by Pitha‐Rowe and colleagues identified that UBE1L, the E1‐activating enzyme of ISGylation, is induced by ATRA treatment in ATRA‐sensitive but not ATRA‐resistant APL cells (Kitareewan et al., 2002). Subsequently, this group identified RARα binding in a domain of the UBE1L promoter, which was repressed by the PML‐RARα oncoprotein (Kitareewan et al., 2002). While our observation that UBE2L6 is induced only in ATRA‐sensitive NB4 APL cells could be explained by direct repression by PML‐RARα, we observe equivalent UBE2L6 expression levels in ATRA‐sensitive and ATRA‐resistant HL60 cells which do not carry the PML‐RARα oncoprotein. We thus speculate that UBE2L6 is activated during the leukemic cell differentiation program with a functional purpose regardless of the presence of fusion oncoproteins.
Equally, ISG15 induction has been reported following ATRA treatment only in differentiating NB4 APL cells and not in ATRA‐resistant NB4R cells (Guo et al., 2010; Pitha‐Rowe et al., 2004). Up until now, a similar finding in HL60 and HL60R cells has not been reported. Our findings again suggest that ISG15 is activated during the cellular differentiation program rather than being regulated by the PML‐RARα oncoprotein. The ISG15 promoter contains a PU.1 binding site in addition to two ISREs, and it is possible that it is activated by this transcription factor during leukemic cell differentiation (Meraro et al., 2002).
The precise cellular functions of ISGylation remain under speculation. Proteomic studies in a range of cell lines have now collectively identified over 300 putative ISGylation targets, with no functional or compartmental class over‐represented among them (Giannakopoulos et al., 2005; Malakhov et al., 2003; Zhao et al., 2005). HERC5, the primary E3 ligase of ISGylation, associates with ribosomes and broadly captures newly synthesized proteins of both endogenous origin and exogenous origin for ISGylation (Durfee et al., 2010). Induced by both type I interferon and lipopolysaccharide, ISGylation is thought to play a role in our defense against viral pathogens, but the exact mechanisms at play remain under investigation (Zhang and Zhang, 2011). While ubiquitination can modulate protein function or promote proteasomal degradation (depending on specific linkages) (Ebner and Versteeg, 2017), it is unclear whether ISGylation has analogous effects on target proteins and whether these effects may be contextual (Zhang and Zhang, 2011). ISGylation can stabilize proteins, competitively preventing their ubiquitination and subsequent degradation, as is seen with interferon regulatory factor 3 (Shi et al., 2010). It may also modify enzymes involved in ubiquitination impeding their function and thus negatively regulating proteasomal degradation of ubiquitin substrates (Takeuchi and Yokosawa, 2005; Zou et al., 2005). ISGylation might also inactivate or destabilize proteins through proteasomal channels. UBE1L‐mediated ISGylation of cyclin D1 in lung cancer cells reduces detectable protein levels with an antiproliferative effect. The reduction in cyclin D1 is reversed with the overexpression of the deISGylating enzyme USP18 (Feng et al., 2008). Further study is warranted into the context‐dependent and possibly tumor‐suppressive actions of ISGylation in physiologic and pathologic settings.
An inhibitory effect on ATRA‐mediated neutrophil differentiation of NB4 APL cells as a direct result of shRNA depletion of either UBE2L6 or ISG15 has not been previously reported. Our findings propose a functional role for these genes in differentiation. We have previously reviewed the effects of ATRA on the PML‐RARα protein in APL cells (Orfali et al., 2014). Briefly, in addition to derepressing transcription, ATRA induces the degradation of the PML‐RARα oncoprotein through caspase‐3‐mediated cleavage, ubiquitin/proteasome‐mediated proteolysis, and lysosomal‐mediated autophagy (Orfali et al., 2014). Investigating a temporal correlation between the reduction in PML‐RARα and an induction of UBE1L in ATRA‐treated NB4 cells, Shah et al. (2008) reported ISGylation of the PML domain of PML‐RARα with subsequent repression. This repression was opposed by the overexpression of USP18. Subsequent work showed that knockdown of USP18 destabilized PML‐RARα and promoted apoptosis in NB4 APL cells but did not have an effect on differentiation (Guo et al., 2010). A similar destabilizing effect of ISGylation on a leukemic oncoprotein is suggested for the BCR‐ABL kinase that drives chronic myeloid leukemia (CML). The expression of BCR‐ABL in mouse bone marrow cells results in splenomegaly and an abnormal myeloproliferation resembling CML. Bone marrow cells harvested from USP18−/− mice and transfected with BCR‐ABL prior to transplantation into wild‐type recipient mice developed a CML‐like state in only 40% of cases, whereas all mice transplanted with USP18+/+ BCR‐ABL expressing cells developed disease (Yan et al., 2007). Degradation of PML‐RARα may be one mechanism through which UBE2L6 and ISGylation contribute to ATRA‐mediated APL cell differentiation as we have observed. However, we speculate that this pathway may have broader functions in this process. This is supported by a proposed role for ISGylation machinery in the differentiation of other hemopoietic cells. High levels of USP18 in murine hematopoietic cells block the cytokine‐induced terminal differentiation of monocytic cells (Liu et al., 1999). ISG15 along with UBE1L and UBCM8 (the murine orthologue of UBE2L6) is induced during erythroid development in mice, and erythroblasts cultured ex vivo from ISG15−/− mice show impaired differentiation (Maragno et al., 2011). Future work will examine the effects of modulating ISGylation in non‐APL models of leukemic differentiation.
Our previous work has proposed that promoting autophagy may enhance the differentiating effects of ATRA on leukemic cells (Orfali et al., 2015). With further study, promoting ISGylation may prove to have similar benefits. Inhibiting USP18, the negative regulator of ISGylation, has been shown in both cell line and in vivo systems to enhance ISG15 conjugation (Ketscher et al., 2015). A small‐molecule inhibitor of this isopeptidase is awaited and will greatly advance study in this arena (Basters et al., 2012).
5. Conclusions
We have identified a novel function of UBE2L6 in the granulocytic differentiation of APL cells, mediated by ISGylation. Our work contributes to the growing area of study of post‐translational protein alteration by ubiquitin‐like modifiers. A greater understanding of the protein handling that occurs during leukemic cell differentiation might allow us to modulate these processes and broaden the application of differentiation therapy for the improved treatment of AML.
Conflict of interest
All authors declare no conflict of interest.
Author contributions
NO, DS‐K, TRO’D, NPM, LJG, MRC, MPT, and SLM conceived, designed, and conducted the experiments; interpreted the results; and wrote the manuscript.
Supporting information
Fig. S1 . ISGylation—in the first step of ISGylation, ISG15 is activated by the E1 enzyme UBE1L in an ATP‐dependent reaction.
Acknowledgements
The authors would like to thank Dr. Michelle J. Nyhan, along with members of the Cork Cancer Research Centre, the Gudas laboratory at Weill Cornell New York, Mongan Laboratory at University of Nottingham, and the Tschan laboratory at the University of Bern, for review of data and critical feedback. MPT, TRO’D, and SLM are members of the TRANSAUTOPHAGY COST ACTION CA15138 and acknowledge support for collaboration.
This research was supported by the National Institutes of Health (CA043796) (LJG) and by Weill Cornell funds to LJG. NO was funded by the Haematology Education and Research Trust (H.E.R.O), and Breakthrough Cancer Research (BCR), with unrestricted educational support from Pfizer, Merck Sharp & Dohme, Bristol Myers Squibb, Novartis, and Amgen. MPT was supported by Swiss Cancer Research (KFS‐3409‐02‐2014) and by the Swiss National Science Foundation (31003A_143739). The financial support of the University of Nottingham to NM is gratefully acknowledged.
References
- Basters A, Ketscher L, Deuerling E, Arkona C, Rademann J, Knobeloch KP and Fritz G (2012) High yield expression of catalytically active USP18 (UBP43) using a Trigger Factor fusion system. BMC Biotechnol 12, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogunovic D, Boisson‐Dupuis S and Casanova JL (2013) ISG15: leading a double life as a secreted molecule. Exp Mol Med 45, e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres‐Cortes JR (2013) Blastic leukaemias (AML): a biologist's view. Cell Biochem Biophys 66, 13–22. [DOI] [PubMed] [Google Scholar]
- Dao CT, Luo JK and Zhang DE (2006) Retinoic acid‐induced protein ISGylation is dependent on interferon signal transduction. Blood Cells Mol Dis 36, 406–413. [DOI] [PubMed] [Google Scholar]
- Durfee LA, Lyon N, Seo K and Huibregtse JM (2010) The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol Cell 38, 722–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebner P and Versteeg GA (2017) Ubiquitin enzymes in the regulation of immune responses. Crit Rev Biochem Mol Biol 52, 425–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Sekula D, Guo Y, Liu X, Black CC, Galimberti F, Shah SJ, Sempere LF, Memoli V, Andersen JB et al (2008) UBE1L causes lung cancer growth suppression by targeting cyclin D1. Mol Cancer Ther 7, 3780–3788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C et al (2013) STRING v9.1: protein‐protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41, D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friend SF, Deason‐Towne F, Peterson LK, Berger AJ and Dragone LL (2014) Regulation of T cell receptor complex‐mediated signaling by ubiquitin and ubiquitin‐like modifications. Am J Clin Exp Immunol 3, 107–123. [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulos NV, Luo JK, Papov V, Zou W, Lenschow DJ, Jacobs BS, Borden EC, Li J, Virgin HW and Zhang DE (2005) Proteomic identification of proteins conjugated to ISG15 in mouse and human cells. Biochem Biophys Res Comm 336, 496–506. [DOI] [PubMed] [Google Scholar]
- Guo Y, Dolinko AV, Chinyengetere F, Stanton B, Bomberger JM, Demidenko E, Zhou DC, Gallagher R, Ma T, Galimberti F et al (2010) Blockade of the ubiquitin protease UBP43 destabilizes transcription factor PML/RARalpha and inhibits the growth of acute promyelocytic leukemia. Can Res 70, 9875–9885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser M (2009) Origin and function of ubiquitin‐like proteins. Nature 458, 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon YJ, Yoo HM and Chung CH (2010) ISG15 and immune diseases. Biochem Biophys Acta 1802, 485–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarjian H and O'Brien S (2010) Questions regarding frontline therapy of acute myeloid leukemia. Cancer 116, 4896–4901. [DOI] [PubMed] [Google Scholar]
- Ketscher L, Hannss R, Morales DJ, Basters A, Guerra S, Goldmann T, Hausmann A, Prinz M, Naumann R, Pekosz A et al (2015) Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc Natl Acad Sci USA 112, 1577–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KI, Giannakopoulos NV, Virgin HW and Zhang DE (2004) Interferon‐inducible ubiquitin E2, Ubc8, is a conjugating enzyme for protein ISGylation. Mol Cell Biol 24, 9592–9600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitareewan S, Pitha‐Rowe I, Sekula D, Lowrey CH, Nemeth MJ, Golub TR, Freemantle SJ and Dmitrovsky E (2002) UBE1L is a retinoid target that triggers PML/RARalpha degradation and apoptosis in acute promyelocytic leukemia. Proc Natl Acad Sci USA 99, 3806–3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D and Rape M (2012) The ubiquitin code. Annu Rev Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Krishna RG and Wold F (1993) Post‐translational modification of proteins. Adv Enzymol Relat Areas Mol Biol 67, 265–298. [DOI] [PubMed] [Google Scholar]
- Liu LQ, Ilaria R Jr, Kingsley PD, Iwama A, van Etten RA, Palis J and Zhang DE (1999) A novel ubiquitin‐specific protease, UBP43, cloned from leukemia fusion protein AML1‐ETO‐expressing mice, functions in hematopoietic cell differentiation. Mol Cell Biol 19, 3029–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malakhov MP, Kim KI, Malakhova OA, Jacobs BS, Borden EC and Zhang DE (2003) High‐throughput immunoblotting. Ubiquitiin‐like protein ISG15 modifies key regulators of signal transduction. J Biol Chem 278, 16608–16613. [DOI] [PubMed] [Google Scholar]
- Malakhov MP, Malakhova OA, Kim KI, Ritchie KJ and Zhang DE (2002) UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J Biol Chem 277, 9976–9981. [DOI] [PubMed] [Google Scholar]
- Maragno AL, Pironin M, Alcalde H, Cong X, Knobeloch KP, Tangy F, Zhang DE, Ghysdael J and Quang CT (2011) ISG15 modulates development of the erythroid lineage. PLoS ONE 6, e26068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraro D, Gleit‐Kielmanowicz M, Hauser H and Levi BZ (2002) IFN‐stimulated gene 15 is synergistically activated through interactions between the myelocyte/lymphocyte‐specific transcription factors, PU.1, IFN regulatory factor‐8/IFN consensus sequence binding protein, and IFN regulatory factor‐4: characterization of a new subtype of IFN‐stimulated response element. J Immunol 168, 6224–6231. [DOI] [PubMed] [Google Scholar]
- Orfali N, McKenna SL, Cahill MR, Gudas LJ and Mongan NP (2014) Retinoid receptor signaling and autophagy in acute promyelocytic leukemia. Exp Cell Res 324, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orfali N, O'Donovan DT, Cahill M, Benjamin D, Nanus D, McKenna S, Gudas L and Mongan NP (2019). All‐trans retinoic acid (ATRA) induced TFEB expression is required for myeloid differentiation in acute promyelocytic leukemia (APL). Eur J Haematol. [Epub ahead of print]. 10.1111/ejh.13367 [DOI] [PubMed] [Google Scholar]
- Orfali N, O'Donovan TR, Nyhan MJ, Britschgi A, Tschan MP, Cahill MR, Mongan NP, Gudas LJ and McKenna SL (2015) Induction of autophagy is a key component of all‐trans‐retinoic acid‐induced differentiation in leukemia cells and a potential target for pharmacologic modulation. Exp Hematol 43, 781–793.e782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitha‐Rowe I, Hassel BA and Dmitrovsky E (2004) Involvement of UBE1L in ISG15 conjugation during retinoid‐induced differentiation of acute promyelocytic leukemia. J Biol Chem 279, 18178–18187. [DOI] [PubMed] [Google Scholar]
- Schlafli AM, Torbett BE, Fey MF and Tschan MP (2012) BIRC6 (APOLLON) is down‐regulated in acute myeloid leukemia and its knockdown attenuates neutrophil differentiation. Exp Hematol Oncol 1, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgorbissa A and Brancolini C (2012) IFNs, ISGylation and cancer: Cui prodest? Cytokine Growth Factor Rev 23, 307–314. [DOI] [PubMed] [Google Scholar]
- Shah SJ, Blumen S, Pitha‐Rowe I, Kitareewan S, Freemantle SJ, Feng Q and Dmitrovsky E (2008) UBE1L represses PML/RAR{alpha} by targeting the PML domain for ISG15ylation. Mol Cancer Ther 7, 905–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi HX, Yang K, Liu X, Liu XY, Wei B, Shan YF, Zhu LH and Wang C (2010) Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol Cell Biol 30, 2424–2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T and Yokosawa H (2005) ISG15 modification of Ubc13 suppresses its ubiquitin‐conjugating activity. Biochem Biophys Res Comm 336, 9–13. [DOI] [PubMed] [Google Scholar]
- Tallman MS and Altman JK (2009) How I treat acute promyelocytic leukemia. Blood 114, 5126–5135. [DOI] [PubMed] [Google Scholar]
- Tang XH and Gudas LJ (2011) Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 6, 345–364. [DOI] [PubMed] [Google Scholar]
- Theilgaard‐Monch K, Jacobsen LC, Borup R, Rasmussen T, Bjerregaard MD, Nielsen FC, Cowland JB and Borregaard N (2005) The transcriptional program of terminal granulocytic differentiation. Blood 105, 1785–1796. [DOI] [PubMed] [Google Scholar]
- Thein MS, Ershler WB, Jemal A, Yates JW and Baer MR (2013) Outcome of older patients with acute myeloid leukemia: an analysis of SEER data over 3 decades. Cancer 119, 2720–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschan MP, Fischer KM, Fung VS, Pirnia F, Borner MM, Fey MF, Tobler A and Torbett BE (2003) Alternative splicing of the human cyclin D‐binding Myb‐like protein (hDMP1) yields a truncated protein isoform that alters macrophage differentiation patterns. J Biol Chem 278, 42750–42760. [DOI] [PubMed] [Google Scholar]
- Yan M, Luo JK, Ritchie KJ, Sakai I, Takeuchi K, Ren R and Zhang DE (2007) Ubp43 regulates BCR‐ABL leukemogenesis via the type 1 interferon receptor signaling. Blood 110, 305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D and Zhang DE (2011) Interferon‐stimulated gene 15 and the protein ISGylation system. J Interferon Cytokine Res 31, 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Beaudenon SL, Kelley ML, Waddell MB, Yuan W, Schulman BA, Huibregtse JM and Krug RM (2004) The UbcH8 ubiquitin E2 enzyme is also the E2 enzyme for ISG15, an IFN‐alpha/beta‐induced ubiquitin‐like protein. Proc Natl Acad Sci USA 101, 7578–7582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Denison C, Huibregtse JM, Gygi S and Krug RM (2005) Human ISG15 conjugation targets both IFN‐induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci USA 102, 10200–10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Papov V, Malakhova O, Kim KI, Dao C, Li J and Zhang DE (2005) ISG15 modification of ubiquitin E2 Ubc13 disrupts its ability to form thioester bond with ubiquitin. Biochem Biophys Res Comm 336, 61–68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 . ISGylation—in the first step of ISGylation, ISG15 is activated by the E1 enzyme UBE1L in an ATP‐dependent reaction.