

Abstract
An essential prerequisite for in vitro biochemical or structural studies is a construct that is amenable to high level expression and purification and is biochemically “well-behaved”. In the field of membrane protein research, the use of Green Fluorescent Protein (GFP) to monitor and optimize the heterologous expression in different hosts has radically changed the ease of streamlining and multiplexing the testing of a large number of candidate constructs. This is achieved by genetically fusing the fluorescent proteins to the N- or C-terminus of the proteins of interest to act as reporters which can then be followed by methods such as microscopy, spectroscopy, or in-gel fluorescence. Nonetheless a systematic study on the effect of GFP and its spectral variants on the expression and yields of recombinant membrane proteins is lacking. In this study, we genetically appended four common fluorescent protein tags, namely mEGFP, mVenus, mCerulean, and mCherry, to the N- or C-terminus of different membrane proteins and assessed their expression in mammalian cells by fluorescence-detection size exclusion chromatography (FSEC) and protein purification. We find that of the four fluorescent proteins, tagging with mVenus systematically results in higher expression levels that translates to higher yields in preparative purifications, thus making a case for switching to this yellow spectral variant as a better fusion tag.
Introduction
Membrane proteins constitute 25–30% of proteins encoded by the human genome [1,2] and make up for more than 50% drug targets [3,4]. Despite their critical roles in important cellular processes such as transport across membranes, signaling and generation of energy – structural studies of membrane proteins have usually lagged behind that of soluble proteins. Currently, the protein data bank (PDB) contains upwards of a hundred thousand coordinates for protein structures of which only two and a half thousand are of membrane proteins; that is membrane proteins constitute only 2.5% of all the structures in the PDB (White, Stephen. http://blanco.biomol.uci.edu/mpstruc/). This is mostly because membrane proteins are difficult to study. As the proteins’ native milieu is a complex hydrophobic lipid bilayer with parts of the protein exposed to aqueous regions on either side of the bilayer, it is difficult to mimic these conditions in vitro to keep the membrane protein stable, let alone crystallize it. However substantial improvements have been made in studying membrane protein structure. Developments in recombinant protein expression [5–7], protein expression and stability screening methods [8–15], detergents [16–19], crystallization conditions mined from literature [20–22], nanodisc/lipodisq technology [23,24], lipid-rich crystallization methods such as lipidic cubic phase [25], HiLide [26] and bicelles [27], nanobodies [28,29], monobodies [30], and fusion partners [31,32] to stabilize membrane proteins and increase ‘crystallizability’, high intensity microfocus x-ray beams with rastering [33,34], and finally the renaissance of high-resolution cryo-electron microscopy [35,36] have all contributed to an exponential increase in membrane protein structures.
Given a target protein, since it is not known a priori which particular sequence variants are the most stable and hence suitable for structural studies – it is usually necessary to screen orthologous proteins from various organisms to find the best candidates. Subsequently, screening engineered variants of chosen orthologs is almost a necessary prerequisite to increase stability for crystallization trials, a route that has been very successful for GPCRs [37–39] and neurotransmitter transporters [40–42]. Traditionally in the field of protein crystallography, a monodisperse size-exclusion chromatographic profile has been generally accepted as a useful predictor for success in crystallization experiments and is thus widely used as a parameter for screening. However, membrane proteins typically do not express at levels as high as soluble proteins. Moreover, reagents needed for extraction and purification are expensive making this initial screening step a bottleneck. The use of green fluorescent protein (GFP) as a fusion protein has emerged as a powerful tool for alleviating this bottleneck. In particular, following the GFPs’ fluorescence-signal during size exclusion chromatography allows one to screen putative candidates for both yield as well as monodispersity in a single experiment from crude lysates of cells [8,9,11]. With autosampler-equipped liquid chromatography systems, one can easily screen upwards of twenty-five constructs per day in a traditional analytical Superdex 200 column with minimal manual intervention. The technique is flexible and has been adapted for various purposes such as thermal denaturation studies and multi-protein complexes [14,43,44]. Recently, the development of anti-GFP nanobodies [45,46] has made it so that the GFP itself can be used as a purification tag thus furthering its attraction as a fusion partner.
During our experiments with the DHHC20 protein acyltransferase [47], a Golgi-localized enzyme with four transmembrane helices that catalyzes protein palmitoylation, we noticed that the auto-palmitoylation activity of cell lysates from HEK 293T cells transiently over-expressing N-terminal mVenus yellow fluorescent protein and mCerulean cyan fluorescent protein tagged DHHC20 were consistently higher than that of enhanced GFP (mEGFP) fused DHHC20 (Figure S1A). mVenus and mCerulean are the yellow and cyan spectral variants of mEGFP obtained by protein engineering [48–50]. We initially hypothesized that an error in molecular biology while making the constructs must have disrupted the promoter or upstream DNA elements; however, sequencing shows no difference (Figure S1C). Confocal microscopy also shows no gross differences in cellular localization between the differently tagged DHHC20 protein (Figure S1B). This suggested that the use of a fluorescent protein tag other than the usual mEGFP might be advantageous. Therefore, we compared the effects of four commonly utilized fluorescent protein fusion tags, namely mVenus, mCerulean, mEGFP, and mCherry [51] on expression and yields of different recombinant integral membrane proteins and report our findings here.
Experimental Procedures
Cloning
Fluorescent proteins coding sequences were PCR amplified using forward primers containing an NdeI restriction site and reverse primers with a XhoI restriction site. The coding sequences of the fluorescent proteins are provided in Table S1 of the supporting information. The PCR products were digested with the NdeI and XhoI enzymes and ligated into a modified pET28b vector using T4 DNA ligase. The modified pET28b has its regular thrombin protease cleavage site replaced by a PreScission protease site. This gives rise to the initial protein sequence MGSSHHHHHHSSGLEVLFQGPHMVSKGEE where the PreScission octapeptide protease cleavage site is underlined.
Our initial protein over-expression construct was a pPICZ decaHis (His10) N- and C-terminal mEGFP tagged DHHC20 (Uniprot Q5W0Z9–4) with a PreScission protease site between the DHHC20 and the mEGFP fusion protein. The DHHC20 cDNA can be swapped out for any other protein of interest using XhoI-EcoRI digestion and T4 DNA ligation (Figure S2). The mEGFP in the pPICZ vector was replaced with the different fluorescent protein cDNAs using overlap extension PCR [52]. Following this, the entire expression cassette composed of the decaHis-tag, the different fluorescent fusion proteins, and DHHC20 was PCR amplified and then inserted downstream of the CMV promoter of a modified pEG-Bacmam vector using overlap extension PCR such that the entire multiple cloning site was replaced. The modified pEG-Bacmam vector has an extra XhoI restriction site in the multiple cloning site downstream of the p10 promoter, incompatible with our expression cassette, removed. To construct fluorescent protein tagged hPORCN (Uniprot Q9H237) and cmCLC (Uniprot M1UVK6–1) over-expression plasmids, the hPORCN and cmCLC cDNA inserts were cut out the pPICZ vector using XhoI and EcoRI and then ligated into equivalent positions in our different fluorescent protein tag expression cassette using T4 DNA ligase. The cmCLC construct has the first 88 residues removed and is the same as the one used for its structure determination [53].
FP-DHHC20 constructs were cloned into a modified pSP vector that used a EF1α promoter instead of a CMV promoter using Gibson assembly [54]. All constructs were sequenced and verified to have the correct fluorescent protein tags. All PCR reactions were done using either Phusion polymerase (NEB, MA) or Pfu polymerase (Agilent). All cloning enzymes or kits were also from NEB. The oligo primers used are listed in Table S2 of the supporting information.
Fluorescent proteins expression and purification
Fluorescent proteins were expressed in BL21(DE3) Gold. Briefly, 2 liters of fresh 2x YT media with kanamycin were directly inoculated with bacterial colonies from overnight plates. The cultures were grown to an OD600 of 0.8 at 37°C. The temperature was then reduced to 25°C and IPTG added to a final concentration of 1 mM from a 1 M stock. Protein induction was carried out overnight. Cells were harvested by centrifugation next morning and the colored pellets re-suspended in 100 ml of 40 mM TrisHCl, pH 7.4, 270 mM NaCl, 10 mM 2-ME, 10 mM MgCl2, DNase (Worthington Biochemicals), lysozyme, 1 mM PMSF, AEBSF, and 5mM BenzamidineHCl. Cells were lysed by sonication on ice. Cell debris was pelleted by centrifugation at 38,000g for 30 min at 4°C. The supernatant was incubated with 2 ml NiNTA HisPur resin for an hr at 4°C on a rotator. The column was poured by gravity and the resin thoroughly washed with 20 ml of 40 mM TrisHCl, pH 7.4, 270 mM NaCl, 10 mM 2-ME containing 10 mM imidazole and then with same buffer containing 50 mM imidazole. Proteins were eluted with 10 ml of 20 mM TrisHCl, pH7.4, 135 mM NaCl, 5 mM 2-ME containing 350 mM imidazole. The eluates were dialyzed against 2 liters of the same buffer without the imidazole overnight at 4°C. The proteins were then flash frozen in liquid nitrogen and stored in −80°C freezer. At time of use, the proteins were thawed overnight in a 4°C fridge, diluted 10- to 20 times and their concentrations determined using a 660nm protein assay kit with BSA standards (Thermo Scientific Pierce). The protein concentrations were converted to μM using calculated molar masses of 29330, 29270, 29436, and 29160 g/mol for mVenus, mCerulean, mEGFP, and mCherry respectively.
Fluorescent protein thermal stability assays
Pseudo-melting curve experiments were done using a thermal cycler with two heating blocks. Purified fluorescent proteins were diluted to 80–120 μg/ml and 50 μl of each aliquoted into 16 PCR tubes. The tubes were briefly spun using a table top mini centrifuge prior to denaturation. To generate a complete pseudo-melting curve, temperature was varied from 60 to 94°C using the Bio-Rad S1000 thermal cycler’s temperature gradient. For this, a 60–84°C gradient and a 70–94°C gradient was simultaneously used with eight tubes per heat block with a 30 min’ incubation followed by cooling at 4°C for 3 min. The samples were once again spun using a table top mini centrifuge and 30 μl transferred to a 96-well black fluorescence plate (Nunc) using a multi-channel pipette. The sample was then diluted two-fold with 30 μl of 20 mM TrisHCl, pH7.4, 135 mM NaCl buffer. The plate was then centrifuged at 1000g for 3 min and read using a plate reader (Tecan Infinite M100 Pro). Excitation/emission wavelengths for mVenus, mCerulean, mEGFP, and mCherry were 515nm/528nm, 433nm/475nm, 488nm/507nm, and 587nm/610nm respectively with the gain auto-optimized for maximal signal. Melting curve data were fitted to a Hill Equation to extract the pseudo melting temperature (Tm).
Isothermal protein denaturation was carried out on AriaMX RealTime PCR machine (Agilent Technologies). In this assay, the fluorescent protein samples are maintained at an elevated temperature while their fluorescence is monitored with time. Fluorescent proteins were diluted to 8–12 μg/ml and two 50 μl aliquots added to a specialized quantitative PCR 8-tube strip. The tubes were briefly spun using a table top mini centrifuge prior to 40 amplification cycles at time intervals of 30 min at 70°C. We empirically determined 70°C as the best temperature at which to observe measurable differences among the four different fluorescent proteins. FAM filter was used for mVenus, mCerulean, and mEGFP. ROX filter was used for mCherry. Data were fit to a single exponential decay although the model does not adequately explain the mEGFP and mCherry data. However, the fluorescence half-life times obtained from the fits were deemed adequate for comparative purposes.
Transient transfection of HEK293T adherent cells
HEK293T cells were maintained in DMEM media supplemented with 10% FBS, 2 mM glutamine, and 100 U/ml penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2. The cells were transiently transfected using polyethyleneimine (PEI) [55]. The evening before the day of transfection, cells from T-75 flasks or 150 mm cell culture dishes were passaged and ~ 0.25 ×106 cells seeded into 12-well plates with 1 ml media per well. The next morning, 1 μg DNA was diluted into 50 μl of DMEM media. A separate tube contained 3 ug of PEI diluted into 50 μl of DMEM media. The two solutions were mixed, pipetted, and incubated for 15–20 min at room temperature. Then ~ 100 μl per well of the DNA:PEI complex was added dropwise to the wells, the plate gently agitated and returned to the 37°C incubator. Alternatively, post addition of the DNA:PEI complex, the plate was incubated in a humidified shaker maintained at 30°C with 5% CO2 and the shaking turned off. For hPORCN constructs, we obtained more consistent results in doing the transient transfections in 6-well plates seeded with ~0.75×106 cells and transfected using 2 μg DNA complexed with 6 μg PEI per well. Larger scale transfections for protein purifications were done in 150-mm cell culture dishes. The dishes were initially seeded with ~10×106 cells and transfected the next day with 30 μg DNA complexed with 90 μg PEI in a final volume of 2 ml DMEM media.
Cells were maintained for another 48 hr post-transfection. For harvesting, cells were re-suspended in the culture media by mechanical disruption using a pipette or a cell-scraper. The cell pellets were collected by centrifugation, washed once with phosphate buffer saline, and stored at – 20°C until later use.
Baculovirus mediated transduction of HEK293S GnTi− suspension cell cultures
Baculovirus were generated using modifications to established protocols [56,57]. Briefly, the pEG-Bacmam protein expression plasmids were used to generate bacmid DNA using DH10bac cells according to published protocols [7]. Sf9 cells were grown in ESF921 media (Expression Systems) to a density of 3 million/ml at 28°C and 130 rpm. On the day of the transfection, the cells were collected by centrifugation and re-suspended at a density of 1.5 million/ml in 50 ml of fresh ESF 921 media supplemented with 3% FBS. 30 μg bacmid DNA was complexed with 360 μg of PEI, mixed vigorously for 10 sec using a vortexer, and added to the Sf9 cells after a 10 min incubation. After a four-day incubation at 28°C and 130 rpm, cells were pelleted and supernatant containing the baculovirus was sterile filtered. These P0 baculovirus stocks were used without any further amplification [57]. Baculovirus stocks were stored at 4°C and in the dark with FBS added to a final concentration of 1% to prevent proteolysis.
HEK293S GnTi− suspension cell cultures were grown in Hyclone CDM4HEK293 (GE Healthcare) media supplemented with 2% FBS and 100 U/ml penicillin/streptomycin in a humidified shaker maintained at 37°C, 5% CO2 and 130 rpm. For protein expression using the baculovirus, cells were grown to a density of ~ 2 million/ml. To 30 ml of cell culture, 3 ml of the P0 baculovirus was added. Depending on the experiment, at this point the culture was either kept in the same incubator or moved to one maintained at 30°C, 5% CO2 and 130 rpm. The next morning, ~ 12–15 hr post virus addition, 0.3 ml of 1 M sodium butyrate was added to give a final concentration of 10 mM. The cultures were continued for 48 hr after which the cells were harvested by centrifugation. The cell pellets were washed with phosphate buffered saline, flash frozen in liquid nitrogen, and stored in a −80°C freezer until later use.
Protein purification from HEK293T and 293S cells
HEK 293T cell pellets were re-suspended in 500 μl of 40 mM TrisHCl, pH 7.4, 270 mM NaCl, 10 mM MgCl2, 10 mM 2-ME, Dnase, and protease inhibitors. 100 μl of 0.3M DDM stock was added and the tubes rotated at 4°C for 1.5 hr to extract the protein. Cell debris was removed by centrifugation for 10 min at 21,000g and 4°C. The clarified supernatant was then applied to ~ 100 μl of TALON resin (Clontech) equilibrated with the same buffer as above minus the DNase and protease inhibitors and incubated for 1.5 hr on a rotator at 4°C. The resin was first washed with 1 ml of same buffer containing 5 mM imidazole and then with 500 μl of same buffer containing 25 mM imidazole. Finally, the bound proteins were eluted with 100 μl of buffer containing 400 mM imidazole. Proteins from HEK 293S cell pellets were purified in a similar fashion. However, the overall volumes were all scaled up by ~ 4 to 5-fold to account for a denser cell pellet as well as better protein expression. All eluates were analyzed by SDS-PAGE. Gel band intensities were analyzed using FIJI [58].
Anti-GFP nanobody (Nb) resin was generated by coupling Nb with CNBr activated sepharose 4B (GE Healthcare). Cells were lysed and the proteins extracted using DDM as above in 20 mM TrisHCl, pH 7.4, 136 mM NaCl, 10 mM MgCl2, 10 mM 2-ME buffer. The lysates were applied to ~0.35 ml of Nb resin pre-equilibrated with the same buffer. After one hr of incubation on a rotator at 4°C, the Nb resins were washed with 6 ml of the same buffer twice. Then the Nb resins were re-suspend in an equal volume of the buffer to give a ~ 50% slurry and 60 μl of PreScission protease (~ 60 μg) added. The resin slurry was incubated overnight on a 4°C rotator. The next morning, the flow-through was collected and analyzed by SDS-PAGE. The flow-through contains our protein of interest which has been cleaved off of the Nb resin which still binds the GFP tag. All buffers used for washing and resin equilibration contain 1 mM DDM.
Sample preparation for Fluorescence-detected size exclusion chromatography (FSEC)
HEK293T cell pellets from a 12-well plate were re-suspended in 200 μl of 40 mM TrisHCl, pH 7.4, 270 mM NaCl, 10 mM MgCl2, 10 mM 2-ME, Dnase, and protease inhibitors. Protein was extracted by adding 50 μl of a 0.3 M DDM stock concentration to give a final DDM concentration of ~ 60 mM and incubated at 4°C on a rotator. One hr later, cell debris was removed by a 10-min 21,000g spin at 4°C on a bench top centrifuge. The supernatant was spin filtered (Costar Spin-X) prior to placing 200 μl of the sample into the FSEC autosampler of which 100 μl of the sample is injected. HEK293S GnTi− cell pellets also were processed in the same way. For extraction with Cymal-7, Triton X-100, and Anzergent 3–14, a 10% detergent stock was prepared and 50 μl added to the 200 μl of re-suspended cells to give a final detergent concentration of ~ 2%. The rest of the steps were as for DDM extraction.
Fluorescence-detected size exclusion chromatography (FSEC)
All FSEC experiments were done in a Shimadzu UFLC system equipped with a LC-20AB liquid chromatography pump, SIL-20AC HT autosampler, RF-20A XS fluorescence detector, SPD-20A UV-VIS detector, in-line with a 10mm × 300 mm Superdex 200 increase 10/300 GL column (GE Healthcare). The mVenus, mCerulean, mEGFP, and mCherry fluorophores were excited at 515nm, 433nm, 488nm, and 587nm with emission at 528nm, 475nm, 507nm, and 610nm respectively. Fluorescence detector was set to medium sensitivity with a 4-fold gain with mCherry being an exception, for which a 16-fold gain was used. These settings were kept constant. The size exclusion column was run at 0.5 ml/min flow-rate with the buffer usually 20 mM TrisHCl, pH 7.4, 136 mM NaCl, 10 mM 2-ME, and 0.5 mM dodecyl maltoside (DDM). For hPORCN, the NaCl and DDM concentrations were increased to 350 mM and 1 mM respectively.
For FSEC of purified fluorescent proteins, solutions of 0.2 μM were prepared by diluting with the FSEC buffer. The protein solutions were spin filtered prior to injection into the Shimadzu UFLC system. Usually multiple chromatograms were obtained for injections ranging from 10 μl to 100 μl over the course of 24–30 hr in a single experiment. We also varied the order in which the fluorescent proteins were injected. FSEC sample preparation for HEK expressed proteins has been described above.
FSEC chromatograms were exported as X-Y tables into Excel. The average value of the first 10 min of the chromatogram was used to adjust the baseline to zero. The peak value of the chromatogram for the mVenus fluorescent protein was found using the ‘maximum’ function in Excel. This value was used to divide all other chromatograms to give normalized FSEC traces with the mVenus peak height being 1. All data analysis and graphing were done using Origin Lab.
Confocal microscopy
HEK293T cells were seeded on 8-well glass-slide bottom imaging chamber (LabTek) the evening before transfection. Cells were transfected with 100 ng of DNA using PEI as described above. 48 hr post-transfection, cells were imaged using a Zeiss LSM780 microscope with a Plan-Apochromat 63x/1.40 Oil objective lens. Cells expressing mVenus, mCerulean, mEGFP, and mCherry tagged DHHC20 were imaged using the instrument’s EYFP, ECFP, Alexa 488, and Alexa Fluor 568 fluorophore default settings respectively. Cells were maintained at 37°C in a humidified chamber with CO2 during imaging.
Results
FSEC of purified mVenus, mCerulean, mEGFP, and mCherry
To compare the effects of the GFP variants on membrane protein expression, it was first necessary to generate a quantitative relationship between the different GFP fluorescent signals in our FSEC system. For this, we purified all four GFP variants expressed in E. coli using metal affinity chromatography (Figure 1A) and injected equimolar amounts of the proteins into our FSEC set-up. Normalizing with respect to the maximum peak signal from mVenus allowed us to establish a fractional relationship for the peak intensity between the fluorescent protein variants (Figure 1B). Therefore, all things being equal, and assuming that appending the fluorescent proteins to our proteins of interest does not affect their spectral properties, if mVenus, mCerulean, mEGFP, and mCherry tagged proteins express equally well, then the relationship of the FSEC chromatogram peak intensity is ~1.0:0.25(±0.01):0.65(±0.03):0.1(±0.01) respectively. We also tested the thermal stability of the four fluorescent proteins. The pseudo-melting curve (Figure 1C) suggests that mCherry is the most stable with a melting temperature (Tm) of 94±7°C and eGPF the least with 70.5±0.3°C, while mVenus and mCerulean are intermediate with Tm of 77.8±0.4 and 79.6±0.3°C respectively. However, isothermal denaturation assay (Figure 1D) shows mVenus and mCerulean to be the most stable with a half-lifetime (t1/2) of 16.7±0.9 and 12.9±0.3 hr, respectively. In this assay, mCherry has a t1/2 of 2.46±0.06 hr whereas once again the least stable is mEGFP with a t1/2 of only 1.68±0.05 hr. The discrepancy for mCherry between the two thermal stability assays is most likely due to a metastable intermediate. This is clearly observed in the isothermal stability assay as the fractional fluorescence of mCherry never decreases to zero. Nonetheless both thermal stability assays show that of the four fluorescent proteins, mEGFP is the least stable.
Figure 1.
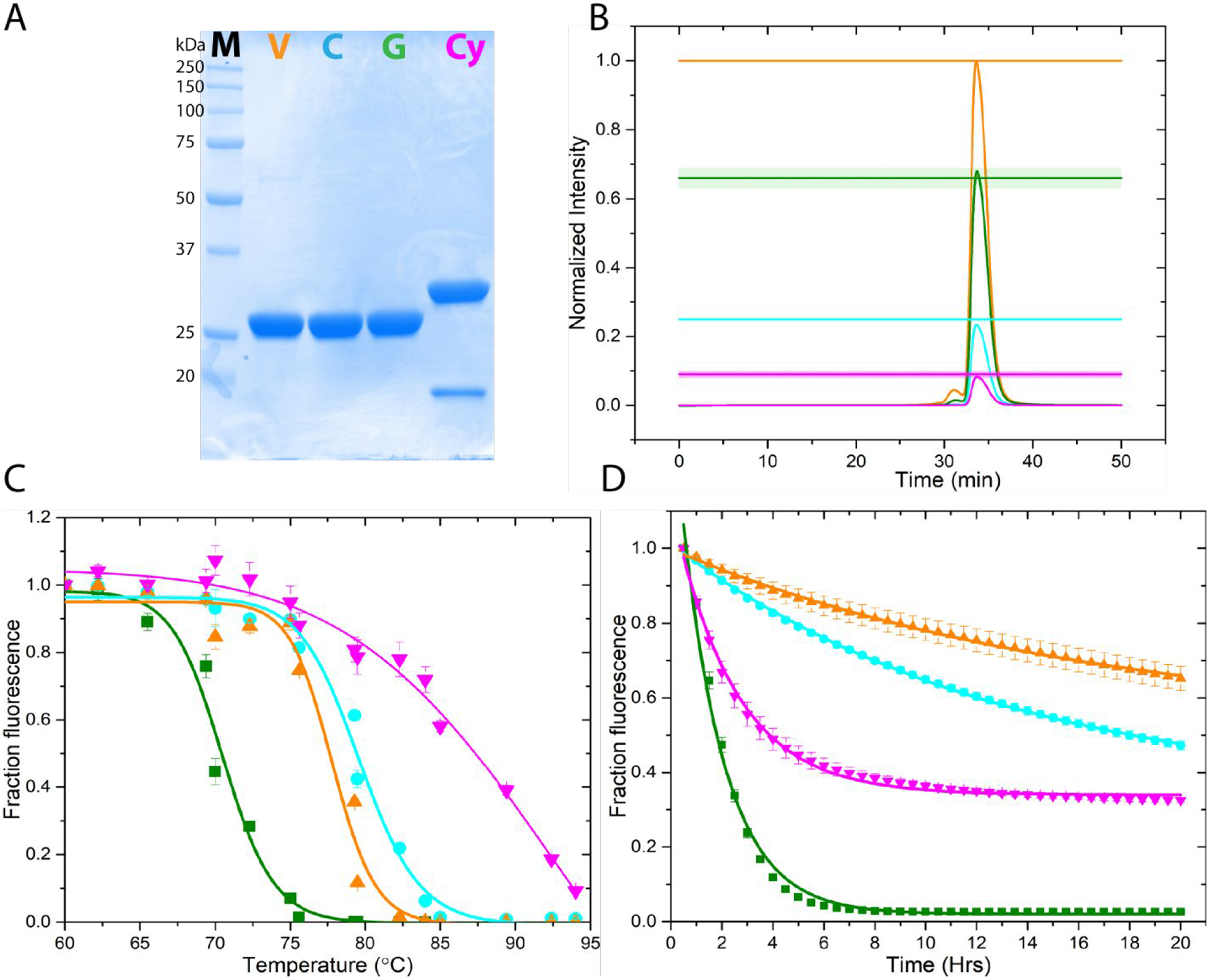
Characterization of the different tag fluorescent proteins. (A) SDS-PAGE analysis of mVenus (V, orange), mCerulean (C, cyan), mEGFP (G, green), and mCherry (Cy, magenta) expressed in E. coli and purified using metal affinity chromatography. The smaller band observed for mCherry is due to an intramolecular protein cleavage during sample denaturation by heating. (B) Establishment of a quantitative FSEC relation between the fluorescent proteins by injecting equimolar concentrations of purified fluorescent proteins into the size exclusion column and normalizing to the brightest protein (mVenus, orange). The solid lines are the mean of six independent experiments. The shaded region is the SEM. The relationship between mVenus:mCerulean:mEGFP:mCherry is ~ 1.0:0.25±0.01:0.65±0.03:0.1±0.01. (C) Pseudo-melting curve assessing thermal stability of the fluorescent proteins. Data were fit to a Hill equation to obtain the pseudo melting temperature (Tm). The data are mean±sem of at least three independent experiments. mVenus, orange; mCerulean, cyan; mEGFP, green; mCherry, magenta (D) Isothermal stability of the various fluorescent proteins measured at 70°C. Data were fit to a single exponential decay to extract half-life. All data are mean±sem of at least three independent experiments. Symbols as in (C).
Effect of fluorescent protein (FP) tags on DHHC20 enzyme expression
Next, we assessed the FSEC profiles of DHHC20 protein transiently over-expressed in adherent HEK293T cells with the different fluorescent protein tag on the N-terminus. It is clear from the FSEC data that while mVenus, mCerulean, and mCherry tagged DHHC20 express to similar amounts, the mEGFP tagged DHHC20 expression is considerably reduced (Figure 2A). This result is independent of the detergent used for membrane protein extraction (Figure S3). The expression of mEGFP tagged DHHC20 also depends on the terminus to which it is fused; DHHC20 tagged at the C-terminus with mEGFP expresses much better than one tagged on the N-terminus (Figure 2C). Nonetheless, even the DHHC20-mEGFP construct still expresses approximately two-fold less than DHHC20 tagged with mVenus and mCerulean. Because mEGFP is the least thermally stable of the four fluorescent proteins, we next asked if lowering the cell culture temperature can rescue protein expression levels of mEGFP tagged DHHC20. Consistent with our expectations, lowering the culture temperature from the usual 37°C to 30°C results in an approximately two-fold increase in the expression of mEGFP tagged DHHC20 (Figure 2B, 2D). Decreasing the incubation temperature of cells to 30°C has previously been used to increase the expression of mEGFP-tagged proteins [7]. Interestingly, expression of the mVenus or mCerulean tagged DHHC20 does not seem to be affected by lowering the temperature to 30°C (Figure S4). To ensure that the effect of the different fluorescent tags on DHHC20 protein expression is real and not just an artifact of FSEC, we purified the tagged DHHC20 proteins by metal affinity chromatography and analyzed the eluates by electrophoresis. The results are consistent with that obtained from FSEC where the expression levels of eGPF tagged DHHC20 are significantly less than of DHHC20 tagged with mVenus, mCerulean, or mCherry (Figure 2E, F, G).
Figure 2.
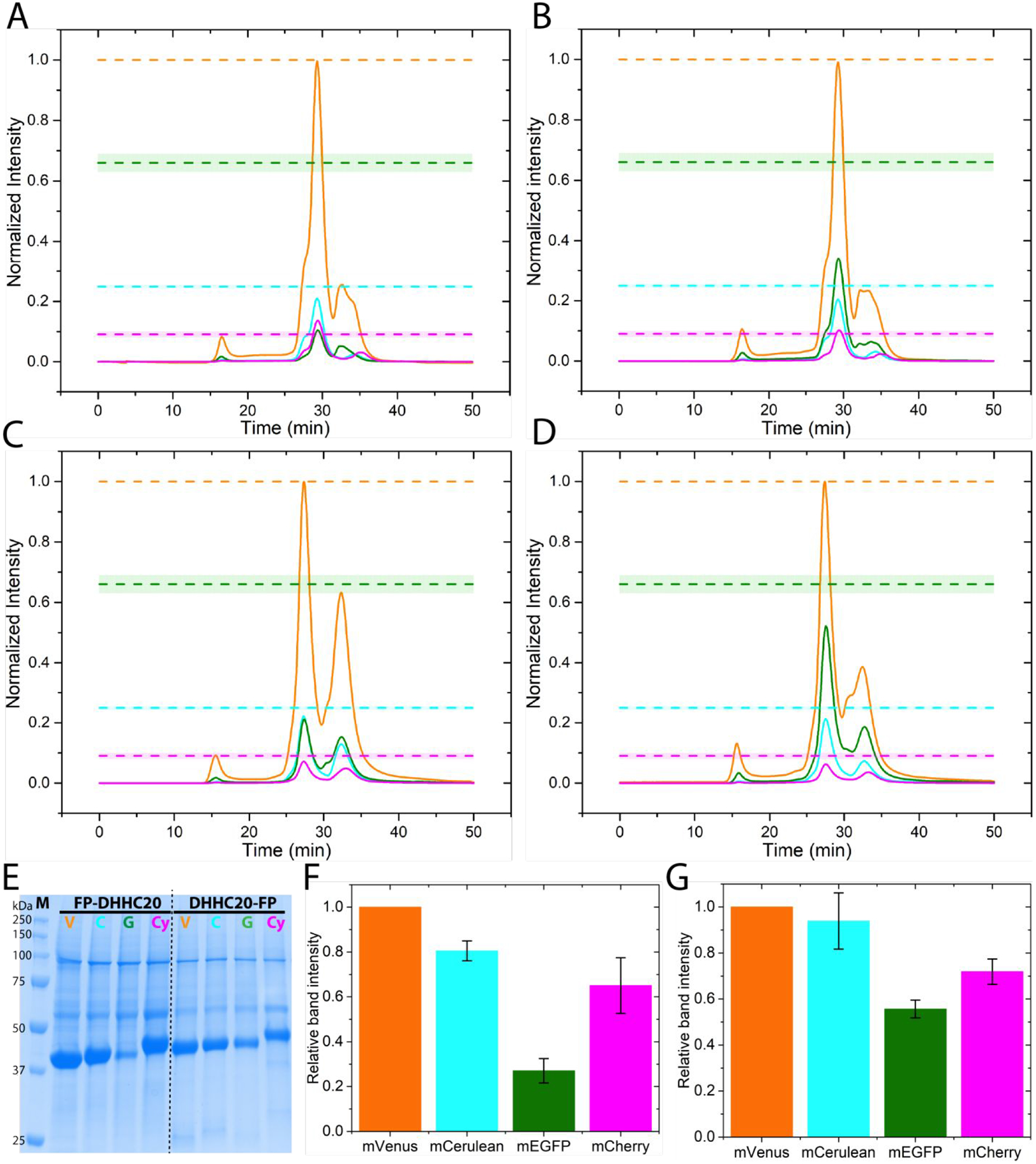
Over-expression of human DHHC20 tagged with the different fluorescent proteins (FP). (A) FSEC traces of N-terminal FP tagged DHHC20 transiently over-expressed in HEK293T cells at 37°C, mVenus-DHHC20 (orange), mCerulean-DHHC20 (cyan), mEGFP-DHHC20 (green), mCherry-DHHC20 (magenta). (B) The same as (A) but at an incubation temperature of 30°C. (C) FSEC traces of C-terminal FP tagged DHHC20 transiently over-expressed in HEK293T cells at 37°C. Color code as in (A). (D) The same as (C) but at 30°C. The dashed lines indicate the expected peak heights for mCerulean(cyan), mEGFP (green), and mCherry (magenta) relative to the mVenus (orange) tagged protein if they all expressed equally well. All chromatograms are averages from two independent experiments. (E) SDS-PAGE analysis of metal affinity purified N- and C-terminal FP tagged DHHC20. mVenus, V; mCerulean, C; mEGFP, G; mCherry, Cy. (F) Quantitation of relative expression levels for N-terminal FP tagged DHHC20. (G) Quantitation of relative expression levels for C-terminal FP tagged DHHC20. Color code as in (A). Values in bar graphs are mean±sd of two independent experiments.
We next looked at the expression levels of FP-DHHC20 using another well-known and used promoter, the elongation factor 1 alpha (EF1α), in HEK293T cells. Compared to the CMV promoter, EF1α is found to be stronger in a variety of cell lines [59]. Our results on the expression of FP-DHHC20 driven by the EF1α promoter are virtually indistinguishable from that obtained with the CMV promoter (Figure 3A,B). As large-scale protein overexpression in mammalian cells is usually done using baculovirus mediated transduction in suspension HEK293S cells [7,60,61], we asked if our results would persist if done in this manner. Irrespective of the use of sodium butyrate to induce higher protein expression, our results match that obtained from adherent cell cultures using polyethyleneimine (PEI) mediated transient transfection (Figure 3C,D) of adherent cells. As expected, lowering the temperature increases the expression levels of the mEGFP-DHHC20 by almost two-fold (Figure 3C,D). To ensure that our results are not solely due to our protein purification method, we used an anti-GFP nanobody resin to purify the proteins. The result is similar to that obtained using metal affinity resin (Figure 3E) and also confirms experimentally that the mVenus and mCerulean fluorescent proteins are compatible with anti-GFP nanobody resin. The mCherry-DHHC20 is not compatible with the anti-GFP nanobody resin because although structurally similar to GFP, it is evolutionarily divergent [46,62].
Figure 3.
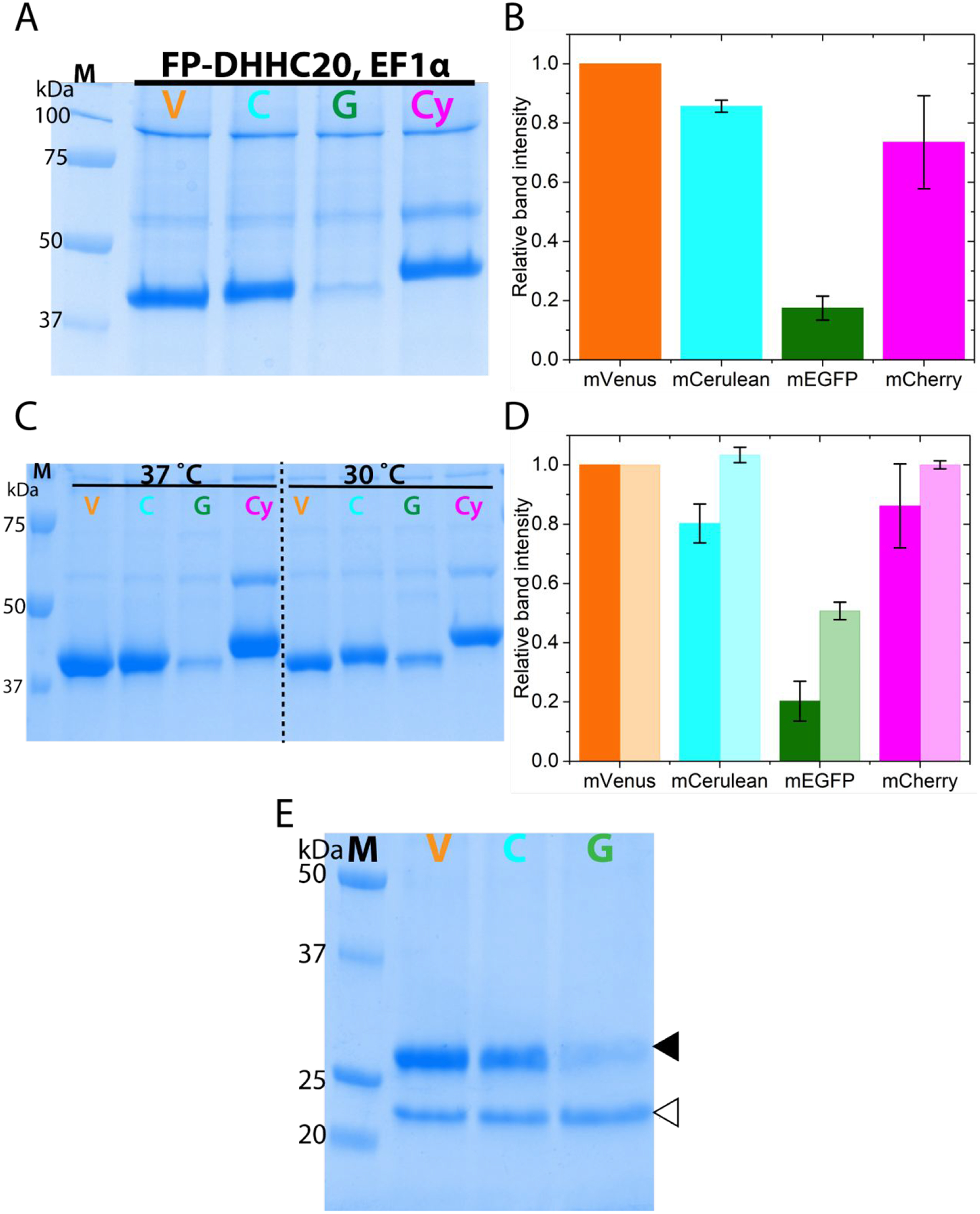
N-terminally fluorescent protein tagged DHHC20 expression under different promoter and using baculovirus mediated over-expression (A) SDS-PAGE analysis of FP-DHHC20 over-expression driven by the elongation factor 1 alpha (EF1α) promoter in HEK293T cells (B) Quantitation of the relative protein levels of FP-DHHC20 expressed using EF1α promoter. (C) SDS-PAGE analysis of metal affinity purified N-terminal FP tagged DHHC20 expressed using baculovirus in HEK 293S GnTi− cells at 37 and 30°C. (D) Quantitation of relative expression levels for N-terminal tagged DHHC20 at 37 (dark fill) and 30°C (light fill). (E) SDS-PAGE analysis of N-terminal FP tagged DHHC20 expressed at 37°C purified using an anti-GFP nanobody resin. The DHHC20 protein (filled triangle) is eluted from the nanobody resin by cleaving it off from the fluorescent protein by the PreScission protease (empty triangle). mVenus-DHHC20, V; mCerulean-DHHC20, C; mEGFP-DHHC20, G; mCherry-DHHC20, Cy. Values in bar graphs are mean±sd of two independent experiments.
Effect of fluorescent protein (FP) tags on Porcupine and CLC proteins
To assess whether the effect of mEGFP fusion tag on DHHC20 can be generalized, we chose two very different membrane proteins to test. Human Porcupine (hPORCN) is an endoplasmic reticulum resident protein that catalyzes the palmitoleoylation of the Wnt proteins [63,64]. The structure of hPORCN is not known but it is predicted to have 8–11 transmembrane helices. Conversely the structure of the Cyanidioschyzon merolae CLC (cmCLC) chloride-ion transporter is known [53]. It consists of 17 transmembrane helices and forms a homodimer. FSEC traces show that hPORCN tagged at the C-terminus with mEGFP gives results similar to DHHC20 and that the levels of expression can be increased by lowering the culture temperature from 37°C to 30°C (Figure 4A,C). Similarly, cmCLC protein tagged at the N-terminus with mEGFP expresses substantially less than the mVenus, mCerulean, and mCherry versions (Figure 4B,D). Although mEGFP-cmCLC expression can be increased by lowering the culture temperature to 30°C, the FSEC profile shows the appearance of a shoulder. As observed for DHHC20 protein, metal affinity purified hPORCN and cmCLC both show reduced expression for the mEGFP tagged versions of the proteins (Figure 4, E–H). Therefore, the beneficial effects of the mVenus and mCerulean tags over the mEGFP tag in protein expression seems to be independent of the target protein of choice.
Figure 4.
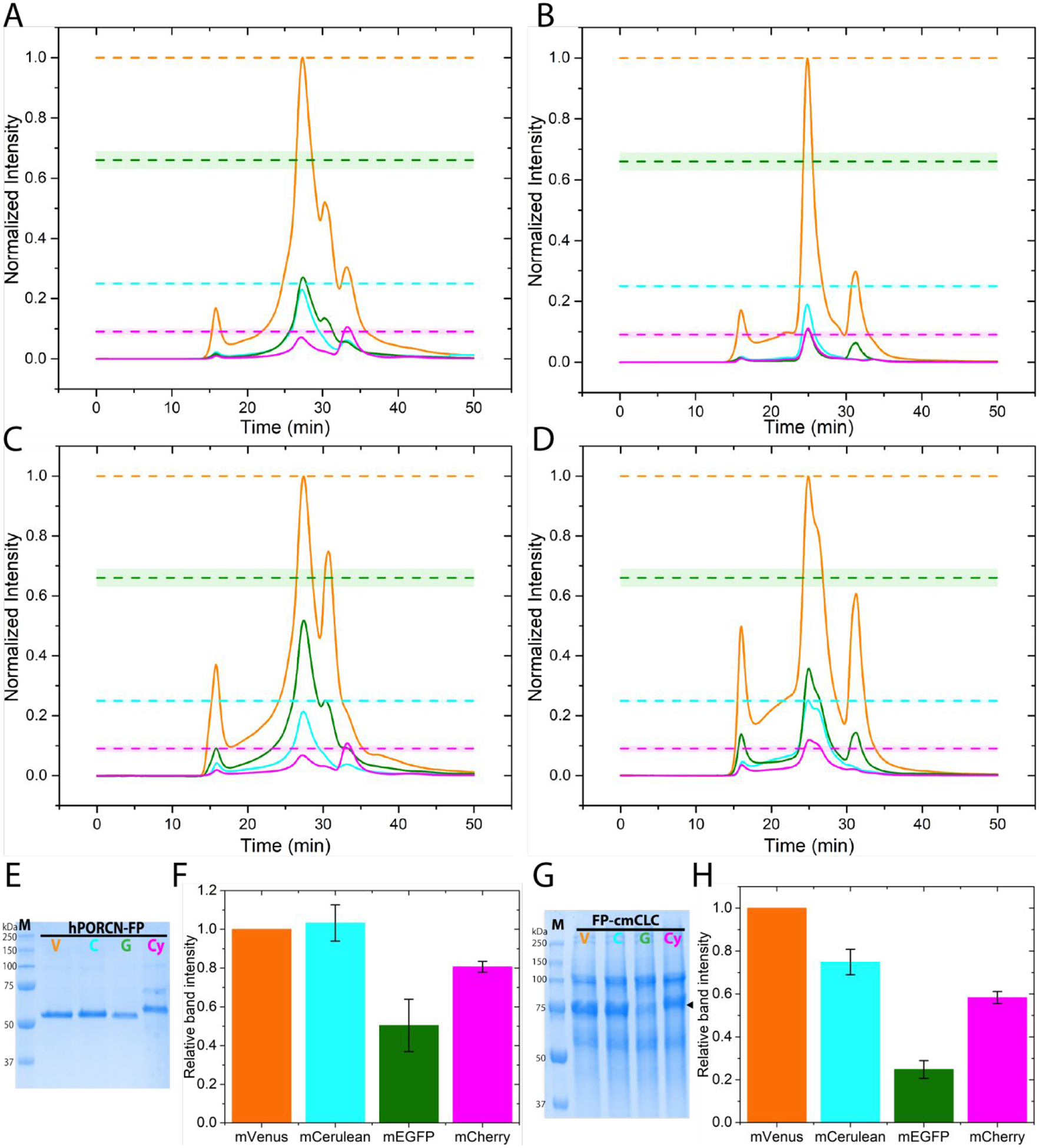
Effect of different FP tags on human Porcupine (hPORCN) and cmCLC expression (A) FSEC traces of hPORCN-mVenus (orange), hPORCN-mCerulean (cyan), hPORCN-mEGFP (green), and hPORCN-mCherry (magenta) transiently over-expressed in HEK293T cells at 37°C. (B) FSEC traces of mVenus-cmCLC (orange), mCerulean-cmCLC (cyan), mEGFP-cmCLC (green), and mCherry-cmCLC (magenta) transiently over-expressed in HEK293T cells at 37°C. (C) The same as (A) but at 30°C. (D) The same as (B) but at 30°C. The dashed lines indicate the expected peak heights for mCerulean(cyan), mEGFP (green), and mCherry (magenta) relative to the mVenus (orange) tagged protein if they all expressed equally well. Chromatograms are averages from two independent experiments. (E) SDS-PAGE analysis of C-terminal tagged hPORCN expressed in HEK 293S GnTi− suspension cells using baculovirus purified by metal affinity chromatography. hPORCN-mVenus, V; hPORCN-mCerulean, C; hPORCN-mEGFP, G; hPORCN-mCherry (Cy) (F) Quantitation of relative expression levels for C-terminal tagged hPORCN at 37°C expressed in HEK293S GnTi− suspension cells using baculovirus. (G) SDS-PAGE analysis of N-terminal tagged cmCLC expressed in HEK 293T adherent cells purified by metal affinity chromatography. mVenus-cmCLC, V; mCerulean-cmCLC, C; mEGFP-cmCLC, G; mCherry-cmCLC (Cy). (H) Quantitation of relative expression levels for N-terminal tagged cmCLC at 37°C. Values in bar graphs are mean±sd of two independent experiments.
Discussion
Our findings demonstrate that the expression and yield in HEK293 cells of membrane proteins tagged with mVenus or mCerulean is substantially better over mEGFP. Yields of membrane proteins tagged with mVenus and mCerulean at the N-terminal are increased by ~ 4–5-fold compared to mEGFP. Even for C-terminal tagged proteins, there is a 2-fold increase. Structural studies of eukaryotic membrane proteins are often severely hampered by the limit imposed on the amount of final purified protein in hand and this increase in expression/purification can substantially hasten up screening and optimization of conditions. The effect seems to be generalizable and observed with three very different proteins: human DHHC20, human Porcupine, and a thermophilic protein cmCLC. Use of mCherry as a tag instead of mEGFP is also advantageous but not to the same extent as mVenus and mCerulean.
We found a correlation between the thermal stability of the fluorescent protein and the expression levels of proteins tagged with it. However, it is unclear whether this can entirely explain the observed effect. Even the least stable fluorescent protein mEGFP still has a pseudo-Tm of 70°C – a value substantially higher than the temperature at which the cells are maintained and grown. Furthermore, Cyanidioschyzon merolae is a red thermophilic alga and therefore the cmCLC protein is thermally more stable. However, this does not ameliorate the negative effects of mEGFP versus mVenus and mCerulean. Therefore, it is more likely that mEGFP tagged membrane proteins express poorly due to another reason. Our initial hypothesis was that fluorescent protein maturation may contribute to the observed effect. However, data from a recent paper that extensively measured the maturation time of fluorescent proteins inside bacterial cells [65] suggest that for the expression levels to be related to maturation time, mCherry tagged proteins would have to express the least because it has the slowest maturation time followed by mEGFP. This is not what we observed. Moreover, maturation rates increase with temperature. This is contrary to our observations where reducing temperatures leads to increase in mEGFP tagged protein expression (Figure 2, 3, 4). Alternately, mEGFP folding may be the problem. As chromophore maturation and protein folding are not necessarily related, and the reduced protein expression effect is most pronounced with the N-terminal tagged proteins, this seems a reasonable hypothesis.
Regardless of the exact mechanism of how mVenus and mCerulean tagged proteins express better than ones tagged with mEGFP, the switch from an mEGFP to a mVenus/mCerulean tag offers a simple method to increase yields of recombinant membrane proteins in HEK293 cells. We did not test the effect of mVenus versus mEGFP for soluble proteins. It would be interesting to see if the beneficial effect persists. The use of mVenus as the fluorescent protein tag instead of mEGFP also makes incubation at 30°C to increase protein yields unnecessary. While seemingly trivial to a well-equipped and funded lab, the ability to maintain all incubators at the same temperature of 37°C can be advantageous to a smaller and modest lab. These results should also provide some guidance to choosing and expressing differentially FP-tagged proteins for multi-component protein complexes in mammalian cells [44]. With the development of anti-GFP and anti-mCherry nanobodies [45,46], the use of a fluorescent protein tag is doubly advantageous. Apart from following and optimizing protein expression using gel-based assay or FSEC, it also allows one to easily purify it in a single step. Both mVenus and mCerulean are compatible with the anti-GFP Nb resin (Figure 3E), although mCherry is not.
In conclusion, based on our results where we observed a 2 to 5-fold increase in protein expression and yields upon using mVenus instead of mEGFP as the fluorescent protein tag of choice, we recommend that laboratories use mVenus as a fluorescent protein fusion tag for expressing recombinant membrane proteins in mammalian cells.
Supplementary Material
Acknowledgment
The authors thank Dr. Chul-jin Lee (NIH) for the initial human Porcupine construct, Dr. Robbins Puthenveetil (NIH) for help with the HEK 293S GnTi− cells and Veronica Roberts (NCI) for guidance on baculovirus preparation. The pSP mammalian vector with a EF1α promoter was a gift from Dorien Roosen in the lab of Dr. Mark Cookson (NIH). The pEG-Bacmam was a gift from Dr. Eric Gouaux (Oregon Health and Science University, Portland). The cmCLC protein construct was used with permission from the lab of Dr. Roderick MacKinnon (The Rockefeller University, New York).
Funding
This work was funded by an NIH intramural grant HD008928-05 to AB.
Footnotes
The authors declare no competing financial interest.
Supporting information. The Supporting Information is available free of charge on the ACS Publications website. Table S1, Fluorescent protein tags coding sequences; Table S2, Primer list; Figure S1, DHHC20 enzyme assay, CMV promoter sequence, confocal images; Figure S2, Construct design; Figure S3, Protein extraction from membrane by different detergents; Figure S4, Temperature independence of mVenus and mCerulean tagged proteins.
References
- 1.Almén MS, Nordström KJ, Fredriksson R, Schiöth HB. Mapping the human membrane proteome: a majority of the human membrane proteins can be classified according to function and evolutionary origin. BMC Biol. 2009. August 13;7:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fagerberg L, Jonasson K, Heijne G von, Uhlén M, Berglund L. Prediction of the human membrane proteome. PROTEOMICS. 2010. March 1;10(6):1141–9. [DOI] [PubMed] [Google Scholar]
- 3.Terstappen GC, Reggiani A. In silico research in drug discovery. Trends Pharmacol Sci. 2001. January 1;22(1):23–6. [DOI] [PubMed] [Google Scholar]
- 4.Yıldırım MA, Goh K-I, Cusick ME, Barabási A-L, Vidal M. Drug—target network. Nat Biotechnol. 2007. October;25(10):1119–26. [DOI] [PubMed] [Google Scholar]
- 5.Parker JL, Newstead S. Method to increase the yield of eukaryotic membrane protein expression in Saccharomyces cerevisiae for structural and functional studies. Protein Sci Publ Protein Soc. 2014. September;23(9):1309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahmad M, Hirz M, Pichler H, Schwab H. Protein expression in Pichia pastoris: recent achievements and perspectives for heterologous protein production. Appl Microbiol Biotechnol. 2014. June 1;98(12):5301–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goehring A, Lee C-H, Wang KH, Michel JC, Claxton DP, Baconguis I, Althoff T, Fischer S, Garcia KC, Gouaux E. Screening and large-scale expression of membrane proteins in mammalian cells for structural studies. Nat Protoc. 2014. November;9(11):2574–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawate T, Gouaux E. Fluorescence-Detection Size-Exclusion Chromatography for Precrystallization Screening of Integral Membrane Proteins. Structure. 2006. April 1;14(4):673–81. [DOI] [PubMed] [Google Scholar]
- 9.Newstead S, Kim H, von Heijne G, Iwata S, Drew D. High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2007. August 28;104(35):13936–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drew D, Lerch M, Kunji E, Slotboom D-J, de Gier J-W. Optimization of membrane protein overexpression and purification using GFP fusions. Nat Methods. 2006. April;3(4):303–13. [DOI] [PubMed] [Google Scholar]
- 11.Drew D, Newstead S, Sonoda Y, Kim H, von Heijne G, Iwata S. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat Protoc. 2008;3(5):784–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hammon J, Palanivelu DV, Chen J, Patel C, Minor DL. A green fluorescent protein screen for identification of well-expressed membrane proteins from a cohort of extremophilic organisms. Protein Sci Publ Protein Soc. 2009. January;18(1):121–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H, Shaffer PL, Huang X, Rose PE. Rapid screening of membrane protein expression in transiently transfected insect cells. Protein Expr Purif. 2013. March 1;88(1):134–42. [DOI] [PubMed] [Google Scholar]
- 14.Hattori M, Hibbs RE, Gouaux E. A Fluorescence-Detection Size-Exclusion Chromatography-Based Thermostability Assay for Membrane Protein Precrystallization Screening. Structure. 2012. August 8;20(8):1293–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moreau MJJ, Morin I, Askin SP, Cooper A, Moreland NJ, Vasudevan SG, Schaeffer PM. Rapid determination of protein stability and ligand binding by differential scanning fluorimetry of GFP-tagged proteins. RSC Adv. 2012;2(31):11892–900. [Google Scholar]
- 16.Chae PS, Rasmussen SGF, Rana R, Gotfryd K, Chandra R, Goren MA, Kruse AC, Nurva S, Loland CJ, Pierre Y, Drew D, Popot J-L, Picot D, Fox BG, Guan L, Gether U, Byrne B, Kobilka B, Gellman SH. Maltose-neopentyl glycol (MNG) amphiphiles for solubilization, stabilization and crystallization of membrane proteins. Nat Methods. 2010. December;7(12):1003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chae PS, Rana RR, Gotfryd K, Rasmussen SGF, Kruse AC, Cho KH, Capaldi S, Carlsson E, Kobilka B, Loland CJ, Gether U, Banerjee S, Byrne B, Lee JK, Gellman SH. Glucose-Neopentyl Glycol (GNG) Amphiphiles for Membrane Protein Solubilization, Stabilization and Crystallization. Chem Commun Camb Engl. 2013. March 21;49(23):2287–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chae PS, Bae HE, Ehsan M, Hussain H, Kim JW. New ganglio-tripod amphiphiles (TPAs) for membrane protein solubilization and stabilization: implications for detergent structure–property relationships. Org Biomol Chem. 2014. October 9;12(42):8480–7. [DOI] [PubMed] [Google Scholar]
- 19.Chae PS, Cho KH, Wander MJ, Bae HE, Gellman SH, Laible PD. Hydrophobic variants of ganglio-tripod amphiphiles for membrane protein manipulation. Biochim Biophys Acta BBA - Biomembr. 2014. January 1;1838(1, Part B):278–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Newstead S, Ferrandon S, Iwata S. Rationalizing α-helical membrane protein crystallization. Protein Sci Publ Protein Soc. 2008. March;17(3):466–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parker JL, Newstead S. Current trends in α-helical membrane protein crystallization: An update. Protein Sci Publ Protein Soc. 2012. September;21(9):1358–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker JL, Newstead S. Membrane protein crystallization: Current trends and future perspectives. Adv Exp Med Biol. 2016;922:61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J Am Chem Soc. 2004. March 1;126(11):3477–87. [DOI] [PubMed] [Google Scholar]
- 24.Postis V, Rawson S, Mitchell JK, Lee SC, Parslow RA, Dafforn TR, Baldwin SA, Muench SP. The use of SMALPs as a novel membrane protein scaffold for structure study by negative stain electron microscopy. Biochim Biophys Acta. 2015. February;1848(2):496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caffrey M. Crystallizing Membrane Proteins for Structure Determination: Use of Lipidic Mesophases. Annu Rev Biophys. 2009;38(1):29–51. [DOI] [PubMed] [Google Scholar]
- 26.Gourdon P, Andersen JL, Hein KL, Bublitz M, Pedersen BP, Liu X-Y, Yatime L, Nyblom M, Nielsen TT, Olesen C, Møller JV, Nissen P, Morth JP. HiLiDe—Systematic Approach to Membrane Protein Crystallization in Lipid and Detergent. Cryst Growth Des. 2011. June;11(6):2098–106. [Google Scholar]
- 27.Faham S, Bowie JU. Bicelle crystallization: a new method for crystallizing membrane proteins yields a monomeric bacteriorhodopsin structure11Edited by D. Rees. J Mol Biol. 2002. February 8;316(1):1–6. [DOI] [PubMed] [Google Scholar]
- 28.Manglik A, Kobilka BK, Steyaert J. Nanobodies to Study G Protein-Coupled Receptor Structure and Function. Annu Rev Pharmacol Toxicol. 2017. January 6;57:19–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Löw C, Yau YH, Pardon E, Jegerschöld C, Wåhlin L, Quistgaard EM, Moberg P, Geifman-Shochat S, Steyaert J, Nordlund P. Nanobody Mediated Crystallization of an Archeal Mechanosensitive Channel. PLOS ONE. 2013. October 21;8(10):e77984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stockbridge RB, Kolmakova-Partensky L, Shane T, Koide A, Koide S, Miller C, Newstead S. Crystal structures of a double-barrelled fluoride ion channel. Nature. 2015. September;525(7570):548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, Kunken J, Xu F, Cherezov V, Hanson MA, Stevens RC. Fusion Partner Toolchest for the Stabilization and Crystallization of G Protein-Coupled Receptors. Struct England1993. 2012. June 6;20(6):967–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kobe B, Ve T, Williams SJ. Fusion-protein-assisted protein crystallization. Acta Crystallogr Sect F Struct Biol Commun. 2015. June 27;71(Pt 7):861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cherezov V, Hanson MA, Griffith MT, Hilgart MC, Sanishvili R, Nagarajan V, Stepanov S, Fischetti RF, Kuhn P, Stevens RC. Rastering strategy for screening and centring of microcrystal samples of human membrane proteins with a sub-10 μm size X-ray synchrotron beam. J R Soc Interface [Internet]. 2009. June 17 [cited 2018 May 24]; Available from: http://rsif.royalsocietypublishing.org/content/early/2009/06/10/rsif.2009.0142.focus [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischetti RF, Xu S, Yoder DW, Becker M, Nagarajan V, Sanishvili R, Hilgart MC, Stepanov S, Makarov O, Smith JL. Mini-beam collimator enables microcrystallography experiments on standard beamlines. J Synchrotron Radiat. 2009. March 1;16(Pt 2):217–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng Y. Single-Particle Cryo-EM at Crystallographic Resolution. Cell. 2015. April 23;161(3):450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merk A, Bartesaghi A, Banerjee S, Falconieri V, Rao P, Davis MI, Pragani R, Boxer MB, Earl LA, Milne JLS, Subramaniam S. Breaking Cryo-EM Resolution Barriers to Facilitate Drug Discovery. Cell. 2016. June 16;165(7):1698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Serrano-Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc Natl Acad Sci U S A. 2008. January 22;105(3):877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibata Y, White JF, Serrano-Vega MJ, Magnani F, Aloia AL, Grisshammer R, Tate CG. Thermostabilisation of the neurotensin receptor NTS1. J Mol Biol. 2009. July 10;390(2):262–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghosh E, Kumari P, Jaiman D, Shukla AK. Methodological advances: the unsung heroes of the GPCR structural revolution. Nat Rev Mol Cell Biol. 2015. February;16(2):69–81. [DOI] [PubMed] [Google Scholar]
- 40.Penmatsa A, Wang KH, Gouaux E. X-ray structure of the dopamine transporter in complex with tricyclic antidepressant. Nature. 2013. November 7;503(7474):85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green EM, Coleman JA, Gouaux E. Thermostabilization of the Human Serotonin Transporter in an Antidepressant-Bound Conformation. PLOS ONE. 2015. December 22;10(12):e0145688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coleman JA, Green EM, Gouaux E. X-ray structures and mechanism of the human serotonin transporter. Nature. 2016. April 21;532(7599):334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parcej D, Guntrum R, Schmidt S, Hinz A, Tampé R. Multicolour Fluorescence-Detection Size-Exclusion Chromatography for Structural Genomics of Membrane Multiprotein Complexes. PLOS ONE. 2013. June 25;8(6):e67112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morales-Perez CL, Noviello CM, Hibbs RE. Manipulation of subunit stoichiometry in heteromeric membrane proteins. Struct Lond Engl 1993. 2016. May 3;24(5):797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kirchhofer A, Helma J, Schmidthals K, Frauer C, Cui S, Karcher A, Pellis M, Muyldermans S, Casas-Delucchi CS, Cardoso MC, Leonhardt H, Hopfner K-P, Rothbauer U. Modulation of protein properties in living cells using nanobodies. Nat Struct Mol Biol. 2010. January;17(1):133–8. [DOI] [PubMed] [Google Scholar]
- 46.Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF, Oeffinger M, Nussenzweig MC, Fenyö D, Chait BT, Rout MP. A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods. 2014. December;11(12):1253–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rana MS, Kumar P, Lee C-J, Verardi R, Rajashankar KR, Banerjee A. Fatty acyl recognition and transfer by an integral membrane S-acyltransferase. Science. 2018. January 12;359(6372):eaao6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A. A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol. 2002. January;20(1):87–90. [DOI] [PubMed] [Google Scholar]
- 49.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004. April;22(4):445–9. [DOI] [PubMed] [Google Scholar]
- 50.Kremers G-J, Goedhart J, van Munster EB, Gadella TWJ. Cyan and Yellow Super Fluorescent Proteins with Improved Brightness, Protein Folding, and FRET Förster Radius,. Biochemistry. 2006. May 1;45(21):6570–80. [DOI] [PubMed] [Google Scholar]
- 51.Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004. December;22(12):1567–72. [DOI] [PubMed] [Google Scholar]
- 52.Bryksin AV, Matsumura I. Overlap extension PCR cloning: a simple and reliable way to create recombinant plasmids. BioTechniques. 2010. June 1;48(6):463–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010. October 29;330(6004):635–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gibson DG, Young L, Chuang R-Y, Venter JC, Iii CAH, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009. May;6(5):343–5. [DOI] [PubMed] [Google Scholar]
- 55.Longo PA, Kavran JM, Kim M-S, Leahy DJ. Transient Mammalian Cell Transfection with Polyethylenimine (PEI). Methods Enzymol. 2013;529:227–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roest S, Kapps-Fouthier S, Klopp J, Rieffel S, Gerhartz B, Shrestha B. Transfection of insect cell in suspension for efficient baculovirus generation. MethodsX. 2016. January 1;3:371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Scholz J, Suppmann S. A new single-step protocol for rapid baculovirus-driven protein production in insect cells. BMC Biotechnol. 2017. November 16;17(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012. July;9(7):676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qin JY, Zhang L, Clift KL, Hulur I, Xiang AP, Ren B-Z, Lahn BT. Systematic Comparison of Constitutive Promoters and the Doxycycline-Inducible Promoter. PLOS ONE. 2010. May 12;5(5):e10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dukkipati A, Park HH, Waghray D, Fischer S, Garcia KC. BacMam System for High-Level Expression of Recombinant Soluble and Membrane Glycoproteins for Structural Studies. Protein Expr Purif. 2008. December;62(2):160–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scott MJ, Modha SS, Rhodes AD, Broadway NM, Hardwicke PI, Zhao HJ, Kennedy-Wilson KM, Sweitzer SM, Martin SL. Efficient expression of secreted proteases via recombinant BacMam virus. Protein Expr Purif. 2007. March 1;52(1):104–16. [DOI] [PubMed] [Google Scholar]
- 62.Shagin DA, Barsova EV, Yanushevich YG, Fradkov AF, Lukyanov KA, Labas YA, Semenova TN, Ugalde JA, Meyers A, Nunez JM, Widder EA, Lukyanov SA, Matz MV. GFP-like Proteins as Ubiquitous Metazoan Superfamily: Evolution of Functional Features and Structural Complexity. Mol Biol Evol. 2004. May 1;21(5):841–50. [DOI] [PubMed] [Google Scholar]
- 63.Takada R, Satomi Y, Kurata T, Ueno N, Norioka S, Kondoh H, Takao T, Takada S. Monounsaturated Fatty Acid Modification of Wnt Protein: Its Role in Wnt Secretion. Dev Cell. 2006. December 1;11(6):791–801. [DOI] [PubMed] [Google Scholar]
- 64.Lee C-J, Rana M, Bae C, Li Y, Banerjee A. In vitro reconstitution of Wnt acylation reveals structural determinants of substrate recognition by the acyltransferase human Porcupine. J Biol Chem. 2018. November 12;jbc.RA118.005746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balleza E, Kim JM, Cluzel P. Systematic characterization of maturation time of fluorescent proteins in living cells. Nat Methods. 2018. January;15(1):47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.