

Abstract
The combined loss of CHD1 and MAP3K7 promotes aggressive prostate cancer (CaP) by unknown mechanisms. Because these genes are both lost genetically in CaP, they cannot be directly targeted. We applied an established computational systems pharmacology approach (TRAP) to identify altered signaling pathways, and associated druggable targets. We compared gene expression profiles of CaP with co-loss of CHD1 & MAP3K7 with CaP diploid for these genes using TCGA patient samples. This analysis prioritized druggable target genes that included CDK1 and CDK2. We validated that inhibitors of these druggable target genes, including the CDK1/ CDK2 inhibitor dinaciclib, had anti-proliferative and cytotoxic effects selectively on mouse prostate cells with knockdown of Chd1 and Map3k7. Dinaciclib had stronger effects on prostate cells with suppression of Map3k7 independent of Chd1 and also compared to cells without loss of Map3k7. Dinaciclib treatment reduced expression of homologous recombination (HR)-repair genes such as ATM, ATR, BRCA2, and RAD51, blocked BRCA1 phosphorylation, reduced RAD51 foci formation, and increased γH2AX foci selectively in prostate cells with suppression of Map3k7, thus inhibiting HR repair of chromosomal double strand breaks. Dinaciclib-induced HR disruption was also observed in human prostate cells with knockdown of MAP3K7. Co-treatment of dinaciclib with DNA damaging agents or PARP inhibitor resulted in a stronger cytotoxic effect on prostate cells with suppression of MAP3K7 compared to those without loss of MAP3K7, or to each single agent.
Keywords: TAK1, CHD1, prostate cancer, non-homologous end joining, dinaciclib, docetaxel, PARP, olaparib
Introduction
Prostate cancer (CaP) is characterized by large genomic rearrangements including translocations, copy number losses and gains (1). MAP3K7 is deleted in 30–40% of primary prostate cancer (homozygous and heterozygous deletion combined), (2,3). Loss of the Chromodomain Helicase DNA-binding protein 1 (CHD1) gene (located on 5q21.1), which occurs in 20–30% of CaP (4,5), is associated with an increased frequency of copy number alterations (CNAs) and massive genomic rearrangements, termed chromothripsy (4). Not all of the CNAs associated with loss of CHD1 are random (4). CHD1 loss is significantly associated with loss of MAP3K7 (located on 6q15.1), which encodes TGF-β activated kinase 1 (TAK1) (4) (p < 0.001; Fisher’s exact test). Homozygous or heterozygous deletion of MAP3K7 in primary CaP is significantly correlated with high Gleason grade tumors (2,3). We previously demonstrated that genomic deletion of CHD1 and MAP3K7 correlates with their respective mRNA expression in multiple patient cohorts and that low mRNA expression is correlated with poor disease free survival (5). We recently demonstrated that MAP3K7 is a prostate tumor suppressor and that co-loss with CHD1 promotes aggressive CaP (3,5). The CHD1 gene encodes a histone-associated protein that is required for DNA double strand break repair and CHD1 depletion enhances PARP inhibitor therapy (6). MAP3K7 encodes MAP kinase kinase kinase 7 that is immediately downstream of cytokine and stress response pathways. Loss of these two tumor suppressors represents a molecularly defined sub-type of aggressive CaP with unknown therapeutic sensitivities. Because these genes are both lost genetically in CaP, they cannot be directly targeted. Therefore, the pathways that are altered must be characterized to identify therapeutic vulnerabilities.
To identify potential signaling pathways altered in CaP with loss of CHD1 & MAP3K7 we applied an established computational systems pharmacology approach, Transcriptional Regulatory Association with Pathways (TRAP) (7), to gene expression data in The Cancer Genome Atlas (TCGA) (8). Through this approach, we identified a number of altered signaling pathways and a set of predicted druggable targets. We investigated several targets in prostate cells with suppression of CHD1 & MAP3K7 expression and identified several promising drugs, including dinaciclib, a multiple cyclin dependent kinase (CDK) inhibitor. We showed that loss of MAP3K7 was the main contributing alteration driving increased sensitivity to these drugs, and that treatment with a CDK inhibitor paired with either PARP inhibitor or DNA damaging agents resulted in potent cell death in cells with loss of MAP3K7.
Materials and Methods
Agents
All drugs were purchased from Selleckchem except for docetaxel which was purchased from Sigma-Aldrich.
Cell Culture
All cell lines were routinely tested for mycoplasma contamination by DAPI staining and PCR. LNCaP cells were obtained from American Type Culture Collection and used below passage 50. LNCaP cells were validated by spectral karyotyping. Generation and maintenance of WFU3 mouse prostate epithelial progenitor/stem cells were described previously (9). Wild-type WFU3 cells generate benign ductal structures and represent a clean model to dissect the contributions of specific genetic alterations to therapeutics and signaling (5,9). LNCaP cells were maintained as described previously (5). Generation of shControl, shMAP3K7, shCHD1, and shDouble cell lines in WFU3 and LNCaP was described previously (5). BHPrE1 cells, a spontaneously immortalized benign prostate cell line that generates benign ductal structures in vivo, were a generous gift from Simon Hayward (10). As there is no record of prior spectral karyotyping, we validated that BHPrE1 cells had a unique karyotype. BHPrE1 cells were maintained and cultured in DMEM/F12 with 1% insulin-transferrin selenium (Gibco, #41400045), 1% bovine pituitary extract (Hammond Cell Tech, #1078-NZ),10 μg/mL EGF (Corning, #354001), 1% Penicillin/ Streptomycin (Thermo Fisher Scientific, #15140122) and 5% fetal bovine serum (Gemini Bio-Products, #100–106). Generation of shControl and shMAP3K7 BHPrE1 cell lines was performed as follows. For knockdown (KD) of MAP3K7, cells were subjected to lentiviral transduction, infection with shMAP3K7-puro or shControl-puro lentiviruses. Production of lentivirus for these shRNA plasmids was described previously (5). Cells were infected with lentivirus in growth media containing 8 μg/ml polybrene and selected with 12 μg/ml puromycin (Sigma, #9620) after 48h. Cells were maintained in medium with 12 μg/ml puromycin.
Cell Proliferation and Cell Death Assay
Cells were cultured at 37°C and 5% CO2 and seeded at 1000 cells (WFU3), 4000 cells (BHPrE1 and LNCaP) per well in a 96-well plate (Costar). Cells were cultured at 37°C and 5% CO2 in an IncuCyte Zoom Live Cell Analysis system (Essen BioScience). Cells were exposed to drugs for up to 48hr for WFU3, 36hr for LNCaP, and 72hr for BHPrE1 in normal growth medium with or without 125 ng/ml of Annexin V (CF488® Annexin V, Biotium #29005 for WFU3, or CF®633 Annexin V, Biotium #29008 for LNCaP and BHPrE1) and/or 1.0μg/ml of Propidium iodide (PI) (Biotium, #40016), and images were captured at 2-, 3-, or 4-hr intervals using a 4x objective. Each experiment was done in triplicate unless otherwise indicated. After monitoring in the Incucyte Zoom, cell proliferation assay for BHPrE1 was performed using CellTiter-Glo® Luminescent Cell Viability Assay (Promega G7572) following the manufacturer’s protocol and luminescence was read using a Modulus Microplate reader. For WFU3 and LNCaP, cell proliferation was determined by the percent confluence of each well using Incucyte Zoom scanning. The proportion of cells per treatment group was normalized to control wells when indicated. Annexin V and PI positive cell counts were normalized to percent confluence in each well.
Cell cycle analysis and cell sorting
Staining of cells for cell cycle analysis was performed as described previously (11). Briefly, WFU3 (2–5 ×105) and BHPrE1 cells (1×105) were seeded in a six-well plate and incubated overnight. WFU3 cells were exposed to vehicle, olaparib 10μM, dinaciclib 60nM, or the combination of these drugs for 24hr prior to collection. BHPrE1 cells were exposed to vehicle, docetaxel 1nM, dinaciclib 4nM, or the combination of these drugs for 24hr prior to collection. Cells were washed once with PBS, pelleted, and vortexed before adding 1 ml Krishan stain solution (11). Cells were vortexed again and incubated overnight at 4°C before analysis with a Gallios instrument using CXP software (Beckman Coulter, Brea, CA). 10,000 events were acquired for each sample and the percent of cells in G1, S, and G2/M phases was determined with ModFit LT v4.0.5 software (Verity Software House, Topsham, ME)
Cell sorting for the cell cycle phases was performed as follows. WFU3 cells (0.3–2×106) were seeded in a 15cm dish and incubated overnight. Cells were exposed to vehicle, or the combination of dinaciclib and olaparib for 36hr prior to collection. Cells were washed once with PBS, pelleted, and vortexed before adding Hoechst33342 (Thermo Fisher Scientific, #H3570). Cells were incubated in medium containing 60 μg/ml of Hoecht33342 for 45 minutes with rocking in an incubator. Around four million cells in each condition were sorted based on G1, S, and G2/M phases, respectively, using UV laser with MoFlo XDP70 (Beckman Coulter, Brea, CA). Collected cells were harvested and proteins were isolated.
Immunoblotting
Standard techniques were used for protein isolation, quantification, and immunoblotting. Antibodies used were: MAP3K7 (Cell Signaling Technology, #4505), CHD1 (Bethyl Laboratories, #A301–218A), BRCA2 (Thermo Fisher Scientific, #MA5–23942), Phospho-BRCA1 (Ser 1524) (Thermo Fisher Scientific, #PA5–17503), RAD51 (Abcam, #ab88572), Anti-phospho-Histone H2A.X (Ser139) (Millipore, #JBW201), p53 (Santa Cruz Technology, #sc-6243), Phospho-ATR (Ser428) (Cell signaling Technology, #2853), ATR (Thermo Fisher Scientific, #PA1–450), ATM (Cell signaling Technology, #2873), Caspase 3 (Cell signaling Technology, #9662), Cleaved caspase 3 (Asp175) (Cell signaling Technology, #9661), βActin (Cell signaling Technology, #3700), α-tubulin (Cell Signaling Technology, #2144), goat anti-rabbit-HRP (Santa Cruz, #sc-2004), and goat anti-mouse-HRP (Santa Cruz, #sc-2005). Where appropriate, bands were quantified using ImageJ (NIH, Bethesda, MD).
Annexin V and PI assay using Flow Cytometry
WFU3 cells were seeded at 2–5 × 105 in a six-well plate or 15 × 105 in a 60mm dish and incubated overnight. Cells were exposed to either vehicle, or the combination of these drugs for 48hr prior to collection. Cells were harvested and stained using Annexin V-APC Detection kit from BioTool (#B32117) according to manufacturer’s instructions. Flow data was acquired on a Gallios (Beckman Coulter) and analyzed using Summit v5.1 (Beckman Coulter, Fort Collins, CO).
Immunofluorescence assay
Methods for immunofluorescence assay were performed as described previously (12). WFU3 cells (1–6 ×105) and BHPrE1 cells (3–6×105) were seeded on coverslips in a twelve-well plate and incubated overnight. WFU3 cells were exposed to vehicle, olaparib 5μM, dinaciclib 60nM, or the combination of these drugs for 12hr for the RAD51 foci assay and 36hr for the γH2AX foci assay prior to harvest. BHPrE1 cells were exposed to vehicle, docetaxel 0.5nM, dinaciclib 4nM, or the combination of these drugs for 24hr prior to harvest. Immunoreaction was performed using the RAD51 antibody (Santa Cruz Technology, #sc-398587, 1:100) overnight at 4°C or Anti-phospho-Histone H2A.X (Ser139) (Millipore, #JBW301. 1:500) for 1hr at room temperature (RT). Cells were washed three times with PBS before incubating with secondary antibody, Alex Fluor594, goat anti-mouse anti-mouse IgG (H+L) (Thermo Fisher Scientific, # A11005, 1:250 for RAD51 and 1:500 for ɤH2AX), for 1hr at RT. After washing three times, the coverslip was mounted onto a microscope slide using VECTASHIELD with DAPI (Vector Laboratories, #H-1200). Images were acquired with a Nikon DS-Ri2 confocal laser-scanning microscope with a 40 × objective lens. For quantitative analysis, software for automatic focus counting was used to count ɤH2AX and RAD51 foci as described previously (13). More than 100 cells from each group were chosen at random, and nuclei were counted to determine the percent positive for RAD51 or ɤH2AX based on a threshold of 5 or greater foci per nucleus.
TRAP analysis
TRAP is an algorithm that was first described by Kwong, Costello and colleagues (7) to identify gene-to-pathway transcriptional regulatory relationships using mouse data. Here, we developed a separate application of TRAP to evaluate human data. We first compiled a human gene expression compendium of 3,531 human gene expression profiles on the Affymetrix HG133plus2.0 platform. Raw, human gene expression files (.CEL) were downloaded from either Gene Expression Omnibus (GEO) (https://www.ncbi.nlm.nih.gov/pubmed/23193258) or ArrayExpress (https://academic.oup.com/nar/article/47/D1/D711/5144130). We preferentially selected experiments with multiple samples per condition and sampled a diversity of treatment conditions, cell lines and tissues. Arrays were batch normalized using RMAexpress (14) with background adjustment, quantile normalization and median polish summarization. Probesets were mapped to HGNC gene symbols using Ensembl (v81) annotations from the BioMart online resource (https://www.ensembl.org/biomart). Multiple probesets mapping to the same gene were averaged. This final gene expression compendium matrix, M, is the input to define the TRAP network. Druggable genes were defined using DGIdb (downloaded Oct 16, 2016) (15); these genes were selected from M to define M1. Canonical pathways were defined using the C2.CP gene set collection from MSigDB (v6.0) (15). The average expression of genes in each gene set was calculated using the M - M1 matrix. The new pathway expression matrix M2 was then concatenated with M1 to create M′, where the columns are samples and the rows are druggable genes or canonical pathways. We calculated mutual information between all druggable gene/canonical pathway pairs in M′. Each value, xi,j, in row i was z-score normalized () to create . Each value, xi,j, in row j was z-score normalized () to create . These two matrices were subsequently combined to create the TRAP matrix, MT, where the z-scored value for the interaction between druggable gene, i, and canonical pathway, j, from , was combined with the z-scored value for the interaction between druggable gene, i, and canonical pathway, j, from , using Stouffer’s method (i.e. ). We set the combined z-score threshold at 4 to arrive at a final set of significant relationships that define the TRAP network (7846 edges and 1636 nodes: 691 pathways, 945 druggable genes; supplemental Table 1). The TRAP network established the background for which we used to identify druggable targets specific to patients with primary prostate tumor defined by the loss of CHD1 & MAP3K7.
To prioritize druggable targets, we first identified patient tumor samples from TCGA (n=499) that had co-loss (homozygous or heterozygous deletion) of CHD1 & MAP3K7 and patient tumors that were diploid at CHD1 & MAP3K7 [8]. Additionally, since the co-loss of CHD1 & MAP3K7 is mutually exclusive of ETS transcription factor fusions, we only considered tumors that were negative for ETS fusions. In total, we identified 69 patients with loss of CHD1 & MAP3K7 (and were ETS fusion negative) and 111 patients that were diploid for CHD1 & MAP3K7 (and were ETS fusion negative). Transcript counts were downloaded from Gene Expression Omnibus (accession number GSE62944) (16) and normalized using the calcNormFactors function in the limma R package (17). Genes were ranked according to their log-fold change between the two patient groups (supplemental Table 2). We ran GSEA in the preranked mode on this ranked list of genes using the C2.CP gene sets (v6.0) (supplemental Table 3) (18,19). The significantly up-regulated pathways (enriched in co-loss patients compared to diploid; FDR < 0.05) were mapped to the TRAP network and the first neighbors were selected to define a subnetwork of druggable genes-to-differentially up-regulated pathways (supplemental Table 4). Using this subnetwork and the full TRAP network, weighted degree centrality was calculated for both networks according to Opsahl et al. (20). To briefly summarize, the weighted average of the edges connecting each node (weighted degree centrality) in the subnetwork is calculated and then compared to the weighted degree centrality in the full TRAP network. The difference from the subnetwork to the full network defines the gain in centrality, which is the measure used to prioritize gene targets (supplemental Table 5). In addition to comparing patients with co-loss of CHD1 & MAP3K7 to patients diploid at those loci, we performed GSEA analysis on patients that had loss of CHD1 alone (n=14) and loss of MAP3K7 alone (n=53), compared to patients diploid at both loci (n=111) following the same procedure previously described.
RNAseq in WFU3
1.5 million shControl, shChd1, shMap3k7 or shDouble WFU3 cells were plated onto 10cm dishes in complete growth medium. Twenty-four hours later RNA was isolated using 5prime Perfect RNA Cell kit (Fischer Scientific) according to the manufacturer’s protocol. After quality control, RNA samples were sequenced by Novagene using the Illumina Novoseq 6000. PE150 reads were generated at an estimated 20 million reads per sample. Raw reads (fastq files) were mapped to the Ensembl v90 transcriptome of the Mus musculus genome build GRCm38 using Bowtie2 with the parameter settings ‘--sensitive --dpad 0 --gbar 99999999 --mp 1,1 --np 1 --score-min L,0,−0.1 --no-mixed --no-discordant’; transcripts were quantified using the rsem-calculate-expression function in RSEM (21,22). A summary and details of sequence report are in supplemental Tables 6–8. Genes that had an average count of less than 5 across all replicates were removed. The log-fold change was calculated for each gene using the voom workflow in the limma R package (17). Using the same C2.CP gene set collection (v6.0) as was used in the analysis of TCGA patient samples, we performed GSEA on the shDouble, shChd1, or shMap3k7 compared to WFU3 shControl cells. For this analysis, mouse gene symbols were mapped to human gene symbols using the vertebrate homology mapping supplied by the Mouse Genome Informatics resource (http://www.informatics.jax.org/downloads/reports/index.html).
Statistical analyses
Statistical analyses were performed with GraphPad Prism V7 (GraphPad). Two-way ANOVA with Sidak’s multiple comparisons test, Tukey’s multiple comparisons test, or Dunnet’s multiple comparisons test was used to analyze two different categorical independent variables. IC50 was determined using the four-parameter logistic model and comparison of curves was performed using extra sum-of-square F test of logIC50 and hill slope. The synergism between the two drugs was assessed using CalcuSyn V2 (Biosoft). Two-tailed p <0.05 was considered statistically significant. Data are shown by mean ± SE unless otherwise indicated.
Results
Identification of Therapeutic Targets using TRAP
Because both CHD1 and MAP3K7 are genetically deleted in prostate cancer, they can’t be directly targeted. Therefore, we used a computational systems pharmacology approach (TRAP) to identify targetable pathways that are modulated in prostate cancer with loss of CHD1 and MAP3K7. Fig 1A provides an overview of TRAP (7) with details described in the Methods section. The goal of TRAP is to prioritize druggable genes highly connected to pathways that are dysregulated in a given experiment. A compendium of 3,531 diverse human gene expression profiles was compiled and strength of connections between druggable genes and canonical pathways calculated using mutual information, where significant edges define the TRAP network (supplemental Table 1). This network is the background for which we compare the following CHD1 & MAP3K7 expression profile. Because CHD1 & MAP3K7 are mutually exclusive of the ERG, ETV1, ETV4, or FLI1 transcription factor fusions (5), we compared gene expression profiles from TCGA patient tumors with co-loss of CHD1 & MAP3K7 and were ETS transcription factor fusion negative (n = 69) to tumors diploid for CHD1 & MAP3K7 and were also ETS fusion negative (n=111) (supplemental Table 2), then used GSEA to identify dysregulated pathways (supplemental Table 3) (18,19). A total of 65 pathways were significantly differentially up-regulated (FDR < 0.05). We mapped these pathways to the TRAP network and by selecting the directly connected neighbors, we created a subnetwork of druggable targets connected to the 65 pathways specific to patients with loss of CHD1 & MAP3K7 (Fig 1B and supplemental Table 4). By identifying the most central genes in this subnetwork, we were able to calculate a prioritization score to each gene target (supplemental Table 5). The top 20 druggable gene targets identified by TRAP are shown in Fig 1C, along with the fold changes in gene expression for the associated genes from TCGA patient tumors (supplemental Table 2) and the WFU3 cells (supplemental Table 8) that we describe next to evaluate target hits.
Figure 1: TRAP analysis and validation in WFU3 cells.
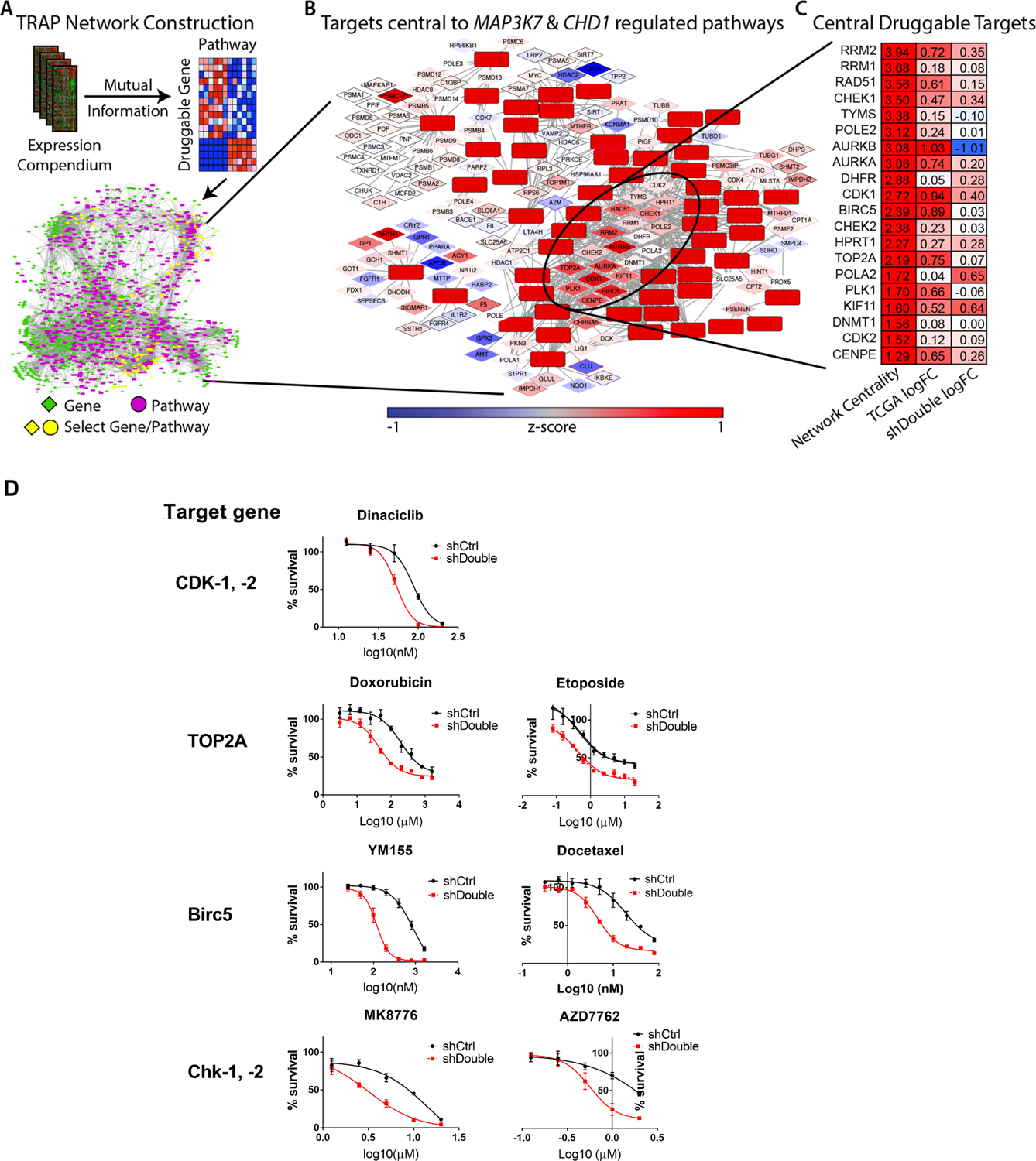
A: The TRAP network construction workflow is shown. A human gene expression compendium of over 3,500 expression profiles was compiled. Druggable genes were defined using DGIdb and canonical pathways using the C2.CP gene sets from MSigDB. Mutual Information was calculated for all druggable genes/canonical pathway pairs; significant relationships define the edges in the TRAP network, which is the background model that defines how druggable genes are connected to pathways. See Methods section for more detail.
B: TCGA patient tumors with co-loss of CHD1 & MAP3K7 were used to identify significantly up-regulated pathways. These pathways and their direct druggable gene neighbors were selected from the TRAP network and the most centrally located genes were identified..
C: The top 20 druggable target genes are shown. Network centrality from the TRAP analysis, the log-fold change between TCGA patients with co-loss of CHD1 & MAP3K7 and those diploid at CHD1 & MAP3K7, and the log-fold change between WFU3 shDouble and shCtrl..
D: Dose-response curves of candidate drugs in cell growth of WFU3 shControl (black) versus shDouble (red). Cells were treated with each drug for 48hr and cell growth was assessed by % confluence of each well in IncuCyte Zoom. Data are shown by mean and SE (n=3–5 per group). These results are representative of three independent experiments.
In vitro validation of TRAP predictions
To model CaP with loss of CHD1 and MAP3K7 expression, we used a previously characterized mouse prostate stem cell line (WFU3) (9) with stably expressing shRNAs against Chd1 and Map3k7 (5). These cells have been extensively characterized in vitro and in vivo for their tumorigenic phenotypes (5). We compared WFU3 cells with shRNAs targeting both Chd1 & Map3k7 (shDouble) to control cells with scrambled shRNAs (shControl). The knockdown status of Map3k7 and Chd1 is shown in supplemental Fig 1A. To assess if similar pathways are modulated in WFU3 cells with suppression of Chd1 and Map3k7 to those of patient samples, we generated RNAseq data from WFU3 shDouble and shControl cells (supplemental Table 7 and 8) and ran GSEA to identify differentially expressed pathways (supplemental Table 9). Of the 65 differentially up-regulated pathways identified from TCGA data, 63 were found in the mouse results, and 56 of the 63 pathways were also up-regulated in the shDouble WFU3 cells compared to WFU3 shControl cells. In total, 18 gene sets were significantly up-regulated (FDR < 0.05) comparing shDouble to shControl in the WFU3 cells with 12 overlapping the 63 pathways (p = 1.7e-11; Fisher’s exact test) from the human data (supplemental Table 3 and 9). This comparison of human and mouse expression profiles supports the WFU3 cells as being a strong model of the human prostate tumor biology, and thus a representative model to assess therapeutic targeting.
We initially selected a CDK-1/ −2 inhibitor for testing because CDK1 and CDK2 are central to the TRAP network and are also upregulated in primary patient tumors with co-loss of CHD1 & MAP3K7, as well as in WFU3 shDouble cells (Fig 1C). Additionally, we screened several selective inhibitors for target genes among the top TRAP hits (Fig 1C). We also screened olaparib, a PARP inhibitor, because this drug has been shown to directly affect the responsiveness of CaP cells with loss of CHD1 (6). As shown in Fig 1D and supplemental table 10, CDK-1/ −2 inhibitor (dinaciclib [IC50 86.80 vs 52.85nM in WFU3 shControl vs shDouble, p<0.0001]), Chk-1/ −2 inhibitors (MK8776 [IC50: 13.99 vs 3.177μM, p<0.0001] and AZD7762 [IC50: 3.629 vs 0.5672μM, p=0.0010]), BIRC5 inhibitor (YM155 [IC50: 880.6 vs 118.6nM, p<0.0001]), docetaxel (IC50 18.84 vs 4.627nM, p<0.0001), TOP2A-inhibitors (doxorubicin [IC50: 193.1 vs 44.43nM, p<0.0001] and etoposide [IC50: 0.5596 vs 0.3712μM, p=0.1329], and PARP inhibitor (olaparib [IC50: 31.53 vs 15.01μM, p=0.0002]) had increased potency on WFU3 shDouble compared to shControl. As negative controls, we assessed the efficacy of cisplatin and temozolomide, both of which were not predicted in the TRAP analysis and showed no effect on WFU3 shDouble compared to shControl (Supplemental Table 10).
Loss of MAP3K7, but not CHD1, sensitized the effect of dinaciclib through disruption of homologous recombination
We next asked if loss of either Map3k7 or Chd1 individually was the main driver to increase the sensitivity to dinaciclib in WFU3. Cells with knockdown of Chd1 alone did not show any increase in sensitivity to dinaciclib relative to shControl cells (Fig 2A). In contrast, cells with knockdown of Map3k7 were more sensitive to dinaciclib, similar to that of shDouble cells (Fig 2A). Importantly, knockdown of Chd1 did not alter/reverse the sensitizing effects of knockdown of Map3k7 (shDouble cells respond similar to shMap3k7 cells). Dinaciclib is a multiple cyclin dependent kinase (CDK) inhibitor, and recent studies have revealed a critical role for CDKs in regulating homologous recombination (HR) (23). We tested whether dinaciclib disrupted HR in WFU3 cells. Dinaciclib decreased BRCA1 phosphorylation and the expression of ataxia-telangiectasia mutated kinase (ATM), BRCA2 and RAD51 in shMap3k7 and shDouble cells compared to shControl and shChd1 cells (Fig 2B and supplemental Table 10A). The decrease in HR-related proteins was accompanied by induction of ɤH2AX and p53 in shMap3k7 and shDouble cells but not in shControl and shChd1 cells (Fig 2B and supplemental Table 11A). These results are consistent with the stronger effects of dinaciclib on cells with loss of Map3k7 (Fig 2A). The time dependent decrease of BRCA2 and RAD51 started by 3 to 6hr after dinaciclib treatment in WFU3 shMap3k7 while p53 induction started by 3hr and ɤH2AX induction was observed at 24hr (supplemental Fig 2A and 2B). The knockdown of Chd1 alone showed increased sensitivity to doxorubicin, etoposide, and olaparib compared to control; however the drugs’ sensitizing effects were greater with suppression of Map3k7 compared to Chd1 suppression (supplemental Fig 3). In addition, the knockdown of Map3k7 sensitized WFU3 cells to docetaxel, but the knockdown of Chd1 did not have an effect (supplemental Fig 3).
Figure 2: Loss of MAP3K7 sensitizes prostate cells to dinaciclib through disruption of homologous recombination in WFU3 cells.
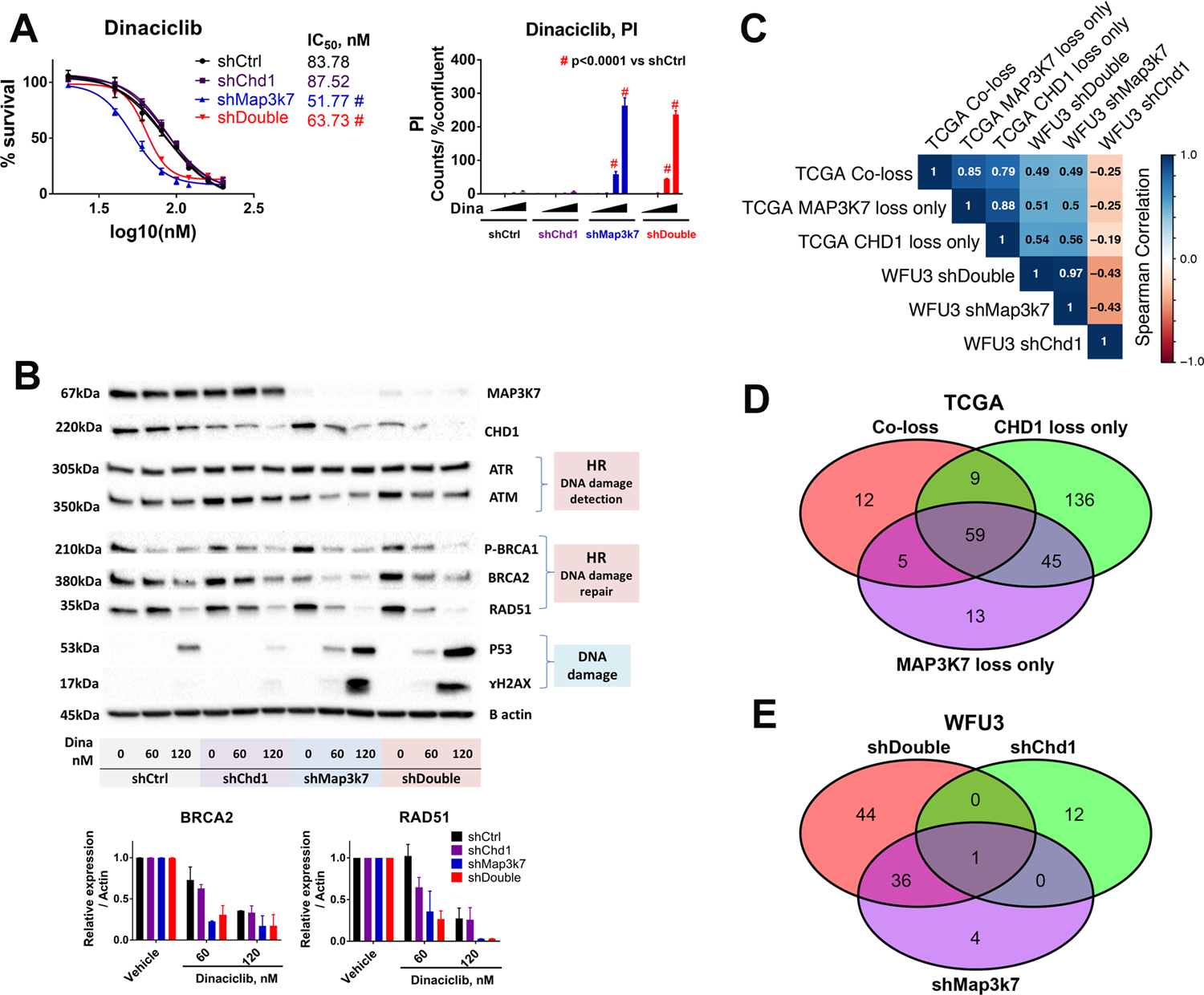
A: Dose response curves of dinaciclib in cell growth of WFU3 shControl (black), shChd1 (purple), shMap3k7 (blue), and shDouble (red) are shown. Cells were treated with vehicle or dinaciclib (25–100nM) for 48hr. Cell growth was assessed by % confluence of each well and PI positive cells were normalized to % confluence in IncuCyte Zoom. Data are shown by mean and SE (n=3 per group). The comparison of dose-response curve fit between the two cells was performed using extra sum-of-square F test. Two different categorical independent variables were analyzed using two-way ANOVA with Sidak’s multiple comparisons test. * p<0.05, ** p<0.01, *** p<0.001, and # p<0.0001 vs shCtrl. These results are representative of three independent experiments.
B: Reduction of ATM, BRCA2, BRCA1 phosphorylation and RAD51, but not ATR, by dinaciclib accompanied by ɤH2AX and p53 induction in WFU3 cells. Cells were treated with vehicle or dinaciclib (60 or 120nM) for 24hr. The results are representative (immunoblot), and mean and SE (bar graphs) of two independent experiments
C: Spearman correlation of normalized enrichment scores of the C2.CP gene sets from MSigDB for TCGA primary prostate tumor with loss of CHD1, MAP3K7 and both, and WFU3 shChd1, shMap3k7 and shDouble as determined by GSEA.
D, E: The overlap of significant C2.CP gene sets from GSEA (FDR < 0.05) among the TCGA primary prostate tumor with loss of CHD1, MAP3K7 and both (D), and WFU3 shChd1, shMap3k7 and shDouble (E).
We found a strong similarity of drug sensitivity and dinaciclib-induced HR disruption in WFU3 shMap3k7 and shDouble, but differences comparing the WFU3 shChd1 cells to the shDouble and shMap3k7 cells. To further investigate these differences, we compared gene expression patterns across these cells and also across TCGA primary prostate tumor with loss of CHD1, MAP3K7 and the combination. We found a strong correlation in altered pathways between TCGA co-loss and MAP3K7 loss alone (r = 0.85) and between WFU3 shDouble and shMap3k7 (r = 0.97) whereas altered pathways between WFU3 shChd1 and shDouble were quite different (Fig 2C). We found strong correlations between TCGA co-loss with WFU3 shDouble (r = 0.49) and with shMap3k7 (r = 0.49) (Fig 2C, and supplemental Table 2 and 8), further supporting the WFU3 cells as a reliable model of human disease. We also found a higher frequency of overlaps of up-regulated pathways between TCGA co-loss and MAP3K7 loss alone, and between WFU3 shDouble and WFU3 shMap3k7 compared to that between TCGA co-loss and CHD1 loss alone, and between WFU3 shDouble and shChd1 (Fig 2D and 2E). The expression patterns show that the shDouble and shMap3k7 are very similar, while the shChd1 expression pattern is quite different. These data suggest that the expression patterns and drug sensitivities are driven by loss of Map3k7.
We next considered whether disruption of HR by dinaciclib was observed in human prostate cells as well. To this end, we used the BHPrE1 non-tumorigenic human prostatic epithelial cell line. BHPrE1 cells display epithelial intermediate/transit amplifying cell characteristics and were generated without external DNA or viral modification (10). The knockdown of MAP3K7 was done using previously validated shRNA targeting MAP3K7 (5). We compared the shMAP3K7 cells to cells stably expressing a non-targeting control shRNA (shControl) (supplemental Fig 1B). Loss of MAP3K7 by itself caused a slight increase in Annexin V and PI positive cells in the condition without drugs (supplemental Fig 1C). Suppression of MAP3K7 augmented the anti-proliferative effects and potency of dinaciclib in BHPrE1 (IC50: 6.567 vs 5.881nM in BHPrE1 shControl vs shMAP3K7, p<0.05, Fig 3A). The IC50 of dinaciclib in BHPrE1 shMAP3K7 (5.881nM) was much lower than that in WFU3 shMap3k7 (51.77nM). Suppression of MAP3K7 by itself increased the HR-related proteins (Fig 3B, supplemental Table 11B, and supplemental Fig 4). Dinaciclib decreased phosphorylation of Ataxia Telangiectasia and Rad3-related protein (ATR) and BRCA1. Dinaciclib also decreased expression of ATR, ATM, BRCA2, and RAD51, and this decrease in ATR, ATM and BRCA2 was more prominent in BHPrE1 shMAP3K7 compared to shControl. These results were accompanied by an increased induction of ɤH2AX (Fig 3B and supplemental Table 11B). Because p53 was already stabilized in BHPrE1 (10), p53 did not increase in response to dinaciclib. The prominent decreases of HR-related proteins, with induction of ɤH2AX in BHPrE1 shMAP3K7 compared to shControl were mostly consistent with WFU3 cells, with some differences between BHPrE1 and WFU3 cells: e.g. dinaciclib-induced RAD51 decrease was similar in BHPrE1 shControl and shMAP3K7 in contrast to the prominent decrease of RAD51 in WFU3 with knockdown of Map3k7 compared to WFU3 shControl. The decrease of ATR expression by dinaciclib was observed in BHPrE1, but not in WFU3 cells.
Figure 3: Loss of MAP3K7 sensitizes prostate cells to dinaciclib through disruption of homologous recombination in BHPrE1.
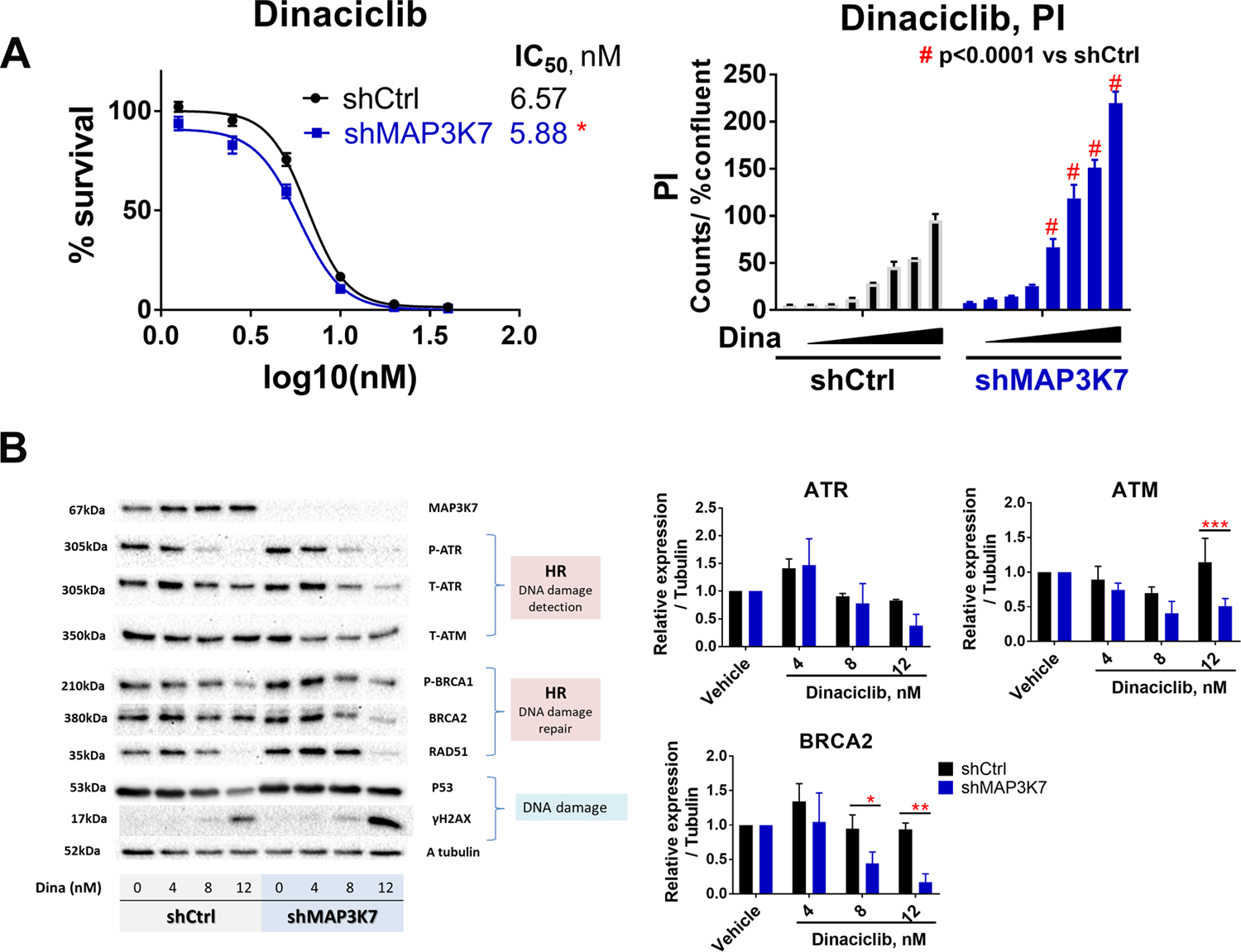
A: Dose-response curves of dinaciclib in BHPrE1 shControl (black) versus shMAP3K7 (blue). Cells were treated with vehicle or dinaciclib (1.25–40nM) for 72hr and cell growth was assessed using CellTiterGlo. Dinaciclib-induced cytotoxicity was assessed by PI assay in IncuCyte Zoom. Cells were treated with dinaciclib (1–16nM) for 48hr. Data are shown by mean and SE (n=3 per group). The results are representative of three independent experiments.
B: Dose response reduction of HR-related proteins and induction of ɤH2AX in response to dinaciclib, which was more prominent, especially BRCA2 and ATR reduction, in BHPrE1 shMAK3K7 than shControl. Cells were treated with vehicle or dinaciclib (4–12nM) for 24h. The results are representative (immunoblot) and mean ± SE (bar graph, n=3 per group) of three independent experiments.
Data were analyzed using two-way ANOVA with Dunnett’s or Sidak’s multiple comparisons test. The comparison of dose-response curve fit between the two cells was performed using extra sum-of-square F test. * p<0.05, ** p<0.01, *** p<0.001, and # p<0.0001 vs shCtrl
Dinaciclib augmented the effect of DNA damaging agents in prostate cells with loss of MAP3K7
In the absence of HR, DNA double strand breaks persist ultimately resulting in cell death unless repaired by error-prone non-homologous end joining mechanisms (24–26). We asked whether dinaciclib-induced disruption of HR sensitizes the cells to the effects of DNA damaging agents including inhibition of PARP. Dinaciclib augmented cytotoxic and anti-proliferative effects of doxorubicin, etoposide, docetaxel, and olaparib in WFU3 shMap3k7 whereas this effect of dinaciclib on each DNA damaging agent was less evident in WFU3 shControl (Fig 4A–B, supplemental Fig 5 and supplemental table 12A–B). In BHPrE1, dinaciclib increased the cytotoxic effects of doxorubicin, etoposide, docetaxel, and olaparib. Dinaciclib’s augmentation of the cytotoxic effect of docetaxel, doxorubicin, and olaparib, but not etoposide, was more prominent in BHPrE1 shMAP3K7 compared to BHPrE1 shControl (Fig 4C–D, Supplemental Table 12 C–D). The combination of dinaciclib with olaparib had less cytotoxicity compared to dinaciclib single agent in BHPrE1 (Fig 4C–D, Supplemental Table 12 C–D), which was inconsistent with the WFU3 results in which the combination therapy induced an obvious increase of cytotoxicity (Fig 4A–B, Supplemental table 12A–B).
Figure 4: Dinaciclib augments effects of DNA damaging agents and olaparib in prostate cells with loss of MAP3K7.
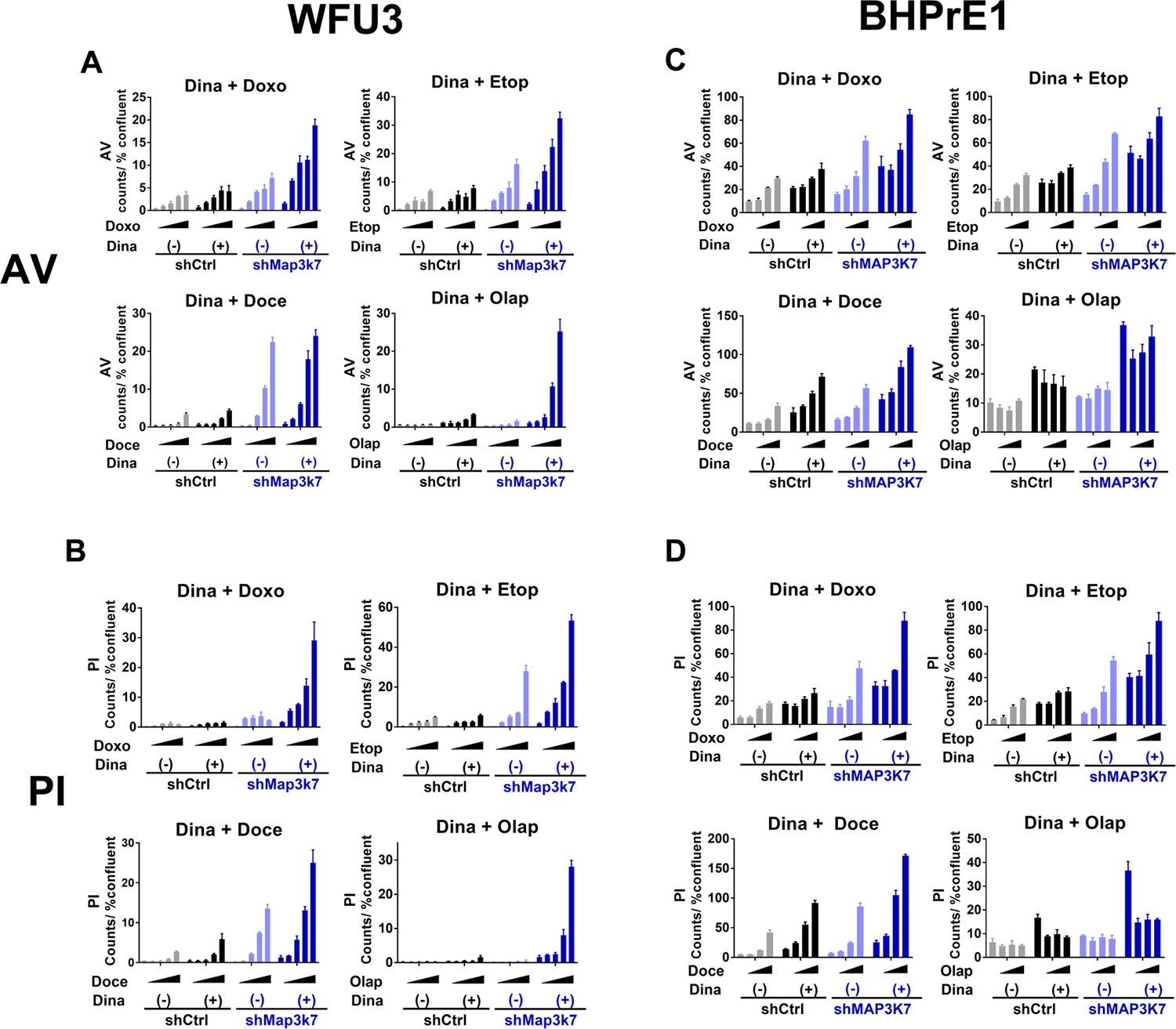
A, B: Annexin V (A) and PI positive cells (B) in response to each DNA damaging agent or olaparib with or without dinaciclib in WFU3 shControl versus shMap3k7. Cells were treated with vehicle or each DNA damaging agent, doxorubicin 50–400nM, etoposide 0.63–5.0μM, docetaxel 1.25–10nM, or olaparib 1.25–10.0μM, with or without dinaciclib 60nM for 48hr. Annexin V and PI positive cells were assessed using IncuCyte Zoom and normalized to % confluence.
C, D: Annexin V (C) and PI positive cells (D) in BHPrE1 shControl versus shMAP3K7 in response to vehicle or each DNA damaging agent, doxorubicin 50–200nM, etoposide 1.0–4.0μM, docetaxel 0.25–1nM or olaparib 2.5–10.0μM with or without dinadicilib 4nM are shown. Cells were treated with these drugs for 48hr. Annexin V and PI positive cells were assessed using IncuCyte Zoom and normalized to % confluence.
Data were analyzed using two-way ANOVA and F ratio of column factor (each DNA damaging agent/ olaparib vs dinaciclib + each DNA damaging agent/olaparib) and p-value are also shown (Supplemental Table 12). Data are shown by mean and SE (n=3 per group). These results are representative of three independent experiments.
Synergistic cytotoxic and anti-proliferative effects between dinaciclib and olaparib
We focused on the combination of dinaciclib and olaparib because there were obvious synergistic anti-proliferative and cytotoxic effects between the two drugs in WFU3 cells with loss of Map3k7 (Fig 4A–B, 5A, supplemental Fig 5D, and supplemental Table 13A). In cell cycle analysis, the combination of dinaciclib and olaparib increased G2/M in WFU3 shMap3k7 compared to WFU3 shControl (2.5- vs 1.8-fold increase compared to vehicle in WFU3 shMap3k7 vs shControl) as well as each single agent (% of G2/M phase: 20.69 ± 2.023, 24.00 ± 2.103, 28.34 ± 1.037, and 51.05 ± 2.256% in vehicle, olaparib, dinaciclib and the combination treatment in WFU3 shMap3k7, respectively) while it decreased S phase (Fig 5B). The combination therapy induced a strong cytotoxic effect in WFU3 with loss of Map3k7, but not loss of Chd1 (Fig 5C). The increase of Annexin V and PI positive cells by the combination of dinacilib and olaparib in WFU3 with loss of Map3K7 was confirmed by flow cytometry analysis (% of Annexin V and PI positive cells: 14.50 ± 0.9613 vs 77.82 ± 1.507% in WFU3 shControl vs shMap3k7 in the combination therapy, p<0.0001, supplemental Fig 6A and 6B).
Figure 5: The combination therapy of dinaciclib and olaparib in WFU3.
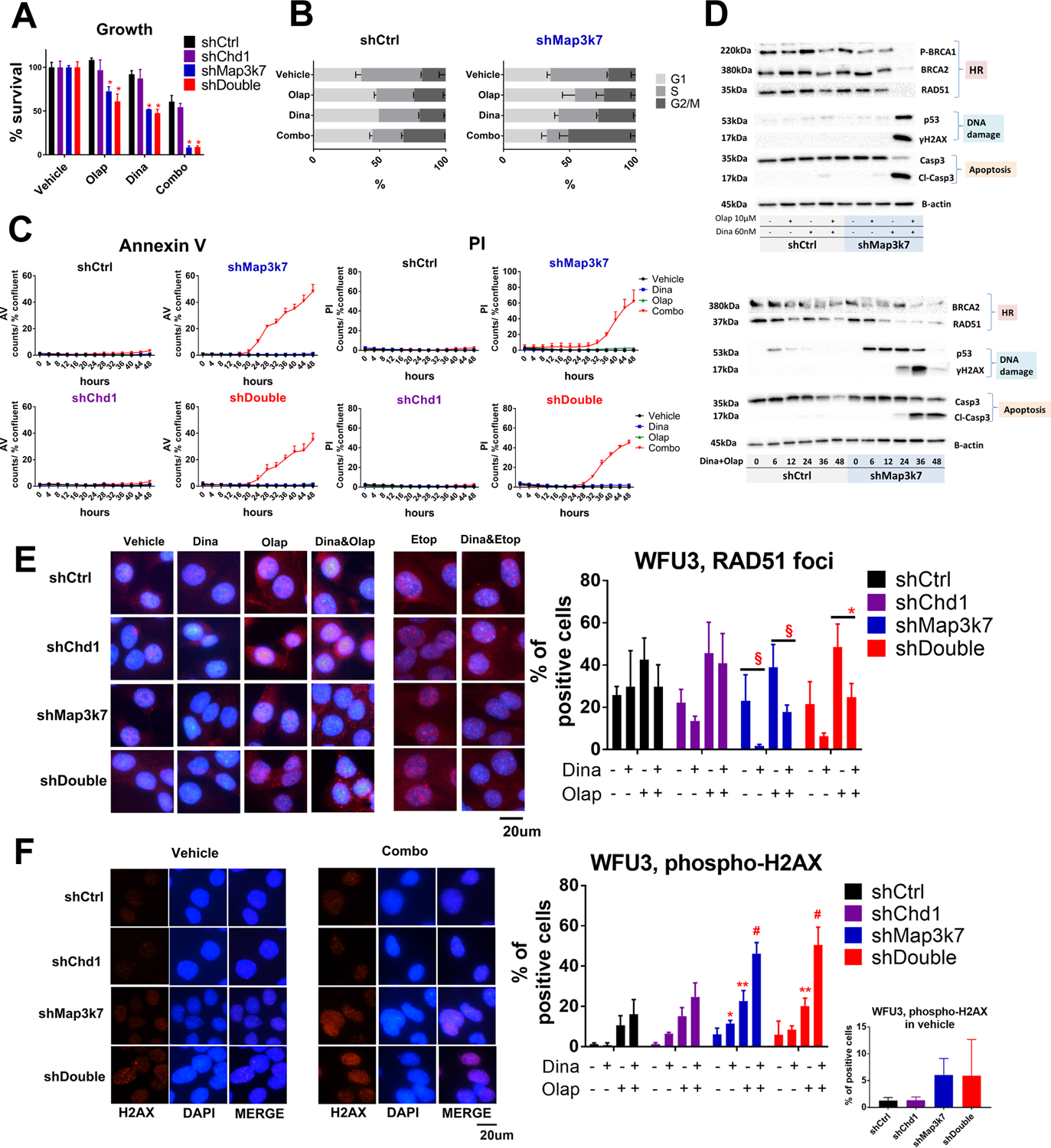
A: Anti-proliferative effects in response to vehicle, dinaciclib (60nM), olaparib (10μM) or the combination of dinaciclib and olaparib in WFU3 shControl, shChd1, shMap3k7 and shDouble. The combination of dinaciclib and olaparib inhibited the cell growth of WFU3 with suppression of Map3k7 without regard to Chd1 status. Cells were treated with these drugs for 48hr. * p<0.0001 compared to WFU3 shControl by two-way ANOVA with Dunnett’s multiple comparisons test. Data are shown by mean and SE (n=3 per group). The results are representative of three independent experiments.
B: The results of cell cycle analysis in response to vehicle, dinaciclib (60nM), olaparib (10μM) or the combination of dinaciclib and olaparib in WFU3 shControl and shMap3k7. Cells were treated with these drugs for 24hr. Data are shown by mean and SE (n=3 per group). The results are representative of two independent experiments.
C: Time course of Annexin V and PI positive cells in response to vehicle (black), dinaciclib (60nM; blue), olaparib (10μM; green), or the combination (red) in WFU3 shControl, shMap3k7, shChd1, versus shDouble. Cells were treated with these drugs for up to 48hr. Data are shown by mean and SE (n=3 per group). The results are representative of three independent experiments.
D: HR-related proteins were selectively suppressed in WFU3 shMap3k7 in response to the combination of dinaciclib (60nM) and olaparib (10μM), which was accompanied by induction of ɤH2AX, p53, and cleaved caspase-3. Cells were treated with drugs for 36hr. Time dependent reduction of BRCA2 and RAD51 accompanied by increases in p53, ɤH2AX and cleavage caspase-3 in response to the combination dinaciclib and olaparib are also shown. The results are representative of at least two independent experiments.
E: RAD51 foci were selectively suppressed in WFU3 shMap3k7 and shDouble in response to the dinaciclib (60nM) with or without olaparib (10μM)/ etoposide (10μM). Cells were treated with drugs for 12hr. Pictures are representative of merged images of RAD51 foci with DAPI. § p<0.1, * p<0.05 by two-way ANOVA with Tukey’s multiple comparisons test. Data are shown by mean and SE (n=3 per group).
F: ɤH2AX foci were more observed in WFU3 shMap3k7 and shDouble in vehicle condition (small graph) as well as in response to the dinaciclib (60nM) and/or olaparib (10μM) (large graph) compared to WFU3 shControl. Cells were treated with drugs for 36hr. Pictures are representative of images of ɤH2AX foci, DAPI, and merge. * p<0.05, ** p<0.01, # p<0.0001 vs shControl by two-way ANOVA with Dunnet’s multiple comparisons test. Data are shown by mean and SE (n=3 per group).
Dinaciclib did not decrease HR-related proteins in WFU3 shControl while the combination of olaparib and dinaciclib slightly decreased HR-related proteins (Fig 5D and supplemental Table 14A). In contrast, in WFU3 shMap3k7, dinaciclib decreased the expression of HR-related proteins and the combination therapy caused nearly complete losses of BRCA1 phosphorylation, BRCA2, and RAD51, accompanied by the strong induction of ɤH2AX, p53, and caspase-3 cleavage (Fig 5D and supplemental Table 14A). The time dependent decrease of BRCA2 and RAD51 started by 6–12hr and the increase of p53 and ɤH2AX started by 6 and 24hr, respectively (Fig 5D and supplemental Table 14B). The decrease in BRCA2 and RAD51 in response to dinaciclib and olaparib was observed in all of cell cycle phases in WFU3 shMap3k7 (supplemental Fig 7), demonstrating that the decrease in these proteins was not reflective of a decrease in S phase cells.
We next used immunofluorescence to detect the formation of RAD51 foci following olaparib treatment in all four types of WFU3 (Fig 5E). The formation of RAD51 foci in the nucleus was suppressed by dinaciclib with or without olaparib in WFU3 shMap3k7 and shDouble, while the decrease of RAD51 foci formation was not significant in WFU3 shControl and shChd1 (relative decrease in RAD51 foci positive cells in the combination therapy with olaparib: 0.458 and 0.510 in WFU3 shMap3k7 and shDouble vs. 0.700 and 0.896 in WFU3 shChd1 and shDouble, respectively). The combination therapy induced perinuclear RAD51 foci formation in WFU3 shMap3k7 and shDouble while this was rarely observed in WFU3 shControl and shChd1 (Fig 5E). Dinacilib also decreased the RAD51 foci formation in response to etoposide treatment in WFU3 shMap3k7 and shDouble while perinuclear RAD51 formation was rarely observed in these combinations (Fig 5E). ɤH2AX foci were increased 4 to 5-fold in WFU3 shMap3k7 and shDouble in the vehicle condition compared to WFU3 shControl or shChd1 (Fig 5F). WFU3 cells treated with the combination of dinaciclib and olaparib displayed greater induction of ɤH2AX compared to those treated with each single agent (Fig 5F). ɤH2AX foci formation was significantly more induced by these drugs in WFU3 shMap3k7 and shDouble, but not in WFU3 shChd1, compared to WFU3 shControl.
We next asked whether the combination of these two drugs had similar effects on human prostate cancer cells. To this end, we used LNCaP, androgen-sensitive human prostate adenocarcinoma cells. The combination of dinaciclib and olaparib caused stronger cytotoxic and anti-proliferative effects compared to each single agent in both LNCaP shControl and shDouble (supplemental Fig 8A). Dinaciclib decreased HR-related proteins with induction of p53 and ɤH2AX in both LNCaP shControl and shDouble. When dinaciclib was combined with olaparib it caused more induction of ɤH2AX and p53 accompanied by an increase of caspase 3 cleavage (supplemental Fig 8B).
Combined cytotoxic and anti-proliferative effects between dinaciclib and docetaxel in BHPrE1
Loss of MAP3K7 augmented the anti-proliferative and cytotoxic effects in response to docetaxel and the combination of dinaciclib and docetaxel in BHPrE1 (Fig 6A and 6B). This combination therapy induced an additive anti-proliferative effect (supplement Table 13B) and slightly increased the proportion of cells in S phase of the cell cycle in both BHPrE1 shControl and shMAP3K7 (1.2- and 1.2-fold increase compared to vehicle in BHPrE1 shControl and shMAP3K7, respectively, Fig 6C). Docetaxel increased HR-related proteins in BHPrE1 shControl, likely a compensation of DNA damage by docetaxel, while the increase of those proteins by docetaxel was not observed in BHPrE1 shMAP3K7 (Fig. 6D and supplemental Table 15). Dinaciclib slightly suppressed HR-related proteins in BHPrE1 shMAP3K7 while the combination of dinaciclib and docetaxel further suppressed them (Fig 6D and supplemental Table 15). The decreases in ATR, ATM and BRCA2 by the combination therapy were selectively observed in BHPrE1 shMAP3K7, and were accompanied by greater induction of ɤH2AX and caspase-3 cleavage (Fig 6D). Similar to what we observed in WFU3 cells (Fig 5F), ɤH2AX foci formation was 5-fold more prevalent in BHPrE1 shMAP3K7 compared to BHPrE1 shControl in the vehicle condition (Fig 6E) while it was also significantly more induced in response to dinaciclib and docetaxel in BHPrE1 shMAP3K compared to BHPrE1 shControl.
Figure 6: The combination therapy of dinaciclib and docetaxel in BHPrE1.
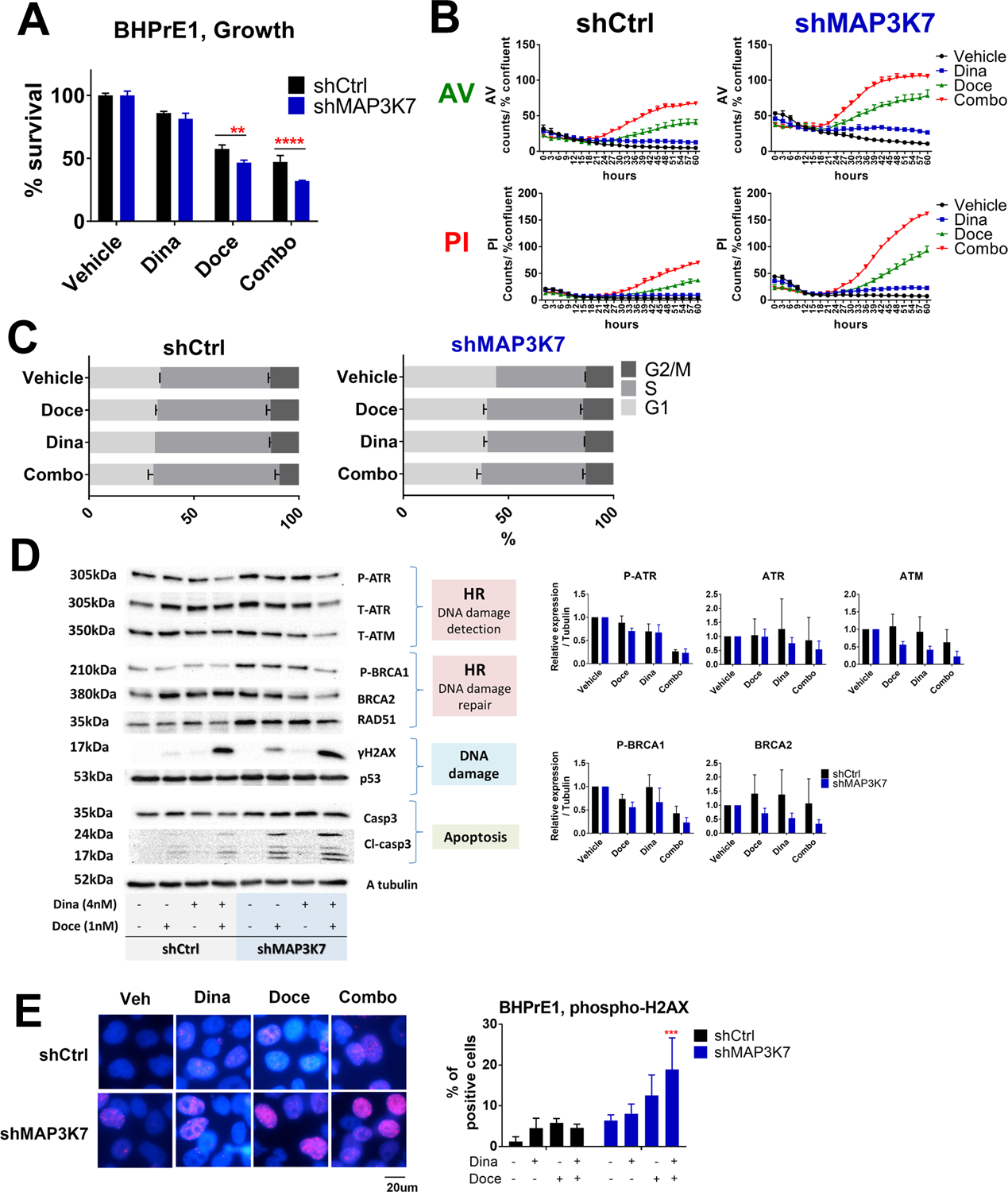
A: Cell growth assay in response to vehicle, dinaciclib (4nM), docetaxel (1nM), or the combination of dinaciclib and docetaxel in BHPrE1 shControl versus shMAP3K7 are shown. Cells were treated with these drugs for 72hr and cell growth was assessed using CellTiterGlo. ** p<0.01 and **** p<0.0001 by two-way ANOVA with Sidak’s multiple comparisons test. Data are shown by mean and SE (n=3 per group). These results are representative of three independent experiments.
B: Time course of Annexin V and PI positive cells in BHPrE1 shControl versus BHPrE1 shMAP3K7 in response to vehicle (black), Dinaciclib (4nM; blue), docetaxel 1nM (green) or the combination of dinaciclib and docetaxel (red). Cells were treated with these drugs for up to 60hr. Data are shown by mean and SE (n=3 per group). These results are representative of three independent experiments.
C: Cell cycle analyses in BHPrE1 shControl and shMAP3K7 in response to vehicle, dinaciclib (4nM), docetaxel (1nM) or the combination of dinaciclib and docetaxel. Cells were treated with these drugs for 24hr. Data are shown by mean and SE (n=3 per group). These results are representative of two independent experiments.
D: Cells were treated with vehicle, dinaciclib (4nM), docetaxel (1nM), or the combination of dinaciclib and docetaxel for 48hr before harvesting. HR-related proteins were suppressed by the combination therapy compared to each single agent, and this was accompanied by induction of ɤH2AX and cleaved caspase-3. These results are representative (bands) and mean and SE (bar graphs) of three independent experiments.
E: ɤH2AX foci increased in BHP shMAP3K7 in vehicle condition as well as in response to the dinaciclib (4nM) and docetaxel (1nM). Cells were treated with drugs for 24hr. Pictures are representative of merged images of ɤH2AX foci with DAPI. The results are representative of two independent experiments (n=3 per each group). *** p<0.001 vs BHPrE1 shControl by two-way ANOVA with Dunnet’s multiple comparisons test. Data are shown by mean and SE (n=3 per group).
Discussion
We applied an established computational systems pharmacology approach (TRAP) to gene expression profile data in TCGA to identify potential signaling pathways altered in CaP with loss of CHD1 & MAP3K7, and we prioritized druggable target genes, including CDK1, CDK2, CHEK1, CHEK2, TOP2A, and BIRC5 (Fig 1C). We also validated a subset of the TRAP predictions using mouse prostate epithelial/stem cells (WFU3) and demonstrated that CDK1/2 inhibitor, CHEK1/2 inhibitors, TOP2A inhibitors, and BIRC5 inhibitors caused a 1.5 – 7.4-fold decrease in IC50 in WFU3 with co-suppression of Chd1 and Map3k7 compared to WFU3 without suppression of these genes (Fig 1D and 1E). In contrast, several anti-cancer drugs not predicted to be effective by TRAP analysis showed no effect.
Dinaciclib inhibits multiple CDKs, including CDK-1, −2, −5, −9, −12 activity in in vitro kinase assays with IC50 values of 3, 1, 1, 4, and 58 nmol/L, respectively (27,28). Dinaciclib has been mainly studied in chronic lymphocytic leukemia, breast cancer, and multiple myeloma in single agent or combination treatments (12,27,29,30), but, to the best of our knowledge, has not been studied in prostate cancer. Although CDKs are established cell cycle regulators, multiple lines of evidence suggest a highly diverse and complex role for CDKs in the cellular response to DNA damage (23). CDKs play crucial roles in both the activation of DNA damage checkpoint signaling and HR-dependent DNA repair. (31–34). CDK-1, −2, and −5 directly regulate ATR and ATM, both of which function as DNA damage checkpoints and mediate the initiation of HR (25). CDK1 also regulates BRCA1 phosphorylation and subsequent recruitment to sites of DNA damage, which is associated with RAD51 and BRCA2 recruitment, and HR DNA repair (23). Previous studies have shown that Dinaciclib disrupts HR-dependent decreases in BRCA1 phosphorylation, RAD51 and BRCA2 (12,27). We demonstrated for the first time that Dinaciclib decreased phosphorylation and expression of ATR and expression of ATM, while also disrupting the HR DNA repair as shown by decreases in BRCA1 phosphorylation, BRCA2 and RAD51 as well as decreased RAD51 foci formation in prostate cells (Fig 2B, 3B, and 5E). Dinaciclib also induced an accumulation of DNA damage as shown by induction of ɤH2AX and p53 as a single agent (Fig 2B, 3B, 5F, and 6E). Taken together, dinaciclib disrupts HR, most likely through CDK inhibition leading to the accumulation of DNA damage in prostate cells.
CHD1 is a member of the SNF2-like family of helicase-related enzymes, and part of a subgroup of CHD proteins that contain tandem chromodomains (35). Recent studies have demonstrated that targeted disruption of CHD1 in human cells leads to a defect in early double-strand break repair via HR, resulting in hypersensitivity to ionizing radiation as well as PARP and PTEN inhibition (6,36,37). Although, in the present study, suppression of Chd1 somewhat sensitized WFU3 cells to doxorubicin, etoposide, and olaparib; the drug sensitizing effect of Chd1 suppression was not as strong as that of Map3k7 suppression (supplemental Fig 3). Furthermore, suppression of Chd1 alone did not sensitize mouse prostate cells to dinaciclib, docetaxel or the combination of dinaciclib and olaparib (Fig 2A, 4A, 4B, and supplemental Fig 3). Although we observed down-regulation of CHD1 expression in response to dinaciclib independent of MAP3K7 status the suppression of CHD1 did not affect the dinaciclib-induced disruption of HR and its sensitivity (Fig 2A, 2B and supplemental Table 11A). The mechanisms of down-regulation of CHD1 by dinaciclib remain unknown.
MAP3K7 encodes a MAP kinase kinase kinase that is immediately downstream of cytokine and stress response pathways. MAP3K7 is lost in 30–40% of primary prostate tumors (2,3). About half of CaP samples with loss of MAP3K7 have co-loss of CHD1 (5). We demonstrated that suppression of MAP3K7 sensitized cells to the effects of dinaciclib through the augmented disruption of HR in both mouse and human prostate cells (Fig 2A, 2B, 3A, and 3B). Suppression of MAP3K7 also affected the anti-tumor effects of DNA damaging agents such as doxorubicin, etoposide, docetaxel, and olaparib (supplemental Fig 3). The association of MAP3K7 with HR has not been previously demonstrated. We hypothesize that prostate cells with loss of MAP3K7 likely have defects in DNA-repair pathways leading to especially high dependence on HR activities while normal cells have intact DNA-repair pathways and accumulate less unrepaired DNA damage. In support of this hypothesis, suppression of MAP3K7 by itself in the absence of any drug treatment increased ɤH2AX foci formation in WFU3 as well as BHPrE1 cells (Fig 5F and 6E). Additionally, prostate cells with suppression of MAP3K7 with or without suppression of CHD1 increased HR-related gene expression, including RAD51, CHEK1, CHEK2, ATM, ATR, and/or BRCA2 in TCGA data, WFU3, and/or BHPrE1(Fig 1C, 3B, supplemental Fig 4, supplemental Table 2). Docetaxel increased the HR-related proteins in BHPrE1 without loss of MAP3K7, whereas HR-related proteins were already upregulated in BHPrE1 with loss of MAP3K7 with no additional increase of these proteins upon docetaxel treatment (Fig 6D and supplemental Table 15). Together, our results support the hypothesis that loss of MAP3K7 results in repair defects in CaP. Although we previously reported single loss of MAP3K7 is not an independent prognostic contributor (5), findings in the present study suggest that loss of MAP3K7, not loss of CHD1, is the main contributor of response to the drugs we tested, which suggests MAP3K7 could be used as a predictive marker of treatment response.. The prevalence of prostate tumors with single loss of MAP3K7 is twice as that with dual loss of MAP3K7 and CHD1 (5) and therefore these findings would open the therapeutic window for prostate cancer treatment.
Although we found that dinaciclib alone significantly decreased cell viability in prostate cells with suppression of MAP3K7, a greater effect occurs when combined with DNA damaging agents such as doxorubicin, etoposide and docetaxel, or a PARP inhibitor (Fig 4). Previous studies have also shown that dinaciclib enhances the antitumor effects of the PARP inhibitors, doxorubicin and gemcitabine (12,27). In WFU3 with suppression of Map3k7, the combination of dinaciclib and olaparib strongly disrupted HR compared to each single agent, and this was associated with accumulation of DNA damage and the striking synergistic anti-proliferative and cytotoxic effects (Fig 5A, 5C–F, and supplemental Table 13A and 14). WFU3 with suppression of Map3k7 displayed perinuclear RAD51 foci formation in response to the combination of dinaciclib and olaparib for 12 hours while this was rarely observed in each single agent or the combination of dinaciclib with etoposide (Fig 5E). Perinuclear RAD51 formation might reflect RAD51 foci formed with damaged DNA in the cytoplasm of dying cells. However, in cell death assays, there were few Annexin V and PI positive cells at 12 hours of treatment (Fig 5C), suggesting another mechanism may be responsible for these observations. The exact mechanisms of perinuclear RAD51 formation by the combination of dinaciclib and olaparib remain unknown. The combination of dinaciclib and docetaxel strongly disrupted HR compared to each single agent, leading to accumulation of DNA damage and an additive anti-proliferative effect in BHPrE1 with suppression of MAP3K7 (Fig 6A, 6C–E, and supplemental Table 13B and 15). Importantly, these effects in BHPrE1 were obtained with very low concentration of drugs, docetaxel 1nM and dinaciclib 4nM, which would open the therapeutic window for prostate cancer treatment. However, it has yet to be determined how MAP3K7 is associated with DNA damage response including HR.
Dinaciclib decreased the expression and phosphorylation of HR-related proteins to a similar degree in LNCaP with suppression of CHD1 and MAP3K7 and those without loss of these genes. This resulted in similar anti-proliferative effects, yet still stronger cytotoxic effects on LNCaP with suppression of these genes (supplemental Fig 8A−B). Because LNCaP cells already have hemizygous deletion of MAP3K7, and thus lower expression of MAP3K7 (2,38,39), additional suppression of MAP3K7 expression might have little effect on augmenting disruption of HR. Accordingly, increased cytotoxicity with the combination of dinaciclib and olaparib was observed in LNCaP with or without knockdown of CHD1 and MAP3K7 (supplement Fig 8A−B).
Previous studies also have shown that MAP3K7 functions as a critical mediator of the genotoxic stress response and is required for NF-ĸB, p38 MAPK, and JNK activation (40,41). MAP3K7 silencing results in an impaired NF-ĸB-dependent anti-apoptotic response and increased sensitivity to DNA damage. MAP3K7 was also demonstrated to be a prosurvival mediator in colon cancer cells displaying hyperactive KRAS-dependent Wnt signaling (42). Therefore, loss of MAP3K7 could modulate these pathways and affect sensitivity to anti-cancer drugs in prostate cells.
In conclusion, dinaciclib selectively disrupts HR in prostate cells with decreased MAP3K7 expression. The combination of dinaciclib and DNA damaging agents causes an accumulation of DNA damage which results in synergistic or additive antitumor effects on prostate cells with loss of MAP3K7 (supplemental Fig 9).
Supplementary Material
Acknowledgements:
This project was supported by grants from the NIH (R21-CA187354 and R01-CA199741) to S.D.C., Cancer League of Colorado Grant to S.D.C and J.C.C., American Cancer Society (126572-PF-14-223-01-CCE) to L.C.R., Boettcher Foundation Webb-Waring grant to J.C.C., and Paul Calabresi Award for Clinical Oncology 5K12CA086913 to E.T.L. The University of Colorado Anschutz Medical Campus Cancer Center Cores used in this study were supported in part by the NIH/NCI Cancer Center Support Grant P30CA046934. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Footnotes
Conflict of interest: Nothing to declare
Implications: These findings demonstrate that loss of MAP3K7 is a main contributing factor to drug response through disruption of homologous recombination in CaP.
Data and materials availability: Accession codes: The RNA-Seq data is deposited in GEO with accession number GSE132664.
Reference
- 1.Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, et al. Punctuated evolution of prostate cancer genomes. Cell 2013;153(3):666–77 doi 10.1016/j.cell.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu W, Chang BL, Cramer S, Koty PP, Li T, Sun J, et al. Deletion of a small consensus region at 6q15, including the MAP3K7 gene, is significantly associated with high-grade prostate cancers. Clin Cancer Res 2007;13(17):5028–33. [DOI] [PubMed] [Google Scholar]
- 3.Wu M, Shi L, Cimic A, Romero L, Sui G, Lees CJ, et al. Suppression of Tak1 promotes prostate tumorigenesis. Cancer research 2012;72(11):2833–43 doi 10.1158/0008-5472.CAN-11-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu W, Lindberg J, Sui G, Luo J, Egevad L, Li T, et al. Identification of novel CHD1-associated collaborative alterations of genomic structure and functional assessment of CHD1 in prostate cancer. Oncogene 2012;31(35):3939–48 doi 10.1038/onc.2011.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodrigues LU, Rider L, Nieto C, Romero L, Karimpour-Fard A, Loda M, et al. Coordinate loss of MAP3K7 and CHD1 promotes aggressive prostate cancer. Cancer Res 2015;75(6):1021–34 doi 10.1158/0008-5472.CAN-14-1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kari V, Mansour WY, Raul SK, Baumgart SJ, Mund A, Grade M, et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO reports 2016;17(11):1609–23 doi 10.15252/embr.201642352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nature Medicine 2012;18(10):1503–10 doi 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cancer Genome Atlas Research N. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015;163(4):1011–25 doi 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barclay WW, Axanova LS, Chen W, Romero L, Maund SL, Soker S, et al. Characterization of adult prostatic progenitor/stem cells exhibiting self-renewal and multilineage differentiation. Stem Cells 2008;26(3):600–10 doi 10.1634/stemcells.2007-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang M, Strand DW, Fernandez S, He Y, Yi Y, Birbach A, et al. Functional remodeling of benign human prostatic tissues in vivo by spontaneously immortalized progenitor and intermediate cells. Stem Cells 2010;28(2):344–56 doi 10.1002/stem.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krishan A Rapid flow cytofluorometric analysis of mammalian cell cycle by propidium iodide staining. J Cell Biol 1975;66(1):188–93 doi 10.1083/jcb.66.1.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alagpulinsa DA, Ayyadevara S, Yaccoby S, Shmookler Reis RJ. A Cyclin-Dependent Kinase Inhibitor, Dinaciclib, Impairs Homologous Recombination and Sensitizes Multiple Myeloma Cells to PARP Inhibition. Mol Cancer Ther 2016;15(2):241–50 doi 10.1158/1535-7163.MCT-15-0660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ivashkevich AN, Martin OA, Smith AJ, Redon CE, Bonner WM, Martin RF, et al. gammaH2AX foci as a measure of DNA damage: a computational approach to automatic analysis. Mutat Res 2011;711(1–2):49–60 doi 10.1016/j.mrfmmm.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003;19(2):185–93 doi 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 15.Griffith M, Griffith OL, Coffman AC, Weible JV, McMichael JF, Spies NC, et al. DGIdb: mining the druggable genome. Nat Methods 2013;10(12):1209–10 doi 10.1038/nmeth.2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rahman M, Jackson LK, Johnson WE, Li DY, Bild AH, Piccolo SR. Alternative preprocessing of RNA-Sequencing data in The Cancer Genome Atlas leads to improved analysis results. Bioinformatics 2015;31(22):3666–72 doi 10.1093/bioinformatics/btv377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34(3):267–73 doi 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 19.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102(43):15545–50 doi 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Opsahl T, Agneessens F, Skvoretz J. Node centrality in weighted networks: Generalizing degree and shortest paths. Social Networks 2010;32:245–51. [Google Scholar]
- 21.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9(4):357–9 doi 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323 doi 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson N, Shapiro GI. Cyclin-dependent kinases (cdks) and the DNA damage response: rationale for cdk inhibitor-chemotherapy combinations as an anticancer strategy for solid tumors. Expert Opin Ther Targets 2010;14(11):1199–212 doi 10.1517/14728222.2010.525221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005;434(7035):913–7 doi 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 25.Helleday T The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 2011;5(4):387–93 doi 10.1016/j.molonc.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel AG, Sarkaria JN, Kaufmann SH. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc Natl Acad Sci U S A 2011;108(8):3406–11 doi 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, et al. CDK12 Inhibition Reverses De Novo and Acquired PARP Inhibitor Resistance in BRCA Wild-Type and Mutated Models of Triple-Negative Breast Cancer. Cell Rep 2016;17(9):2367–81 doi 10.1016/j.celrep.2016.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parry D, Guzi T, Shanahan F, Davis N, Prabhavalkar D, Wiswell D, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther 2010;9(8):2344–53 doi 10.1158/1535-7163.MCT-10-0324. [DOI] [PubMed] [Google Scholar]
- 29.Ghia P, Scarfo L, Perez S, Pathiraja K, Derosier M, Small K, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017;129(13):1876–8 doi 10.1182/blood-2016-10-748210. [DOI] [PubMed] [Google Scholar]
- 30.Mita MM, Joy AA, Mita A, Sankhala K, Jou YM, Zhang D, et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer 2014;14(3):169–76 doi 10.1016/j.clbc.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 31.Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004;431(7011):1011–7 doi 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006;8(1):37–45 doi 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 33.Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, et al. Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 2009;35(3):327–39 doi 10.1016/j.molcel.2009.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scully R, Xie A. In my end is my beginning: control of end resection and DSBR pathway ‘choice’ by cyclin-dependent kinases. Oncogene 2005;24(17):2871–6 doi 10.1038/sj.onc.1208609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou J, Li J, Serafim RB, Ketchum S, Ferreira CG, Liu JC, et al. Human CHD1 is required for early DNA-damage signaling and is uniquely regulated by its N terminus. Nucleic Acids Res 2018;46(8):3891–905 doi 10.1093/nar/gky128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shenoy TR, Boysen G, Wang MY, Xu QZ, Guo W, Koh FM, et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann Oncol 2017;28(7):1495–507 doi 10.1093/annonc/mdx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian B, Yang Q, Mao Z. Phosphorylation of ATM by Cdk5 mediates DNA damage signalling and regulates neuronal death. Nat Cell Biol 2009;11(2):211–8 doi 10.1038/ncb1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 2015;33(3):306–12 doi 10.1038/nbt.3080. [DOI] [PubMed] [Google Scholar]
- 39.Kluth M, Hesse J, Heinl A, Krohn A, Steurer S, Sirma H, et al. Genomic deletion of MAP3K7 at 6q12–22 is associated with early PSA recurrence in prostate cancer and absence of TMPRSS2:ERG fusions. Mod Pathol 2013;26(7):975–83 doi 10.1038/modpathol.2012.236. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001;412(6844):346–51 doi 10.1038/35085597. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y, Xia F, Hermance N, Mabb A, Simonson S, Morrissey S, et al. A cytosolic ATM/NEMO/RIP1 complex recruits TAK1 to mediate the NF-kappaB and p38 mitogen-activated protein kinase (MAPK)/MAPK-activated protein 2 responses to DNA damage. Mol Cell Biol 2011;31(14):2774–86 doi 10.1128/MCB.01139-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Singh A, Sweeney MF, Yu M, Burger A, Greninger P, Benes C, et al. TAK1 inhibition promotes apoptosis in KRAS-dependent colon cancers. Cell 2012;148(4):639–50 doi 10.1016/j.cell.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.