

Abstract
Rapid, specific, and sensitive detection of pathogenic bacteria in drink, food, and clinical samples is an important goal for public health. In addition, rapid characterization of antibiotic susceptibility could inform clinical choices and improve antibiotic stewardship. We previously reported a straightforward, inexpensive strategy to detect Gram-negative bacterial pathogens, including Pseudomonas aeruginosa, Vibrio cholerae, and Escherichia coli, taking advantage of the high affinity and specificity of phages for their bacterial hosts. Chimeric phages targeted different bacterial pathogens, and thiolation of the phages induced aggregation of gold nanoparticles (AuNPs), leading to a visible colorimetric response in the presence of at least ∼100 cells of the target bacteria. Here, we apply this strategy to complex biological samples (milk, urine, and swabs from a porcine ex vivo model of P. aeruginosa infection). We also show that this assay can be used to identify the antibiotic susceptibility profile based on detection of bacterial growth in the presence of different antibiotics. The prospect for using phage-conjugated AuNPs to detect bacterial pathogens in clinical samples and guide antibiotic choice is discussed.
Keywords: bacteriophage, bacterial detection, gold nanoparticles, antibiotic sensitivity, antibiotic susceptibility profile, biosensor
Infectious bacterial pathogens represent a major threat to human health worldwide.1 The CDC’s 2019 Antibiotic Resistance Threats report cites diagnostics as a critical area for development, including antibiotic susceptibility testing. Rapid diagnostic testing is associated with improved clinical outcomes and reduced hospital costs.2,3 Accurate diagnosis at the point of care is also important for antibiotic stewardship,4 as an estimated 30% of antibiotic prescriptions in the outpatient setting are inappropriate.5 Current biodetection methods include culture and cell counting of bacteria,6 antibody-based detection (e.g., enzyme-linked immunosorbent assay (ELISA)),7,8 nucleic acid-based detection (e.g., polymerase chain reaction (PCR)),9,10 and other methods such as mass spectrometry and bioluminescence.11−14 Each of these methods has recognized drawbacks for an outpatient setting, such as reliance on specialized equipment or longer detection times (hours to days).15 Additional drawbacks that particularly impact low-resource settings include the susceptibility of enzymes16,17 and antibodies to aggregation and loss of activity under harsh conditions18−21 and the sensitivity of reactions (e.g., PCR) to complex samples. Improved strategies for diagnosing bacterial infections are needed.
In addition to identifying the causative organisms, characterizing the antibiotic susceptibility profiles (ASPs) of an infectious bacterial strain is an important but typically slow task.22 Methods can be either based on phenotype, requiring observation of bacterial growth (or lack thereof) in the presence of the antibiotic, or genotype, requiring knowledge of gene sequences causing antibiotic resistance. The classic phenotypic antibiotic susceptibility test involves culturing bacteria with and without antibiotics and typically yields results in a few days. This delay means that clinicians often must make a best guess at the causative organism and its ASP on the basis of epidemiological and other factors. In critical care settings, the time to appropriate antibiotic treatment is an important determinant of clinical outcomes, leading to a tendency to prescribe broad-spectrum antibiotics and thus suboptimal antibiotic stewardship.23 Although genotypic methods can be faster and more sensitive than phenotypic methods, they are prone to both false positives, as they detect the gene for resistance rather than the phenotypic expression, and to false negatives, if genetic mechanisms of resistance are not fully known. Thus, development of rapid phenotypic diagnostics for ASPs is an area in considerable need for improvement.
Bacteriophages (phages) are a natural source of molecular diagnostics for bacteria, as phages have evolved in uncontrolled environments over billions of years to attach to and infect targeted bacterial cells. Unlike antibodies, the interaction between phages and their host bacteria can be quite robust in suboptimal environments.24 The life cycle of lytic phages25 has previously been exploited in bacterial detection.26 For example, colorimetric detection of Escherichia coli cells can be achieved using T7 phage engineered to produce β-galactosidase (β-gal).27 Cell lysis released β-gal, which hydrolyzed p-aminophenyl β-d-galactopyranoside to produce p-aminophenol (PAP). Reduction of silver ions by PAP yielded a silver shell on the surface of gold nanorods, resulting in a blue-shift of the surface plasmon resonance (SPR) peak and a color change. This strategy requires development of an engineered phage capable of infection and gene expression on the targeted host cell. Alternatively, another detection method used lytic phage isolated from the environment to cause targeted cell lysis.28 The activity of adenylate kinase released by the cells caused conversion of adenosine diphosphate into adenosine triphosphate, which was detected by a bioluminescent assay. This strategy requires knowledge of the conditions for phage lysis, and the possibility of contamination cannot be ignored. On the other hand, nonlytic phages29 can also be utilized, such as the filamentous phage M13, if the readout does not depend on cell lysis.30,31 For example, Belcher et al. reported single-wall carbon nanotubes functionalized by M13 phage for in vivo bacterial imaging.32 Although not suitable for point-of-care or resource-limited situations, this work demonstrates the utility of combining the targeting specificity of M13 with nanoscale structures for detection.
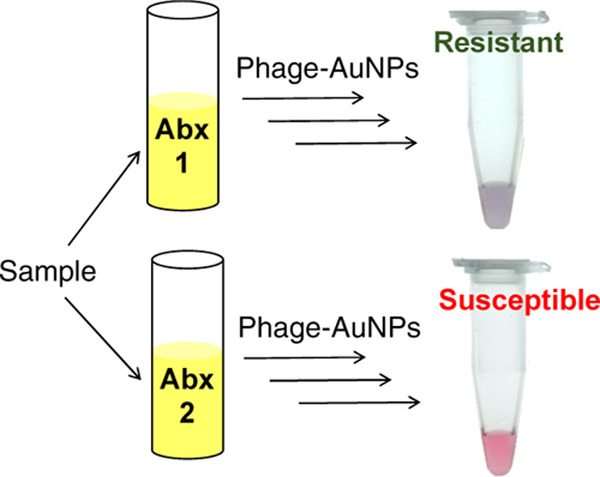
We recently reported a phage-based strategy for rapid, sensitive, and specific detection of bacteria using engineered M13, in which the receptor-binding protein (RBP) domain of g3p had been swapped for the corresponding RBP domain from another filamentous phage (e.g., Pf1), causing the chimeric phage to attach to the host of the other phage (e.g., Pseudomonas aeruginosa; Text S1).33 The chimeric phage cannot (and need not) complete an infection cycle in E. coli, due to lack of attachment, or on the alternative host, due to lack of compatible machinery for downstream infection and propagation, and thus serves essentially as an affinity reagent for the bacteria. The chimeric phages are also thiolated, so they bind to and induce aggregation of gold nanoparticles (AuNPs), which results in a visible shift of the SPR absorbance spectrum. AuNP aggregation has been used similarly for other biosensing applications.34−36 In the assay, the sample containing bacteria is mixed with the chimeric phage and the cells (with or without phages attached) are spun down (Figure 1). When the pellet is resuspended with AuNPs, the color of the solution indicates either free AuNPs (red, i.e., undetectable bacterial cells) or AuNPs assembled onto phages (purple, i.e., more than ∼100 bacterial cells). Because only the binding between phages and bacteria is necessary for detection, both live and dead bacteria should be detectable as long as the host receptor protein is still able to bind the RBP. This simple assay is rapid, inexpensive, extensible to several bacterial species including P. aeruginosa, an important human pathogen,37,38 and potentially compatible with low-resource settings.
Figure 1.

Scheme for bacterial detection by phage AuNPs. The thiolated phages were added to a sample containing bacteria recognized by the phage (a) or not recognized by the phage (b). The cell–phage complexes (a) or nonhost cells (b) were separated from free phage by centrifugation. Resuspension of cell pellets containing thiolated phage-induced aggregation of AuNPs (a), producing a color change from red (AuNPs) to purple (aggregates of AuNPs), while nonhost cells do not cause aggregation of AuNPs and thus cause no color change (b).
In the current work, we verify that the phage–AuNP assay is tolerant to complex biological media, including milk, urine, and swabs of a model of P. aeruginosa infection, and we study the robustness of the assay to the size and functionalization of the gold nanoparticles. Furthermore, we demonstrate how this assay could be used to rapidly determine the phenotypic antibiotic susceptibility profile of the targeted bacteria.
Experimental Section
Materials
Reagents were obtained from the following sources: gold(III) chloride trihydrate (HAuCl3, 99.9%, Sigma), sodium borohydride (NaBH4, 98%, Fisher Scientific), trisodium citrate dehydrate (99.9%, Sigma), P. aeruginosa (Schroeter) Migula (ATCC25102), Vibrio cholerae 0395 (donation from Prof. Michael J. Mahan, UCSB), M13KE phage (NEB), M13-NotI-Kan construct,39 sodium chloride (NaCl, 99%, Fisher BioReagents), tryptone (99%, Fisher BioReagents), yeast extract (99%, Fisher BioReagents), E. coli ER2738 (NEB), N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride (EDC, 99%, Sigma), N-hydroxysuccinimide (NHS, 98%, Sigma), cysteamine (98%, Sigma), poly(ethylene glycol) (PEG-8000, Sigma), dialysis kit (MWCO 3500 Da, Spectrum Laboratories), tetracycline (Sigma), thiol-PEG-acid (HOOC-PEG-SH, PEG average Mn 5000 Da, Sigma), kanamycin sulfate (Sigma), ampicillin sodium salt (Fisher BioReagents), Mix and Go competent cells (Zymo Research), QIAprep Spin Miniprep kit (Qiagen), QIAquick Gel Extraction kit (Qiagen), KpnI-HF/NotI-HF restriction enzyme and T4 DNA ligase (NEB), and QuickDetect E. coli Protein (ECP) ELISA kit (BioVision).
Chimeric Phages
The construction of the chimeric phages used here (M13-g3p(CTXϕ) and M13-g3p(Pf1)) was previously reported.33 Phages were propagated and quantified by real-time PCR, as previously described.33 See Supporting Methods for more details.
Thiol Functionalization of Phages
At least three solvent-accessible carboxylic acids are present near the N-terminus of g8p of M13 (Glu2, Asp4, and Asp5), which can be used for chemical modification.40 To increase the level of thiolation, EDC chemistry was performed in a gradient of pH conditions. The phage solution and other reagents were purged with dry nitrogen for 30 min to remove oxygen. A total of 1012 phages were reacted with 1 mM EDC, 1 mM NHS, and 1 mM cysteamine in a volume of 2 mL with gentle stirring at room temperature and mildly acidic condition (pH 5.5) to facilitate the reaction of carboxylic groups and EDC. The same amount of EDC was added 2 more times at time intervals of 30 min; the number of EDC additions was based on a previously published protocol and was not further optimized here.40 1 h after the last addition of EDC, the pH of the reaction solution was adjusted to 7.5 using 2 M NaOH solution to improve the reaction efficiency between the intermediates and cysteamine40 and the reaction was continued overnight. The phages were purified with two rounds of PEG/NaCl precipitation and extensive dialysis through regenerated cellulose dialysis tubing (molecular weight cutoff of 3500 Da) to remove trace amounts of cysteamine.
The concentration of chemically incorporated thiol groups was determined by Ellman’s assay,41 while that of phage particles was quantified by real-time PCR. Additional characterization was performed by attenuated total reflection Fourier transform infrared (ATR-FTIR) and zeta potential measurement.
Gold Nanoparticle Preparation
Four sizes of monodisperse gold nanoparticles were synthesized according to the seed-mediated gold nanoparticle growth procedure reported by Bastus et al.42 The smallest AuNPs were used as seed for the larger particles. To synthesize AuNP seeds, 15 mL of sodium citrate (2.2 mM) was heated to reflux under vigorous stirring for 15 min. Then, 0.1 mL of HAuCl4 (25 mM) was injected and the solution was stirred for another 10 min, resulting in AuNPs of 7 nm diameter.
For seeded growth synthesis of larger AuNPs, after the solution of Au seeds was cooled down to 90 °C, 0.1 mL of a HAuCl4 solution (25 mM) was injected without stirring. After 30 min, 0.1 mL of a HAuCl4 solution (25 mM) was added again to react for another 30 min. After that, the sample was diluted by extracting 5.5 mL of the sample and adding 5.3 mL of water and 0.2 mL of 60 mM sodium citrate. The above growth process was repeated one more time to obtain 20 nm gold nanoparticles. Gold nanoparticles of size 50 and 85 nm were synthesized by repeating the above process for six more and nine more times, respectively.
Modification of AuNPs with PEG
The ligand exchange of citrate with HOOC-PEG was performed according to a previous report.43 Ten microliters of HCOOH-PEG-SH solution (1 mM) was added to 1 mL of citrate-capped AuNPs with stirring. The solution was stirred at room temperature for 1 h. The excess HOOC-PEG-SH was removed by centrifugation (15 000 rpm, 45 min), and the modified AuNPs were resuspended in Milli-Q water.
Detection of Bacterial Cells Using Chimeric Phage and AuNPs in Complex Aqueous Samples
A single colony of E. coli ER2738 was grown overnight by the standard protocol (see Supporting Methods). The cells were diluted to the desired concentrations in different media: tap water (from a drinking water fountain at UCSB), human urine (from healthy donors; filtered through 0.22 μm filters before use), and fat-free bovine milk (from local market; filtered through 0.22 μm filters before use). Urine was collected from volunteers in accordance with the study protocol 5-19-0937 approved by the UCSB Human Subjects Committee. Informed consent was obtained and documented by written signature. One milliliter of cell suspension was mixed with 200 μL of phage (1012 PFU/mL) and incubated for 30 min at room temperature. The cells along with the attached phage were collected by centrifugation (5000 rpm for 10 min at 4 °C). The supernatant was discarded and the pellet was carefully washed with Milli-Q water twice. Hundred microlitre of AuNP solution was added to resuspend the pellet. The color change was captured by a digital camera (Canon PowerShot ELPH 360 HS) and the absorbance of the solution was recorded by UV–vis spectroscopy.
Detection of Bacterial Cells Using the Enzyme-Linked Immunosorbent Assay (ELISA) Kit in Complex Aqueous Samples
Bacterial detection using the ELISA method was performed with the QuickDetect E. coli Protein (ECP) ELISA kit (BioVision, Inc.) to compare with the phage-based method. The ECP of samples with different ER2738 concentrations (106, 105, 104, 103, 102, and 10 CFU/mL) in tap water was collected according the manufacturer’s instructions. Briefly, the cells were destroyed to release ECP by repeated freezing and thawing cycles (10 times). The ECP in the supernatants was collected by centrifuging at 3000 rpm for 20 min and measured by ELISA according to the manufacturer’s instruction, to obtain the relationship between bacterial concentration (CFU/mL) and absorbance at 450 nm.
Specificity of Bacterial Detection
The specificity of the assay was assessed by detection in a mixture of host cells (a mixture of E. coli ER2738, V. cholerae 0395, and P. aeruginosa (ATCC25102)), using the method described above. The same assay was also performed in the absence of the expected host of the chimeric phage.
Detection of Bacterial Cells from Swabs
Swabs mimicking clinical samples were prepared by immersing sterile cotton swabs (FLOQ Swabs) into 0.2 mL of bacterial solutions (PBS buffer) containing 102 CFU, 104 CFU, or 106 CFU cells. Swabs were then immersed in 1 mL of PBS and vortexed for 1 min and then incubated in a shaker (150 rpm) at room temperature for 30 min to resuspend the attached cells. Bacterial detection in the PBS solution was performed using the above method. A sterile cotton swab with no bacterial cells was used as a negative control.
Detection of P. aeruginosa from Swabbing of Biofilm Grown on the ex Vivo Porcine Lung Tissue
To mimic swabbing from a biological tissue, the P. aeruginosa biofilm was grown on the ex vivo porcine lung tissue by following a reported protocol.44 Briefly, cubes of approximately 5 mm3 were dissected from the ventral surface of the lung with a sterile blade and washed with PBS buffer for three times. P. aeruginosa was cultured overnight in LB broth, collected, and washed twice with PBS buffer before resuspending in synthetic cystic fibrosis sputum medium (SCFM).45 The cubes were inoculated with 104 washed cells from the overnight culture (resuspended in 50 μL of synthetic SCFM) and incubated in a shaker for 24 h at 37 °C (250 rpm). A control sample was prepared in the same conditions with no P. aeruginosa in the solution. After incubation, cubes were rinsed with PBS buffer to remove loosely adhering cells and P. aeruginosa bacterial cells were isolated as follows.46 Swabs were used to capture cells by wiping or scratching the cubes back and forth 20 times. The cotton swabs were then immersed in 1 mL of PBS buffer and subjected to vortexing for 1 min before incubation in a shaker at room temperature for 30 min to release the attached cells. Then, bacterial cells were separated by centrifugation for 5 min at 5000 rpm and the supernatant was discarded. The cells were carefully washed with PBS buffer and suspended in 1 mL of PBS buffer. Then, bacterial detection in the PBS solution was performed using the aforementioned methods.
Phenotypic Antibiotic Sensitivity Test
A single colony of E. coli ER2738 was grown to an optical density (OD600nm) of 0.2 with the standard protocol. The bacterial solution was diluted 20 times in the growth medium to obtain a 2.6 × 106 CFU/mL suspension (OD600nm ∼ 0.01). The cell solution was then transferred to three aliquots (1 mL). Then, 1 μL of tetracycline solution (10 mg/mL), kanamycin (10 mg/mL), or ampicillin (100 mg/mL) was added to each solution. The aliquots were incubated in a shaker at 37 °C for another 2 h to allow cell growth. Then, a series of 1 mL dilution samples (10-, 102-, 103-, 104-, 105-, 106-, and 107-fold diluted) were prepared. The bacterial detection assay with thiolated M13KE phage and AuNPs was performed with these samples.
Minimal Inhibitory Concentration (MIC) Determination
The MICs of different antibiotics of E. coli ER2738 in LB media were determined according to reported procedures47,48 based on optical density and the developed phage-based method. Briefly, in a 96-well plate, each well was filled with 200 μL of media containing serial dilutions of antibiotics (ampicillin, kanamycin, and tetracycline). To each well, 105 CFU of cells were added and incubated overnight at 37 °C. LB media with only antibiotics and LB media with the same amount of E. coli ER2738 without antibiotics were used as controls. The optical density at 600 nm (OD600 nm) was measured using a plate reader (Tecan Infinite M200 PRO, Tecan Group Ltd., Switzerland). The MIC tests based on the phage-based method reported here were performed as described for the phenotypic antibiotic sensitivity test. Antibiotics of different concentrations were used in the assay.
Transmission Electron Microscopy (TEM)
TEM was performed on a Tecnai FEI G2 Sphera microscope. Five microliter of the phage sample was pipetted onto the TEM grids and left for 5 min. The residual solution was removed by a filter paper and the sample was stained using 0.5% uranyl acetate for 30 s (negative stain). Excess stain was removed by the filter paper and the sample was rinsed with Milli-Q water and air-dried before measurement.
Dynamic Light Scattering (DLS) and ζ Potential
DLS and ζ potential were measured with a Malvern Zetasizer Nano ZSP running software v7.11, using a 4 mW He–Ne laser at 633 nm. Samples were equilibrated for 2 min at 25 °C before measurement. All of the results are averages of a minimum of three individual samples; each sample was measured 5 times, with each measurement consisting of 10 runs. The measured sizes are reported as intensity-weighted diameters.
Attenuated Total Reflection Infrared (ATR-FTIR) Spectra
ATR-FTIR spectra were measured with a Nicolet iS10 FTIR using a MCT detector and a Harrick Scientific Corporation GATR accessory (Materials Research Laboratory (MRL) at UCSB).
Ultraviolet–Visible Spectra
UV–vis spectra were recorded on a Shimadzu UV-1800 UV–vis spectrophotometer with a quartz spectrasil UV–vis cuvette, using direct detection at a slit width of 2 nm (California Nanosystems Institute (CNSI) at UCSB).
Results
Detection of Bacteria in Complex Aqueous Media
Two chimeric phages were previously constructed using an M13KE scaffold in which the N-terminal domain of g3p had been replaced by the homologous domain from phage CTXϕ, yielding M13-g3p(CTXϕ), and phage Pf1, yielding M13-g3p(Pf1), to target V. cholerae and P. aeruginosa, respectively.33 Here, these phages, along with M13KE (targeting E. coli), were used with AuNPs (∼7 nm in diameter as measured by TEM, Figure S1) for detection of their respective host bacterial species. Thiolated phages induce the aggregation of AuNPs, causing a red-shift in the absorbance spectrum and a visible color change from red to purple. We modified our previously published procedure33 to increase the thiolation efficiency and probe a variety of AuNP sizes. In general, thiolated phages (Figure S2, Table S1) were incubated with samples for 30 min and cells (with any attached phage) were spun down, washed twice with Milli-Q water, and resuspended in the solution containing AuNPs. We first validated the assay using tap drinking water as the medium (Figures 2a,d and S3–S6). Samples to which the host bacterial species had been added showed an immediate change in the UV–vis absorption spectrum as expected. Negative control samples containing the host bacterial species but using nonthiolated phage did not induce a change in absorbance. Indeed, the zeta potential ζ of the AuNPs was measured to be −25.2 mV, indicating a negatively charged surface. AuNPs did not aggregate on nonthiolated phage (ζ of M13-g3p(Pf1): −39.7 mV) or bacteria (ζ of P. aeruginosa: −15.7 mV) (Figure S7), consistent with electrostatic repulsion. The limit of detection was on the order of 102 CFU, with ∼40 CFU being detectable (Figure S3). Specificity was also validated by exposure of each thiolated phage to nonhost bacteria; no cross-reactivity was observed (Figures S8 and S9). The sensitivity and specificity of detection of E. coli, V. cholerae, and P. aeruginosa were similar when using the respectively targeted phages (Figures S4, S5, S8, and S9).
Figure 2.

Detection of P. aeruginosa with thiolated M13-g3p(Pf1) and AuNPs in (a, d) tap drinking water, (b, e) commercially purchased nonfat bovine milk, and (c, f) human urine. Digital photos (top row) and UV–vis spectra (bottom row) are shown. Samples from the left to right in each photo are AuNPs alone (no bacteria or phages), AuNPs with unmodified M13-g3p(Pf1) phage and 106 CFU P. aeruginosa, and AuNPs with thiolated M13-g3p(Pf1) phage with P. aeruginosa at 102, 104, and 106 CFU, respectively.
We then tested the ability of the assay to detect these three bacterial species in two complex aqueous settings: nonfat bovine milk and human urine. Incubations in both media yielded a detectable colorimetric response and corresponding spectral shift in the presence of 100 CFU or greater (Figure 2, Figures S4 and S5). In Figure 2, the nonfat bovine milk was filtered through 0.22 μm filters before use,49 to remove any existing microbes before testing the assay’s ability to detect a known amount of added bacteria. We also performed the bacterial detection assay with nonfiltered nonfat bovine milk, since real samples would not be filtered. Detection in nonfiltered and filtered milk yielded similar results (Figure S10). These results indicate that the phage–AuNP detection assay would be suitable for detecting bacteria existing in nonfiltered samples.
The phage–AuNP detection technique was compared with a commercially available bacterial detection kit. The QuickDetect E. coli Protein ELISA kit (BioVision, CA) was used to detect E. coli ER2738 in tap water. A standard curve of bacterial concentration and absorbance at 450 nm was obtained (Figure S11). With no bacteria added, A450nm = 0.035 (standard deviation = 0.002), and at 6 × 103 CFU/mL, A450nm = 0.053 (standard deviation = 0.019). The 95% confidence intervals for the measurement of the 6 × 103 CFU/mL sample overlap with that for the sample having no bacteria, indicating that the limit of detection (LOD) with this kit was >6 × 103 CFU/mL. Thus, this LOD is at least 2 orders of magnitude higher than the LOD of the phage–AuNP method,33 i.e., the ELISA kit is substantially less sensitive. Moreover, because the ELISA kit detects protein, additional steps are required to extract the protein by a repeated freeze–thaw process.
Effect of AuNP Size and Colloidal Stabilization
The colloidal stability of AuNPs can be significantly influenced by properties such as size, stabilizer, and surface charge.50 To determine how robust the bacterial detection assay was to alterations in these properties, we synthesized monodispersed citrate-stabilized AuNPs with larger sizes by a seed-mediated growth method.42 The sizes measured by TEM were 20, 50, and 85 nm in diameter, with the hydrodynamic diameter measured by DLS being 28, 69, and 106 nm, respectively (Figure S1). The ζ potentials of the AuNPs in water were −27.2, −25.8, and −27.4 mV, respectively, indicating similar highly negatively charged surfaces in solution.51 Despite having similar ζ potentials to the 7 nm AuNPs, these larger AuNPs appeared to have greater colloidal stability in that no spectral shift indicating aggregation was observed even in the presence of 106 CFU bacteria with thiolated phages (Figure S12). In addition, AuNPs modified with a stronger stabilizer (PEG-COOH) prevented detection (Figure S13).
Detection of P. aeruginosa Swabbed from Biofilm Grown on the ex Vivo Porcine Lung Tissue
In addition to detection in liquid media, detection of bacteria from swab samples is of clinical interest for diagnosing tissue infections. We first tested whether bacteria applied directly to cotton swabs could be released and detected by this method. P. aeruginosa cells in different amounts (106, 104, and 102 CFU) were adsorbed on sterile cotton swabs and then released in the solution by vortexing for 1 min and shaking in PBS buffer at room temperature for 30 min. The detection assay was performed in the solution as described above. The color change from red to purple and red-shift of SPR peaks were clearly observed (Figure 3), indicating successful detection of P. aeruginosa released from swabs.
Figure 3.
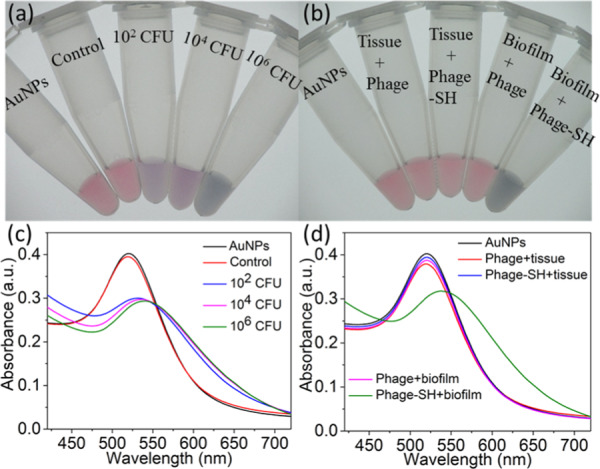
Detection of P. aeruginosa with thiolated M13-g3p(Pf1) and AuNPs from (a, c) P. aeruginosa directly adsorbed to cotton swabs and (b, d) swabs of P. aeruginosa biofilm grown ex vivo on the porcine lung tissue. (a, b) Digital photos and (c, d) UV–vis spectra are shown. Samples from left to right in (a) are AuNPs with no bacteria or phages, thiolated M13-g3p(Pf1) phage with the sample from a sterile cotton swab with no bacterial cells added (control), thiolated M13-g3p(Pf1) and samples from swabs contaminated with 102, 104, and 106 CFU P. aeruginosa, respectively. Corresponding spectra are shown in (c). Samples from left to right in (b) are AuNPs with no bacteria or phages, unmodified M13-g3p(Pf1) and swab samples from the porcine lung tissue with no P. aeruginosa (“phage + tissue”), thiolated M13-g3p(Pf1) and swab samples from the same control (“phage-SH + tissue″), unmodified M13-g3p(Pf1) and swab samples from P. aeruginosa biofilm grown ex vivo on the porcine lung tissue (“phage + biofilm”), and thiolated M13-g3p(Pf1) and swab samples from the same ex vivo biofilm model (“phage-SH + biofilm”).
Having verified that this technique can detect bacteria from P. aeruginosa-contaminated swabs, we tested the assay in a scenario meant to mimic swabbing of an infected tissue. We adopted a previously validated model of P. aeruginosa biofilm grown ex vivo on 5 mm3 cubes of porcine lung tissue.44 The biofilm-containing cubes were rinsed with PBS buffer to remove loosely adhering cells and the cubes were swabbed to capture cells. The cotton swabs were then treated as above to release the attached cells, which were spun down, washed, and resuspended in 1 mL of PBS buffer. The detection assay was performed as described above. Colorimetric response and spectral red-shift only occurred in the sample containing P. aeruginosa, while the control sample (porcine lung tissue with no bacteria added) showed no detectable change in the color or absorbance spectrum (Figure 3), confirming the compatibility of the assay with a swab obtained from a biofilm grown on mammalian tissue.
Rapid Determination of Antibiotic Sensitivity
Assessing the growth of bacteria in the presence of antibiotics is a standard approach to determining antibiotic resistance. Since the phage-AuNP technique can rapidly detect small amounts of bacteria, we explored its application to identify the antibiotic resistance profile of E. coli (Figure 4). The E. coli strain ER2738 is tetracycline-resistant but sensitive to ampicillin and kanamycin. A solution of E. coli ER2738 was diluted to 2.6 × 106 CFU/mL (OD600nm ∼ 0.01) in the growth medium and then aliquoted into tubes containing one of the three antibiotics. The bacterial cultures were incubated in a shaker at 37 °C for 2 h. A 10-fold dilution series of each culture was assayed with thiolated M13KE phage and AuNPs, and the dilution at which bacteria became detectable was noted. At this dilution, ∼100 CFU was presumed to be present in 1 mL of solution, allowing approximate (order-of-magnitude) inference of the concentration of bacteria in the culture grown with each antibiotic. Thus, if the microorganism is susceptible to the antibiotic, a low density of cells will be inferred (similar to that of the starting aliquot). If the microorganism is resistant to that antibiotic, a higher density of cells will be observed (corresponding to the exponential growth of the starting aliquot).
Figure 4.
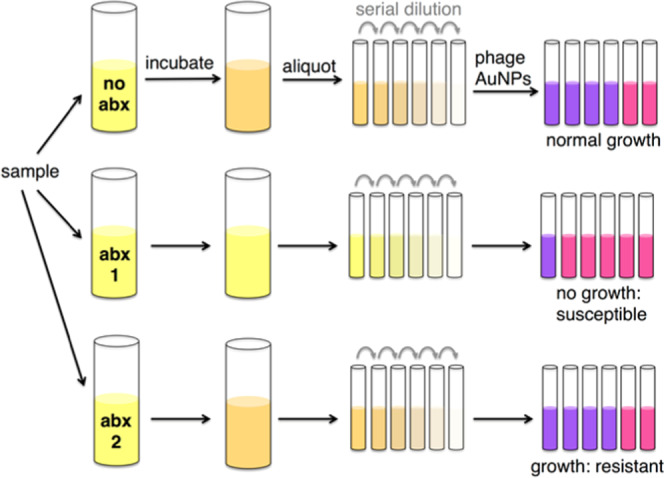
Scheme for phage–AuNP detection of bacteria coupled to a growth assay for characterization of the antibiotic susceptibility profile. A sample (e.g., swab, fluid) is incubated under growth conditions in the presence and absence of the antibiotic (abx) for several doubling times (e.g., 2 h for E. coli). Whether growth occurred is determined by addition of phage–AuNPs to a dilution series (purple indicates >100 cells; pink indicates <100 cells), allowing inference of susceptibility to the antibiotic.
As shown in Figure 5, in the presence of kanamycin and ampicillin, the 105-, 106-, and 107-fold dilutions do not show a red-shift, indicating bacterial detection, but less-dilute samples, including the 104-fold dilution, do show a red-shift. Given the LOD of the assay, this indicates ∼102 cells present in the 104-fold dilution or a cell density of ∼106 cells/mL after the 2 h incubation in ampicillin or kanamycin. This density is similar to the starting concentration, indicating arrested growth in the presence of ampicillin or kanamycin. In contrast, in the presence of tetracycline, the most dilute sample at which a red-shift could be detected was the 106-fold dilution, indicating a cell density of ∼108 cells/mL after 2 h of growth in tetracycline. This is consistent with an ∼40-fold increase in cell number after 2 h of growth in the presence of tetracycline. Assuming a doubling time of 20 min, 2 h of growth at 37 °C is expected to correspond to 6 doublings of E. coli ER2738 or a 64-fold increase. This indicates that growth was not substantially affected by the presence of tetracycline, i.e., the bacterial strain is resistant to tetracycline. The phage–AuNP technique can therefore be used to determine phenotypic antibiotic susceptibility. The total time of the assay was approximately 2.5 h (2 h of growth followed by approximately 30 min of detection time).
Figure 5.

Determination of growth in the presence of antibiotics using thiolated M13KE phage and AuNPs. (a–c) Digital photos and (d–f) UV–vis spectra are shown. Samples in (a, d), (b, e), and (c, f) were grown with ampicillin, kanamycin, or tetracycline, respectively. Samples from left to right in each photo are AuNPs with no bacteria or phages, control (106 CFU cells with unmodified M13KE phage and AuNPs), and thiolated M13KE phage and AuNPs with the bacterial sample at the following dilutions: 1-, 10-, 102-, 103-, 104-, 105-, 106-, and 107-fold.
The minimum inhibitory concentrations (MIC) of ampicillin, kanamycin, and tetracycline against E. coli ER2738 were determined using phage–AuNPs or OD600 nm to assess bacterial growth in culture (Figure 6). E. coli ER2738 grew well in the presence of up to 100 μg/mL of tetracycline, confirming the resistance of this strain to tetracycline. The MICs of ampicillin and kanamycin, to which ER2738 is sensitive, were defined as the lowest concentration of antibiotic resulting in no detectable growth. The MICs of both ampicillin and kanamycin were found to be 11 μg/mL by both assays. However, the phage-based assay required less time, labor, and instrumentation.
Figure 6.
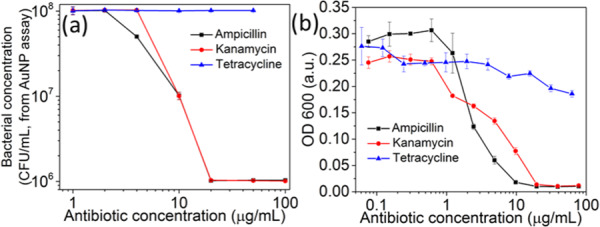
Determination of minimal inhibitory concentrations of ampicillin, kanamycin, and tetracycline against E. coli ER2738 in the LB medium, using the two methods to assess growth: (a) phage–AuNPs and (b) optical density. Each point represents three triplicates.
Discussion
The conventional method (“culture and sensitivity”) to identify organisms, still widely used in clinical pathology laboratories, involves culturing the sample on different selective media, followed by biochemical testing with antibiotic susceptibility determined by culturing on selective media.52 Culture-based methods generally take days to yield results,53,54 and other methods have disadvantages for point-of-care or resource-limited applications. We previously reported a detection strategy based on recognition of specific bacterial strains by chimeric, thiolated phages and aggregation of AuNPs onto these phages,33 which tolerated conditions including seawater and serum. Unlike antibodies, which may be similarly employed to recognize specific bacterial antigens, the activity of phages is preserved over a wider range of conditions.24 In our assay, the supernatant (including free phages) is discarded and the bacteria–phage pellets are resuspended in the AuNP solution to induce a colorimetric response. Therefore, opaque suspensions, such as milk, and colored solutions, such as urine, should interfere minimally with the readout of the assay. Milk is often associated with the foodborne outbreaks,55 while analysis of urine samples is important for diagnosis of urinary tract infections (UTIs), one of the most common infectious diseases worldwide.56−58 Indeed, we found that the phage–AuNP assay maintains sensitivity and specificity for detection of bacteria in these biological media. In addition, the assay was effective for swabs of a bacterial biofilm formed on the ex vivo tissue. Note that the cell amounts detected (102 CFU) on the swabs are likely considerably lower than the amounts present in infected wounds (∼105 CFU/cm2 59), suggesting the assay could be useful in assessing such wounds. The results indicate that the phage–AuNP assay is tolerant to these types of clinical or food safety samples.
In contrast to the robustness of the assay to different sample media, our attempts to alter nanoscale features, namely, increasing the AuNP size and altering the surface coating, resulted in severe loss of sensitivity. Aggregation is a critical phenomenon that arises from the interplay between attractive van der Waals interactions and repulsive electrostatic interactions among the AuNPs.60,61 The AuNPs used here were stabilized electrostatically against aggregation by a citrate coating. However, if the particles are overly stable, aggregation will not occur even in the presence of bacteria. It has been previously suggested that the surfaces of small AuNPs (5–10 nm dia) are too high in curvature for citrate to be well packed,62 leading to defects in the coating and a tendency toward aggregation when combined with increased proximity due to phage association. On the other hand, larger AuNPs, having less surface curvature, may be efficiently coated by citrate, leading to stable colloids.63 We also investigated whether PEG coating could be tolerated on the AuNPs, since polymer brushes, particularly PEG, can increase the bioavailability of nanoparticles.64 However, PEG coating is also known to decrease aggregation. Indeed, ligand exchange to coat the AuNPs with PEG stabilized the particles against aggregation, to the extent that they were inactive for bacterial detection. Thus, small (<10 nm dia), citrate-coated nanoparticles appear to be most suitable for this assay.
An important potential application of bacterial detection technology is phenotypic determination of antibiotic susceptibility. Phenotypic ASP determination consists of two stages: growth (or nongrowth) in media containing antibiotic, followed by assessment of growth. Improved assays can reduce the time of ASP determination in two ways (1) highly sensitive assays reduce the time necessary to produce detectable growth and (2) rapid assays reduce the time needed to assess growth. We demonstrated that the phage–AuNP assay could be used for ASP determination, allowing approximately six doubling times for growth (∼2 h for E. coli) and ∼30 min for the detection assay. The number of doublings required depends on the quantitative accuracy of the assay; in this case, we used a series of 10-fold dilutions, such that a >10-fold growth would be required to generate a reliable difference between growth with and without the antibiotic (if susceptible). A more finely divided dilution series could lower the amount of growth necessary and further shorten the growth time, although it is not likely to be reduced by more than an hour. More slowly growing organisms would require correspondingly more time for any phenotypic ASP assay, including this one.
Conclusions
The phage–AuNP detection assay relies on the high affinity and specificity of the phage–bacteria interaction. The phages used here are chimeras of viruses in the Inoviridae family, which targets Gram-negative organisms, a group of increasing concern for rapid diagnosis.2 The use of chimeric g3p allows rational design of phages with a specific target within known Inovirus hosts, a potential advantage over antibodies, which must be developed for the intended specificity. The rapid, sensitive, environmentally robust, and inexpensive assay for detection and antibiotic susceptibility assessment of specific bacteria in biosamples may be of further interest in point-of-care or resource-limited situations.
Acknowledgments
Financial support from the NIH (DP2 GM123457-01) and the Institute for Collaborative Biotechnologies (Contract W911NF-09-0001 from the US Army Research Office) is acknowledged. We thank M. Mahan for bacterial strains and Conan Zhao, John Varga, and Sam Brown for advice on the ex vivo model. We thank the Biological Nanostructures Laboratory (UV–vis) within the California NanoSystems Institute, supported by the University of California, Santa Barbara, and the University of California, Office of the President, and the MRL Shared Experimental Facilities (attenuated total reflection–FTIR, TEM, XPS), supported by the Materials Research Science and Engineering Center Program of the NSF under Award DMR 1720256, a member of the NSF-funded Materials Research Facilities Network (https://www.mrfn.org/).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssensors.0c00654.
Supporting text, TEM images of gold nanoparticles and phage, DLS data, FTIR spectra, detection of P. aeruginosa and V. cholerae with thiolated phage and AuNPs in drinking water, fat-free milk, and urine, standard calibration curve of concentration of E. coli ER2738 vs absorbance at 450 nm determined by the QuickDetect E. coli Protein (ECP) ELISA kit, cross-reactivity and specificity test, bacterial detection with phage and larger gold nanoparticles, standard curves of real-time PCR of M13 phages and Ellman’s assay, and number of thiol groups per chimeric phage after chemical modification (PDF)
The authors declare the following competing financial interest(s): Patent pending: UC Case 2018-758-0.
Supplementary Material
References
- Dye C. After 2015: infectious diseases in a new era of health and development. Philos. Trans. R. Soc., B 2014, 369, 20130426. 10.1098/rstb.2013.0426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer K. A.; Perez K. K.; Forrest G. N.; Goff D. A. Review of Rapid Diagnostic Tests Used by Antimicrobial Stewardship Programs. Clin. Infect. Dis. 2014, 59, S134–S145. 10.1093/cid/ciu547. [DOI] [PubMed] [Google Scholar]
- Barlam T. F.; Cosgrove S. E.; Abbo L. M.; MacDougall C.; Schuetz A. N.; Septimus E. J.; Srinivasan A.; Dellit T. H.; Falck-Ytter Y. T.; Fishman N. O.; Hamilton C. W.; Jenkins T. C.; Lipsett P. A.; Malani P. N.; May L. S.; Moran G. J.; Neuhauser M. M.; Newland J. G.; Ohl C. A.; Samore M. H.; Seo S. K.; Trivedi K. K. Implementing an Antibiotic Stewardship Program: Guidelines by the Infectious Diseases Society of America and the Society for Healthcare Epidemiology of America. Clin. Infect. Dis. 2016, 62, 1197–1202. 10.1093/cid/ciw217. [DOI] [PubMed] [Google Scholar]
- Dobson E. L.; Klepser M. E.; Pogue J. M.; Labreche M. J.; Adams A. J.; Gauthier T. P.; Turner R. B.; Su C. P.; Jacobs D. M.; Suda K. J. Outpatient antibiotic stewardship: Interventions and opportunities. J. Am. Pharm. Assoc. 2017, 57, 464–473. 10.1016/j.japh.2017.03.014. [DOI] [PubMed] [Google Scholar]
- Fleming-Dutra K. E.; Hersh A. L.; Shapiro D. J.; Bartoces M.; Enns E. A.; File T. M.; Finkelstein J. A.; Gerber J. S.; Hyun D. Y.; Linder J. A.; Lynfield R.; Margolis D. J.; May L. S.; Merenstein D.; Metlay J. P.; Newland J. G.; Piccirillo J. F.; Roberts R. M.; Sanchez G. V.; Suda K. J.; Thomas A.; Woo T. M.; Zetts R. M.; Hicks L. A. Prevalence of Inappropriate Antibiotic Prescriptions Among US Ambulatory Care Visits, 2010-2011. J. Am. Med. Assoc. 2016, 315, 1864–1873. 10.1001/jama.2016.4151. [DOI] [PubMed] [Google Scholar]
- Högmander M.; Paul C. J.; Chan S.; Hokkanen E.; Eskonen V.; Pahikkala T.; Pihlasalo S. Luminometric Label Array for Counting and Differentiation of Bacteria. Anal. Chem. 2017, 89, 3208–3216. 10.1021/acs.analchem.6b05142. [DOI] [PubMed] [Google Scholar]
- Sandström G. E.; Wolfwatz H.; Tarnvik A. Duct Elisa for Detection of Bacteria in Fluid Samples. J. Microbiol. Methods 1986, 5, 41–47. 10.1016/0167-7012(86)90022-9. [DOI] [Google Scholar]
- Zhu L. J.; He J.; Cao X. H.; Huang K. L.; Luo Y. B.; Xu W. T. Development of a double-antibody sandwich ELISA for rapid detection of Bacillus Cereus in food. Sci. Rep. 2016, 6, 16092 10.1038/srep16092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlsson P.; Evander M.; Petersson K.; Mellhammar L.; Lehmusvuori A.; Karhunen U.; Soikkeli M.; Seppä T.; Tuunainen E.; Spangar A.; von Lode P.; Rantakokko-Jalava K.; Otto G.; Scheding S.; Soukka T.; Wittfooth S.; Laurell T. Integrated Acoustic Separation, Enrichment, and Microchip Polymerase Chain Reaction Detection of Bacteria from Blood for Rapid Sepsis Diagnostics. Anal. Chem. 2016, 88, 9403–9411. 10.1021/acs.analchem.6b00323. [DOI] [PubMed] [Google Scholar]
- Petralia S.; Conoci S. PCR Technologies for Point of Care Testing: Progress and Perspectives. ACS Sensors 2017, 2, 876–891. 10.1021/acssensors.7b00299. [DOI] [PubMed] [Google Scholar]
- Sauer S.; Kliem M. Mass spectrometry tools for the classification and identification of bacteria. Nat. Rev. Microbiol. 2010, 8, 74–82. 10.1038/nrmicro2243. [DOI] [PubMed] [Google Scholar]
- von Wintzingerode F.; Bocker S.; Schlotelburg C.; Chiu N. H. L.; Storm N.; Jurinke C.; Cantor C. R.; Gobel U. B.; van den Boom D. Base-specific fragmentation of amplified 16S rRNA genes analyzed by mass spectrometry: A tool for rapid bacterial identification. Proc. Natl. Acad. Sci. USA 2002, 99, 7039–7044. 10.1073/pnas.102165899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong T.; Zhao X. Y. Rapid Identification and Susceptibility Testing of Uropathogenic Microbes via Immunosorbent ATP-Bioluminescence Assay on a Microfluidic Simulator for Antibiotic Therapy. Anal. Chem. 2015, 87, 2410–2418. 10.1021/ac504428t. [DOI] [PubMed] [Google Scholar]
- Gregor C.; Gwosch K. C.; Sahl S. J.; Hell S. W. Strongly enhanced bacterial bioluminescence with the ilux operon for single-cell imaging. Proc. Natl. Acad. Sci. USA 2018, 115, 962–967. 10.1073/pnas.1715946115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M. Z.; Liu Y.; Jiang X. Y. Barcoded point-of-care bioassays. Chem. Soc. Rev. 2019, 48, 850–884. 10.1039/C8CS00303C. [DOI] [PubMed] [Google Scholar]
- Thompson S. A.; Paterson S.; Azab M. M. M.; Wark A. W.; de la Rica R. Light-Triggered Inactivation of Enzymes with Photothermal Nanoheaters. Small 2017, 13, 1603195 10.1002/smll.201603195. [DOI] [PubMed] [Google Scholar]
- Hernández A.; Cano M. P. High-pressure and temperature effects on enzyme inactivation in tomato puree. J. Agric. Food Chem. 1998, 46, 266–270. 10.1021/jf970455g. [DOI] [PubMed] [Google Scholar]
- Zhou J. H.; Rossi J. Aptamers as targeted therapeutics: current potential and challenges. Nat. Rev. Drug Discovery 2017, 16, 181–202. 10.1038/nrd.2016.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn M. R.; Jimenez R. M.; Chaput J. C. Analysis of aptamer discovery and technology. Nat. Rev. Chem. 2017, 1, 1304. 10.1038/s41570-017-0076. [DOI] [Google Scholar]
- Lowe D.; Dudgeon K.; Rouet R.; Schofield P.; Jermutus L.; Christ D. Aggregation, Stability, and Formulation of Human Antibody Therapeutics. Adv. Protein Chem. Struct. Biol. 2011, 84, 41–61. 10.1016/B978-0-12-386483-3.00004-5. [DOI] [PubMed] [Google Scholar]
- Sormanni P.; Aprile F. A.; Vendruscolo M. Third generation antibody discovery methods: in silico rational design. Chem. Soc. Rev. 2018, 47, 9137–9157. 10.1039/C8CS00523K. [DOI] [PubMed] [Google Scholar]
- Maugeri G.; Lychko I.; Sobral R.; Roque A. C. A. Identification and Antibiotic-Susceptibility Profiling of Infectious Bacterial Agents: A Review of Current and Future Trends. Biotechnol. J. 2019, 14, 1700750 10.1002/biot.201700750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyt C. E.; Brechot N.; Trouillet J. L.; Chastre J. Antibiotic stewardship in the intensive care unit. Crit. Care 2014, 18, 480. 10.1186/s13054-014-0480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.; Borg R. E.; Dow L. P.; Pruitt B. L.; Chen I. A. Controlled phage therapy by photothermal ablation of specific bacterial species using gold nanorods targeted by chimeric phages. Proc. Natl. Acad. Sci. USA 2020, 201913234 10.1073/pnas.1913234117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Toit A. Phage induction in different contexts. Nat. Rev. Microbiol. 2019, 17, 126–127. 10.1038/nrmicro3445. [DOI] [PubMed] [Google Scholar]
- Chen J.; Andler S. M.; Goddard J. M.; Nugen S. R.; Rotello V. M. Integrating recognition elements with nanomaterials for bacteria sensing. Chem. Soc. Rev. 2017, 46, 1272–1283. 10.1039/C6CS00313C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. H.; Jackson A. A.; Rotello V. M.; Nugen S. R. Colorimetric Detection of Escherichia coli Based on the Enzyme-Induced Metallization of Gold Nanorods. Small 2016, 12, 2469–2475. 10.1002/smll.201503682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squirrell D. J.; Price R. L.; Murphy M. J. Rapid and specific detection of bacteria using bioluminescence. Anal. Chim. Acta 2002, 457, 109–114. 10.1016/S0003-2670(01)01495-7. [DOI] [Google Scholar]
- Howard-Varona C.; Hargreaves K. R.; Abedon S. T.; Sullivan M. B. Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. 10.1038/ismej.2017.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunderland K. S.; Yang M. Y.; Mao C. B. Phage-Enabled Nanomedicine: From Probes to Therapeutics in Precision Medicine. Angew. Chem., Int. Ed. 2017, 56, 1964–1992. 10.1002/anie.201606181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen E. V.; Sorokulova I. B.; Petrenko V. A.; Chen I. H.; Barbaree J. M.; Vodyanoy V. J. Affinity-selected filamentous bacteriophage as a probe for acoustic wave biodetectors of Salmonella typhimurium. Biosens. Bioelectron. 2006, 21, 1434–1442. 10.1016/j.bios.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Bardhan N. M.; Ghosh D.; Belcher A. M. Carbon nanotubes as in vivo bacterial probes. Nat. Commun. 2014, 5, 4918 10.1038/ncomms5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H.; Chen I. A. Rapid Colorimetric Detection of Bacterial Species through the Capture of Gold Nanoparticles by Chimeric Phages. ACS Nano 2019, 13, 1244–1252. 10.1021/acsnano.8b06395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. H.; Wang Y.; Chen P.; McCadden A.; Palaniappan A.; Zhang J. L.; Liedberg B. Peptide Functionalized Gold Nanoparticles with Optimized Particle Size and Concentration for Colorimetric Assay Development: Detection of Cardiac Troponin I. ACS Sensors 2016, 1, 1416–1422. 10.1021/acssensors.6b00493. [DOI] [Google Scholar]
- Xia F.; Zuo X. L.; Yang R. Q.; Xiao Y.; Kang D.; Vallee-Belisle A.; Gong X.; Yuen J. D.; Hsu B. B. Y.; Heeger A. J.; Plaxco K. W. Colorimetric detection of DNA, small molecules, proteins, and ions using unmodified gold nanoparticles and conjugated polyelectrolytes. Proc. Natl. Acad. Sci. USA 2010, 107, 10837–10841. 10.1073/pnas.1005632107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang L. H.; Liu X. F.; Liang Z. Q.; Song S. P.; Li W. X.; Li G. X.; Fan C. H. A gold nanoparticle-based aptamer target binding readout for ATP assay. Adv. Mater. 2007, 19, 3943–3946. 10.1002/adma.200602256. [DOI] [Google Scholar]
- Kerr K. G.; Snelling A. M. Pseudomonas aeruginosa: a formidable and ever-present adversary. J. Hosp. Infect. 2009, 73, 338–344. 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention, HAI Data and Statistics. https://www.cdc.gov/hai/data/index.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fhai%2Fsurveillance%2Findex.html.
- Lin A.; Jimenez J.; Derr J.; Vera P.; Manapat M. L.; Esvelt K. M.; Villanueva L.; Liu D. R.; Chen I. A. Inhibition of Bacterial Conjugation by Phage M13 and Its Protein g3p: Quantitative Analysis and Model. PLoS One 2011, 6, e19991 10.1371/journal.pone.0019991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yacoby I.; Bar H.; Benhar I. Targeted drug-carrying bacteriophages as antibacterial nanomedicines. Antimicrob. Agents Chemother. 2007, 51, 2156–2163. 10.1128/AAC.00163-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellman G. L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- Bastus N. G.; Comenge J.; Puntes V. Kinetically Controlled Seeded Growth Synthesis of Citrate-Stabilized Gold Nanoparticles of up to 200 nm: Size Focusing versus Ostwald Ripening. Langmuir 2011, 27, 11098–11105. 10.1021/la201938u. [DOI] [PubMed] [Google Scholar]
- Rahme K.; Chen L.; Hobbs R. G.; Morris M. A.; O’Driscoll C.; Holmes J. D. PEGylated gold nanoparticles: polymer quantification as a function of PEG lengths and nanoparticle dimensions. RSC Adv. 2013, 3, 6085–6094. 10.1039/C3RA22739A. [DOI] [Google Scholar]
- Harrison F.; Muruli A.; Higgins S.; Diggle S. P. Development of an Ex Vivo Porcine Lung Model for Studying Growth, Virulence, and Signaling of Pseudomonas aeruginosa. Infect. Immun. 2014, 82, 3312–3323. 10.1128/IAI.01554-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K. L.; Aye L. A.; Whiteley M. Nutritional cues control Pseudomonas aeruginosa multicellular Behavior in cystic fibrosis sputum. J. Bacteriol. 2007, 189, 8079–8087. 10.1128/JB.01138-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dortet L.; Poirel L.; Nordmann P. Rapid Identification of Carbapenemase Types in Enterobacteriaceae and Pseudomonas spp. by Using a Biochemical Test. Antimicrob. Agents Chemother. 2012, 56, 6437–6440. 10.1128/AAC.01395-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews J. M. Determination of minimum inhibitory concentrations. J. Antimicrob. Chemother. 2001, 48, 5–16. 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Reimer L. G.; Stratton C. W.; Reller L. B. Minimum inhibitory and bactericidal concentrations of 44 antimicrobial agents against three standard control strains in broth with and without human serum. Antimicrob. Agents Chemother. 1981, 19, 1050–1055. 10.1128/AAC.19.6.1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. X.; Wu L. N.; Huang T. X.; Wang S.; Yan X. M. Rapid detection and enumeration of total bacteria in drinking water and tea beverages using a laboratory-built high-sensitivity flow cytometer. Anal. Methods 2015, 7, 3072–3079. 10.1039/C4AY02919D. [DOI] [Google Scholar]
- Phan H. T.; Haes A. J. What Does Nanoparticle Stability Mean?. J. Phys. Chem. C 2019, 123, 16495–16507. 10.1021/acs.jpcc.9b00913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feick J. D.; Velegol D. Electrophoresis of spheroidal particles having a random distribution of zeta potential. Langmuir 2000, 16, 10315–10321. 10.1021/la001031a. [DOI] [PubMed] [Google Scholar]
- Laupland K. B.; Valiquette L. The changing culture of the microbiology laboratory. Can. J. Infect. Dis. Med. 2013, 24, 125–128. 10.1155/2013/101630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. N.; Chang A. C. Y.; Hsu L. Nonchromosomal Antibiotic Resistance in Bacteria - Genetic Transformation of Escherichia-Coli by R-Factor DNA. Proc. Natl. Acad. Sci. USA 1972, 69, 2110. 10.1073/pnas.69.8.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste R.; Davies J. Mechanisms of Antibiotic-Resistance in Bacteria. Annu. Rev. Biochem. 1973, 42, 471–506. 10.1146/annurev.bi.42.070173.002351. [DOI] [PubMed] [Google Scholar]
- Creamer L. K.; Pearce L. E.; Hill J. P.; Boland M. J. Milk and Dairy Products in the 21st Century Prepared for the 50th Anniversary of the Journal of Agricultural and Food Chemistry. J. Agric. Food Chem. 2002, 50, 7187–7193. 10.1021/jf020711b. [DOI] [PubMed] [Google Scholar]
- Hooton T. M. Uncomplicated Urinary Tract Infection REPLY. N. Engl. J. Med. 2012, 367, 185. 10.1056/NEJMc1205507. [DOI] [PubMed] [Google Scholar]
- Wolfe A. J.; Toh E.; Shibata N.; Rong R. C.; Kenton K.; FitzGerald M.; Mueller E. R.; Schreckenberger P.; Dong Q. F.; Nelson D. E.; Brubaker L. Evidence of Uncultivated Bacteria in the Adult Female Bladder. J. Clin. Microbiol. 2012, 50, 1376–1383. 10.1128/JCM.05852-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvatore S.; Salvatore S.; Cattoni E.; Siesto G.; Serati M.; Sorice P.; Torella M. Urinary tract infections in women. Eur. J. Obstet. Gynecol. Reprod. Biol. 2011, 156, 131–136. 10.1016/j.ejogrb.2011.01.028. [DOI] [PubMed] [Google Scholar]
- Raahave D.; Friismoller A.; Bjerrejepsen K.; Thiisknudsen J.; Rasmussen L. B. The Infective Dose of Aerobic and Anaerobic-Bacteria in Postoperative Wound Sepsis. Arch. Surg. 1986, 121, 924–929. 10.1001/archsurg.1986.01400080070012. [DOI] [PubMed] [Google Scholar]
- van Oss C. J. The Extended Dlvo Theory. Interface Sci. Technol. 2008, 16, 31–48. 10.1016/S1573-4285(08)00203-2. [DOI] [Google Scholar]
- Schäfer B.; Hecht M.; Harting J.; Nirschl H. Agglomeration and filtration of colloidal suspensions with DVLO interactions in simulation and experiment. J. Colloid Interface Sci. 2010, 349, 186–195. 10.1016/j.jcis.2010.05.025. [DOI] [PubMed] [Google Scholar]
- Esfahani M. R.; Pallem V. L.; Stretz H. A.; Wells M. J. M. Extinction, emission, and scattering spectroscopy of 5-50 nm citrate-coated gold nanoparticles: An argument for curvature effects on aggregation. Spectrochim. Acta, Part A 2017, 175, 100–109. 10.1016/j.saa.2016.11.052. [DOI] [PubMed] [Google Scholar]
- Chegel V.; Rachkov O.; Lopatynskyi A.; Ishihara S.; Yanchuk I.; Nemoto Y.; Hill J. P.; Ariga K. Gold Nanoparticles Aggregation: Drastic Effect of Cooperative Functionalities in a Single Molecular Conjugate. J. Phys. Chem. C 2012, 116, 2683–2690. 10.1021/jp209251y. [DOI] [Google Scholar]
- Smith C. A.; Simpson C. A.; Kim G.; Carter C. J.; Feldheim D. L. Gastrointestinal Bioavailability of 2.0 nm Diameter Gold Nanoparticles. ACS Nano 2013, 7, 3991–3996. 10.1021/nn305930e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.