

Summary
Cytosolic DNA acts as a universal danger-associated molecular pattern (DAMP) signal; however, the mechanisms of self-DNA release into the cytosol and its role in inflammatory tissue injury are not well understood. We found that the internalized bacterial endotoxin lipopolysaccharide (LPS) activated the pore forming protein Gasdermin D, which formed mitochondrial pores and induced mitochondrial DNA (mtDNA) release into the cytosol of endothelial cells. mtDNA was recognized by the DNA sensor cGAS and generated the second messenger cGAMP, which suppressed endothelial cell proliferation by downregulating YAP1 signaling. This indicated that the surviving endothelial cells in the penumbrium of the inflammatory injury were compromised in their regenerative capacity. In an experimental model of inflammatory lung injury, deletion of cGas in mice restored endothelial regeneration. The results suggest that targeting the endothelial Gasdermin D activated cGAS-YAP signaling pathway could serve as a potential strategy for restoring endothelial function following inflammatory injury.
Keywords: cGAS, endothelial regeneration, lung injury, mitochondrial DNA, Gasdermin D
Graphical Abstract
eTOC Blurb
Sepsis induces profound vascular injury by promoting endothelial cell death but the role of mitochondria in vascular endothelial injury is not well understood. Huang et al. demonstrate that the pore-forming molecule Gasdermin D releases mitochondrial DNA into the cytosol which sets into motion an amplified response resulting in suppression of endothelial regeneration mediated by the cytosolic DNA sensor cGAS.
Introduction
The endothelium lining all blood vessels plays an essential role in tissue homeostasis, maintenance of fluid balance, and as a “gate-keeper” for infiltration of inflammatory cells and flux of plasma proteins and water (Komarova et al., 2017). Endothelial death and barrier injury are central pathogenic features of many inflammatory diseases including acute lung injury (ALI) as well as reperfusion injury following myocardial ischemia (Hausenloy and Yellon, 2013). Endothelial repair secondary to regeneration of endothelial cells (EC) is required for resolution of inflammatory in injury and tissue homeostasis (Lee and Slutsky, 2010; Liu et al., 2019). Defective endothelial regeneration induces uncontrolled neutrophil infiltration, vascular hyperpermeability, and progressively intractable inflammation and tissue injury (Lee and Slutsky, 2010; Liu et al., 2019; Nourshargh and Alon, 2014), and is a hallmark of multiple acute and chronic inflammatory diseases (Castellon and Bogdanova, 2016).
The bacterial endotoxin lipopolysaccharide (LPS) induces inflammation in large part due to its toxic effects on the endothelium (Deng et al., 2018; Mandal et al., 2018). It has recently been shown that intracellular breaching by LPS initiates endothelial pyroptosis and the loss of vascular integrity secondary to activation of inflammatory caspases, specifically caspases 4/5 in humans and the homolog caspase-11 in mice, which function as intracellular LPS receptors (Cheng et al., 2017). These inflammatory caspases cleave Gasdermin D (GSDMD), leading to insertion N-terminal GSDMD domain in the plasmalemma membrane, and resulting in formation pores that cause pyroptotic cell death (Liu et al., 2016). The cleaved GSDMD is also enriched in the mitochondrial membrane where it triggers mitochondrial reactive oxygen species (ROS) generation and mitochondrial injury (Platnich et al., 2018). Another key feature of mitochondrial injury is release of mtDNA into the cytosol (Galluzzi et al., 2016; Nakahira et al., 2011; Platnich et al., 2018). Cyclic GMP-AMP Synthase (cGAS) as a DNA sensor in cells detects both self-DNA and non-self-DNA, to induce cell injury (Cai et al., 2014). After binding to cytosolic double-stranded DNA (dsDNA), cGAS induces synthesis of cyclic GMP–AMP (cGAMP) by consuming adenosine 5´-triphosphate (ATP) and guanosine 5´-triphosphate (GTP) (Ablasser et al., 2013). The second messenger cGAMP in turn binds to stimulator of interferon genes (STING) which induces is a critical activator of inflammation by increasing the generation of type 1 interferon (Ahn et al., 2012; Burdette and Vance, 2013).
Pyroptotic death occurs in a relatively small number of endothelial cells but is nevertheless sufficient to mediate tissue and vascular injury (Cheng et al., 2017), thus indicating that the inflammatory injury propagated by internalized LPS extends beyond the induction of pyroptotic cell death. Here we addressed whether surviving endothelial cells were modified in any way and interfered with resolution of tissue integrity. We observed that activation of caspase-11 by internalized LPS induced GSDMD cleavage and the release of mtDNA into the cytosol, thus leading to activation of cGAS-STING pathway. Activation of cGAS/STING signaling in turn induced phosphorylation and inactivation of the cell cycle regulatory transcription factor YAP1 and impaired transcription of cyclin D genes. Thus, endothelial cells surviving pyroptotic death remained severely defective due to suppression of YAP signaling and impairment in proliferation capacity; and thereby prevented full resolution of inflammatory lung injury.
Results
Mitochondrial DNA (mtDNA) release induced by intracellular LPS is dependent on caspase-11 and Gasdermin D
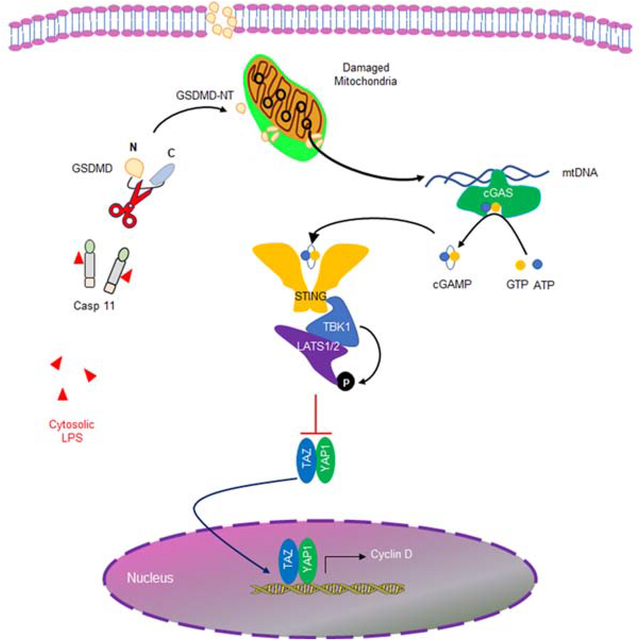
We first studied the time course of pyroptotic death in endothelial cells (EC) by determing lactate dehydrogenase (LDH) release following LPS transfection (LPS(T)) which emulates LPS breaching by bacterial vesicles and increases intracellular LPS (Cheng et al., 2017). While all EC internalized LPS, we observed that only 14% of cells died at 16h, the peak death response, which dissipated within 48h (Fig. S1A). In control experiments in which LPS did not breach the plasma membrane, we did not observe any pyroptosis during the 48h study period (Fig. S1A). Mitochondrial membrane potential (MMP) determined with the potentiometric mitochondrial dye TMRM showed a marked decrease after LPS(T) at 6h and recovery by 48h, whereas there was no change in the absence of LPS(T) exposure (Fig. S1B). The decreases of MMP seen in WT EC in response to LPS(T) were not seen in EC obtained from Casp11−/− and Gsdmd−/− mice that were similarily exposed to LPS(T) (Fig. 1A) indicating the requirement for caspase-11 and GSDMD in mediating mitochondrial depolarization. We also observed that LPS(T) induced dsDNA release into the cytosol in a caspase-dependent manner as the response was abolished by pretreatment of Z-VAD, a pan caspase inhibitor (Fig. 1B). Next, we investigated the source of cytosolic DNA using real-time qPCR, and observed that the LPS(T) induced dsDNA release in cytosol was exclusively from mtDNA with no nuclear DNA (nDNA) contribution (Fig. 1C). Importantly, deletion of either caspase-11 or GSDMD prevented LPS(T) -induced mtDNA release (Fig. 1D & E).
Fig. 1. Intracellular LPS induces mtDNA release through caspase11 and Gasdermin D.
(A) Quantification of MMP in mLMVECs from WT, Casp11−/− and GSDMD−/− mouse lungs transfected with LPS. mLMVECs were primed with LPS (LPS (E), 0.2 μg/ml, 3h), followed with medium change and transfection with or without LPS (LPS (T), 0.5 μg/ml, 0–24 h). Values are shown as mean ± SD; data were obtained from three independent experiments (n= 15–25). (B) Percentage of cells with cytosol dsDNA in LPS (T) treated hLMVECs were analyzed by staining dsDNA. hLMVECs were pretreated with (0, 50 μM, 30 min) Z-VAD-FMK and followed with priming with LPS (0.2 μg/ml, 3h) and treated with LPS (T) (0, 0.5 g/ml, 0–24h). Values are shown as mean ± SD; data were obtained from three independent experiments (n= 5–10). (C) RT-qPCR analysis of cytosolic nuclei DNA (nDNA) and mitochondrial DNA (mtDNA) in hLMVECs after LPS (T) (0.5 g/ml, 24h), n =5–6. (D & E) RT-qPCR quantification of cytosolic mtDNA in mLMVECs ((D) WT vs. Casp11−/−, n=7; (F) WT vs. GSDMD−/−, n=5; after LPS (T) (0, 0.5 g/ml, 24h). (F) LPS (T) induced endogenous GSDMD-NT formation and trans-localizes to mitochondrial membrane. Primed hLMVECs (LPS, 0.2 μg/ml, 3h) were transfected with LPS (0.5 μg/ml, 16h). The mitochondrial fractions were isolated using mitochondrial isolation kit, and the expression of protein were analyzed by immunoblotting, n = 4. (G-I) GSDMD-NT localizes to mitochondrial membrane, decreases MMP, induces cell pyroptosis and cytosolic release of mtDNA. Vehicle plasmid and the GSDMD-NT plasmid (3 μg/ml) were transfected into hLMVECs for 24h, the LDH released into the medium was assayed. The mitochondrial membrane potential was analyzed using the potentiometric dye TMRM, and the release of mtDNA into cytosolic fraction was analyzed by RT-qPCR. (G) Fractionation analysis of hLMVECs with or without overexpression of GSDMD-NT (24h), n = 3. (H) LDH release from hLMVECs with or without GSDMD-NT overexpression, n = 5. (I) TMRM staining (100 nM, 15 min) and analysis of hLMVECs with or without overexpression of GSDMD-NT. Quantification of MMP in hLMVECs with or without GSDMD-NT transfection (24h). Values are shown as mean ± SD, data were obtained from three independent experiments, n = 15–19. (J) RT-qPCR quantification of cytosolic nDNA and mtDNA from hLMVECs with or without GSDMD-NT overexpression, n = 4–6. * P < 0.05, * * P < 0.01, * * * P < 0.001, two-tailed t-test. Values are mean ± SD. Please also see Figure S1.
As the formation of membrane pores by GSDMD occurs downstream of caspase-11 activation by intracellular LPS (Kayagaki et al., 2015), we next investigated whether the decreased MMP and released mtDNA were the result of GSDMD-induced mitochondrial pore formation because intracellular LPS induces EC pyroptosis via formation of GSDMD N-terminal fragment (GSDMD-NT) (Cheng et al., 2017). We observed that the endogenous active GSDMD-NT fragment localized to mitochondria following LPS transfection (Fig. 1F). Overexpression of the active GSDMD-NT fragment in EC also induced GSDMD-NT localization to mitochondria (Fig. 1G) and pyroptosis in these cells (Fig. 1H) that was accompanied by decrease in MMP (Fig. 1I) and release of mtDNA but not nuclear DNA (nDNA) into the cytosol (Fig. 1J). Thus, intracellular LPS induced mtDNA generation in the cytosol through activation of caspase-11 and generation of GSDMD-NT.
Defective repair and proliferation of endothelial cells surviving pyroptotic cell death
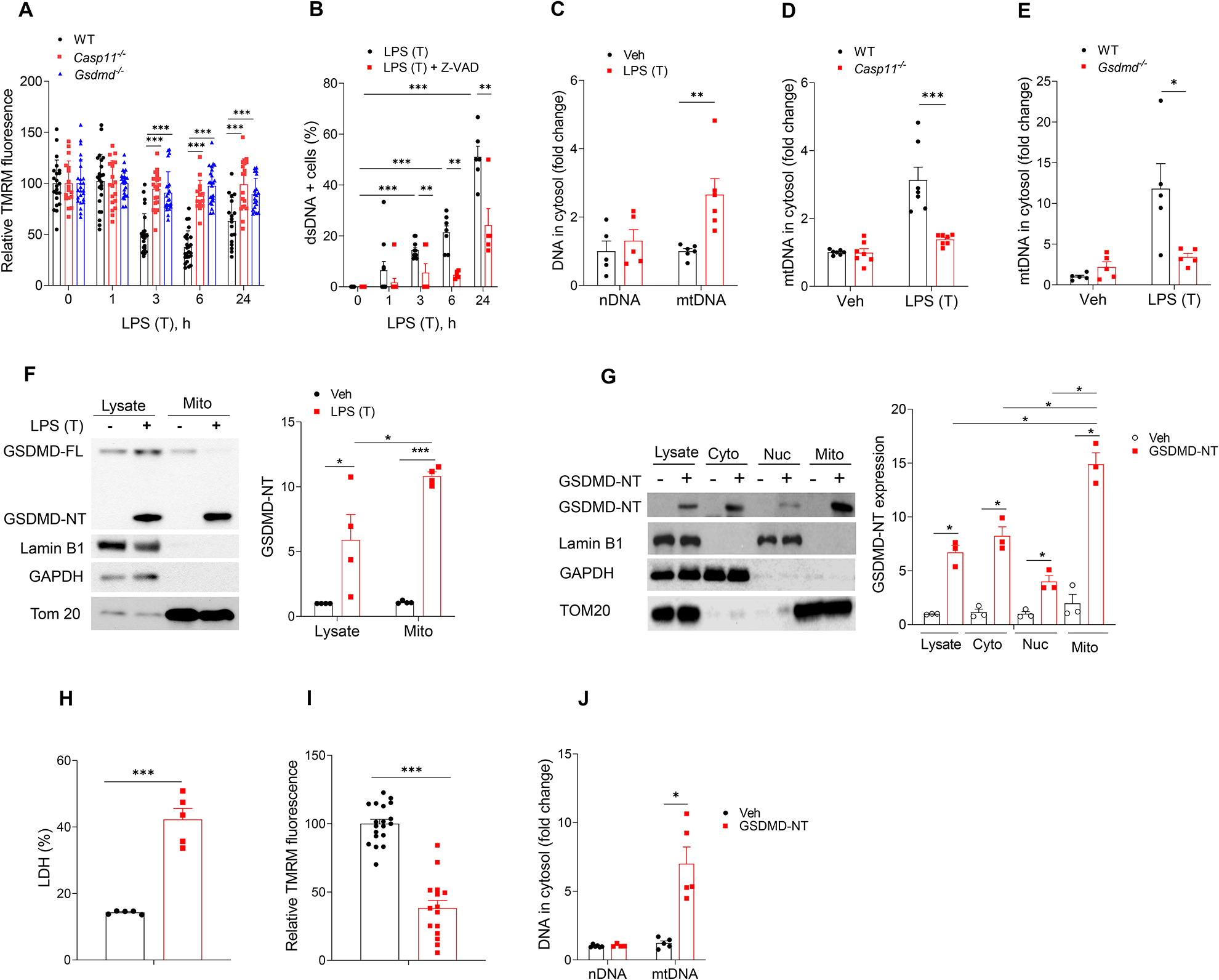
Next we determined whether the ECs surviving from pyroptosis showed any defects in their reparative capacity (Fig. S1A & B). Here we determined whether EC-specific deletion of the intracellular LPS receptor caspase-11 showed any differences in EC proliferation as measured by BrdU incorporation. We used the cecal ligation puncture (CLP) model to induce polymicrobial sepsis because this model is known to induce entry of LPS into the cytosol of endothelial cells (Cheng et al., 2017). Casp11fl/fl x Cdh5-CreERT2 mouse lungs exhibited marked increases in EC proliferation post-CLP (Fig. 2A & B) and -LPS (Fig. 2C) as compared to control Casp11fl/fl lungs. We also determined the restoration of endothelial integrity by measuring Evans blue-albumin (EBA) transendothelial flux. As shown in Fig. 2D, EC barrier integrity was restored at 72h after CLP challenge in Casp11fl/fl x Cdh5-CreERT2 lungs whereas Casp11fl/fl lung EC exhibited persistent injury, suggesting EC deletion of Casp11 promoted recovery of lung injury during sepsis-induced lung injury. Staining of mouse lung tissue for cytosolic dsDNA and EC marker CD31 showed markedly increased intracellular dsDNA in lung EC from Casp11fl/fl in contrast to Casp11fl/fl x Cdh5-CreERT2 mice (Fig. 2E & F).
Fig. 2. Caspase11-Gasdermin D pathway induces mtDNA release, impairs endothelial cell proliferation, and resolution of inflammatory lung injury following polymicrobial sepsis and endotoxemia.
(A-G) EC specific deletion of Casp11 (Casp11fl/fl x Cdh5-CreERT2) in mice promotes endothelial regeneration and recovery from inflammatory lung injury. Casp11fl/fl mice were crossed with Cdh5-CreERT2 mice to delete caspase 11 in endothelial cells. Mice underwent either CLP or LPS injection (10 mg/kg, i.p). (A-C) Proliferation of endothelial cells as determined by the number of BrdU+ endothelial cells in lungs of Casp11fl/fl and Casp11ECKO mice post CLP and LPS challenge. (A) Representative images of proliferating lung endothelial cells from three independent experiments following CLP challenge for 72 h. Green, anti-BrdU; Red, anti–CD31; Blue, DAPI. Arrows indicate proliferating endothelial cells. Scale bar, 100 m. Br indicates bronchia; V, vessel. (B & C) Quantification of BrdU+ endothelial cell from Casp11fl/fl x Cdh5-CreERT2 and Casp11fl/fl mice lung post CLP (B) or LPS (C) challenge for 72 h, n = 5. (D) Lung transvascular albumin permeability in Casp11ECKO and Casp11fl/fl mice with or without CLP challenge, n = 5–6. (E & F) Representative images from three independent experiments (E) and quantification (F) of endothelial cells with cytosolic dsDNA in Casp11fl/fl and Casp11fl/fl x Cdh5-CreERT2 lungs post LPS (10 mg/kg, i.p, 24 h) challenge, n = 5. (G-K) GSDMD deletion (GSDMD−/−) in mice promoted endothelial regeneration and recovery from inflammatory lung injury in response to CLP. (G & H) Proliferation of endothelial cells as determined by the number of BrdU+ endothelial cells in lungs from WT and GSDMD−/− mice post CLP challenge. (G) Representative images of proliferating lung endothelial cells from three independent experiments following CLP challenge for 72 h. Green, anti-BrdU; Red, anti–CD31; Blue, DAPI. Arrows indicate proliferating endothelial cells. Scale bar, 200 m. Br indicates bronchia; V, vessels. (H) Quantification of BrdU+ endothelial cells from WT and GSDMD−/− mice lung post CLP for 72 h, n = 5. (I) Lung transvascular albumin permeability in WT and GSDMD−/− mice with or without CLP challenge, n = 5–6. (J & K) Representative images from three independent experiments (J) and quantification (K) of endothelial cells with cytosolic dsDNA in WT and GSDMD−/− mice lungs post-LPS (10 mg/kg, i.p, 24 h) challenge, n = 5. * P < 0.05, * * P < 0.01, two-tailed t-test. Values are shown as mean ± SD.
We next used Gsdmd−/− mice to determine the role of GSDMD in preventing EC proliferation as GSDMD is a downstream target of caspase-11. The number of proliferating ECs post CLP challenge was higher in Gsdmd−/− lungs as compared to WT (Fig. 2G & H), suggesting that GSDMD activation inhibited EC proliferation. As shown in Fig. 2I, EC integrity returned to basal at 72h after CLP challenge in Gsdmd−/− lungs as compared to WT; thus, the Gsdmd−/− EC survivors showed enhanced recovery of lung injury due to increased proliferation of ECs as compared to WT EC. Deletion of the caspase-11 substrate GSDMD in mice also prevented dsDNA release in EC after LPS (Fig. 2J & K). These data together show that ECs surviving pyroptotic death are markedly defective in EC proliferation due to dsDNA release into the cytosol primarily derived from mtDNA via the Casp11-GSDMD pathway.
mtDNA activation of cGAS signaling suppresses endothelial proliferation
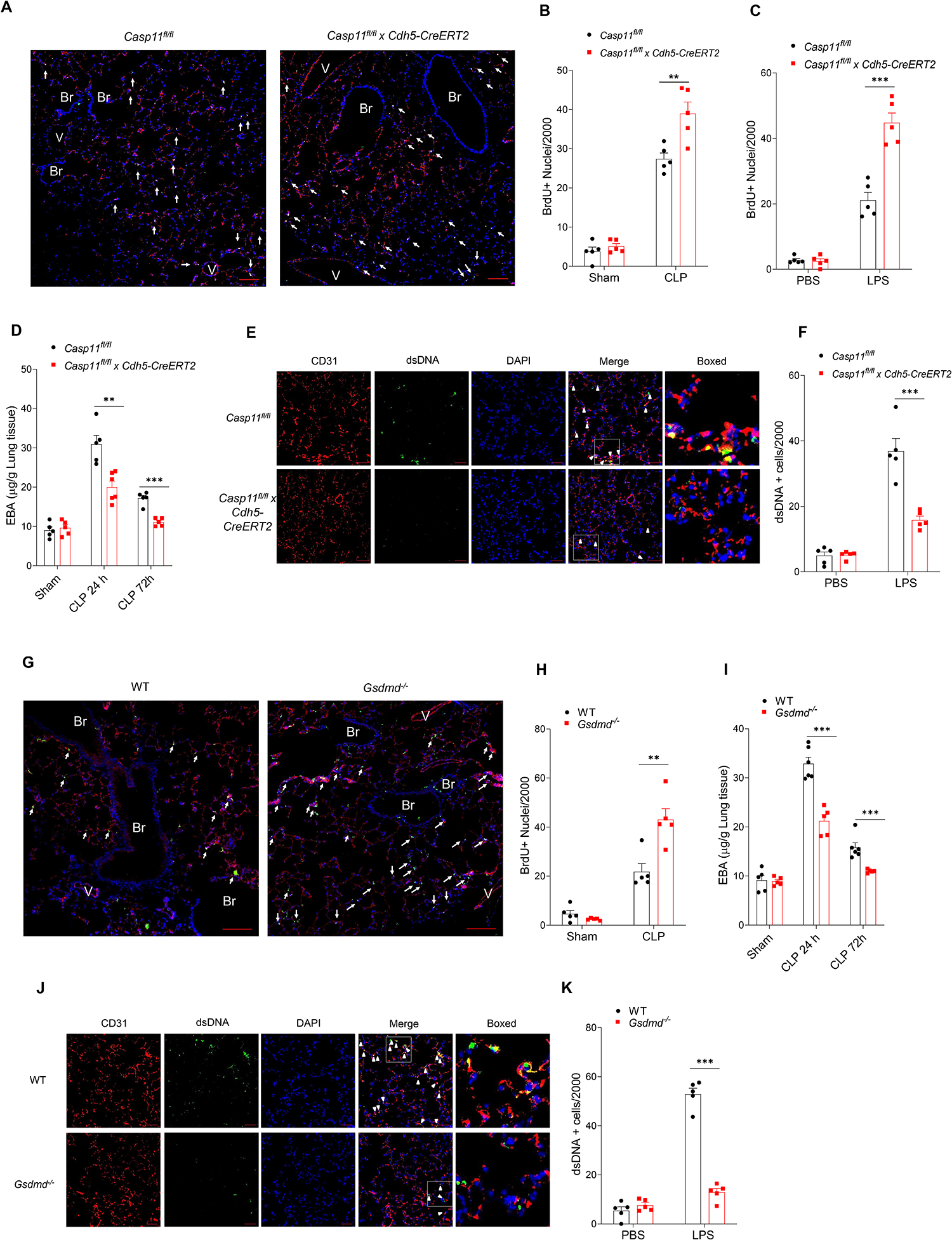
To study the role of cytosolic DNA-sensing cGAS-STING pathway in preventing endothelial recovery, we first quantified changes in cytosolic cGAMP concentrations using LC/MS/MS because generation of the second messenger cGAMP by cGAS is a key effector of this DNA sensing pathway. LPS induced time-dependent increases in cGAMP with a peak at 6h (Fig. 3A) whereas deletion of Caspase-11 in EC (Casp11fl/fl x Cdh5-CreERT2) prevented cGAMP generation (Fig. 3B). Compared to control mice, Casp11fl/fl x Cdh5-CreERT2 or Gsdmd−/− also showed reduced generation of circulating IFN-1β (Fig. 3C &D), cGAS dependent phosphorylation of the TBK1 kinase and IRF3 transcription factor (Fig. S2 A &B) as well as decreased expression of multiple downstream cGAS target genes (Fig. 3E & F) in lung ECs post LPS. In cell culture studies using human lung microvascular endothelial cells (hLMVECs), we observed that depletion of GSDMD reduced the phosphorylation of TBK1 and IRF3 induced by LPS transfection (Fig. S2 C) prior to induction of pyroptosis (Fig. S2D). Similar to the transfection of the positive control plasmid DNA (Fig. S3), transfection of mtDNA in hLMVECs induced cGAS pathway activation (Fig. 3G and Fig. S3). Quantification of cGAMP in hLMVEC showed that both plasmid DNA and mtDNA transfection induced cGAMP generation (Fig. 3H). Finally, we observed that intracellular mtDNA (Fig. 3I) and cGAMP (Fig. 3J) prevented EC proliferation. These data together show that caspase-11 and GSDMD activation suppress EC proliferation secondary to cGAS activation.
Fig. 3. Intracellular dsDNA activates cGAS-STING pathway and induces defective endothelial cell proliferation.
(A) Time course of cGAMP concentration in mouse lung tissues post-LPS (10 mg/kg, i.p,) challenge, n = 4–11. (B) cGAMP concentration in lung tissues from Casp11fl/fl and Casp11fl/fl x Cdh5-CreERT2 mice post-LPS challenge (10 mg/kg, i.p, 6 h), n = 5–8. (C &D) circulating IFN-1β concentration in LPS treated mice (10 mg/kg, i.p, 6h). (C) Casp11fl/fl vs Casp11fl/fl x Cdh5-CreERT2 mice, n = 5–8; (D) WT vs Gsdmd−/− mice, n =5. (E & F) Expression of cGAS/STING responsive genes (CXCL10, IFIT1 and IFIT3) in lung ECs isolated from Casp11fl/fl and Casp11fl/fl x Cdh5-CreERT2 mice (E) or WT and Gsdmd−/− mice (F) post LPS challenge (10 mg/kg, 6 h, i.p), n = 5–8. (G) Immuno blot analysis of protein expression in hLMVECs transfected with mtDNA (3 μg/ml, 6h), n =3. (H) Intracellular cGAMP concentration in hLMVECs transfected with the positive control plasmid DNA or mtDNA (3 μg/ml, 6h), n = 4. (I & J) hLMVECs transfected with mtDNA (0–10 μg/ml), n = 5; (I) or cGAMP (0–12 μg/ml), n = 8, (J) for 20 h followed by serum treated (10%, 8 h). Endothelial cell proliferation (BrdU+ cells) was analyzed by proliferation kit. Data are shown as mean ± SD obtained from at least three independent experiments. * P < 0.05, * * P < 0.01, * * * P < 0.001, two-tailed t-test. Please also see Figure S2 & 3.
cGAS-STING pathway activation suppresses endothelial proliferation and vascular repair
Next, we determined whether cGAS signaling also inhibited EC regeneration in vivo. Here we observed that the number of proliferating EC in cGas−/− mice after CLP increased when compared to those observed in control WT mice (Fig. 4A). To assess the role of cGAS signaling in inflammatory lung injury, we determined EC integrity by measuring lung endothelial permeability in cGas−/− and WT mice. EC integrity returned to basal at 72h after CLP challenge in cGas−/− lungs as compared to the persistent loss of barrier integrity in WT mice as assessed by transvascular albumin leak (Fig. 4B). H&E staining revealed that perivascular infiltration of leukocytes in cGas−/− mouse lungs during the repair phase (eg, 72 hours after CLP) was lower than that observed in WT lungs (Fig. 4C). At the peak of lung vascular injury (24 hours post CLP), cGas−/− mouse lungs exhibited similar myeloperoxidase (MPO) activity to that of WT (Fig. 4D). However, during the repair phase (72 hours post CLP), the MPO activity in cGas−/− lungs returned to basal (Fig. 4D) whereas WT lungs demonstrated persistent inflammation. Similar to Casp11fl/fl x Cdh5-CreERT2 and GSDMD−/−, cGas−/− also decreased LPS-induced circulating IFN-1β concentrations (Fig. 4E). We also studied the role of cGAS deletion in human endothelial cells and found that depletion of cGAS in hLMVECs rescued the decrease in EC proliferation induced by LPS transfection (Fig. S4 A) without modulating its effects on mitochondrial depolarization (Fig. S4 B) and cell pyroptosis (Fig. S4C).
Fig. 4. cGAS-STING pathway impairs endothelial regeneration and recovery from inflammatory injury.
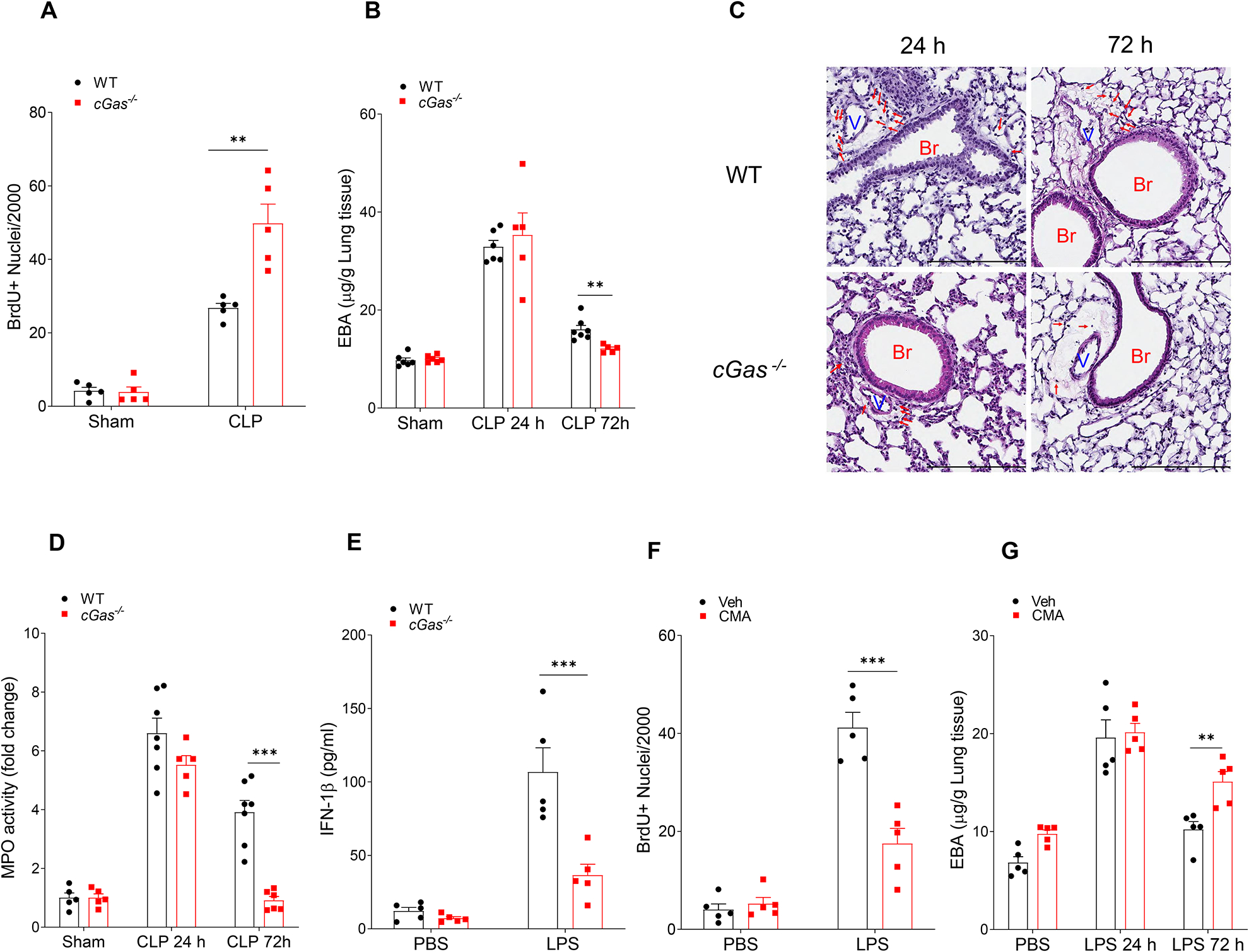
(A) Quantification of BrdU+ lung endothelial cell from WT and cGas−/− mice post CLP for 72 h, n = 5. (B) Lung transvascular albumin permeability in WT and cGAS−/− mice with/without CLP challenge, n = 5–7. (C) Representative H&E staining of lung tissue from three independent experiments (WT and cGas−/−) post CLP challenge (24 and 72 h). Red arrows indicate infiltrated immune cells. Scale bar, 200 m. Br indicates bronchia; and V, vessels. (D) Myeloperoxidase (MPO) activity in mouse lungs after CLP challenge, n = 5–7. (E) IFN-1β concentration in serum from LPS treated WT and cGas−/− mice (10 mg/kg, i.p, 6h), n = 5. (F) Quantification of BrdU+ lung endothelial cells from WT mice post-LPS exposure (10 mg/kg, i.p) with or without treatment of the STING agonist, CMA, (10 mg/kg, i.p, every 24h from 24h-72h post LPS challenge) for up to 72h post-LPS, n = 5. (G) Lung transvascular albumin permeability in LPS treated mice with or without CMA treatment, n = 5. Data are shown as mean ± SD. * P < 0.05, * * P < 0.01, two-tailed t-test. Please also see Figure S4.
We next investigated whether activation of cGAS/STING pathway was involved in the impairment of cell repair program. Here we used the STING agonist, 10-carboxymethyl-9-acridanone (CMA) which activates the STING pathway independent of cytosolic DNA or cGAMP (Cavlar et al., 2013; Gaidt et al., 2017; Haag et al., 2018). We observed that STING activation impaired endothelial proliferation post LPS challenge by using the BrdU incorporation assay (Fig. 4F), suppressed restoration of EC integrity and recovery of lung injury (Fig. 4G).
cGAS/STING activation inhibits endothelial proliferation through suppression of YAP signaling
To identify downstream effectors of cGAS/STING pathway on endothelial proliferation and lung injury repair, we focused on the role of mtDNA and cGAMP in the YAP pathway, which is known to regulate cell proliferation (LaCanna et al., 2019). We observed that mtDNA (Fig. 5A) and cGAMP (Fig. 5B) increased YAP phosphorylation at S127 and S397, as well as the phosphorylation of the large tumor suppressor kinase (LATS1), a kinase which directly induces YAP phosphorylation (Meng et al., 2016). Depletion of cGAS in hLMVEC suppressed mtDNA induced increase in phosphorylation of LATS1, phosphorylation and degradation of YAP, and activation of cGAS signaling (Fig. S5 A).
Fig. 5. mtDNA and cGAMP prevent endothelial proliferation through inhibition of YAP.
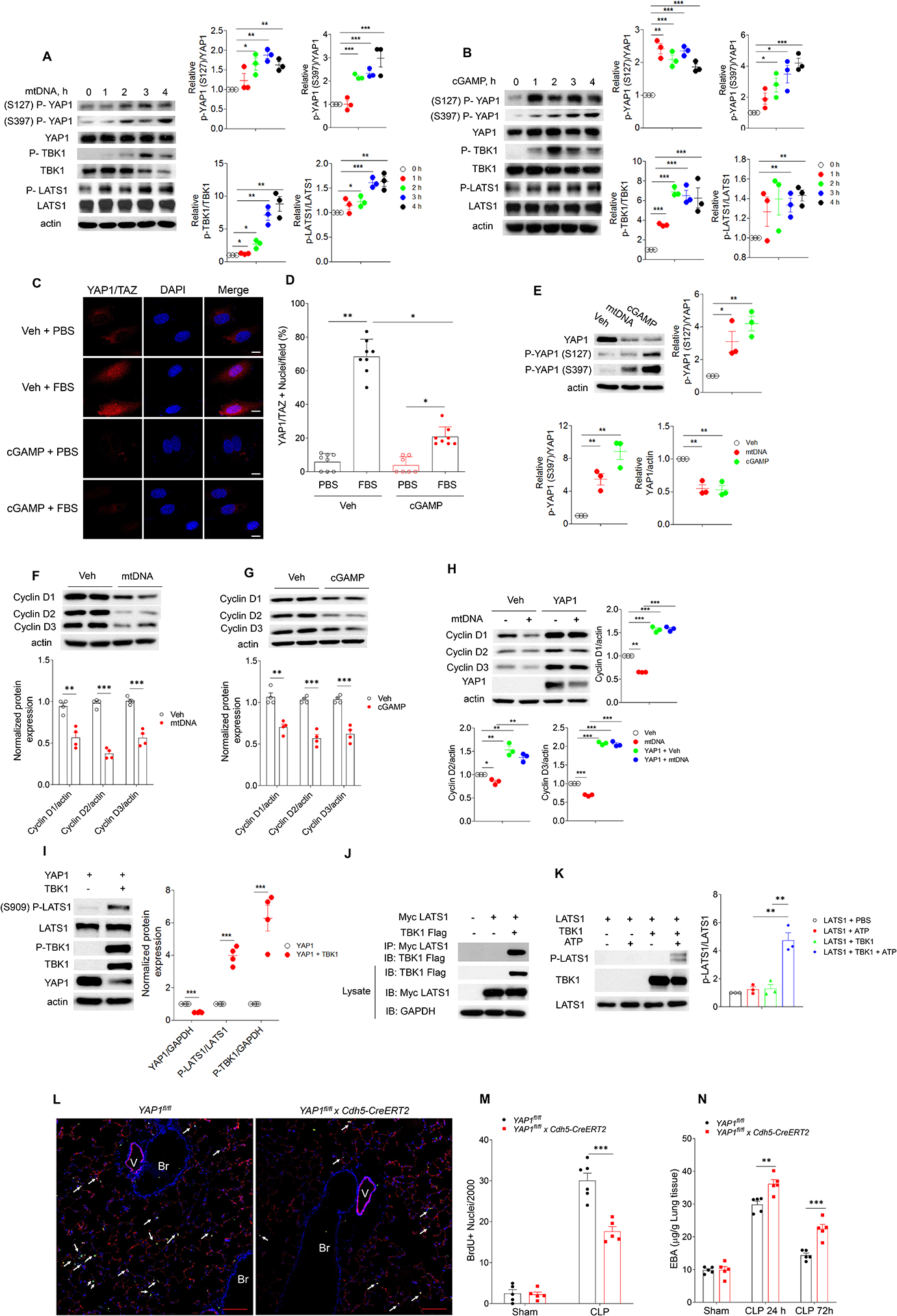
(A, B) Immuno blot of protein expression in hLMVECs with transfection of mtDNA (3 μg/ml, 0–4h, A) or cGAMP (3 μg/ml, 0–4h, B), n = 3. (C, D) hLMVECs with cGAMP (3 μg/ml, 20h) transfection were challenged with 10% FBS for 1 h, and the YAP1-TAZ nuclear translocation in hLMVECs were analyzed by Immunofluorescence staining. (C) Representative image and (D) percentage of hLMVECs with YAP1-TAZ nuclear trans-localization with FBS challenge. Values are shown as mean ± SD; data were obtained from three independent experiments, n= 7–8. (E) Immuno blot analysis YAP1 phosphorylation and YAP1 expression in hLMVECs with transfection of mtDNA (3 μg/ml, 20h) or cGAMP (3 μg/ml, 20h), n=3. (F, G) Expression of cyclin D1,D2, D3 in hLMVECs transfected with mtDNA (F) or cGAMP (G) (3 μg/ml, 20h), n = 3. (H) YAP1 restores mtDNA induced decrease of cyclin D1, 2, 3 expression in hLMVECs. Control plasmid or YAP1 plasmid (3 μg/ml, 48h) transfected hLMVECs were transfected with mtDNA (3 μg/ml) for 20h, the expression of cyclin D1, 2, 3 were analyzed by immunoblotting, n = 3. (I) Transfection of TBK1 induces LATS1 phosphorylation. HEK293T cells were co-transfected with plasmids encoding TBK1 and YAP1 (3 μg/ml, 48h) before immunoblotting analysis, n = 4. (J) HEK293 cells were co-transfected with myc-tagged LATS1 (myc-LATS1) and either vector control (Vector), Flag-tagged TBK1 (Flag-TBK1). LATS1 was immunoprecipitated to assess the binding proteins by immunoblotting. Input lysates were used to blot protein and verify the transfected protein expression. Representative images from three independent experiments, n = 3. (K) IP-purified myc-LATS1 (Myc-LATS1) from myc-LATS1 transfected HEK293 cells was co-incubated with IP-purified Flag-TBK1 from Fag-TBK1 transfected HEK293 cells. Purified proteins were used for in vitro kinase assay. Phosphorylated LATS1 (S909) was analyzed via immunoblotting and quantitated via densitometry, n = 3. (L-N) Endothelial cell specific deletion of Yap1 (Yap1fl/fl x Cdh5-CreERT2) in mice prevents endothelial regeneration and recovery from lung injury. Yap1fl/fl mice were crossed with Cdh5-CreERT2 to delete YAP1 in endothelial cells. After tamoxifen induced deletion of Yap1, mice were used for CLP. (L) Representative micrographs from three independent experiments showing endothelial cell (EC) proliferation in lungs from Yap1fl/fl and Yap1fl/fl x Cdh5-CreERT2 mice at 72h post CLP. Green, anti-BrdU; Red, anti–CD31; Blue, DAPI. Arrows indicate proliferating endothelial cells. Scale bar, 200 m. Br indicates bronchia; V, vessel. (M) Graphic presentation of increased proliferating endothelial cells in Yap1fl/fl as compared to Yap1fl/fl x Cdh5-CreERT2 lungs post-CLP, n = 5. (N) Lung transvascular permeability measurement following CLP showed defective recovery in Yap1fl/fl x Cdh5-CreERT2 as compared to Yap1fl/fl mice, n = 5. Data are shown as mean ± SD. * P < 0.05, * * P < 0.01, * * * P < 0.001, two-tailed t-test. Please also see Figure S2, 4 & 5.
We next addressed whether mtDNA, cGAMP or increase in intracellular LPS regulated translocation of the YAP-TAZ complex to nucleus. We observed that in control EC FBS induced YAP-TAZ nuclear translocation whereas cGAMP prevented this response (Fig. 5C,D). Similarly, LPS transfection or mtDNA transfection also inhibited YAP-TAZ nuclear translocation in ECs transfected with control si-RNA (Fig. S4D). However, depletion of cGAS in hLMVECs rescued the decrease in YAP-TAZ nuclear translocation following transfection of LPS or mtDNA, but not cGAMP (Fig. S4D). mtDNA and cGAMP also induced degradation of YAP1 in hLMVECs (Fig. 5E). As IKKε kinase is known to phosphorylate YAP and trigger its degradation during viral infections (Wang et al., 2017), we assessed the potential role of IKKε in regulating the mtDNA-induced suppression of YAP signaling. We observed that mtDNA phosphorylated and thus activated IKKε in hLMVECs, whereas depletion of IKKε reduced YAP1 phosphorylation (Fig. S5 B & C).
In LPS challenged mice, the isolated lung ECs from Casp11fl/fl x Cdh5-CreERT2 (Fig. S2A) or Gsdmd−/− (Fig. S2B) showed decreased LPS induced phosphorylation of YAP1 and prevented of YAP1 degradation. We also observed that siRNA silencing of GSDMD (Fig. S2C) or cGAS (Fig. S4E) in hLMVECs reduced the LPS(T) induced phosphorylation of TBK1 and IRF3, and phosphorylation and degradation of YAP1. In addition, both mtDNA and cGAMP reduced the expression of cyclin D1, D2 and D3 (Fig. 5 F, G), known as the downstream gene of YAP1 signaling, and overexpression of YAP1 restored the mtDNA and cGAMP induced decreases of cyclin D1, D2 and D3 expression in hLMVECs (Fig. 5H). siRNA silencing of cGAS in hLMVECs also restored LPS(T) induced decrease in cyclin D1 and 2 expression (Fig. S4E). These data collectively show that LPS(T) decreased EC proliferation via the cGAS regulated YAP1 pathway.
We observed that TBK1 was also concomitantly activated by both mtDNA and cGAMP (Fig. 5A,B), suggesting that TBK1 regulation of YAP phosphorylation prevented YAP nuclear translocation, and promoted its degradation. Thus, we next investigated whether TBK was responsible for YAP1 degradation. Here upon overexpression of YAP1 and TBK1 in HEK293 cells, we observed (Fig. 5I) that TBK1 induced LATS1 phosphorylation at S909, resulting in phosphorylation and degradation of YAP1. Immunoprecititation of LATS1 from HEK293 cells overexpressing both TBK1 and LAST1 showed binding of LATS1 with TBK1 (Fig. 5J). The kinase assay showed that TBK1 directly phosphorylated LATS1 (Fig. 5K). Thus, mtDNA induced activation of TBK1 was responsible for YAP1 degraduation through the activation of LATS1.
To address the effects of YAP1 on EC proliferation and repair, we used Yap1fl/fl x Cdh5-CreERT2 mice. We observed that EC proliferation was reduced in Yap1fl/fl x Cdh5-CreERT2 mice as compared to Yap1fl/fl mice post CLP challenge (Fig. 5L & M). Furthermore, transendothelial albumin flux (with an Evans Blue Albumin tracer) measurements were used to assess the recovery of endothelial barrier integrity. We observed that Yap1fl/fl x Cdh5-CreERT2 mice demonstrated prolonged endothelial injury and slower recovery (Fig. 5N) as compared to control Yap1fl/fl mice following CLP injury. These results together show that mtDNA-cGAS signaling induces YAP phosphorylation and thereby prevents downstream YAP-mediated endothelial proliferation and repair.
Discussion
Here we have identified an additional function of the pore-forming Gasdermin D (GSDMD) in releasing mitochondrial DNA into the cytosol in inflammatory injury and activating DNA-sensing cGAS/STING pathway, and thereby suppressing endothelial cell proliferation and vascular repair. cGAS/STING signaling functioned by inhibiting dephosphorylation and nuclear translocation of the transcription factor YAP1, and inhibited cyclin D-mediated cell proliferation. We showed that intracellular LPS triggered mitochondrial membrane localization of active N-terminal GSDMD (GSDMD-NT) fragment resulting in severe mitochondrial injury. Mitochondrial injury decreased mitochondrial membrane potential and stimulated mtDNA release into endothelial cytosol which in turn was recognized by cGAS and induced generation of the second messenger cGAMP and activation of the pro-inflammatory STING pathway.
Studies have identified the GSDMD as a critical downstream effector of inflammatory caspases mediating pyroptosis of macrophages (Kayagaki et al., 2015; Shi et al., 2015) and endothelial cells (Cheng et al., 2017). Inflammatory caspases-1, −4, −5 and −11 cleave GSDMD to form GSDMD-NT which is the pore forming domain (Broz, 2015; Cheng et al., 2017; Liu et al., 2016). The pores formed by oligomerization of GSDMD-NT induce loss of cytoplasmic content and pyroptotic cell death (Ding et al., 2016), and transport of pro-inflammatory cytokine IL-1β across the plasma membrane (Evavold et al., 2018). GSDMD-NT binding is not only restricted to the plasmalemmal membrane, but also to mitochondrial membrane and induce release of mitochondrial reactive oxygen species (ROS) (Platnich et al., 2018). We observed that mitochondrial membrane translocation of GSDMD-NT induced collapse of mitochondrial membrane potential and release of mtDNA demonstrating an additional mitochondrial injury mechanism.
Endothelial pyroptosis has been shown to serve as a key mechanism of inflammatory vascular injury as it resulted in endothelial death and release of IL-1β (Cheng et al., 2017). Importantly, endothelial cells have the capacity to respond to cell death by compensatory proliferation (Liu et al., 2019). However, it is not known whether inflammatory injury undermines this crucial homeostatic regenerative mechanism in the large number surviving endothelial cells. Here we found that intracellular LPS induced Caspase-11-GSDMD-mediated release of mtDNA into the cytosol which in turn activated the pro-inflammatory cGAS-STING pathway and suppressed cell proliferation of the surviving ECs which did not undergo pyroptosis. These findings demonstrate that the severe vascular injury induced by intracellular LPS is based on at least three distinct processes: 1) pyroptotic cell death in a subset of ECs, 2) suppressed cell proliferation due to activation of the cGAS/STING cascade which prevents surviving cells from adaptively compensating for pyroptotic cell loss and 3) release of inflammatory mediators such as the type I interferon-1β by the surviving endothelial cells. Both Casp11fl/fl x Cdh5-CreERT2 and Gsdmd−/− mice promote endothelial regeneration through suppression the LPS induced mtDNA release into lung ECs. The greater protection seen in Gsdmd−/− mice when compared to Casp11fl/fl x Cdh5-CreERT2 mice is likely due to the fact that GSDMD is the downstream effector of multiple pathways, including Casp11 and Casp1 (Ding et al., 2016; Rathinam et al., 2019).
Previous studies of cGAS signaling primarily focused on how self-dsDNA (genomic DNA fragments or mtDNA) released into the cytosol are recognized by cGAS, trigger cGAMP synthesis, and activats STING mediated pro-inflammatory transcriptional programs (Gao et al., 2015; Gluck et al., 2017; Rongvaux et al., 2014). Activating mutations in STING have also been identified in patients with autoimmune disease (Gaidt et al., 2017; Jeremiah et al., 2014), underscoring the role of dsDNA-cGAS-STING pathway in mediating inflammation. The protective effects of cGas deletion in terms of reducing endothelial barrier leakiness at 24h in the polymicrobial CLP model as assessed by the Evans blue-albumin (EBA) tracer flux assay were weaker than those seen with endothelial Casp11 or Gsdmd deletion. This likely reflected the fact that Caspase-11 and Gasdermin D promote injury at this earlier stage of injury via several downstream mediators including the release of pro-inflammatory cytokines whereas cGAS activation plays a more dominant role during the subsequent repair phase at 72h when it suppresses endothelial regeneration. Endothelial cell proliferation and regeneration is critical to the restoration of lung endothelial integrity post sepsis (Huang et al., 2016; Liu et al., 2019). Additionally, it has also become apparent that cGAS signaling inhibits cell proliferation by promoting premature cellular senescence (Gluck et al., 2017; Yang et al., 2017). We cannot rule out that additional DNA sensing pathways such as the AIM2 inflammasome may also contribute to the sensing of mtDNA (Dang et al., 2017), however the near complete protection at 72 h post CLP we observed in the cGas−/− mice does indicate that cGAS serves as the primary DNA sensor in ECs regeneration and recovery of lung injury during polymicrobial sepsis.
The present work identifies the key role of YAP signaling as a mechanism by which cGAS-STING signaling suppresses cell proliferation and regeneration in the penumbral zone of pyroptotic cell death. YAP1-TAZ signaling serves as a critical regulator of cell proliferation during tissue regeneration (He et al., 2018; LaCanna et al., 2019). YAP1-TAZ are effectors of the Hippo pathway which comprises a signaling cascade involving the mammalian ste20-like kinases 1 and 2 (MST1 and MST2), the large tumor suppressor kinases 1 and 2 (LATS1 and LATS2), and transcription co-activators YAP1 and TAZ (Meng et al., 2016). Activation of Hippo pathway induces phosphorylation of MSTs which phosphorylate LATSs (Lim et al., 2019). Activated LATSs in turn induces phosphorylation and ubiquitin-proteasome mediated degradation of YAP, which is exported from the nucleus and undergoes degradation, thus preventing YAP1-TAZ mediated cell proliferation (Hansen et al., 2015). Mechanical forces mediated by actin cytoskeleton, integrin signaling, and cell-cell contact activate the Hippo pathway and suppress YAP1-TAZ signaling, and thus induce contact inhibition in intact endothelial monolayers to avoid hyper-proliferation (Seo and Kim, 2018; Sun and Irvine, 2016). During cell injury or death, mechanical cues from neighboring cells are lost, thus leading to nuclear translocation of YAP1-TAZ to initiate compensatory cell proliferation (Hong et al., 2016). We found that mtDNA-induced increases in cGAMP concentrations during inflammatory injury actively suppressed YAP1-TAZ signaling via stimulating LATS dependent the YAP1 phosphorylation and degradation. As IKKε is a known trigger YAP1 degradation during viral infections (Wang et al., 2017), we assessed the potential role of IKKε in mediating mtDNA-induced suppression of YAP1 signaling. mtDNA also activated IKKε in hLMVECs, whereas depletion of IKKε reduced YAP1 phosphorylation. These findings suggest that mtDNA regulates ECs regeneration through cGAS-YAP1 pathway, which is regulated by LATS and IKKε dependent mechanism. Despite endothelial injury during severe inflammatory lung injury (Cheng et al., 2017; LaCanna et al., 2019), which should activate the regenerative YAP1-TAZ pathway and cell cycle genes (Liu et al., 2019), we observed that cGAS/STING activation suppressed this key adaptive endothelial proliferation pathway. Thus, endotoxemia not only induced endothelial death (Cheng et al., 2017) but also suppressed regenerative pathways responsible for restoring vascular homeostasis. Our findings help to explain why endotoxemic and septic acute lung injury are so devastating with mortality rates of 20–40% (Cochi et al., 2016).
mtDNA-cGAMP-STING cascade amplified endothelial inflammation and suppressed endothelial regeneration raises an intriguing question about the teleological role for this anti-regenerative response. It is possible that cells face a “fight-or-fix” dilemma during inflammatory injury in which amplified inflammation is prioritized over tissue regeneration to enhance host defense function of the organism. Our work also identifies several new possibilities for shifting the cellular responses on the “fight-or-fix” continuum towards regeneration and reduced inflammation. These include preventing GSDMD activation using inhibitory peptides targeting GSDMD (Rathkey et al., 2018) and inactivation of cGAS (Vincent et al., 2017), STING (Haag et al., 2018) and Hippo signaling (Johnson and Halder, 2014). Conversely, activation of endothelial proliferation by targeting pro-regenerative endothelial transcription factors such as FoXM1 (Huang et al., 2016), Sox17 (Liu et al., 2019), or ER71 (Park et al., 2016) may be an additonal therapeutic approaches that counterbalance the anti-proliferative effects of mtDNA release. In summary, we have identified a fundamental role for mtDNA in activating a self-injurious and anti-regenerative response in the endothelium mediated by the DNA sensor cGAS.
STAR Methods
MATERIALS AND METHODS
Mice.
Casp11–/– and Casp11fl/fl mice were provided by Dr. Vishva Dixit (Genentech, South San Fransciso, CA); Gsdmd−/− mice were obtained from Dr Thirumala-Devi Kanneganti (St. Jude Children’s Research Hospital, Memphis, TN); and cGas−/− mice were obtained from Dr Herbert W. Virgin (Washington University School of Medicine, St. Louis, MI). Yap1fl/fl (Stock Number: 027929) mice were obtained from Jackson laboratory.Mice with Casp-11 and Yap1 EC-specific genetic deletion were generated by backcrossing Casp11fl/fl or Yap1fl/fl mice with Cdh5-CreERT2 mice (provided by Dr. Ralf Adams) respectively. Mice (male, 8–12 weeks) were used for all experiments. Littermate wild-type (WT) mice (C57BL/6J background) were used as controls. All mice were bred and maintained in the University of Illinois animal facility under pathogen–free conditions according to NIH guidelines and all animal experiments were performed according to protocols approved by the institutional Animal Care and Use Committee.
LPS administration and cecal ligation puncture (CLP) in mice.
To induce endothelial specific deletion of genes, Casp11fl/fl x Cdh5-CreERT2 or Yap1fl/fl x Cdh5-CreERT2 mice were injected with tamoxifen (2 mg/mouse in corn oil, i.p., once per day) for 5 consecutive days followed by a 4wk rest period. To evaluate lung injury and endothelial proliferation, mice were challenged with sub-lethal dosage of LPS (10 mg/kg, i.p.) resulting in peak injury at 24h post LPS challenge; this was followed by monitoring mice and their sacrifice at different time points for tissue harvesting.
CLP was also used to induce polymicrobial sepsis as previously described (Cheng et al., 2017). Mice were anesthetized with inhaled isofluorane (2.5% mixed with room air) and subjected to either CLP involving two 21-gauge punctures or sham control conditions (laparotomy surgery). When mice failed to respond to paw pinch, buprenex (0.1 mg/kg) was administered subcutaneously prior to sterilization of the skin with providone iodine, after which a midline abdominal incision was performed. The cecum was exposed and ligated with a 3.0 silk tie 0.5 cm from the tip and the cecal wall was perforated with a 21-gauge needle. Sham control mice underwent anesthesia, laparotomy, and wound closure but no CLP. Immediately following the procedure, 500 μl of warmed PBS solution was administered subcutaneously. Mice received a second dose of buprenex at 6–8 h post-surgery subcutaneously.
Silencing of cGAS and GSDMD.
siRNAs targeting human cGAS, GSDMD and a siRNA negative control (non-targeting pool siRNA) were obtained from Dharmacon. Transient transfections of these siRNAs (300 nM) in human lung microvasculae endothelial cells (hLMVECs) were performed with the Amaxa human endothelial nucleofector kit (VPI-1001, Lonza) according to the manufacturer’s instructions. To evaluate the efficiency of protein suppresion, protein expression was examined via immunoblotting in cells 2–3 days after transfection.
Immunoblotting.
Cell lysates were prepared in lysis buffer containing EDTA-free complete protease inhibitors, followed by centrifugation at 10,000 g for 10 min, and boiled with Laemmli sample buffer for 5 min. Cell lysates (20 μg protein) were separated on 10% or 4–20% SDS-PAGE, transferred to PVDF membranes, and blocked with TBST containing 5% BSA prior to incubation with primary antibodies (1:1000 dilution) overnight, and secondary antibodies (1:3000 dilution) for 2h at room temperature. Blots were developed using an ECL chemiluminescence kit. Densitometry analysis was performed using ImageJ software (NIH), and data were normalized against housekeeping proteins.
LDH cytotoxicity assay.
Cells were cultured in 96-well plates. The supernatants of untreated and treated hLMVECs were collected for analysis of LDH activity using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega). Briefly, 50 l of supernatant samples were transferred to a 96-well plate and mixed with 50 l of the CytoTox 96 reagent and incubated at room temperature for 30 min. After stoping the reaction by adding 50 l of stopping buffer, the LDH activity was measured and analyzed according to the manufacturer’s instructions.
ELISA.
The Type 1 interferon (IFN-1β) serum concentrations were measured using the Quantikine sandwich ELISA kit (R&D Systems) according to the manufacturer’s instructions and the standard curve was calculated by GraphPad Prism linear regression analysis.
Myeloperoxidase (MPO) assay.
MPO content was measured by using the MPO assay kit according to the manufacturer’s instructions. Briefly, after homogenization in 1 ml of ice cold PBS with proteinase inhibitors, lung tissues (~100 mg) were centrifuged at 4°C (8,000 g, 10 min). The pellets were suspended in 1mL of lysate buffer with 0.5% hexadecyltrimethylammonium bromide incubated at 4°C for 1 h, and the solution was centrifuged at 4°C (10,000 g, 10 min). The supernat ant was then collected and mixed 1/30 (vol/vol) with assay buffer (0.2 mg/mL o-dianisidine hydrochloride and 0.0005% H2O2). The change in absorbance was measured at 460 nm for 3 min, and MPO activity was calculated as change in absorbance over time.
Evans blue-albumin tracer measurement of transendothelial flux.
Evans blue-albumin (EBA) tracer flux assay was performed to determine lung vascular injury as described (Cheng et al., 2017). EBA was injected in mice (20 mg/kg BW, i.v.) at 30 min before tissue collection. Lungs were perfused with blood-free PBS, blotted dry, weighed, and snap frozen in liquid nitrogen. The lung was homogenized in 1 ml PBS, and Evans blue dye was extracted by mixing with 2 ml formamide and incubating at 60°C for 18h. After centrifugation (5,000 g, 30 min), the supernatant was collected and dye concentration was determined at 620 nm and 740 nm. EBA uptake in lung tissue was quantified as micrograms of Evans blue dye per g lung tissue. The EBA value post LPS or CLP was used an indicator of lung injury.
Endothelial cell proliferation assay.
Endothelial cell (EC) proliferation in mice lungs was determined by staining BrdU labeled lung ECs as previously described (Liu et al., 2019). Briefly, BrdU (i.p., 150 mg/kg, BW; Sigma B5002) was injected in mice 14 h before sacrifice. Mouse lung cryosections (5 m thick) were stained with mouse anti-BrdU antibody (1:25, BD Biosciences), rat Anti-CD31 antibody (1:50, BD Biosciences) and DAPI. EC number was analyzed by using ImageJ software (NIH). The number of ECs with positive staining of BrdU was normalized to the total number of ECs. In vitro EC proliferation was also assayed in cultured ECs using the proliferation kit obtained from Abcam Inc (ab126556, Cambridge, United Kingdom).
Culture of primary microvascular endothelial cells.
Human lung microvascular ECs (hLMVECs) were obtained from Lonza and cultured in EGM2 medium (Lonza; Basel, Switzerland). Mouse lung microvascular ECs (mLMVECs) were isolated and cells were cultured in mouse endothelial culture medium (Cheng et al., 2017). hLMVECs and mLMVECs at passages 3–7 were used for all experiments.
Mouse EC isolation and pull down assay.
To remove blood and circulating immune cells from the lung tissue, the mouse lungs were flushed with 5 ml of PBS from the right ventricle. Then, lungs were minced and digested with 5 ml Type 1 collagenase I (2 mg/ml in PBS, 37 °C, 1 h), then the cell tissue mixtures were pipett ed through a 40 m disposable cell strainer. After treatment with red blood cell lysis buffer (Qiagen) for 5 min on ice, isolated cells were incubated with 5 μg CD31 antibody (4 °C, 20 min, millpore sigma, CBL1337), and followed with pulldown with pre-washed 25 μl of Dynabeads (4 °C, 25 min, Invitrogen #11035) at with gentle tilting and rotation.
Transfection of LPS, mtDNA, plasmids and cGAMP.
LPS, mtDNA plasmids and cGAMP were transfected into cells via lipofectamine 2000 according to the manufacturer’s instructions. For transfecting LPS into hLMVECs, cells were first primed with (0–0.2 μg/ml) extracellular LPS for 3 h, and then transfected with LPS (0–0.5 μg/ml) to enable LPS internalization.
Co-immunoprecipitation assay (Co-IP).
Co-IP was performed as in described before (Lim et al., 2019). Cell lysates from HEK293 cells transfected with 2 μg/ml of myc-LATS1 and flag-TBK1 for 48 h using lipofectamine 2000 transfection reagent. Cells were lysated with lysate buffer including proteinase inhibitor and incubated with either mouse anti–myc antibody, or an equal amount of normal mouse IgG, and then with protein A– or protein G–conjugated Sepharose beads, followed by Immuno blot analysis.
In vitro kinase assay.
In vitro kinase assay was performed as described before (Lim et al., 2019). Briefly, HEK293 cells transfected with either FLAG-TBK1 or myc-LATS1 were lysed in ice cold cell lysis buffer with protease inhibitor and phosphatase inhibitor cocktail. Immunoprecipitation with anti-FLAG or anti-myc antibodies was performed as described above. Immunoprecipitated pellets were washed with ice-cold cell lysis buffer and ice-cold in vitro kinase assay buffer with protease inhibitor cocktail. Then, mixed the pellets containing immunoprecipitated FLAG-TBK1 or Myc-LATS1 were incubated at 30°C for 30 min with or without 500 μM ATP. The rea ctions were terminated by adding 4X sample buffer and incubating at 95°C for 5min. L ATS phosphorylation was analyzed by immunoblotting.
Immunofluorescence staining.
hLMVECs were fixed (3.7% paraformaldehyde, 10 min), permeabilized (4 min in Tris-buffered saline (TBS) containing 0.25% Triton X-100) and incubated with primary antibodies (1:200 dilution in blocking buffer) for 1h, followed by 3 rinses (15 min each) in TBST and stained with Alexa Fluor secondary antibodies (1:200 dilution in blocking buffer; Life Technologies, Grand Island, NY) for 1h. To determine mitochondrial membrane potential, cells were stained with TMRM (100 nM) and Hoechst dye (1 μM) for 15 min, and then washed with EBM2 medium (3×5 mins) and imaged. Slides and stained cells were examined using a confocal microscope system (LSM880; Carl Zeiss, Inc) equipped with a 63×1.2 NA objective lens (Carl Zeiss, Inc.).
Staining and analysis of cytosolic dsDNA.
Lung tissues were flushed with 5 ml of PBS from the right ventricle to remove blood. Then, lungs were infused with 70% OCT intratracheally, placed in a cassette with OCT for freezing on dry ice. After storage for 48h at −80°C, lungs were sectioned into 5 μm frozen slices. Cryosections and cells were fixed with 4% paraformaldehyde for 15 min, permeabilized by incubation with Triton-X 100 (0.02%, 10 min at room temperature) (Winton et al., 2008), and overnight incubated with anti-dsDNA antibody (1:100, Millipore) and anti-CD31 (1:40, BD Biosciences) at 4°C, and incubated with donkey anti -rabbit and anti-mouse secondary antibodies (1:200, invitrogen) at room temperature for 2 h. Stained sections were imaged with a confocal microscope system LSM880 (Zeiss) and analyzed by Zen software (Zeiss). EC number was determined using ImageJ software (NIH), and the number of ECs positive for intracellular dsDNA was normalized to the total EC number.
RT-qPCR analysis.
Total RNA was isolated from cells using TRIZOL reagent (Life Technology, Rockville, MD), and RNA (1 μg) was reversed transcripted using cDNA synthesis kit (Bio-Rad). Quantitative real-time PCR (RT-qPCR) analysis was then performed with the sequence detection system (ABI Prism 7000; Applied Biosystems/Invitrogen) with a SYBR Green 1-step kit (Invitrogen). Mouse gene expression was normalized to mouse GAPDH. Thermal cycling conditions were as follows: 10 s at 95°C, 40 cycles of 5 s at 95°C, an d 30 s at 60°C.
Subcellular fractionation.
Cell fractions were isolated using the Cell Fractionation Kit (Abcam, ab109719) according to the manufacturer’s instructions. The purity of fraction was analyzed by immunoblotting.
Mitochondrial DNA isolation and quantification.
Mitochondria were purified from mouse liver and cultured cells using the mitochondrial isolation kit (Abcam, ab110168). Freshly isolated mitochondria were used to isolate mtDNA by using the mitochondrial DNA isolation kit (Abcam) following manufacturer’s instruction. Total DNA in cytosolic fraction was isolated using the DNeasy Blood and Tissue Kit from Qiagen Inc, according to manufacturer’s manual (Nakahira et al., 2013). For quantitative real-time polymerase chain reaction (qPCR) assay, DNA solution was further diluted 10x with nuclease-free deionized distilled H2O. The primer sequences were as follows: human NADH dehydrogenase 1 gene (mtDNA): forward 5’-ATACCCATGGCCAACCTCCT-3’, reverse 5’-GGGCCTTTGCGTAGTTGTAT-3’; human b-globin (nuclear DNA): forward 5’-GTGCACCTGACTCCTGAGGAGA-3’, reverse 5’-CCTTGATACCAACCTGCCCAG-3’. Mouse NADH dehydrogenase 1 gene (mtDNA) forward primer: 5′-TATCTCAACCCTAGCAGAAA-3′; Reverse primer: 5′-TAACGCGAATGGGCCGGCTG - 3′. Then RT-qPCR analysis was performed with a sequence detection system (ABI Prism 7000; Applied Biosystems/Invitrogen) with a SYBR Green 1-step kit (Invitrogen). The thermal cycling conditions for both genes were pre-denatured at 95°C for 10min, followed by 40 cycles of 95°C for 15 s and 58°C for 1 min with data collection.
Determining cGAMP concentration.
Cells were lysed in cold 80% methanol, followed by adding 0.45 pmol of cyclic-di-AMP as internal control. Lung tissue (~300 mg) was homogenized in 1 ml of ice cold 80% methanol. After removing cell debris by centrifugation, samples were dried by flowing nitrogen, and subjected to solid phase extraction using HyperSep Aminopropyl columns (Thermo Fisher), which was pre-activated by using 80% methanol. After loading samples, the columns were washed twice with a solution of 2% (v/v) acetic acid/80% (v/v) methanol, and final elution was performed using a solution of 4% (v/v) ammonium hydroxide/80% (v/v) methanol. Samples were dried under flowing nitrogen and resuspended in 40 μl H2O for analysis by liquid chromatography and mass spectrometry (LC-MS) as described before (Almine et al., 2017).
Statistical analysis.
Each data point represents an in vitro biological replicate or an individual animal. Data are obtained from at least three independent experiments and are presented as mean ± standard deviation as indicated in the figure legends. Results were subjected to a two-tailed Student t test when comparing two groups (GraphPad Prism 8, GraphPad Software, San Diego, CA). Values of P < 0.05 were considered significant.
Supplementary Material
Table 1.
Key resource
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-GSDMD | Abcam | ab209845 |
| Rabbit monoclonal anti-pIRF3 (Ser396) | Cell signaling Technology | 29047S |
| Rabbit monoclonal anti-pIRF3 (Ser386) | Abcam | Ab76493 |
| mouse monoclonal anti-IRF3 | Santa Cruz | sc-33641 |
| Caspase-11 Antibody (17D9) | Novus | NB-120-10454 |
| Alexa Fluor® 488 Mouse anti-BrdU | BD Bioscience | 558599 |
| Mouse Anti-GAPDH antibody | Santa Cruz | Sc-47724 |
| Mouse anti-beta-actin antibody | Sigma | A5316 |
| Rabbit Anti-TBK1 | Cell signaling Tech | 3504T |
| Rabbit Anti-pTBK1 (Ser172) | Cell signaling Tech | 5483S |
| Rabbit monoclonal Anti-STING | Cell signaling Tech | 13647 |
| Rabbit monoclonal Anti-pSTING (Ser365) | Cell signaling Tech | 72971 |
| Phospho-YAP/TAZ Antibody Sampler Kit | Cell signaling Tech | 52420T |
| Rabbit monoclonal Anti-LATS1 | Cell signaling Tech | 3477S |
| Rabbit monoclonal Anti-MST 1 | Cell signaling Tech | 14946S |
| Rabbit anti-pLATS1 (Ser909) | Cell signaling Tech | 9157S |
| Rabbit monoclonal Anti-pMST1 (Thr183)/MST2 (Thr180) | Cell signaling Tech | 49332S |
| Mouse Anti-BrdU antibody | BD Biosciences | 347580 |
| Mouse Monoclonal Anti-β-Actin antibody | Sigma | A5441 |
| Rabbit Ployclonal anti-cGAS | mybiosource | MBS8291689 |
| Cyclin Antibody sample kit | Cell signaling Tech | 9869 |
| Mouse monoclonal anti-Flag | Sigma | F1804 |
| AF488 donkey anti-rat antibody | Invitrogen | A-21208 |
| AF594 donkey anti-rat antibody | Invitrogen | A-21209 |
| AF594 donkey anti-mouse antibody | Invitrogen | A-21203 |
| AF594 donkey anti-rabbit antibody | Invitrogen | A-21207 |
| AF488 donkey anti-mouse antibody | Invitrogen | A-11001 |
| Rabbit monoclonal Anti-TAZ | Cell signaling Tech | 83669S |
| Rat anti-CD31 | BD Bioscience | 550274 |
| Rat anti-CD31 | millpore sigma | CBL1337 |
| Mouse Anti-dsDNA Antibody | Millipore | MAB1293 |
| Rabbit anti-Tom20 Antibody | Santa Cruz | sc-11415 |
| Rabbit anti-IKKε antibody | Cell signaling Tech | 2905T |
| Rabit anti-pIKKε antibody (ser172) | Cell signaling Tech | 8766S |
| Mouse anti-Lamin B1 antibody | Santa Cruz | sc-56143 |
| Chemicals and Recombinant Proteins | ||
| 9-Oxo-10(9H)-acridineacetic acid (CMA) | Sigma | 17927 |
| 2′,3′-cGAMP sodium salt | Sigma | SML1229 |
| LPS | Sigma | L2630 |
| LPS | InvivoGen | tlrl-3pelps |
| BrdU | Sigma | B5002 −1G |
| Mitochondrial DNA Isolation Kit | abcam | Ab65321 |
| DNeasy Blood & Tissue Kits | QIAGEN | 69506 |
| Tetramethylrhodamine (TMRM) | ThermoFisher scientific | T668 |
| ProLong™ Gold Antifade Mountant | ThermoFisher scientific | P10144 |
| Z-VAD-FMK | Selleckchem | S7023 |
| Protease Inhibitor Cocktail | Sigma | P8340 |
| BrdU Cell Proliferation ELISA Kit | abcam | ab126556 |
| Evans Blue | Sigma | E2129 |
| iScript cDNA synthesis kit | BioRad | 1708897BUN |
| SYBR Green PCR Master Mix | ThermoFisher scientific | 4312704 |
| Phosphatase Inhibitor Cocktail | VWR | 80501-130 |
| Endothelial growth medium 2 (EBM-2) | Lonza | CC-3162 |
| Non-Radioactive Cytotoxicity Assay Kit | Promega | G1780 |
| Lipofectamine 2000 | ThermoFisher scientific | 11668019 |
| TRIZOL reagent | ThermoFisher scientific | 15596026 |
| BrdU Cell Proliferation ELISA Kit | Abcam | ab126556 |
| ECL prime immuno blotting reagent | Fisher Scientific | RPN2232 |
| RIPA Buffer | Sigma | R0278 |
| HyperSep Aminopropyl columns | ThermoFisher scientific | 60108-364 |
| Mitochondria Isolation Kit for cell | Abcam | ab110170 |
| Mitochondria Isolation Kit for Tissue | Abcam | ab110169 |
| pClneoMyc-LATS1 | Addgene | 66851 |
| pWZL Neo Myr Flag TBK1 | Addgene | #20648 |
| pcDNA4/HisMaxB-YAP1 | Addgene | #18978 |
| Flag-Gsdmd | Addgene | #80950 |
| Flag-Gsdmd-NT | Addgene | #80951 |
| Fetal Bovine Serum | Sigma | F2442 |
| Amaxa human endothelial nucleofector kit | Lonza | VPI-1001 |
| Human si-cGAS | Dharmacon | E-015607-00-0005 |
| sc-RNA negative control | Dharmacon | D-001810-10-20 |
| Human si-GSDMD | Dharmacon | E-016207-00-0005 |
| Human si-IKKε | Dharmacon | E-oo3723-00-0005 |
| LDH assay kit | Promega | G1780 |
| Tamoxifen | Sigma | T5648-1g |
| MPO assay kit | Cell Biologics | CB6937 |
| Adenosine 5′ -triphosphate disodium salt | Sigma | A2383 |
| Cell Fractionation Kit | Abcam | ab109719 |
| Kinase assay buffer | Cell signaling Tech | 9802 |
| Dynabeads Sheep Anti-Rat IgG | Invitrogen | 11035 |
| RBC Lysis buffer | Qiagen | 158904 |
| Triton-X 100 | Fisher Scientific | 151-100 |
| Mouse INF-beta duoset ELISA kit | R&D systems | DY8234-05 |
| Cells | ||
| Human lung microvascular ECs (hMVECs) | Lonza | Lonza™ CC-2527 |
| HEK293T | ATCC | Cat# CRL-3216 |
Table 2.
Primers for RT-qPCR.
| Primer | Sequence |
|---|---|
| Human NADH 1 forward | 5’-ATACCCATGGCCAACCTCCT-3’ |
| Human NADH 1 reverse | 5’-GGGCCTTTGCGTAGTTGTAT-3’ |
| Human b-globin forward | 5’-GTGCACCTGACTCCTGAGGAGA-3’ |
| Human b-globin reverse | 5’-CCTTGATACCAACCTGCCCAG-3’ |
| Mouse NADH1 forward | 5′-TATCTCAACCCTAGCAGAAA-3′ |
| Mouse NADH1 reverse | 5′-TAACGCGAATGGGCCGGCTG-3′ |
| Mouse IFIT1 forward | 5′-TCTAAACAGGGCCTTGCAG-3′ |
| Mouse IFIT1 reverse | 5′-GCAGAGCCCTTTTTGATAATGT-3′ |
| Mouse IFIT3 forward | 5′-T GAACT G CT CAGCCCACA-3′ |
| Mouse IFIT3 reverse | 5′-TCCCGGTTGACCTCACTC-3′ |
| Mouse CXCL10 forward | 5′-GCCGTCATTTTCTGCCTCA-3′ |
| Mouse CXCL10 reverse | 5′-CGTCCTTGCGAGAGGGATC-3′ |
| Mouse GAPDH forward | 5′-AGGTCGGTGTGAACGGATTTG-3′ |
| Mouse GAPDH reverse | 5′-GGGGTCGTTGATGGCAACA-3 |
Highlights.
Activated Gasdermin D forms mitochondrial pores in the endothelium
Mitochondrial pore formation releases mitochondrial DNA into the cytosol
Released mitochondrial DNA activates cGAS signaling and suppresses endothelial regeneration
Deletion of cGAS in an experimental model of polymicrobial sepsis reduces tissue injury
Acknowledgments
The studies were supported by NIH grants R01-HL45638 (to ABM), P01-HL60678 (to ABM and JR), T32-HL007829 (to ABM), R01-HL118068 (to JR and ABM), R01-HL90152 (to JR and ABM). Cdh5-CreERT2 mice were provided by Dr. Ralf Adams.
Abbreviations:
- LPS
popolysaccharide
- mtDNA
mitochondrial DNA
- DAMP
danger-associated molecular pattern
- ALI
acute lung injury
- EC
endothelial cells
- GSDMD
Gasdermin D
- cGAS
Cyclic GMP-AMP Synthase
- dsDNA
double-stranded DNA
- cGAMP
cyclic GMP–AMP
- ATP
adenosine 5´-triphosphate
- GTP
guanosine 5´-triphosphate
- nDNA
nuclear DNA
- hLMVECs
Human lung microvascular ECs
- mLMVECs
mouse lung microvascular ECs
- TBS
Tris-buffered saline
- RT-qPCR
Quantitative real-time PCR
- NT-GSDMD
GSDMD N-terminal fragment
- EBA
Evans blue-albumin
- ROS
reactive oxygen species
- LC-MS
liquid chromatography and mass spectrometry
- LPS (T)
intracellular LPS
- CMA
10-carboxymethyl-9-acridanone
- WT
wild-type
- LATS1/2
large tumor suppressor kinases ½
- LPS (E)
extracellular LPS
- MMP
mitochondrial membrane potential
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing financial interests.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, and Hornung V (2013). cGAS produces a 2’−5’-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, and Barber GN (2012). STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A 109, 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almine JF, O’Hare CA, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, et al. (2017). IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun 8, 14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P (2015). Immunology: Caspase target drives pyroptosis. Nature 526, 642–643. [DOI] [PubMed] [Google Scholar]
- Burdette DL, and Vance RE (2013). STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol 14, 19–26. [DOI] [PubMed] [Google Scholar]
- Cai X, Chiu YH, and Chen ZJ (2014). The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54, 289–296. [DOI] [PubMed] [Google Scholar]
- Castellon X, and Bogdanova V (2016). Chronic Inflammatory Diseases and Endothelial Dysfunction. Aging Dis 7, 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavlar T, Deimling T, Ablasser A, Hopfner KP, and Hornung V (2013). Species-specific detection of the antiviral small-molecule compound CMA by STING. EMBO J 32, 1440–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KT, Xiong S, Ye Z, Hong Z, Di A, Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al. (2017). Caspase-11-mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 127, 4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochi SE, Kempker JA, Annangi S, Kramer MR, and Martin GS (2016). Mortality Trends of Acute Respiratory Distress Syndrome in the United States from 1999 to 2013. Ann Am Thorac Soc 13, 1742–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, McDonald JG, Russell DW, and Cyster JG (2017). Oxysterol Restraint of Cholesterol Synthesis Prevents AIM2 Inflammasome Activation. Cell 171, 1057–1071 e1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng M, Tang Y, Li W, Wang X, Zhang R, Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al. (2018). The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 49, 740–753 e747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. [DOI] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, and Kagan JC (2018). The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O’Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124 e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Lopez-Soto A, Kumar S, and Kroemer G (2016). Caspases Connect Cell-Death Signaling to Organismal Homeostasis. Immunity 44, 221–231. [DOI] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, and Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112, E5699–5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, and Ablasser A (2018). Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. [DOI] [PubMed] [Google Scholar]
- Hansen CG, Moroishi T, and Guan KL (2015). YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol 25, 499–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausenloy DJ, and Yellon DM (2013). Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Bao Q, Zhang Y, Liu M, Lv H, Liu Y, Yao L, Li B, Zhang C, He S, et al. (2018). Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ Res 122, 591–605. [DOI] [PubMed] [Google Scholar]
- Hong AW, Meng Z, and Guan KL (2016). The Hippo pathway in intestinal regeneration and disease. Nat Rev Gastroenterol Hepatol 13, 324–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Dai Z, Cai L, Sun K, Cho J, Albertine KH, Malik AB, Schraufnagel DE, and Zhao YY (2016). Endothelial p110gammaPI3K Mediates Endothelial Regeneration and Vascular Repair After Inflammatory Vascular Injury. Circulation 133, 1093–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, et al. (2014). Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 124, 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, and Halder G (2014). The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov 13, 63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. [DOI] [PubMed] [Google Scholar]
- Komarova YA, Kruse K, Mehta D, and Malik AB (2017). Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ Res 120, 179–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaCanna R, Liccardo D, Zhang P, Tragesser L, Wang Y, Cao T, Chapman HA, Morrisey EE, Shen H, Koch WJ, et al. (2019). Yap/Taz regulate alveolar regeneration and resolution of lung inflammation. J Clin Invest 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, and Slutsky AS (2010). Sepsis and endothelial permeability. N Engl J Med 363, 689–691. [DOI] [PubMed] [Google Scholar]
- Lim S, Hermance N, Mudianto T, Mustaly HM, Mauricio IPM, Vittoria MA, Quinton RJ, Howell BW, Cornils H, Manning AL, and Ganem NJ (2019). Identification of the kinase STK25 as an upstream activator of LATS signaling. Nat Commun 10, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Zhang L, Marsboom G, Jambusaria A, Xiong S, Toth PT, Benevolenskaya EV, Rehman J, and Malik AB (2019). Sox17 is required for endothelial regeneration following inflammation-induced vascular injury. Nat Commun 10, 2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P, Feng Y, Lyons JD, Berger SB, Otani S, DeLaney A, Tharp GK, Maner-Smith K, Burd EM, Schaeffer M, et al. (2018). Caspase-8 Collaborates with Caspase-11 to Drive Tissue Damage and Execution of Endotoxic Shock. Immunity 49, 42–55 e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, and Guan KL (2016). Mechanisms of Hippo pathway regulation. Genes Dev 30, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahira K, Kyung SY, Rogers AJ, Gazourian L, Youn S, Massaro AF, Quintana C, Osorio JC, Wang Z, Zhao Y, et al. (2013). Circulating mitochondrial DNA in patients in the ICU as a marker of mortality: derivation and validation. PLoS Med 10, e1001577; discussion e1001577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nourshargh S, and Alon R (2014). Leukocyte migration into inflamed tissues. Immunity 41, 694–707. [DOI] [PubMed] [Google Scholar]
- Park C, Lee TJ, Bhang SH, Liu F, Nakamura R, Oladipupo SS, Pitha-Rowe I, Capoccia B, Choi HS, Kim TM, et al. (2016). Injury-Mediated Vascular Regeneration Requires Endothelial ER71/ETV2. Arterioscler Thromb Vasc Biol 36, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platnich JM, Chung H, Lau A, Sandall CF, Bondzi-Simpson A, Chen HM, Komada T, Trotman-Grant AC, Brandelli JR, Chun J, et al. (2018). Shiga Toxin/Lipopolysaccharide Activates Caspase-4 and Gasdermin D to Trigger Mitochondrial Reactive Oxygen Species Upstream of the NLRP3 Inflammasome. Cell Rep 25, 1525–1536 e1527. [DOI] [PubMed] [Google Scholar]
- Rathinam VAK, Zhao Y, and Shao F (2019). Innate immunity to intracellular LPS. Nat Immunol 20, 527–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, et al. (2018). Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159, 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo J, and Kim J (2018). Regulation of Hippo signaling by actin remodeling. BMB Rep 51, 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. [DOI] [PubMed] [Google Scholar]
- Sun S, and Irvine KD (2016). Cellular Organization and Cytoskeletal Regulation of the Hippo Signaling Network. Trends Cell Biol 26, 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, et al. (2017). Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat Commun 8, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xie F, Chu F, Zhang Z, Yang B, Dai T, Gao L, Wang L, Ling L, Jia J, et al. (2017). YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKvarepsilon-mediated phosphorylation. Nat Immunol 18, 733–743. [DOI] [PubMed] [Google Scholar]
- Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, and Lee VM (2008). Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem 283, 13302–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc Natl Acad Sci U S A 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.