

Abstract
Single‐cell transcriptomics have revolutionized our understanding of the cell composition of tumors and allowed us to identify new subtypes of cells. Despite rapid technological advancements, single‐cell analysis remains resource‐intense hampering the scalability that is required to profile a sufficient number of samples for clinical associations. Therefore, more scalable approaches are needed to understand the contribution of individual cell types to the development and treatment response of solid tumors such as esophageal adenocarcinoma where comprehensive genomic studies have only led to a small number of targeted therapies. Due to the limited treatment options and late diagnosis, esophageal adenocarcinoma has a poor prognosis. Understanding the interaction between and dysfunction of individual cell populations provides an opportunity for the development of new interventions. In an attempt to address the technological and clinical needs, we developed a protocol for the separation of esophageal carcinoma tissue into leukocytes (CD45+), epithelial cells (EpCAM+), and fibroblasts (two out of PDGFRα, CD90, anti‐fibroblast) by fluorescence‐activated cell sorting and subsequent RNA sequencing. We confirm successful separation of the three cell populations by mapping their transcriptomic profiles to reference cell lineage expression data. Gene‐level analysis further supports the isolation of individual cell populations with high expression of CD3, CD4, CD8, CD19, and CD20 for leukocytes, CDH1 and MUC1 for epithelial cells, and FAP, SMA, COL1A1, and COL3A1 for fibroblasts. As a proof of concept, we profiled tumor samples of nine patients and explored expression differences in the three cell populations between tumor and normal tissue. Interestingly, we found that angiogenesis‐related genes were upregulated in fibroblasts isolated from tumors compared with normal tissue. Overall, we suggest our protocol as a complementary and more scalable approach compared with single‐cell RNA sequencing to investigate associations between clinical parameters and transcriptomic alterations of specific cell populations in esophageal adenocarcinoma.
Keywords: cancer‐associated fibroblasts, cell types, esophageal adenocarcinoma, transcriptomics, tumor microenvironment
Little is known about the interactions between the different intratumoral cell types, that is, epithelial tumor cells, cancer‐associated fibroblasts, and immune cells. We developed a scalable and cost‐effective workflow to separate these cell types from esophageal adenocarcinoma (EAC) biopsies using fluorescence‐activated cell sorting and subsequent RNA sequencing. Using this approach, we characterize these cell types in EAC and normal tissue.
Abbreviations
- CAF
cancer‐associated fibroblast
- DEG
differentially expressed gene
- DMSO
dimethyl sulfoxide
- EAC
esophageal adenocarcinoma
- EMT
epithelial‐to‐mesenchymal transition
- FACS
fluorescence‐activated cell sorting
- FAP
fibroblast activation protein
- FBS
fetal bovine serum
- FDR
false discovery rate
- FITC
fluorescein isothiocyanate
- GO
gene ontology
- LD
life dead
- MMP
matrix metalloproteinase
- NSCLC
non‐small cell lung cancer
- PCA
principal component analysis
- PDGF
platelet‐derived growth factor
- PDGFRα
PDGF receptor α
- PD‐L1
programmed death ligand 1
- RCA
reference component analysis
- scRNA‐seq
single‐cell RNA sequencing
- TGF‐β
transforming growth factor β
- VEGF
vascular endothelial growth factor
1. Introduction
Esophageal cancer is associated with the sixth‐highest mortality rate of cancer‐related deaths and presumed to be one of the most mortal malignancies worldwide (Coleman et al., 2018). Over the past decades, there has been a rapid increase particularly in the incidence of esophageal adenocarcinoma (EAC) in the Western world, but despite improvements in perioperative treatments, there is no sufficient therapeutic strategy for the majority of patients so far (Coleman et al., 2018). Further, prediction of therapy success is poor and response to neoadjuvant therapies varies dramatically ranging from no to complete response (Vallböhmer et al., 2010).
Focus in cancer research has shifted from considering epithelial cancer cells to analyses of their interactions with different components of the tumor microenvironment. Particularly, the immune cell population has revealed an immense effect on tumor progression. Today, immune checkpoint blockage is first‐line therapy in several solid neoplasia such as malignant melanoma and non‐small cell lung cancer (NSCLC) (Incorvaia et al., 2019; Spagnolo et al., 2019). For EAC, several biomarkers in the immune compartment have been detected, and currently, clinical trials are ongoing to evaluate the safety and efficacy of possible immunotherapies (Tanaka et al., 2017).
There is also rising evidence for an important impact of cancer‐associated fibroblasts (CAFs) on tumor biology and disease progression (Shiga et al., 2015). CAFs are a heterogeneous cell population of unknown origin that form the stromal part of solid tumors (Shiga et al., 2015). They release cytokines (e.g., TGF‐β), proteases (e.g., matrix metalloproteinases), and growth factors (e.g., VEGF, PDGF) with various autocrine and paracrine functions that can enhance tumor growth, neovascularization, and migration of cancer cells (Kakarla et al., 2012). In several cancer types, including colorectal, breast, ovarian, and head and neck cancer, the presence of CAFs correlates with poor prognosis (Lai et al., 2012; Marsh et al., 2011; Tsujino et al., 2007; Yamashita et al., 2012). In esophageal cancer, Wang et al. described pleiotropic functions in carcinogenesis, proliferation, angiogenesis, and metastasis (Wang et al., 2016). Although CAFs obviously influence tumor biology, they are still poorly characterized. Since targeting either CAFs or their secreted paracrine factors could improve therapeutic response, characterization of the roles of fibroblasts in EAC can help developing clinically effective treatments (Kakarla et al., 2012).
Single‐cell transcriptome sequencing (scRNA‐seq) has opened new avenues for the understanding of the biological role of cell populations, their origins, and interactions (Ren et al., 2018). Single‐cell approaches – in contrast to bulk tissue sequencing – allow to determine which cell type is responsible for transcriptomic changes, which is of fundamental importance for the understanding of tumor biology. Single‐cell RNA sequencing of colorectal cancers, for example, has shown that epithelial‐to‐mesenchymal transition (EMT) signature genes are upregulated in CAFs and not in epithelial cells (Li et al., 2017), a phenomenon that might have gotten misinterpreted as EMT by bulk sequencing. Hence, a systematic analysis of gene expression profiles in different tumor cell types of EAC in comparison with normal esophageal mucosa will help to understand the role of the interaction between (cancer associated) fibroblasts, immune cells, and epithelial tumor cells in carcinogenesis and disease progression. Single‐cell sequencing, however, is still expensive, limiting the number of clinical samples that are usually profiled per study. More scalable approaches for the analysis of individual cell populations of larger series of tumors are warranted to be able to identify cell type‐specific alterations that are associated with clinical features. It is therefore desirable to develop economic methodologies that allow to analyze cell types separately but not necessarily on a single‐cell level.
To address this need, we developed a protocol to isolate different cell types (epithelial cells, immune cells, fibroblasts) of endoscopically obtained EAC tissue as well as normal esophageal mucosa and to sequence the transcriptome of each cell type separately. By mapping the transcriptomes to reference tissue expression data, we demonstrate the successful separation of the three cell types. Further, we explored the utility of this approach by comparing expression profiles of esophageal normal mucosa with adenocarcinoma tissues. Our protocol provides a scalable tool for the systematic and cost‐effective investigation of the individual cell populations of EAC. It will help to improve our understanding of pathological processes and possibly identify novel therapeutic targets in EAC.
2. Methods
2.1. Patients and tumor samples
Fresh tissue samples from nine patients with histologically confirmed adenocarcinoma of the distal esophagus or the gastroesophageal junction were prospectively collected between June and December 2018. In six of these patients, both corresponding tumor and normal esophageal mucosa were taken, while in one patient only tumor tissue and in two patients normal mucosa had been biopsied. Processing and consecutive separation of these samples led to 31 cell populations after FACS sorting with sufficient yield for further analysis via RNA‐seq.
All patients underwent primary staging including esophagogastroduodenoscopy, endoscopic ultrasound, and spiral contrast‐enhanced computer tomography of thorax and abdomen within the Department of General, Visceral and Cancer Surgery at the University Hospital of Cologne. Written informed consent was obtained from all patients before participating in the analysis, and the study was approved by the local Institutional Review Board (Ethics No. 18‐274). The study's methodologies conformed to the standards set by the Declaration of Helsinki and its later amendments.
Samples were taken either by endoscopic biopsy (six samples) or obtained from surgical specimens (three samples). During endoscopy, standardized biopsies from both tumor and corresponding normal esophageal mucosa at 5‐cm distance were taken and immediately transferred into 1 mL of RPMI 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) at room temperature for further processing.
Standard surgical procedure was laparotomic or laparoscopic gastrolysis and right transthoracic en bloc esophagectomy including two‐field lymphadenectomy of mediastinal and abdominal lymph nodes (Ivor Lewis esophagectomy) as described previously (Plum et al., 2018). Samples obtained from a surgical specimen were also immediately transferred into RPMI 1640 medium at room temperature and further processed as described below.
In case of neoadjuvant treatment (five patients), either neoadjuvant chemoradiation analog CROSS (four patients) or perioperative chemotherapy analog FLOT regimen (one patient) was applied (Al‐Batran et al., 2016; van Hagen et al., 2012).
2.2. Single‐cell dissection
Immediately after endoscopy, corresponding tumor and normal mucosa biopsies were processed separately (Fig. 1). Tissue samples were transferred together with 1 mL of Gibco™ RPMI 1640 Medium (Thermo Fisher Scientific) and 1 mL of PBS (Thermo Fisher Scientific) into Petri dishes and dissected mechanically using two scalpels. After transfer into gentleMACS C Tubes (Miltenyi Biotec, Bergisch Gladbach, Germany), additional 1 mL of each of the following enzymes was added: DNAse I (500 U·mL−1; AppliChem PanReac, Darmstadt, Germany; in PBS), collagenase IV (320 U·mL−1; Thermo Fisher Scientific; in PBS), and dispase II (2 U·mL−1; Sigma‐Aldrich, St. Louis, MO, USA; in PBS). Automated tissue dissociation was performed using the preset human tumor programs 1, 2, and 3 of the gentleMACS™ Dissociator (Miltenyi Biotec). Mechanical dissociation steps 1 and 2 were followed by enzymatic digestion for 20 min while rotating at 37 °C. Following enzymatic‐mechanical tissue dissociation, samples were diluted with PBS and filtered through a Falcon® 100‐µm cell strainer (Corning, New York, NY, USA) to remove larger debris. Filtering was repeated if necessary. Dissociated cells were collected by centrifugation at 405 g for 5 min at room temperature. Cells were resuspended by vortexing within freezing medium including 60% Gibco™ RPMI 1640 medium (Thermo Fisher Scientific), 30% FBS (Capricorn Scientific, Ebsdorfergrund, Germany), and 10% dimethyl sulfoxide (DMSO) (Sigma‐Aldrich). Afterward, cells were frozen at −80 °C for 24 h and then transferred to liquid nitrogen for long‐term storage until fluorescence‐activated cell sorting (FACS). As one freeze/thaw cycle of the single‐cell solutions resulted in hemolysis, no additional erythrocyte lysis was performed.
Fig. 1.
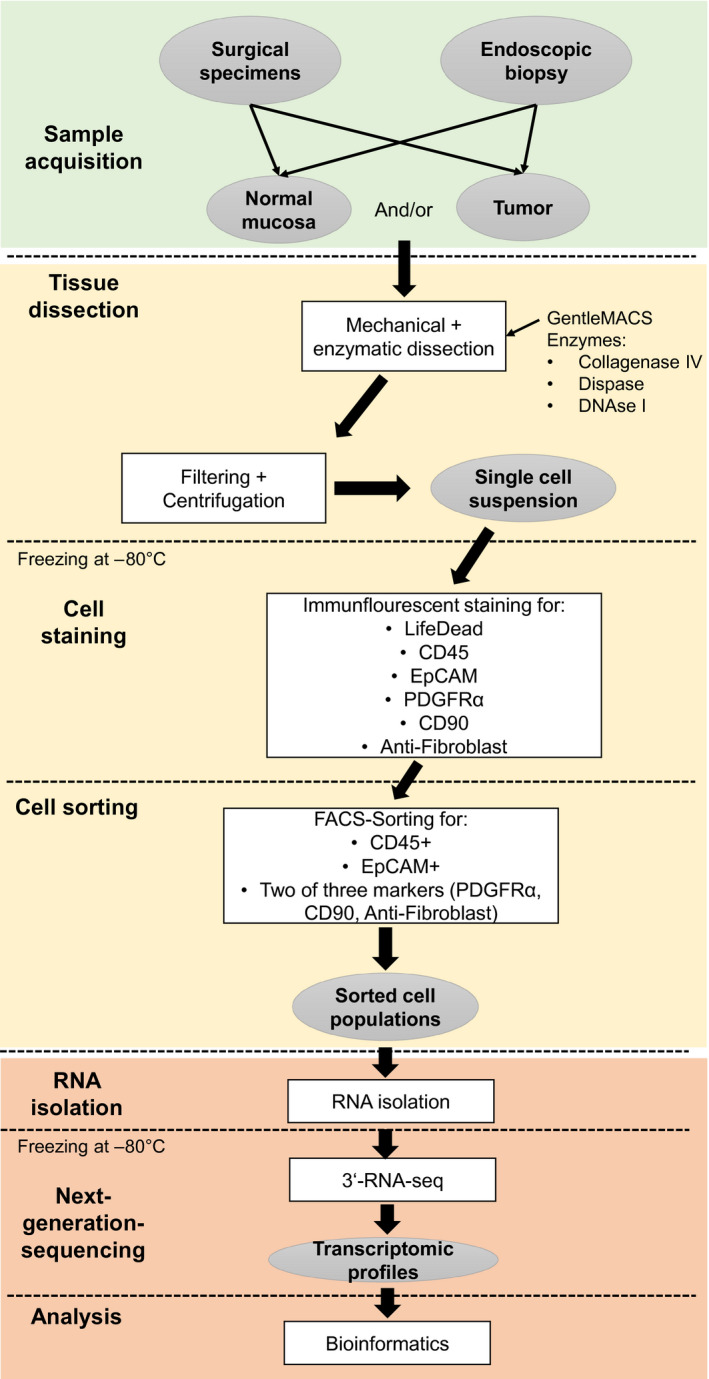
Schematic representation of the workflow.
2.3. Immunofluorescence staining of single‐cell suspension
Frozen single‐cell suspensions were thawed, added to 10 mL of Gibco™ RPMI 1640 Medium (Thermo Fisher Scientific) at 37 °C and vortexed. Before further processing, 500 µL of each sample was separated and mixed with 1 mL of medium for the unstained control. Samples were centrifuged at 450 g for 5 min at room temperature. After centrifugation, all supernatants were discarded. The collected cells were resuspended in 500 µL MACS buffer [PBS (pH 7.2) + 2 mm EDTA + 0.5% BSA] and kept on ice until FACS analysis. The following incubation steps were performed on ice in the dark. Samples were stained consecutively with the following monoclonal anti‐human antibodies: 2 µL Alexa Fluor® 647‐conjugated anti‐PDGF receptor α (PDGFRα; Cell Signaling Technology, Danvers, MA, USA) and 1 µL eBioscience™ Fixable Viability Dye eFluor™ 506 (Thermo Fisher Scientific) for 15 min followed by 5 min of incubation with 1 µL PE/Cy7‐conjugated anti‐CD45 (Biolegend, San Diego, CA, USA). Cells were incubated for 10 min with additional 2 µL FITC‐conjugated anti‐EpCAM (Miltenyi Biotec), 5 µL PE‐conjugated anti‐fibroblast (Miltenyi Biotec), and 2 µL VioBlue‐conjugated anti‐CD90 (Miltenyi Biotec). Additional staining for epithelial cells utilizing 1 µL APC/Fire™ 750‐conjugated anti‐mouse/human CD324 (E‐Cadherin) (Biolegend) was performed in six samples. This additional staining was omitted in subsequent samples as E‐Cadherin did not stain additional epithelial cells that were not stained by EpCAM, including normal esophagus (data not shown). Cells were spun down at 450 g for 5 min at 4 °C. Supernatants were discarded and collected cells resuspended in 500 µL cold MACS buffer.
Simultaneously to the cells, compensation beads were prepared for analysis by flow cytometry utilizing the ArC™ Amine Reactive Compensation Bead Kit for life‐dead (LD) staining (Thermo Fisher Scientific) and the AbC™ Total Antibody Compensation Bead Kit (Thermo Fisher Scientific), respectively, according to the manufacturer's instructions. Immunofluorescent stained cell suspensions and beads were kept on ice until sorting.
2.4. Flow cytometry analysis and sorting
Sorting of the single‐cell suspensions was performed using a BD FACSAria Fusion (BD Biosciences, San Jose, CA, USA) using a 100‐µm nozzle and 20 psi pressure, using aerosol containment. Immediately before analysis, cell suspensions were filtered once again using a 70‐µm CellTrics strainer (Sysmex, Kobe, Japan). Gating strategy was as follows: After viability gating, cells were gated according to the surface expression of CD45 as marker for immune cells (‘immune cell population’). CD45‐negative cells were analyzed for the expression of PDGFRα, fibroblast marker, and CD90. Those cells which were positive for at least two of those markers were defined as fibroblasts (‘fibroblast cell population’). Finally, all other CD45‐negative cells were analyzed for expression of EpCAM (or E‐Cadherin) as marker for tumor cells of epithelial origin (‘epithelial cell population’). Cell subpopulations were sorted into 500 µL cold MACS buffer at 4 °C.
2.5. RNA isolation and next‐generation sequencing
After sorting, cells were kept on ice and RNA isolation was performed using the PicoPure™ RNA Isolation Kit (Thermo Fisher Scientific) according to manufacturer's instructions. Isolated RNA was stored at −80 °C. Libraries for RNA sequencing were prepared using the QuantSeq 3′ mRNA‐Seq Library Prep Kit FWD for Illumina (Lexogen GmbH, Vienna, Austria) according to the low‐input protocol. Libraries were sequenced on a HiSeq 4000 (Illumina) by 1× 50 bases.
2.6. RNA‐seq analysis
Reads were aligned to the human genome (Homo sapiens GRCh38) using star software v. 2.6 (Dobin et al., 2013). Mapped reads were counted with HTSeq, and differential gene expression analysis was conducted using bioconductor r package deseq2 version 1.22.2. (Love et al., 2014). An adjusted P‐value threshold of 0.05 and a log2 fold change ≥ 1 were set to determine differential gene expression. The complete lists of DEGs in a pairwise manner are available in the Table S1.
2.7. Reference component analysis
Clustering of the independent samples was performed using the r package reference component analysis (rca) v. 1.0. (Li et al., 2017) with the default option ‘Global Panel’, and this panel contains a set of featured genes from the reference bulk transcriptomes in the HumanU133A/GNF1H Gene Atlas and the Primary Cell Atlas. The resulting RCA clusters for the different samples were plotted as heat maps or principal component analysis (PCA) as part of the downstream analysis pipeline of the rca package.
2.8. Pseudo‐bulk analysis
Processed RNA‐seq gene counts for all the analyzed cells were obtained from the supplementary datasets in Owen et al. (2018). Single‐cell datasets from four patients were selected (A, D, E, and F), where A and D are patients with Barrett's esophagus and E and F are normal esophageal mucosae of patients without Barrett's esophagus. For each of the four analyzed patients, a sample of 100 single cells was randomly selected and the processed counts from each patient were aggregated by obtaining the mean for each gene across the 100 sampled cells to emulate a pseudo‐bulk dataset. The resulting averaged counts were used as input to perform the RCA.
2.9. Gene ontology
After differential gene expression analysis was performed among the different flow cytometry‐sorted cell populations, genes whose expression was exclusively upregulated in comparison with the remaining cell types were analyzed with the go software (http://geneontology.org/; Ashburner et al., 2000; Carbon et al., 2019) to test for over‐representation of common biological processes using all genes as background. To investigate tumor/normal differential expression, all expressed genes of the respective cell type defined by the sum of normalized expression tag counts > 1 were used as background.
3. Results
3.1. Patients' characteristics
Tumor samples from nine patients were collected including seven male and two female patients. Median age of all patients participating was 65 years (minimum: 57 years, maximum: 83 years). Initial tumor grading was G2 in five patients and G3 in two patients, pT stage was pT1 in one patient, pT2 in three patients, pT3 in two patients, and pT4 in one patient (no information available for 2 patients since those patients did not undergo surgery due to metastasis). Four patients received neoadjuvant chemoradiation analog CROSS, and one patient received perioperative chemotherapy analog FLOT regimen. Full baseline characteristics are provided in Table 1.
Table 1.
Baseline characteristics of patient included.
| Variable | Total (n = 9) | |
|---|---|---|
| Number (n) | Percentage | |
| Age | ||
| Median (min–max) | 65 years (57–83 years) | |
| Gender | ||
| Male | 7 | 77.8 |
| Female | 2 | 22.2 |
| Anatomical localization | ||
| Esophagogastric junction | 8 | 88.9 |
| Gastric | 1 | 11.1 |
| Sample origin | ||
| Endoscopic biopsy | 6 | 66.7 |
| Surgical specimen | 3 | 33.3 |
| Samples | ||
| Tumor | 1 | 11.1 |
| Normal mucosa | 2 | 22.2 |
| Tumor and normal mucosa | 6 | 66.7 |
| Kind of neoadjuvant therapy | ||
| None | 4 | 44.4 |
| Neoadjuvant chemoradiation | 4 | 44.4 |
| Perioperative chemotherapy | 1 | 11.1 |
| pT category | ||
| pT1 | 1 | 11.1 |
| pT2 | 3 | 33.3 |
| PT3 | 2 | 22.2 |
| pT4 | 1 | 11.1 |
| No information a | 2 | 22.2 |
| pN category | ||
| pN0 | 3 | 33.3 |
| pN1 | 2 | 22.2 |
| pN2 | 0 | 0 |
| pN3 | 2 | 22.2 |
| No information a | 2 | 22.2 |
| Grading | ||
| G1 | 0 | 0 |
| G2 | 5 | 55.6 |
| G3 | 2 | 22.2 |
| No information | 2 | 22.2 |
No further information available since one patient had progression after primary staging and lost to follow‐up in another case.
3.2. Workflow
We developed a protocol for isolating the different cell populations (immune cells, fibroblasts, epithelial cells) from EAC and corresponding esophageal mucosa to sequence the transcriptome separately for each cell type. In brief, the workflow consists of a gentle single‐cell dissection, immunofluorescence staining, consecutive flow cytometry sorting, and RNA analysis of each cell fraction (Fig. 1).
Single‐cell suspension was obtained simultaneously by enzymatic dissociation using DNAse I, collagenase IV, and dispase II and simultaneous mechanical dissociation with a gentleMACS™ Dissociator. Antibody staining and subsequent sorting of the single‐cell suspension were implemented on literature‐based antigen selection (see below). An example of flow cytometry sorting strategy is shown in Fig. 2.
Fig. 2.
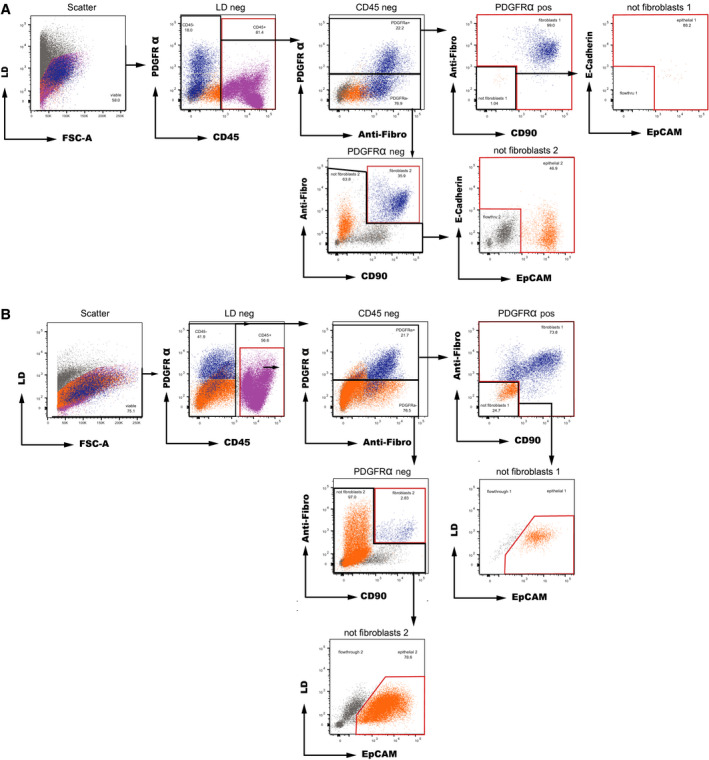
Representative sample processing for separation of EAC cell populations from endoscopic biopsies by flow cytometry sorting with E‐Cadherin co‐staining (A) and without E‐Cadherin co‐staining (B). After initial viability gating, cells were separated into the (a) CD45+ immune cell population (pink); (b) fibroblast cell population (blue), defined as positive for at least two out of three of PDGFRα, anti‐fibroblast, and CD90; and (c) epithelial cell population (dark orange) that were positive for EpCAM and/or E‐Cadherin (A) or simply positive for EpCAM (B). The sorted cell subpopulations are highlighted in red.
For sorting the immune cell population, we selected membrane antigen CD45, a glycoprotein which is expressed on nearly all hematopoietic cells except for mature erythrocytes and platelets (Nakano et al., 1990). CD45 has been revealed as a potent marker to differentiate hematopoietic cells from carcinoma cells in solid and fluid tumor tissue via flow cytometry (Acosta et al., 2016) and serves as a pan‐leukocyte marker (Ruffell et al., 2012), therefore ideal for detection of immune cells.
Epithelial cells were sorted for epithelial cell adhesion molecule (EpCAM), which is exclusively displayed in epithelia and epithelial‐derived neoplasms (Patriarca et al., 2012) and expressed in various carcinomas, including EAC (Sun et al., 2018). EpCAM antibodies are commonly used to detect epithelial cells in flow cytometry, for example, for detecting leptomeningeal metastasis of solid tumors in liquor (Milojkovic Kerklaan et al., 2016) or isolating epithelial cells from normal and tumor tissue (Bantikassegn et al., 2015; Sinha and Lowell, 2016).
Fibroblasts represent a heterogeneous cell population and display an inconsistent expression of surface markers (Lynch and Watt, 2018). Here, isolation of fibroblasts was based on detection of CD90 and PDGFRα and binding of an anti‐fibroblast antibody (Miltenyi, exact antigen is unknown). A cell was defined as ‘fibroblastic’ if positive for at least two of the three surface markers. CD90 has previously been used as a surface marker to isolate (cancer‐associated) fibroblasts in gastrointestinal mucosa and ovarian cancer tissue (Gedye et al., 2014; Kisselbach et al., 2009) and also serves as a widely expressed mesenchymal surface marker (Jiang and Rinkevich, 2018). Likewise, PDGFRα is described as a membrane antigen of mesenchymal cells (Houlihan et al., 2012) and supposed to participate in the recruitment of fibroblasts, pericytes, and endothelial cells during wound healing (Horikawa et al., 2015). There is evidence for a successful enrichment of fibroblasts in flow cytometry using PDGFRα as a surface marker (Pallangyo et al., 2015).
A total of 31 cell populations were successfully separated via flow cytometry and passed RNA‐seq for further analysis. Table 2 illustrates all samples which underwent the complete algorithm.
Table 2.
Origin of all cell populations included for further RNA‐seq.
| Patient No. | Sample origin | Successful RNA‐seq of following cell populations after FACS sorting | |||
|---|---|---|---|---|---|
| Tumor tissue | Sample labeling | Normal tissue | Sample labeling | ||
| 1 | Endoscopic biopsy |
|
|
|
|
| 2 | Endoscopic biopsy |
|
|
– | – |
| 3 | Endoscopic biopsy |
|
|
|
|
| 4 | Endoscopic biopsy |
|
|
|
|
| 5 | Endoscopic biopsy |
|
|
– | – |
| 6 | Surgical specimen | a | a |
|
|
| 7 | Endoscopic biopsy |
|
|
a | a |
| 8 | Surgical specimen | a | a |
|
|
| 9 | Surgical specimen |
|
|
|
|
| Total | N = 20 | N = 11 | |||
No samples taken.
We used gene ontology analysis, PCA, and RCA as well as population‐specific expression of marker genes to demonstrate the successful separation of the three cell types.
3.3. Principal component analysis
To confirm an obvious separation of the cell populations, we used dimensionality reduction (PCA) to summarize the data into two dimensions and then visually identify obvious clusters. Cell types sorted by flow cytometry that have similar expression profiles were clustered together. Based on the PCA plot, there were three clean clusters corresponding to the different expected cell types and the first two principal components explain most of the variability (Fig. 3). The PCA supports a successful separation in three different cell populations.
Fig. 3.
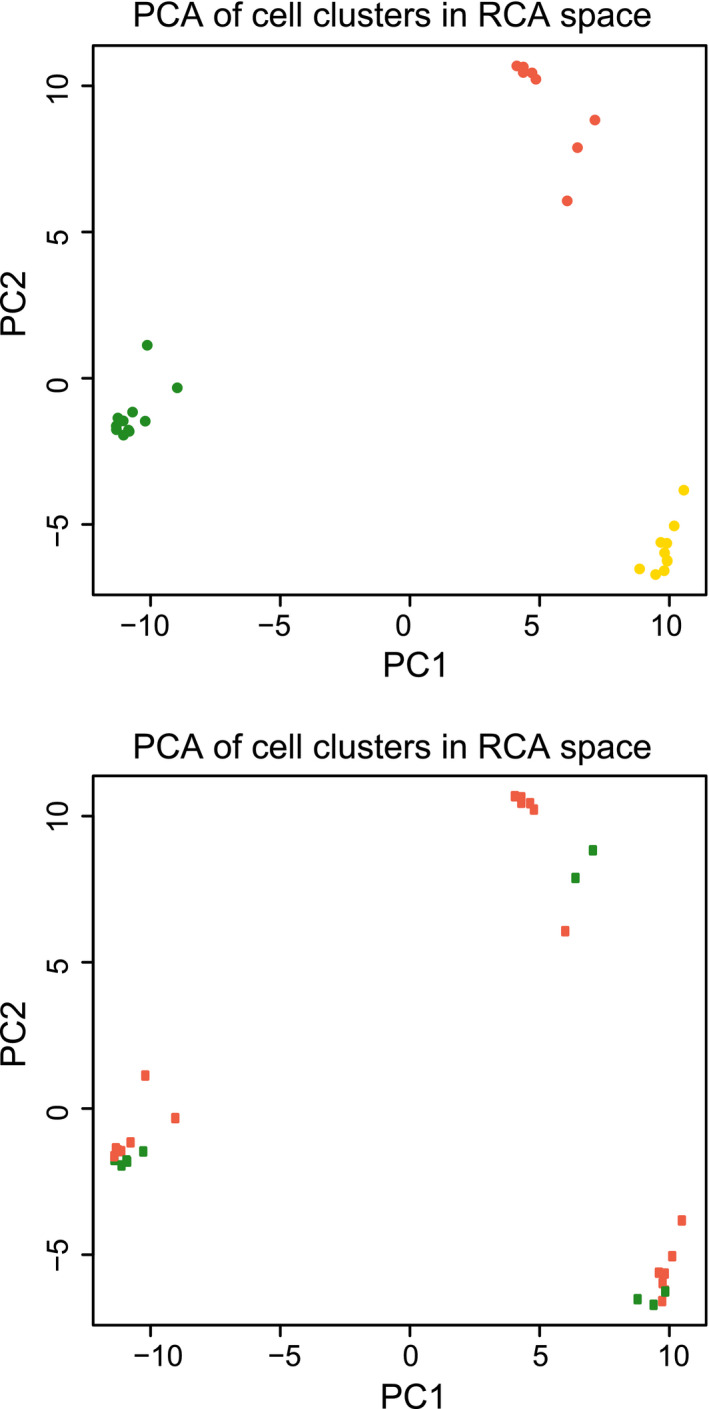
Principal component analysis of RNA‐seq of flow cytometry‐sorted cell fractions. Thirty‐one cell fractions were 3′RNA‐sequenced and plotted by their first two principal components. Top: orange, epithelial cells; yellow, fibroblasts; green, leukocytes. Bottom: orange, tumor cells; green, normal cells.
3.4. Gene ontology analysis
Gene ontology (GO) is a widely used method to structure a large number of genes in certain categories (GO terms) describing biological processes, molecular functions, or cell components. Based on differential gene expression (Table S2) between the three cell populations, we created a list of the top upregulated genes for each cell type and matched them to specific GO terms of biological processes in which they were significantly overrepresented (FDR < 0.05). In the following, we list the top five GO terms for each sorted cell fraction, and for a complete overview, we refer to the Table S2.
Cells sorted for ‘immune cell population’ showed an enrichment in the categories immune system process, immune response, regulation of immune system process, regulation of immune response, and cell activation, whereas sorted cells of the ‘fibroblast cell population’ were enriched for extracellular matrix organization, anatomical structure morphogenesis, extracellular structure organization, multicellular organism development, and anatomical structure development. Those cells of the ‘epithelial cell population’ had highly expressed genes in GO terms tissue development, epithelium development, epidermis development, epithelial cell differentiation, and cornification.
Gene ontology analysis illustrated expected cell type‐specific GO terms of biological processes for the three sorted cell populations, indicating a successful enrichment of the respective target cell types.
3.5. Marker gene analysis
To further confirm the separation of the three cell types, we considered marker genes of our target cell populations and evaluated their distribution within the differently expressed gene lists. Within the immune cell population, there was a significant upregulation of surface proteins CD3 (gene name: CD3D), CD4 (CD4), CD8 (CD8A + CD8B), CD19 (CD19), and CD20 (MS4A1), all established markers for B/T lymphocytes. Furthermore, monocyte marker CD14 (CD14) was significantly upregulated as well as granulocyte marker CD11b (ITGAM).
In the fibroblast cell population, there was a significant increase in fibroblast activation protein (FAP), commonly described as a marker for (myo)fibroblasts (Shiga et al., 2015). FAP is a surface peptidase with mostly unknown substrates which is involved in numerous physiological processes such as inhibition of fibrinolysis (Hamson et al., 2014). In addition, we observed an upregulation of alpha‐smooth muscle actin/alpha SMA (ACTA2) expression, characteristic for fibroblastic cells and which has been widely described in CAFs (Sharon et al., 2013). We further observed in the fibroblast population a significant upregulation of expression of collagen genes (e.g., COL1A1 and COL3A1), responsible for the formation of the extracellular matrix (Yue, 2014).
E‐Cadherin (CDH1) is a key epithelial marker, responsible for an epithelial barrier between neighboring cells via binding to adjacent cadherins and cytoskeleton (Serrano‐Gomez et al., 2016). CDH1 was significantly upregulated in the sorted epithelial cells. Similarly, we found a significant increase in expression of epithelial membrane antigen (EMA, also known as MUC1) in this population. EMA is expressed in various epithelia and responsible for a physical barrier and anti‐adhesive property of the tissue (Nath and Mukherjee, 2014).
In conclusion, cell type‐specific genes showed a significant enrichment in their corresponding target cell populations, supporting the expected cell separation.
3.6. Reference component analysis
Reference component analysis is used to compare gene expression data sets to reference cell lineages. We compared each of the 31 sorted cell fractions by RCA with reference data (Fig. 4). RCA indicated consistent accordance to reference transcriptome data within equally sorted cells. There were three differential mapping (transcriptome correlation) patterns, each belonged to one of the three sorted cell types (‘immune cell population’, ‘fibroblast cell population’, ‘epithelial cell population’), confirming the qualitative sufficiency of the separation process.
Fig. 4.
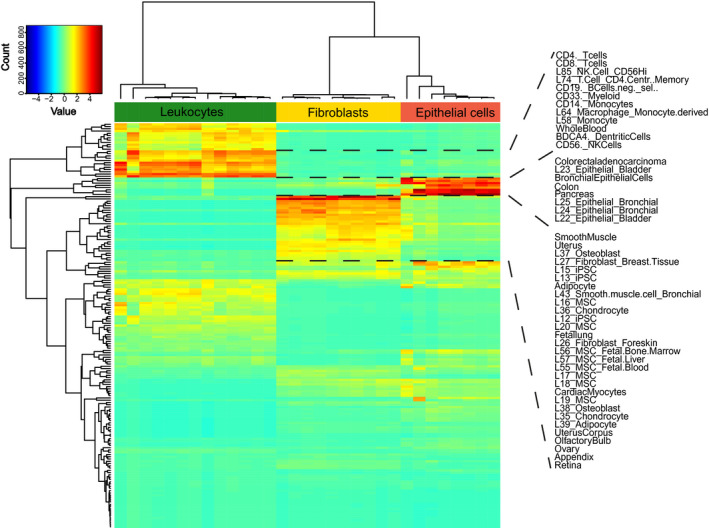
Reference component analysis heat map of the cell populations' transcriptomic profiles correlated with expression profiles of reference tissues and cell lineages. RNA‐seq data of 31 cell fractions plotted in columns with their color‐coded Spearman's correlation values relative to reference expression datasets represented in rows. Cluster color code corresponds to colors in Fig. 3.
Cells sorted as ‘immune cells’ showed a high correlation with reference gene expression of inflammatory cells, for example, natural killer cells, T cells, B cells, monocytes, and macrophages. Cells of the ‘epithelial cell population’ matched to epithelial cell lines of bladder, lung, and colon and non‐specific cells derived from tissue of pancreas and colon. Further correlations were found for trachea and intestinal tissue. Cells sorted as ‘fibroblasts’ revealed correlation with fibroblast cell lines, various mesenchymal stem cell lines, adipocytes, hematopoietic stem cells, endothelial cells, and smooth muscles cells (and organs with high proportion of smooth muscle cells, e.g., uterus). Cancer‐associated fibroblasts (CAFs) have been reported to arise from various origins, for example, normal fibroblasts, adipocytes, bone marrow‐derived cells (including mesenchymal stem cells), and endothelial cells (CD31+) (Wang et al., 2016). Correlations with transcriptome data of these differential cell types might reflect the heterogeneous origin of the fibroblastic cells.
We explored how similar our cell type bulk sequencing was compared with scRNA‐seq and co‐analyzed esophagus epithelial scRNA‐seq data of Owen et al. (2018) with epithelial cell types of our analyses. We observed largely congruent correlations with reference transcriptomes (Figs S1 and S2) indicating that the cell type bulk sequencing is pure enough for a representative snapshot of these cells. As expected, smaller cell populations with individual expression profiles can only be seen in scRNA‐seq. Of note, the cell type‐specific sequencing detected more genes per sample compared with scRNA‐seq per cell (average 12 142 vs. 3260 to 4391 genes with ≥ 1 read).
3.7. Differently expressed genes between EAC and normal esophageal mucosa
Although the number of tumor–normal pairs does not allow general interpretations, we exploratively compared differentially expressed genes between EAC tissue and normal esophageal mucosa for each sorted cell population (Table S3). We first used GO analysis to categorize generic changes. Only the tumor/normal comparison of the fibroblast cell population showed significant enrichment of GO categories. Interestingly, blood vessel development, angiogenesis, and vasculature development were among the top 10 significantly enriched categories of biological processes (Table S4). These categories remained significant when restricting the analysis to upregulated genes, while downregulated genes were strongly enriched for the establishment of protein localization to endoplasmic reticulum, cotranslational protein targeting to membrane, and regulation of developmental process (Table S4). When focusing on individual genes with reported functions, the fibroblastic population showed strong upregulation of matrix metalloproteinase 11 (MMP11) in tumors (adjusted P = 1.6 × 10−15, Fisher's exact test). MMP11 serves as an endopeptidase‐degrading extracellular matrix (Gómez‐Macías et al., 2018). MMP11 overexpression in CAFs and cancer cells has previously been described to correlate with an aggressive cancer profile and promotion of metastasis (González de Vega et al., 2019; Peruzzi et al., 2009). Further, an upregulation of glycoprotein CD38 was detected in the sorted immune cells from EAC tumors (P = 8.6 × 10−7, Fisher's exact test). CD38, usually expressed on plasma cells and other lymphoid and myeloid cell populations (Morandi et al., 2018), has been revealed to mediate immunosuppression as a tumor escape mechanism, and there is evidence for an unfavorable CD38 influence on tumor progression in esophageal cancer (Chen et al., 2018; Karakasheva et al., 2015).
4. Discussion
In an effort to create a scalable approach for cell type‐specific comparison of tumors, we have developed a protocol to dissociate fresh biopsies of EACs and corresponding esophageal mucosa, and sort leukocytes (CD45+), epithelial cells (EpCAM+), and fibroblasts (positive for at least two out of PDGFRα, CD90, anti‐fibroblast), followed by 3′RNA sequencing. This workflow allows us to investigate the transcriptomic changes in the three cell populations in EAC when compared to normal esophageal mucosa. Importantly, this approach is significantly more economic and scalable for investigating transcriptomes of different cell types compared with single‐cell RNA sequencing (scRNA‐seq) and can therefore serve as a cost‐effective alternative to broaden understanding of tumor biology in EAC. In our local setting, the costs for the analysis of a tumor/normal pair with three cell types each by scRNA‐seq (hashing for tumor and normal) would be 3.2 times higher compared with cell type‐specific RNA‐seq.
In order to prove the successful workflow of this approach, we demonstrate a clear separation of our three target cell populations (fibroblasts, immune cells, and epithelial cells) and performed 3′RNA sequencing of these sorted cell types. We used different strategies to verify the identity of the cells. GO analysis elucidated functionally compatible and expected biological processes for the respective cell populations. We provide further evidence for a successful separation by comparing transcriptomic profiles with reference expression data using RCA and obtained three different expression correlation patterns, each specific for the respective target population: Cells sorted for epithelial cell compartment showed high correlation with epithelial cell lineages, the immune cell compartment with reference data of differential lymphocytes, monocytes, and granulocytes and fibroblasts with fibroblasts, various mesenchymal cell lines, and cell lineages that can be considered plausible to serve as origins of CAFs. Literature‐based marker gene analysis within differentially expressed genes further supported a qualitative separation into the three cell compartments.
We focused on optimizing the protocol for small amounts of tissue, allowing to obtain the samples endoscopically at primary staging. Up‐scaling of our protocol will enable us to compare biopsies of treatment‐naïve EAC tissue with normal esophageal mucosa of patient cohorts at moderate cost. Such systematic cell type‐specific analysis of treatment‐naïve, endoscopically obtained samples and correlation with clinical features will allow to find new cell type‐specific pathway alterations, responsible, for example, for treatment response or relapse.
Although we show a potential application of our protocol for biopsies and surgical specimens in normal esophageal mucosa and EAC tissue, the protocol can be further improved. Especially, freezing of dissociated single‐cell suspensions, necessary due to processing logistics, was responsible for a substantial loss of living cells. A continuous workflow without freezing will improve the cell viability and quantity of RNA for sequencing. Since the different samples have varying proportions of the three cell populations, we were not able to obtain sufficient cells for all sorted cell populations. This may explain the lack of significantly enriched GO categories for the leukocyte and epithelial compartments. An increase in the number of viable cells by a continuous workflow will enhance the chance to robustly obtain sufficient amounts of RNA for subsequent 3′RNA‐seq.
In order to demonstrate the potential utility of our approach, we finally compared differently expressed genes between normal esophageal mucosa and EAC tissue and highlighted cell type‐specific alterations. For instance, CD38 upregulation in immune cells of EAC could establish a possible target of immunotherapy, as suggested in combination with anti‐PD‐L1 therapy (Chen et al., 2018; Mittal et al., 2018). For MMP11, which is usually expressed both in epithelial cells and fibroblasts, we found an upregulation restricted to the fibroblast cell population as described earlier (Pedersen et al., 2009). Although sample numbers used here are limited, it is interesting to note that fibroblasts in the tumor showed upregulation of angiogenesis genes, when compared to normal mucosa. Angiogenesis‐promoting properties of CAFs have been described earlier (reviewed in Wang et al., 2019) and are an area of intense investigation in the search for new angles of intervention. Future implementation of the protocol in a large cohort of patients can therefore help to identify targets for novel therapy concepts and complement our understanding of tumor biology in EAC.
In conclusion, we present a new approach for a cost‐effective and scalable procedure to determine cell type‐specific transcriptome alterations in treatment‐naïve EAC tissue with the potential to obtain treatment‐relevant findings in large cohorts of patients with EAC.
Author contributions
MK, PSP, AQ, HS, S‐HC, and AMH designed the study. MK, PSP, KF‐D, MT, IG‐M, CW, SB, JW, MF, and JA carried out the experiments. PSP, HS, WS, CJB, HA, and S‐HC collected and selected the patients' samples. HL, AQ, and RB performed the histopathological examinations. OVC performed bioinformatic analyses. MK, PSP, OVC, CW, AQ, and AMH interpreted the data. MK, PSP, OVC, and AMH wrote the manuscript with contributions of CW and KF‐D and approval from all authors.
Conflict of interest
MK is supported by the Koeln Fortune Program/Faculty of Medicine, University of Cologne, grant number 410/2018. PSP is fellow of the Else Kröner Forschungskolleg Cologne ‘Clonal Evolution in Cancer’ (2016‐Kolleg‐19). All other authors declare no conflict of interest.
Supporting information
Fig. S1. Joint normal esophageal mucosa RCA of scRNA‐seq and cell type RNA‐seq.
Fig. S2. Normal esophageal mucosa RCA of pseudo‐bulk scRNA‐seq.
Table S1. Complete lists of DEGs in a pairwise manner.
Table S2. List of the top five Gene Ontology terms for each sorted cell fraction.
Table S3. Differentially expressed genes between EAC tissue and normal esophageal mucosa for each sorted cell population (Gene Ontology Analysis).
Table S4. Top ten significantly enriched categories of biological processes between EAC tissue and normal esophageal mucosa for each sorted cell population (Gene Ontology Analysis).
Acknowledgements
We thank the patients for donation of samples. This study was supported by funds of the Medical Faculty of the University Hospital Cologne (to MK and AMH), the Institute of Pathology of the University Hospital Cologne (to AMH) and the Else Kröner‐Fresenius‐Foundation (to PSP).
Max Krämer and Patrick S. Plum contributed equally
Contributor Information
Seung‐Hun Chon, Email: seung-hun.chon@uk-koeln.de.
Axel M. Hillmer, Email: ahillmer@uni-koeln.de.
Data accessibility
The RNA‐seq data have been deposited at the European Genome‐Phenome Archive and can be accessed via the accession number EGAS00001004053.
References
- Acosta M, Pereira J and Arroz M (2016) Screening of carcinoma metastasis by flow cytometry: a study of 238 cases. Cytometry B Clin Cytom 90, 289–294. [DOI] [PubMed] [Google Scholar]
- Al‐Batran S‐E, Hofheinz RD, Pauligk C, Kopp H‐G, Haag GM, Luley KB, Meiler J, Homann N, Lorenzen S, Schmalenberg H et al (2016) Histopathological regression after neoadjuvant docetaxel, oxaliplatin, fluorouracil, and leucovorin versus epirubicin, cisplatin, and fluorouracil or capecitabine in patients with resectable gastric or gastro‐oesophageal junction adenocarcinoma (FLOT4‐AIO): results from the phase 2 part of a multicentre, open‐label, randomised phase 2/3 trial. Lancet Oncol 17, 1697–1708. [DOI] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantikassegn A, Song X and Politi K (2015) Isolation of epithelial, endothelial, and immune cells from lungs of transgenic mice with oncogene‐induced lung adenocarcinomas. Am J Respir Cell Mol Biol 52, 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbon S, Douglass E, Dunn N, Good B, Harris NL, Lewis CJ, Mungall S, Basu RL, Chisholm RJ, Dodson E et al (2019) The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res 47, D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, Villalobos PA, Cascone T, Liu X, Tan L et al (2018) CD38‐mediated immunosuppression as a mechanism of tumor cell escape from PD‐1/PD‐l1 blockade. Cancer Discov 8, 1156–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HG, Xie SH and Lagergren J (2018) The epidemiology of esophageal adenocarcinoma. Gastroenterology 154, 390–405. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye CA, Hussain A, Paterson J, Smrke A, Saini H, Sirskyj D, Pereira K, Lobo N, Stewart J, Go C et al (2014) Cell surface profiling using high‐throughput flow cytometry: a platform for biomarker discovery and analysis of cellular heterogeneity. PLoS ONE 9, e105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez‐Macías GS, Garza‐Rodríguez ML, Garza‐Guajardo R, Monsiváis‐Ovalle D, Ancer‐Rodríguez J, Barrera‐Saldaña HA and Barboza‐Quintana O (2018) Overexpression of the matrix metalloproteinase 11 gene is a potential biomarker for type 1 endometrial cancer. Oncol Lett 16, 1073–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González de Vega R, Clases D, Fernández‐Sánchez ML, Eiró N, González LO, Vizoso FJ, Doble PA and Sanz‐Medel A (2019) MMP‐11 as a biomarker for metastatic breast cancer by immunohistochemical‐assisted imaging mass spectrometry. Anal Bioanal Chem 411, 639–646. [DOI] [PubMed] [Google Scholar]
- Hamson EJ, Keane FM, Tholen S, Schilling O and Gorrell MD (2014) Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl 8, 454–463. [DOI] [PubMed] [Google Scholar]
- Horikawa S, Ishii Y, Hamashima T, Yamamoto S, Mori H, Fujimori T, Shen J, Inoue R, Nishizono H, Itoh H et al (2015) PDGFRα plays a crucial role in connective tissue remodeling. Sci Rep 5, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houlihan DD, Mabuchi Y, Morikawa S, Niibe K, Araki D, Suzuki S, Okano H and Matsuzaki Y (2012) Isolation of mouse mesenchymal stem cells on the basis of expression of Sca‐1 and PDGFR‐α. Nat Protoc 7, 2103–2111. [DOI] [PubMed] [Google Scholar]
- Incorvaia L, Fanale D, Badalamenti G, Barraco N, Bono M, Corsini LR, Galvano A, Gristina V, Listì A, Vieni S et al (2019) Programmed death ligand 1 (PD‐L1) as a predictive biomarker for pembrolizumab therapy in patients with advanced non‐small‐cell lung cancer (NSCLC). Adv Ther 36, 2600–2617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D and Rinkevich Y (2018) Defining skin fibroblastic cell types beyond CD90. Front Cell Dev Biol 6, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarla S, Song XT and Gottschalk S (2012) Cancer‐associated fibroblasts as targets for immunotherapy. Immunotherapy 4, 1129–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasheva TA, Waldron TJ, Eruslanov E, Kim SB, Lee JS, O'Brien S, Hicks PD, Basu D, Singhal S, Malavasi F et al (2015) CD38‐expressing myeloid‐derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res 75, 4074–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselbach L, Merges M, Bossie A and Boyd A (2009) CD90 expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology 59, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai D, Ma L and Wang F (2012) Fibroblast activation protein regulates tumor‐associated fibroblasts and epithelial ovarian cancer cells. Int J Oncol 41, 541–550. [DOI] [PubMed] [Google Scholar]
- Li H, Courtois ET, Sengupta D, Tan Y, Chen KH, Goh JJL, Kong SL, Chua C, Hon LK, Tan WS et al (2017) Reference component analysis of single‐cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat Genet 49, 708–718. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W and Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MD and Watt FM (2018) Fibroblast heterogeneity: implications for human disease. J Clin Invest 128, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh D, Suchak K, Moutasim KA, Vallath S, Hopper C, Jerjes W, Upile T, Kalavrezos N, Violette SM, Weinreb PH et al (2011) Stromal features are predictive of disease mortality in oral cancer patients. J Pathol 223, 470–481. [DOI] [PubMed] [Google Scholar]
- Milojkovic Kerklaan B, Pluim D, Bol M, Hofland I, Westerga J, Van Tinteren H, Beijnen JH, Boogerd W, Schellens JHM and Brandsma D (2016) EpCAM‐based flow cytometry in cerebrospinal fluid greatly improves diagnostic accuracy of leptomeningeal metastases from epithelial tumors. Neuro Oncol 18, 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal D, Vijayan D and Smyth MJ (2018) Overcoming acquired PD‐1/PD‐L1 resistance with CD38 blockade. Cancer Discov 8, 1066–1068. [DOI] [PubMed] [Google Scholar]
- Morandi F, Horenstein AL, Costa F, Giuliani N, Pistoia V and Malavasi F (2018) CD38: a target for immunotherapeutic approaches in multiple myeloma. Front Immunol 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Harada T, Morikawa S and Kato Y (1990) Expression of leukocyte common antigen (CD45) on various human leukemia/lymphoma cell lines. Pathol Int 40, 107–115. [DOI] [PubMed] [Google Scholar]
- Nath S and Mukherjee P (2014) MUC1: a multifaceted oncoprotein with a key role in cancer progression. Trends Mol Med 20, 332–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen RP, White MJ, Severson DT, Braden B, Bailey A, Goldin R, Wang LM, Ruiz‐Puig C, Maynard ND, Green A et al (2018) Single cell RNA‐seq reveals profound transcriptional similarity between Barrett's oesophagus and oesophageal submucosal glands. Nat Commun 9, 4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallangyo CK, Ziegler PK and Greten FR (2015) IKKβ acts as a tumor suppressor in cancer‐associated fibroblasts during intestinal tumorigenesis. J Exp Med 212, 2253–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarca C, Macchi RM, Marschner AK and Mellstedt H (2012) Epithelial cell adhesion molecule expression (CD326) in cancer: a short review. Cancer Treat Rev 38, 68–75. [DOI] [PubMed] [Google Scholar]
- Pedersen G, Saermark T, Kirkegaard T and Brynskov J (2009) Spontaneous and cytokine induced expression and activity of matrix metalloproteinases in human colonic epithelium. Clin Exp Immunol 155, 257–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi D, Mori F, Conforti A, Lazzaro D, De Rinaldis E, Ciliberto G, La Monica N and Aurisicchio L (2009) MMP11: a novel target antigen for cancer immunotherapy. Clin Cancer Res 15, 4104–4113. [DOI] [PubMed] [Google Scholar]
- Plum PS, Hölscher AH, Pacheco Godoy K, Schmidt H, Berlth F, Chon S‐H, Alakus H and Bollschweiler E (2018) Prognosis of patients with superficial T1 esophageal cancer who underwent endoscopic resection before esophagectomy‐A propensity score‐matched comparison. Surg Endosc 32, 3972–3980. [DOI] [PubMed] [Google Scholar]
- Ren X, Kang B and Zhang Z (2018) Understanding tumor ecosystems by single‐cell sequencing: promises and limitations. Genome Biol 19, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Au A, Rugo HS, Esserman LJ, Hwang ES and Coussens LM (2012) Leukocyte composition of human breast cancer. Proc Natl Acad Sci USA 109, 2796–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano‐Gomez SJ, Maziveyi M and Alahari SK (2016) Regulation of epithelial‐mesenchymal transition through epigenetic and post‐translational modifications. Mol Cancer 15, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon Y, Alon L, Glanz S, Servais C and Erez N (2013) Isolation of normal and cancer‐associated fibroblasts from fresh tissues by fluorescence activated cell sorting (FACS). J Vis Exp 71, e4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiga K, Hara M, Nagasaki T, Sato T, Takahashi H and Takeyama H (2015) Cancer‐associated fibroblasts: their characteristics and their roles in tumor growth. Cancers (Basel) 7, 2443–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M and Lowell CA (2016) Isolation of highly pure primary mouse alveolar epithelial type ii cells by flow cytometric cell sorting. Bio Protoc 6, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spagnolo F, Boutros A, Tanda E and Queirolo P (2019) The adjuvant treatment revolution for high‐risk melanoma patients. Semin Cancer Biol 59, 283–289. [DOI] [PubMed] [Google Scholar]
- Sun X, Martin RCG, Zheng Q, Farmer R, Pandit H, Li X, Jacob K, Suo J and Li Y (2018) Drug‐induced expression of EpCAM contributes to therapy resistance in esophageal adenocarcinoma. Cell Oncol (Dordr) 41, 651–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Nakamura J and Noshiro H (2017) Promising immunotherapies for esophageal cancer. Expert Opin Biol Ther 17, 723–733. [DOI] [PubMed] [Google Scholar]
- Tsujino T, Seshimo I, Yamamoto H, Chew YN, Ezumi K, Takemasa I, Ikeda M, Sekimoto M, Matsuura N and Monden M (2007) Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res 13, 2082–2090. [DOI] [PubMed] [Google Scholar]
- Vallböhmer D, Hölscher AH, Demeester S, Demeester T, Salo J, Peters J, Lerut T, Swisher SG, Schröder W, , Bollschweiler E et al (2010) A multicenter study of survival after neoadjuvant radiotherapy/chemotherapy and esophagectomy for ypT0N0M0R0 esophageal cancer. Ann Surg 252, 744–748. [DOI] [PubMed] [Google Scholar]
- van Hagen P, Hulshof MCCM, van Lanschot JJB, Steyerberg EW, Henegouwen MIVB, Wijnhoven BP, Richel DJ, Nieuwenhuijzen GA, Hospers GA, Bonenkamp JJ et al (2012) Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med 366, 2074–2084. [DOI] [PubMed] [Google Scholar]
- Wang F‐T, Sun W, Zhang J‐T and Fan Y‐Z (2019) Cancer‐associated fibroblast regulation of tumor neo‐angiogenesis as a therapeutic target in cancer. Oncol Lett 17, 3055–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhang G, Wang J, Wang L, Huang X and Cheng Y (2016) The role of cancer‐associated fibroblasts in esophageal cancer. J Transl Med 14, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Ogawa T, Zhang X, Hanamura N, Kashikura Y, Takamura M, Yoneda M and Shiraishi T (2012) Role of stromal myofibroblasts in invasive breast cancer: stromal expression of alpha‐smooth muscle actin correlates with worse clinical outcome. Breast Cancer 19, 170–176. [DOI] [PubMed] [Google Scholar]
- Yue B (2014) Biology of the extracellular matrix: an overview. J Glaucoma 23, S20–S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Joint normal esophageal mucosa RCA of scRNA‐seq and cell type RNA‐seq.
Fig. S2. Normal esophageal mucosa RCA of pseudo‐bulk scRNA‐seq.
Table S1. Complete lists of DEGs in a pairwise manner.
Table S2. List of the top five Gene Ontology terms for each sorted cell fraction.
Table S3. Differentially expressed genes between EAC tissue and normal esophageal mucosa for each sorted cell population (Gene Ontology Analysis).
Table S4. Top ten significantly enriched categories of biological processes between EAC tissue and normal esophageal mucosa for each sorted cell population (Gene Ontology Analysis).
Data Availability Statement
The RNA‐seq data have been deposited at the European Genome‐Phenome Archive and can be accessed via the accession number EGAS00001004053.