

Abstract
The present work is an investigation to test hydroxychloroquine as an inhibitor for the COVID-19 main protease. Molecular docking studies revealed a high docking score and interaction energies and decent level of docking within the cavity in protease moiety. Molecular dynamics simulations also lead to the evaluation of conformational energies, average H-bonding distance, RMSD plots etc. Large RMSD fluctuations for the first 2 ns seem to provide the conformational and rotational changes associated with the drug molecule when it comes into the vicinity on the protease matrix. Snapshots of structural changes with respect to time vividly indicates that drug molecule has a profound impact on the binding sites as well as overall geometry of the protease moiety. On the whole, hydroxyxhloroquine confers good inhibitory response to COVID-19 main protease. We hope the present study should help workers in the field to develop potential vaccines and therapeutics against the novel coronavirus.
Keywords: Hydroxychloroquine, COVID-19 main chain protease, Molecular docking, MD-Simulation
Highlights
-
•
Molecular Docking studies reveals that hydroxychloroquine (HCQ) possesses a high docking score and interaction energy compared to other small drug molecule with the COVID-19 main protease.
-
•
Atomistic Molecular Dynamics simulation indicates considerable interaction between HCQ and COVID-19 protease.
-
•
RMSD plot suggests complex formation between HCQ and COVID-19 protease after 2 ns.
-
•
Equilibration of binding occurs between HCQ and COVID-19 protease between 7 and 10 ns.
-
•
Binding with HCQ induced large structural change in COVID-19 protease, hence change in its activity.
1. Introduction
From the last two decades infectious viral diseases like Lassa, Ebola, Zika have created a great burden to human, social and economical health. Along with that, coronaviruses like the severe acute respiratory syndrome CoV (SARS-CoV), Middle East respiratory syndrome CoV (MERS-CoV) and highly pathogenic influenza show their rampage. But somehow mankind recovered from those attacks by use of proper medicines. From December 2019, a deadly virus viz. severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) created an uproar worldwide known as COVID-19. Observing the devastating impact of COVID-19 and its huge mortality rate, the World Health Organization (WHO) has declared this outbreak as a pandemic on 12th March 2020 [1]. Till 12th March 2020, globally 125,048 people were infected and 4613 people were dead [1].
The first coronavirus B814 was found in 1965 in human embryonic tracheal organ cultures [2]. These viruses can cause disease in many animal species like rats, mice, dogs, cats, chickens etc [3]. Severe acute respiratory syndrome (SARS) originating from animal corona viruses emerged in 2002–2003 from southern China, with outbreaks in 29 countries in North and South America, Europe and Asia [[3], [4], [5], [6]]. Coronaviruses have positive sense single-standard RNA which are enveloped within fatty lipid molecules. These are mainly categorised into four groups viz. α-COV, β-COV, γ-COV and δ-COV, while a clear idea about SARS-COV-2 remains unknown.
The spiky ball type SARS-COV-2 first attacks human protein within the surface cell and then increases its activity. In recent days, from December 2019, emergency situation arising from novel coronavirus (SARS-COV-2) outbreaks reportedly started from Wuhan, China and spread throughout the world rapidly [7,8]. Its outbreak has created a pandemic and challenges the scientific community to design and discover the ultimate treatment strategy against this deadly virus (SARS-CoV-2). Till date, there is no antiviral therapy or vaccine available in the market which can efficiently combat the infection caused by this virus. SARS-CoV-2 uses the viral surface spike glycoprotein to bind to the host cell. After infecting the host cell with its RNA, it forces the host cell to bend and leave free copies of the virus created within the cell (a process known as “budding”). So the virus specific molecular interaction with the host cell represents a promising therapeutic target for identifying SARS-CoV-2 antiviral drugs. With the available experimental techniques, such selection is a difficult task as they offer low-resolution data to study the necessary interactions. In this regard potential in silico methods can be applied to select promising drug molecules with fewer trials and errors effectively reducing time and cost of researchers. The purpose of this work is to investigate the potential subunits of vaccines against the novel Coronavirus. Using detailed 3D-structures of relevant biomolecules and drug compounds, available bioinformatic techniques such as molecular docking may be applied to quickly identify promising and potential drug candidates. This can be followed with atomistic molecular dynamics or similar simulation methods in order to fully unravel underlying interactions responsible for drug-receptor binding and resulting structural modifications, leading to change in activity of the biological system. During clinical trials, some known drugs showed potent effect on this disease by curing a few COVID-19 patients [11]. But the actual mechanism or mode of action of those drugs are still unknown. Azithromycine, which can be used for the treatment of Zika virus [9,10], was applied to prevent COVID-19 along with hydroxychloroquine [11]. Chloroquine, an anti-malarial drug, is known to possess antiviral activity [12]. This was also applied to COVID-19 patients [13,14]. Chloroquine phosphate has shown positive activity on COVID-19 [15]. Though both chloroquine and hydroxychloroquine have some toxic effects, and their use in high doses can create major risks to the patients [16], under the present situation there is not enough information to find out other potent drugs with immediate effect.
Though there are few reported works, some of which already referred above, most are still in clinical trials and no results from in-vivo experiments have been reported yet. In the present work, we have examined effect of hydroxychloroquine on the coronavirus main protease (PDB ID: 6LU7) with the help of molecular docking and molecular dynamics simulation studies.
2. Methodology
2.1. Docking of hydroxychloroquine with COVID-19 main protease
The PDB file of COVID-19 main protease (PDB ID: 6LU7) was obtained from the Protein Data Bank. The structures of Chloroquine (CQ) (PubChem CID: 2719), Hydroxychloroquine (HCQ) (PubChem CID: 3652), Lamivudin (LV) (PubChem CID: 60825) and Emtricitabine (EMC) (PubChem SID: 46507606) have been taken from PUBCHEM. The structure of the protease was cleaned by removing all water molecules and by removing hetero groups using UCSF Chimera (ver 1.12) [17] and then the docking simulations were done by selecting best binding sites using AutoDock Vina package [18]. Auto Dock Tools was used to construct the initial structures to run the docking simulations.
2.2. MD simulation of PROTEASE-HCQ complexes
The molecular dynamics (MD) simulation studies were carried out on the minimum energy configurations obtained from the docking studies using GROMACS (Version 2018.5) [19] with the CHARMM36-mar2019 force-field [20] using TIP3P model [21]. The ligand (HCQ) topology and parameters were generated using CHARMM General Force Field server. A cubical box was generated where the protease-HCQ complex was at least 1 nm from the edges of the box to maintain at least 2 nm distance between two successive images of the complex using periodic boundary conditions. Four Na+ ions were added to maintain charge neutrality of the system. First, energy minimization was carried out until the maximum force becomes less than 10 kJ mol−1nm−1. The steepest descent algorithm was used followed by conjugate gradient protocol. Then the system was equilibrated for 100 ps using isochoric-isothermal (NVT) equilibration at 300 K. The time step was taken 2 fs This was followed by equilibration at isothermal-isobaric or NPT ensemble at 300 K for 100 ps Modified Berendsen thermostat was used for the NPT ensemble. Here also the time step was kept 2 fs For both NVT and NPT equilibration, electrostatic and van der Waals interaction cut off were kept at 1.0 nm. Long range interactions were calculated using smooth particle mesh Ewald (PME) method [22]. The equilibrated ensembles were finally subjected to MD simulation for 10 ns, with electrostatic and van der Waals cut off as before. PME method was used to calculate long range electrostatic interactions. A modified Berendsen thermostat and a Parinello-Rahman barostat were used with reference temperature and pressure at 300 K and 1 bar respectively. Snapshots of the trajectory were saved every 1 ns for each case.
2.3. Analysis of MD simulations
For protease-HCQ systems, structural trajectories were calculated using trjconv tool. The trjconv tool of GROMACS was used to recentre the protein and other molecules within the cubical box. van der Waals interaction, electrostatic energy, interaction energy values etc were calculated using gmx energy tool of GROMACS. RMSD plots were done using xmgrace plotting tool.
3. Results and discussion
3.1. Molecular docking studies
Required PDB file for the crystal structure of COVID-19 main protease (PDB ID: 6LU7) was downloaded from protein data bank and the drugs for screening was obtained from PubChem. The drugs which are used for screening of molecular docking are either anti-malarial or anti HIV agents. CQ, HCQ, LV and EMC were used for screening and docking was made with protease of COVID-19. The docking power of drugs on the co-crystal of COVID-19 was evaluated by measuring docking score. We find a docking score of −6.3 for hydroxychloroquine (HCQ) which is quite high compared to the other tested drugs. The docking score of all drugs are shown in Table 1 .
Table 1.
Docking scores and docked structures of the selected drugs used for screening with COVID-19 main protease (PDB ID: 6LU7).
| Drugs which are docked with 6LU7 | Docking Score | Nearest residues | Docked structure |
|---|---|---|---|
| Chloroquine | −5.9 | THR25, THR26, LEU27, HIS41, MET49, PHE140, LEU141, ASN142, GLY143, SER144, CYS145, MET165, HIS164, GLU166, GLN189 | 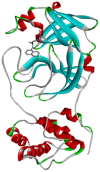 |
| Hydroxychloroquine | −6.3 | PHE140, LEU141, ASN142, GLY143, SER144, CYS145, HIS163, HIS164, MET165, GLU166, HIS172, ARG188, GLN189, THR190, GLN192 | 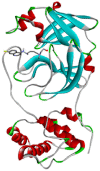 |
| Lamivudin | −5.7 | HIS41, MET49, PHE140, LEU141, ASN142, GLY143, SER144, CYS145, HIS163, HIS164, MET165, GLU166, HIS172 | 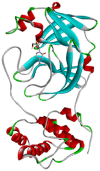 |
| Emtricitabine | −5.8 | HIS41, MET49, PHE140, LEU141, ASN142, GLY143, SER144, CYS145, HIS163, HIS164, MET165, GLU166, HIS172 | 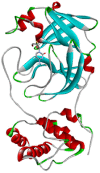 |
Due to high docking score found in the molecular docking studies, we have performed molecular dynamics simulation studies between the HCQ and COVID-19 main protease. The neighbouring groups presents in the crystal structure of COVID-19 protease in complex with inhibitor N3 are LEU, GLY, GLU, MET, HIS. Our drug in the docked structure was also surrounded by the same groups in the same binding sites with a good binding score. The interaction between drug and amino acid residues are mainly electrostatic and van der Waals types. The H-bond distance between the oxygen atom of hydroxyl group present in the drug and the nearest neighbour CYS 145, SER 144, GLY 143 are 3.1, 3.1 and 2.8 Å respectively and the H-bond distance H-atom of secondary amine group and GLN 189 is 2.2 Å. These H-bonding distances show good affinity of drug towards the protease and all the interactions and docked structure are given in Fig. 1 .
Fig. 1.

Docked structure of COVID-19 main protease with HCQ and their nearest neighbours.
3.2. MD simulation studies
MD simulations run with the stable docked structure for a period of 10 ns. Fig. 2 shows the snapshots of docked COVID-19 protease structure. It is clear from the snap shot that there is considerable conformational changes due to the translation and rotational movement of the drug molecule within the protease matrix.
Fig. 2.

Structure of COVID-19 protease and hydroxychloroquine after equilibration.
Fig. 3 A showed the RMSD fluctuation along with the MD simulation time. A considerable structural change is noticed within a short period of time of 2 ns It is also observed that the docked protease structure has reached equilibrium after 8 ns After the equilibration, the drug molecule is about 2.3 Å away with the protease which indicates considerable interactions between the protease and drug. Furthermore, the RMSD of the drug molecule measures not only the conformational changes, but also its translational and rotational movements inside the protease binding site. Evidently due to the less complex geometry of the drug molecule there is a large RMSD fluctuation as compared with the protease. RMSD plot for the protease alone has smaller fluctuation with respect to RMSD of the docked protease as shown in the Fig. 3B. This indicates higher stability of the protease compared to its docked structure. The instability of the docked protease structure implies that the drug molecule has a profound impact on the regular geometry of the protease. The total interaction energy i.e. the sum of average short-range Coulombic interaction energy and short-range Lennard-Jones energy between drug molecule and the protease is found to be −119.15 kJ/mol.
Fig. 3.

Root mean square deviation (RMSD) plot of receptor (protease) and ligand (drug) before docking and of receptor-ligand complex after docking (3a). RMSD plot of receptor alone before and after docking (3b).
Structural fluctuation of main protease was also studied from root mean square fluctuation (RMSF) analysis, shown in Fig. 4 . It is clear from the Fig. 4 that residues ARG60, ARG76, TYR154, GLN189, GLN306 are highly flexible. Their flexibility is near about 2 Å more in docked form compared to protease alone, which makes the protease-HCQ complex flexible, and prone to disruption.
Fig. 4.

Root Mean Square Fluctuations of the protease and its docked form with HCQ.
Table 2 represents the conformational energy and total energy of the systems along with the number of H-bonds and average distance of H-bonds. The value of conformational energy is more in case of composite system which impart more flexibility within the molecule. Again total energy for the protease is more than the composite system after MD simulations, which suggests that the drug makes the system less stable. This is also corroborated by the RMSD plot in Fig. 3A. The number of H-bonds are more in case of protease and drug system and that can be explained by the fact that as the drug enters into the protease cavity it forms additional H-bonds though the average distance of H-bond increases.
Table 2.
The average conformational energy, total energy, number of H-bonds and average distance of H-bond in two simulations. (A) Protease alone surrounded by ions and water molecules and (B) protease with drug surrounded by ions and water.
| System | Conformational Energy (kJ/mol) | Total Energy (kJ/mol) | Number of H-bonds | Average distance (Å) of H-bond |
|---|---|---|---|---|
| Protease (A) | 17203.87 | −1.656 x 106 | 232 | 2.05 |
| Protease + drug (B) | 17394.47 | −1.1649 x 106 | 243 | 2.07 |
Structural changes during the MD-simulation were also captured after every nanosecond. Some are shown in the Fig. 5 . The change in the structures after 1, 3, 5, 7 and 9 ns, as given in Fig. 5, clearly indicates that after 2 ns there is an abrupt change in conformation of the protease as the drug enters.
Fig. 5.

Structural change at different times (Fig. 5a–e), Fig. 5f contains two different structures at t = 0 ns and at t = 10 ns
From Fig. 5a and b it is seen that there is noticeable change in the structure of protease and throughout further simulation process this change is increased. The remarkable changes occurred in the sequence containing ARG 60, ILE 59, LEU 57 and ASP 56 residues as shown in Fig. 5a–e. Again, on the binding site of the drug molecule there is a huge structural reconstruction due to the newly formed H-bonding of the drug molecule with the protease moiety. These structural changes have a profound impact on the LYS 236, MET 235, VAL 233, ALA 266, LEU 262, ASP 229, HIS 246, GLY 251 sequences. Fig. 5f shows the net deviation in the structure of the protease from t = 0 ns to t = 10 ns of the protease backbone.
After the first 3 ns, RMSD plot shows a huge deviation which is reflected in Fig. 5b. Possibly, drug enters into closer proximity of the protease moiety in this period. After 7 ns the RMSD fluctuation appears to stabilize, indicating equilibration of the geometry.
4. Conclusions
The present work involves study of interaction between hydroxychloroquine, a known anti-malarial and a putative anti-SARS-COV-2 drug, and the protease of the latter. Molecular docking study reveals that HCQ docked at the binding site due to high electrostatic interactions, H-bonds etc. MD simulation study shows there is a noticeable change in the main protease of SARS-COV-2. RMSD data shows that there is a remarkable structural change in the protease moiety at 2 ns, around the time the drug begins closer interaction with the protease. The drug-protease system attains equilibrium at around 7 ns The RMSD plot also shows that the main protease backbone gets destabilized following interaction with the drug molecule. It is interesting that the number of H-bonds between the drug and protease increases as drug-protease complex forms, while the average H-bonding distance shows the opposite trend. The present molecular docking and MD simulation studies suggest that the drug hydroxychloroquine may have considerable effect on structure protease of SARS-COV-2, which may lead to significant inhibitory effect on the latter.
Data availability
Data is available on request to the corresponding author.
CRediT authorship contribution statement
Nabajyoti Baildya: Conceptualization, Formal analysis, Writing - review & editing. Narendra Nath Ghosh: Conceptualization, Formal analysis, Writing - review & editing. Asoke P. Chattopadhyay: Supervision, Writing - review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors wish to acknowledge infrastructural support received from the Dept. of Chemistry, University of Kalyani, and from the Dept. of Chemistry, University of Gour Banga, Malda.
This research did not receive any special grant from funding agencies in the public, commercial, or not-for-profit sectors.
References
- 1.Liu C., Jhou Q., Li Y., Garner L.V., Watkins S.P., Carter L.J., Smoot J., Gregg A.C., Daniels A.D., Jervey S., Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tyrrell D.A., Bynoe M.L. Cultivation of viruses from a high proportion of patients with colds. Lancet. 1966;1:76–77. doi: 10.1016/s0140-6736(66)92364-6. [DOI] [PubMed] [Google Scholar]
- 3.Khan J.S., McIntosh K. History and recent advances in coronavirus discovery. Pediatr. Infect. Dis. J. 2005;24:S223–S227. doi: 10.1097/01.inf.0000188166.17324.60. [DOI] [PubMed] [Google Scholar]
- 4.Drosten C., Gunther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 5.Ksiazek T.G., Erdman D., Goldsmith C.S. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 6.Peiris J.S., Lai St, Poon L.L. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai C.C., Shih T.P., Ko W.C., Tang H.J., Hsueh P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and corona virus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Atnimicrob. Agent. 2020 doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang L., Wang Y., Ye D., Liu Q. A review of the 2019 Novel Coronavirus (COVID-19) based on current evidence. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Retallack H., Lullo E.D., Arias C., Knopp K.A., Laurie M.T., Sandoval-Espinosa C., Leon W.R.M., Krencik R., Ullian E.M., Spatazza J., Pollen A.A., Mandel-Brehm C., Nowakowski T.J., Kriegstein A.R., DeRisi J.L. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. Unit. States Am. 2016;113:14408–14413. doi: 10.1073/pnas.1618029113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bosseboeuf E., Aubry M., Nhan T., Pina J.J., Rolain J.M., Raoult D., Musso D. Azithromycin inhibits the replication of Zika virus. J. Antivir. Antiretrovir. 2018;10:6–18. [Google Scholar]
- 11.Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., Dupont H.T., Honor′e S., Colson P., Chabri′ere E., Scola B.L., Rolain J.M., Brouqui P., Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020:105949. doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Biot C., Dahe W., Chavain N., Fandeur T., Khalife J., Dive D., Clercq E.D. Design and synthesis of hydroxyferroquine derivatives with antimalarial and antiviral activities. J. Med. Chem. 2006;49:2845–2849. doi: 10.1021/jm0601856. [DOI] [PubMed] [Google Scholar]
- 13.Colson P., Rolain J.M., Lagier J.C., Brouqui P., Raoult D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents. 2020:105932. doi: 10.1016/j.ijantimicag.2020.105932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Philippe C., Jean-Marc R., Didier R. Chloroquine for the 2019 novel coronavirus. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends. 2020 doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 16.Marmor M.F., Kellner U., Lai T.Y., Melles R.B., Mieler W.F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 revision) Ophthalmology. 2016;123:1386–1394. doi: 10.1016/j.ophtha.2016.01.058. [DOI] [PubMed] [Google Scholar]
- 17.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 18.Trott O., Olson A.J., Vina AutoDock. Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91(1–3):43–56. [Google Scholar]
- 20.Lee S., Tran A., Allsopp M., Lim J.B., Hénin J., Klauda J.B. CHARMM36 united atom chain model for lipids and surfactants. J. Phys. Chem. B. 2014;118:547–556. doi: 10.1021/jp410344g. [DOI] [PubMed] [Google Scholar]
- 21.Boonstra S., Onck P.R., van der Giessen e. CHARMM TIP3P water model suppresses peptide folding by solvating the unfolded state. J. Phys. Chem. B. 2016;120:3692–3698. doi: 10.1021/acs.jpcb.6b01316. [DOI] [PubMed] [Google Scholar]
- 22.Abraham M.J., Gready J.E. Optimization of parameters for molecular dynamics simulation using smooth particle-mesh Ewald in GROMACS 4.5. J. Comput. Chem. 2011;32:2031–2040. doi: 10.1002/jcc.21773. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data is available on request to the corresponding author.