

Abstract
Regulatory T cells (Tregs) are necessary to prevent autoimmune disease. As such, stable FoxP3 expression is required for the proper function of Tregs in the control of autoimmune disease. Different Treg subsets that utilize different mechanisms of suppression have been identified. The T-cell immunoglobulin immunoreceptor tyrosine-based inhibitory motif (TIGIT) is a relatively new Treg cell marker that has a suppressive function. We have previously identified the adenosine 2A receptor (A2Ar) as a requirement for the emergence of Tregs following resolution of autoimmune disease. Using a FoxP3-GFP-Cre reporter mouse, we identify FoxP3 and ‘exFoxP3’ cells, show FoxP3 and not exFoxP3 cells are suppressive. We further show FoxP3 cells express TIGIT, and are induced through A2Ar in healthy volunteers, but not patients with autoimmune disease. Furthermore, we show Tregs emerge in the target tissue at the onset of autoimmune disease in an A2Ar-dependent manner. In summary, we identify a novel subset of TIGIT+ Tregs that are induced through stimulation of the A2Ar.
Keywords: experimental autoimmune uveitis, Tregs, TIGIT, A2Ar
1. Introduction
Regulatory T cells (Tregs) are critical for controlling inflammation [1, 2]. Defects in the the transcription factor, forkhead box P3 (FoxP3), that drives Treg differentiation and function results in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) in humans, and a similar phenotype also occurs in scurfy mice [3, 4]. Because the continued suppressive function of Treg cells is linked with FoxP3 expression, autoimmunity can result if FoxP3 expression is unstable [1, 5]. In order to track FoxP3 and ‘exFoxP3’ cells, a reporter mouse that expresses GFP and Cre recombinase under the FoxP3 promoter (FoxP3-GFP-Cre) has been used [6-8]. FoxP3-GFP-Cre mice have been successfully used to identify exFoxP3 cells, and these cells have been studied in Type 1 diabetes, lymphoproliferative disease, cancer, and experimental autoimmune encephalomyelitis [7-9]. However, the emergence of these cells has not been tracked non-invasively in vivo over the course of autoimmune disease. Because the eye is naturally clear, non-invasive fluorescence detection during inflammation of the retina is possible. As such, we determined when during the course of autoimmune uveitis, in a murine model, Tregs emerged in the target tissue, and the effect of the adenosine 2A receptor (A2Ar) in this process. We also determined if exFoxP3 cells are suppressive, identified T-cell immunoglobulin immunoreceptor tyrosine-based inhibitory motif (TIGIT) expressing Tregs as a distinct Treg population, and showed that stimulation of A2Ar to induce these Tregs was impaired in uveitis patients.
Uveitis is inflammation of the eye and can cause permanent damage to the light-gathering structures of the eye [10, 11]. In Western countries uveitis is the third leading cause of blindness with an incidence between 25.6 – 122 cases per 100,000 a year, and a prevalence of 69 - 623 cases per 100,000 [12-14]. Transient or permanent vision loss occurs in approximately 17.6% of active uveitis patients, and it is estimated that glaucoma will occur in 12.5% of these patients [15]. As with other autoimmune diseases, autoimmune uveitis patients experience relapsing and remitting inflammation, and 33% of anterior uveitis cases become chronic [16]. A better understanding of the immunobiology that contributes to relapsing and remitting intraocular inflammation has the potential to develop novel more effective treatments for autoimmune uveitis.
Experimental autoimmune uveitis (EAU) is the most widely used mouse model of human autoimmune uveitis [17]. In C57BL/6J mice, resolution of EAU occurs at day 75-90 following immunization for EAU without further relapse [18-20], and at this point (post-EAU) regulatory immunity is found in the spleen [21]. This post-EAU regulatory immunity provides resistance to relapse, and suppresses EAU when transferred to recipient EAU mice [21-24]. A critical component of post-EAU regulatory immunity are FoxP3+PD-1+CD25+CD4+ ocular autoantigen-specific regulatory T cells (Tregs) that are dependent on expression of the A2Ar [23-25]. However, it has not been determined if exFoxP3 cells that emerge in the post-EAU spleen are suppressive, and if additional Treg subsets emerge in the post-EAU spleen.
TIGIT is a coinhibitory molecule associated with inhibition of autoimmune disease and is expressed on NK cells, Treg cells, and T effector cells [26, 27]. TIGIT binds CD155 on dendritic cells to prevent IL-12 production and induce IL-10 production [28]. Tregs have also been shown to suppress Th1 and Th17 responses following TIGIT ligation [29]. It has not been shown if post-EAU Tregs express TIGIT and if TIGIT expression is influenced by A2Ar.
In this report we demonstrate that FoxP3 Tregs cells emerge in the eye at the onset of EAU in an A2Ar-dependent manner. The post-EAU Treg cells express FoxP3 and the exFoxP3 cells are not functionally suppressive. Further characterization of the post-EAU Treg cells identified a TIGIT+ Treg subset that is distinct from the previously characterized PD-1+ subset, showing there are multiple subsets. We additionally show that the emergence of post-EAU TIGIT+ Tregs are reduced in A2Ar-deficient mice and the induction of TIGIT+ Tregs through A2Ar stimulation is reduced in uveitis patients.
2. Materials and Methods
2.1. Mice
All mouse procedures described in this study were approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee (OUHSC IACUC) and all mouse study methods were carried out in accordance with the relevant guidelines approved by the OUHSC IACUC. C57BL/6J mice and Adora2a(−/−) (A2Ar(−/−)) mice were purchased from Jackson Laboratories.
2.2. Human Studies
Institutional Review Board Approval was obtained from New England IRB, and the University of Oklahoma Human Research Participant Protection IRB to collect and analyze PBMC from human uveitis patients. All experiments involving human subjects were carried out in accordance with the relevant guidelines approved by the appropriate IRB, and adhered to the Declaration of Helsinki. Prior to the collection of patient samples informed consent was obtained from all human subjects. Patient samples were de-identified and limited patient information was made available to the research staff.
2.3. Experimental Autoimmune Uveoretinitis (EAU)
Mice were immunized for EAU as previously described [24]. Briefly, an emulsion of complete Freund’s adjuvant (CFA) with 5 mg/mL desiccatedM. tuberculosis (Difco Laboratories, Detroit, MI) and 2 mg/ml interphotoreceptor retinoid binding protein (peptides 1-20) (IRBP) (Genscript, Piscataway, NJ) was used to immunize mice for EAU. A volume of 100 μL of the emulsion was injected subcutaneously at two sites in the lower back followed by an intraperitoneal injection of 0.3 μg pertussis toxin. The severity of retinal inflammation during the course of EAU was evaluated every 3-4 days by fundus examination using a slit lamp microscope. Before examining the retina, the iris was dilated with 1% tropicamide, the cornea was numbed with 0.5% proparacaine, and the cornea was flattened with a glass coverslip in order to examine the retina. The clinical signs of observable infiltration and vasculitis in the retina were scored on a 5-point scale as previously described [30]. Both eyes were scored and the higher score was used to represent that mouse for that day, the average score for the group of mice was then calculated.
2.4. In Vitro Stimulation
Spleens were collected into 5% FBS in RPMI supplemented with 10 μg/ml Gentamycin (Sigma), 10 mM HEPES, 1 mM Sodium Pyruvate (BioWhittaker), Nonessential Amino Acids 0.2% (BioWhittaker) and made into a single cell suspension that was depleted of red blood cells using RBC lysis buffer (Sigma, St Louis, MO). The spleen cells were resuspended in serum free media (SFM) and IRBP was added at 50 μg/mL for 48 hours at 37°C and 5% CO2 to reactivate antigen specific T cells. SFM consisted of RPMI-1640 with 1% ITS+1 solution (Sigma) and 0.1% BSA (Sigma). Following the reactivation supernatants were collected and analyzed and/or cells were collected for adoptive transfer into recipient mice.
2.5. In Vivo Imaging
Mouse retinas were imaged using a Micron IV microscope imaging system (Phoenix Technologies, Milpitas, CA) complete with green fluorescence and red fluorescence filters. Mice were anesthetized and the eye was dilated with 1% tropicamide that also served to lubricate the surface of the eye.
2.6. Cytokine Analysis
Cell culture supernatants were assayed using the human Th1/Th2/Th17/Th22/Treg 18-multiplex procartaplex kit, (Invitrogen, Vienna, Austria) was used to assay the PBMC culture supernatants. The Multiplex plate was analyzed with Bio-Rad plate reader (Bioplex system, Hercules, CA). The assay was performed according to manufacturer’s instructions. The TGF-β concentration was measured with the standard Mv1Lu bioassay [31].
2.7. Flow Cytometry
Mouse spleen cells were washed with PBS containing 1% BSA (staining buffer), blocked with mouse IgG in staining buffer, then stained with conjugated antibodies. Antibodies used were anti-PD-1 (clone 29F.1A12, Biolegend, San Diego, CA), anti-TIGIT (clone 1G9, Biolegend), anti-GITR (clone yGITR765, Biolegend), anti-CD25 (clone PC61, Biolegend), anti-PD-L1 (clone 10F.9G2, Biolegend), anti-CTLA4 (clone UC10-4B9, Biolegend), anti-2B4 (clone M2B4(B6)458.1, Biolegend), anti-VISTA (clone MIH63, Biolegend), anti-LAG3 (clone C9B7W, Biolegend), anti-TIM3 (clone RMT3-23, Biolegend), and anti-CD4 (clone RM4-5, Biolegend).
Human PBMCs were stained with anti-CD4 (clone OKT4, Biolegend), anti-CD25 (clone BC96, Biolegend), anti-FoxP3 (clone 236A/E7, Biolegend), anti-PD-1 (clone EH12.2H7, Biolegend), anti-TIGIT (clone A15153G, Biolegend). Prior to anti-FoxP3 staining, the cells were fixed for 18 hours and permeabilized.
2.8. Intracellular cytokine staining
Cells were stimulated phorbol 12-myristate-13-acetate (PMA) (81 nM) and ionomycin (1.3 μM) (Biolegend) for one hour at 37°C and 5% CO2 then monensin (2 μM) (Biolegend) was added without washing the cells for four additional hours. Cells were then washed with staining buffer, stained for surface receptors, then fixed for 20 minutes, and stained for intracellular cytokines in permeabilization buffer. The intracellular antibodies that were used were anti-TNF-α (clone MP6-XT22, Biolegend), anti-IL-6 (clone MP5-20F3, Biolegend), anti-IL-17A (clone TC11-18H10.1, Biolegend), anti-IFN-γ (clone XMG1.2, Biolegend), anti-IL-2 (clone JES6-5H4, Biolegend), anti-IL-4 (clone 11B11, Biolegend), and anti-IL-10 (clone JES5-16E3, Biolegend).
Stained cells were analyzed in the Oklahoma Medical Research Facility (OMRF) Flow Cytometry Core Facility on a BD LSRII or Celesta (BD Biosciences) and data was analyzed using FlowJo Software (Tree Star, Inc., Ashland, OR).
2.9. Statistics
Statistical significance between EAU scores was determined using nonparametric Mann-Whitney U test between groups of mice. Two-way ANOVA was also used to assess significant changes in the tempo of disease between the groups of treated EAU mice. Cytokine concentrations were statistically analyzed by one-way ANOVA with post-test Bonferroni comparison analysis. Statistical significance was determined when P ≤ 0.05 (two-sided). Healthy controls (n = 19) were compared to patients with uveitis (n = 15-17) across a range of selected biomarkers. Each comparison was conducted via a Mann-Whitney U test using a per-test alpha level of 0.05 (overall alpha correction was not applied at this exploratory stage). Nonparametric methods were used because the data were largely right-skewed for each biomarker. All statistical tests used to analyze human samples were conducted in R v3.5.1 and statistical analysis for mouse experiments were analyzed with Graphpad Prism software.
3. Results
3.1. Post-EAU FoxP3 and ExFoxP3 T Cells
We and others have identified ocular autoantigen-specific post-EAU Treg cells in the spleen that provide resistance to EAU [22-24, 32-34]. As such, we asked if both FoxP3 and exFoxP3 T cells emerge in the spleen at the resolution of EAU and what is the suppressive capacity of these populations. In order to address this question, we utilized FoxP3-GFP-Cre mice, previously described [8]. These mice were crossed with Rosa26-loxP-STOP-loxP-TdTomato (R26-Tom) reporter mice that constitutively express TdTomato after the STOP codon is removed by Cre recombinase (Fig 1A) [8]. FoxP3-GFP-Cre; R26-Tom mice were immunized for EAU and the spleen was collected at resolution of disease. Flow cytometry analysis of post-EAU T cells reactivated with IRBPp (1-20) from the spleen showed that both FoxP3 and exFoxP3 cells are present (Fig 1B). In order to further understand a functional role of these populations we asked what cytokines they may be producing. Intracellular staining of these populations revealed a significantly greater percentage of TNF-α, IL-6, IFN-γ, IL-4, IL-2 expressing cells in the exFoxP3 population compared to the FoxP3 population (Fig 1C). While the percentage of IL-17+ and IL-10+ cells was not significantly different between exFoxP3 and FoxP3 cells (Fig 1D), the number of IL-10+ cells in FoxP3 and exFoxP3 was significantly greater than FoxP3− cells (Fig 1D). We next asked which population is functionally suppressive. The sorted the FoxP3 and exFoxP3 cell populations were transferred each into recipient mice immunized for EAU. EAU mice that received FoxP3 cells had significantly suppressed EAU scores and reduced severity compared to EAU mice that did not receive a cell transfer (Fig 1E). In contrast, exFoxP3 cells did not suppress EAU in recipient mice compared to EAU mice that did not receive a cell transfer (Fig 1F) and EAU scores were significantly greater compared to EAU mice that received post-EAU FoxP3 T cells (Fig 1G). These observations demonstrated post-EAU Tregs express FoxP3, whereas the exFoxP3 cells express proinflammatory cytokines and were not functionally suppressive.
Figure 1. Post-EAU exFoxP3 cells fail to suppress uveitis.
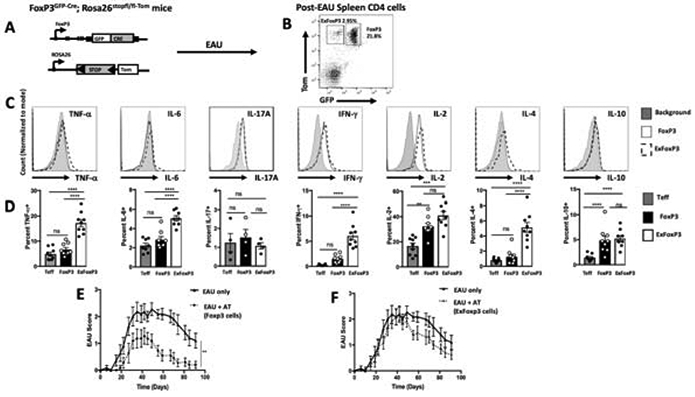
Schematic representation of the lineage tracing reporter mice, FoxP3GFP-Cre; Rosa26stopfl/fl-tdTomato (A). Post-EAU splenocytes from FoxP3GFP-Cre; Rosa26stopfl/fl-tdTomato mice were reactivated with IRBP (1-20) in vitro for 48 hours then stained with anti-CD4 antibody and analyzed by flow cytometry for CD4, GFP, and Tomato. Representative flow plot of 6 independent experiments, with 2-5 mice per experiment (n=16) is shown GFP−Tom+ (exFoxP3) cells, GFP+Tom+ (FoxP3 cells) (B). Splenocytes were activated with PMA/Ionomycin and cytokine secretion was blocked with monensin for 4-5 hrs. Cells were fixed and stained for TNF-α, IL-6, IL-17A, IFN-γ, IL-2, IL-4, and IL-10 and analyzed by flow cytometry. Representative histograms of the intracellular cytokines of cells gated on exFoxP3 (dashed line), FoxP3+ (black line), or background (light gray) are shown (C). Summary data show the mean ± SEM of frequencies with T effector cells as CD4+ (GFP−Tom−) of cytokines from 2 independent experiments with 2-5 mice per experiment (D). Post-EAU splenocytes were sorted into FoxP3 and exFoxP3 groups following reactivation with IRBPp (1-20) and transferred to recipient mice immunized for EAU. Clinical EAU scores obtained from fundus exams of EAU mice are shown. Average scores per group over time for non-recipient control mice (EAU only, n=11) are compared with scores of FoxP3-recipient mice (n=11) (E) or exFoxP3-recipient (n=13) mice (F). The graphs show the mean ± SEM scores on the indicated day per group. Results are from 3 independent experiments with 3-5 mice in each group per experiment, where each symbol on the bar graph represents one mouse. Cytokine results were analyzed with one-way ANOVA **p<0.01, ***p<0.001, ****p<0.0001, ns not significant. EAU results were analyzed with two-way ANOVA with Bonferroni post-test, **p<0.005 n.s. not significant.
3.2. Identification of Multiple Treg Cell Subsets
We next asked if there are multiple post-EAU Treg cell subsets in the spleen. Expression of PD-1, TIGIT, GITR, VISTA, CD25, PD-L1, CTLA4, CD39, CD244 (2B4), LAG3, and Tim3, in the FoxP3 Treg population was compared with the exFoxP3 population following reactivation with IRBPp (1-20). We observed significantly more PD-1, PD-L1, 2B4, CD25, TIGIT, GITR, and LAG3 positive cells in the FoxP3 population compared with the exFoxP3 population (Fig 2A, B). Since PD-1, 2B4, TIGIT, GITR, and LAG3 expression on FoxP3 Tregs was significantly greater than on exFoxP3 T cells we asked if expression on Tregs in other tissues was similar. The percentage of PD-1+ Tregs greater in the eye compared with the spleen and cervical lymph nodes (Fig 3A, 3B). Whereas, TIGIT+ Tregs were significantly greater in the eye compared to the cervical lymph nodes and trended greater compared to the spleen (Fig 3C, 3D). In contrast, GITR expressing Tregs was lower in the eye compared to the spleen and cervical lymph nodes (Fig 3E, 3F). The percentage of 2B4 positive cells was significantly greater in the eye compared to the cervical lymph nodes and spleen (Fig 3G, 3H), and LAG3 positive cells was greater in the eye compared to the cervical lymph nodes but not the spleen (Fig 3I, 3J). These observations suggested that multiple populations of Treg subsets emerged in the spleen, cervical lymph nodes, and eye at resolution of EAU.
Figure 2. Post-EAU exFoxP3 cells lack a Treg phenotype.
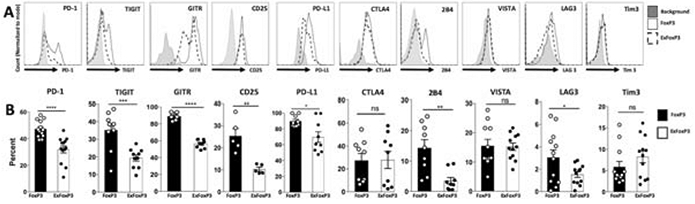
Post-EAU splenocytes from lineage tracing reporter mice were reactivated with IRBPp (1-20) in vitro for 48 hrs. The reactivated cells were stained and analyzed by flow cytometry for various Treg markers. Representative flow plots of PD-1, TIGIT, GITR, CD25, PD-L1, 2B4, VISTA, LAG3, and Tim3 expression on CD4+ GFP+Tom+ (FoxP3 cells, solid line) or CD4+GFP−Tom+ (exFoxP3 cells, dashed line), or background (gray) are shown (A). The mean ± SEM of the frequencies of the Treg markers from 3-6 independent experiments, with 2-5 mice per experiment are shown, where each symbol on the bar graph represents one mouse (B). Summary data were analyzed with unpaired t-test or Mann-Whitney test, * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s. not significant.
Figure 3. TIGIT, PD-1, LAG 3, and 2B4 are expressed on ocular post-EAU Treg cells.
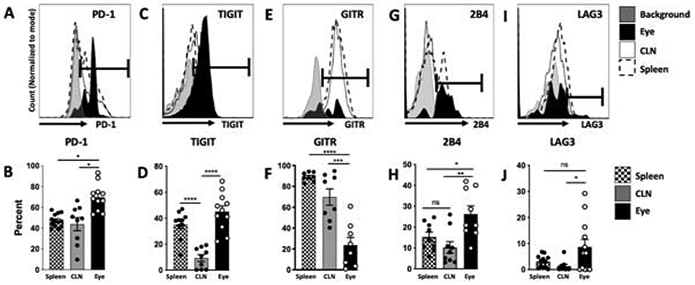
Post-EAU lymphocytes from the spleen, cervical lymph node (CLN), and the eyes were reactivated with IRBPp (1-20) for 48 hrs and analyzed with flow cytometry. The expression of PD-1, TIGIT, GITR, 2B4, and LAG3 is shown in representative histograms of CD4+GFP+Tom+ gated cells from the eye, CLN or spleen (A). Bar graphs show the mean ± SEM of the percentages of the indicated marker from the eye, CLN or spleen for all experiments (B). Data shown is from 3-4 independent experiments with 2-5 mice per experiment, where each symbol on the bar graph represents one mouse. Summary data were analyzed by one-way ANOVA, with Tukey multiple-comparison test, * p<0.05, **p<0.01, ***p<0.001, ****p<0.0001, n.s. not significant.
Of the Treg markers that were significantly elevated in the eye compared to other tissues TIGIT is the only marker that has a role in suppression of other immune cells [26, 28, 29]. Therefore, we next asked if TIGIT+ Tregs are a distinct subset from the previously characterized PD-1+ post-EAU Tregs [33]. Analysis of PD-1 and TIGIT expression on CD4+FoxP3+ Tregs revealed four populations (Fig 4A, 4B) that were PD-1+TIGIT+ (DP), PD-1+TIGIT− (PD-1 SP), PD-1−TIGIT+ (TIGIT SP), and PD-1−TIGIT− (DN). In order to further define each of these populations we asked the expression of each of the Treg markers we stained for above on each of these subsets. We utilized t-distributed Stochastic Neighbor Embedding (t-SNE) to cluster and visualize the Treg markers for each subset. The majority of the populations expressed GITR and PD-L1, but a significant enhancement of CTLA4 cells was found in the PD-1 SP group not found in the TIGIT SP group (Fig 4C). In contrast, the TIGIT SP group was relatively homogeneous with a slight but not significant enhancement of 2B4 expressing cells, the DP group was composed of a mix of populations, and the DN group had significantly fewer PD-L1 expressing cells (Fig 4D). These observations indicated that the PD-1 SP and TIGIT SP Treg groups were two distinct populations that differentially express CTLA4.
Figure 4. TIGIT and PD-1 expression define distinct post-EAU Treg subsets.
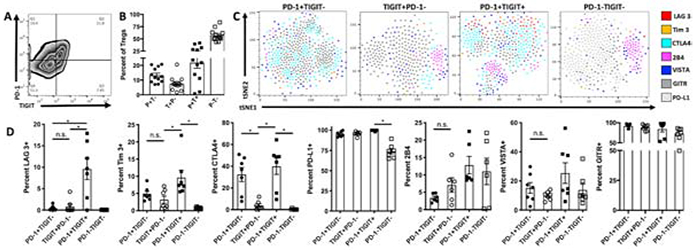
Splenic post-EAU Treg cells were reactivated with IRBPp (1-20), stained for CD4, PD-1, TIGIT, GITR, CD25, PD-L1, 2B4, VISTA, LAG3, and Tim3, and analyzed by flow cytometry. Subsets based on TIGIT and PD-1 expression were analyzed for differences in expression of additional Treg markers. Representative flow plot of TIGIT and PD-1 expression on CD4+FoxP3+ gated cells from the eye is shown (A). The mean ± SEM of the percentages for all experiments of the TIGIT and PD-1 post-EAU Treg subsets are shown (B). Each of the PD-1 and TIGIT Treg subsets were clustered using tSNE (FlowJo) based on the distribution and expression of LAG3, Tim3, CTLA4, 2B4, VISTA, GITR, and PD-L1 markers with each Treg marker color-coded to show the cells that express the indicated marker within the tSNE plot (C). The mean ± SEM of the percentage of LAG3, Tim3, CTLA4, 2B4, VISTA, GITR, and PD-L1 are shown for each PD-1 and TIGIT defined subsets for all experiments (D). The results are representative plots or summary data of 3-4 experiments with 2-4 mice per experiment, where each symbol on the bar graph represents one mouse. Summary data were analyzed by one-way ANOVA, with Tukey multiple-comparison test, * p<0.05, n.s. not significant.
3.3. TIGIT+ Tregs suppress EAU, but to a lesser extent compared to PD-1+ Tregs
We next asked if each the four post-EAU subsets defined by PD-1 and TIGIT expression are functionally suppressive. Post-EAU splenocytes were collected and reactivated in vitro as we have done before [32-34], sorted on CD4 and CD25, then into four subsets based on PD-1 and TIGIT expression, and transferred to recipient mice immunized for EAU. Mice that received PD-1+TIGIT− cells showed a significant suppression of disease (Fig 5A). Mice that received TIGIT+PD-1− cells showed an accelerated resolution (Fig 5B) and mice that received TIGIT+PD-1+ cells also showed a significant suppression of disease with a delayed onset (Fig 5C). In contrast, mice that received TIGIT−PD-1− cells showed no change in disease. These observations demonstrate the TIGIT+ and TIGIT+PD-1+ Tregs are suppressive, but not to the same extent as PD-1+ Treg cells.
Figure 5. TIGIT and PD-1 expression on post-EAU Treg cells defines functional Treg subsets.
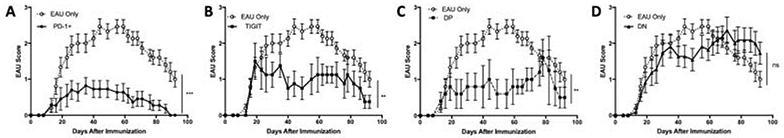
Splenocytes from post-EAU mice were reactivated with IRBP (1-20) in vitro for 48hrs. Total CD4+CD25+ Tregs were FACS sorted based on PD-1 and/or TIGIT expression. Different PD-1 or TIGIT Treg subsets were transferred into recipient EAU mice. The graphs show the clinical EAU scores obtained from fundus exams of recipient EAU mice that received the following post-EAU T cell subsets, PD-1+TIGIT− (n=11) (A), PD-1−TIGIT+ (n=8) (B), PD-1+TIGIT+ (n=5) (C), and PD-1−TIGIT− (n=11) (D). Average EAU scores of the non-recipient control mice (dashed line, EAU only, n=15) and recipient mice (solid line) over the course of the disease are shown. The data illustrate the mean ± SEM scores on the indicated day per group. Data are from 4 independent experiments with 1-4 mice in each group per experiment. Data were analyzed with 2-way ANOVA with Bonferroni post-test, **p<0.005 ***p<0.001 ns not significant.
3.4. A2Ar is necessary for the emergence of Tregs in the eye at the onset of EAU
The FoxP3-GFP reporter mice allow for in vivo real-time tracking of Tregs as disease occurs. Study of ocular inflammation allows us to image and track the GFP expressing Tregs through a minimally invasive procedure that only requires anesthetizing the mouse. We report here for the first time to our knowledge that Tregs migrate into the eye at the onset of EAU, and the Tregs that emerge in the eye persist until resolution (Fig 6A-6D) and reside in the eye for at least 140 days after immunization of EAU (Fig 6D). Because we have previously demonstrated the importance of A2Ar for the induction of post-EAU Treg cells, we asked if the Tregs migrating into the eye during EAU require A2Ar. A2Ar(−/−); FoxP3-GFP mice showed a delay in the emergence of Tregs into the eye until resolution of disease (Fig 6E-6H). The role of the post-EAU Tregs found in the eye and the role of A2Ar-deficient Tregs remains to be examined. However, these results showed that an A2Ar-dependent population of Tregs migrated into the eye at the onset of EAU, and an A2Ar-independent population of Tregs migrated into the eye at resolution of EAU.
Figure 6. A2Ar is necessary for the emergence of Tregs in the eyes during EAU.
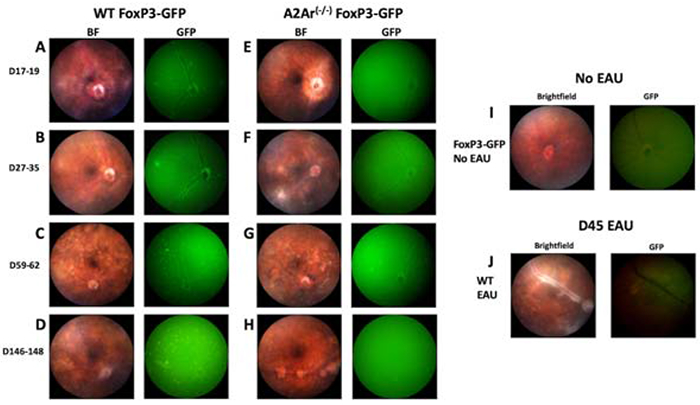
WT FoxP3-GFP and A2Ar(−/−) FoxP3-GFP mice were immunized for EAU and retinal images were obtained with the Micron IV microscope during the disease course. Fundoscopic images were taken with the brightfield (BF) or the green fluorescence (GFP) illumination at the specified days post-immunization. Representative retinal images of EAU WT FoxP3-GFP (n=25) and A2Ar(−/−) FoxP3-GFP (n=9) at disease onset (A, E), disease peak (B, F), chronic phase (C, G), and post-EAU (D, H). The images are representative of 3-10 experiments with 2-3 mice per experiment. Retinal images of unimmunized (naïve) WTFoxP3-GFP under the BF and GFP channels are displayed (I). Fundoscopic images of non-GFP WT mice at disease peak are shown (J).
3.5. Stimulation of A2Ar is required for the emergence of TIGIT+ Tregs in mice and is reduced in uveitis patients
We previously demonstrated that FoxP3 expression is decreased in A2Ar(−/−) post-EAU T cells [33]. The spleens of post-EAU wild-type and A2Ar-deficient mice were collected and restimulated in vitro and stained for CD4, FoxP3, and TIGIT. Here, tSNE analysis based on CD25, PD-1, and TIGIT expression shows the post-EAU T cell populations are different, and the heatmap overlay for FoxP3 (Fig 7A) and MFI (Fig 7B) confirms our previous observation that FoxP3 expression is lower in A2Ar deficient post-EAU mice. Furthermore, within the CD4+FoxP3+ population we observed significantly fewer TIGIT+ and PD-1+ Tregs in post-EAU A2Ar(−/−) mice compared with post-EAU WT mice (Fig 7C, 7D). When we examined each of the four populations we found no significant difference between the percentage of TIGIT+PD-1+ in post-EAU WT mice compared with post-EAU A2Ar(−/−) mice, but there was a significantly greater percentage of double-negative cells in the post-EAU A2Ar(−/−) mice (Fig 7F). The tSNE distribution of each of the four populations showed different patterns between the post-EAU WT and post-EAU A2Ar(−/−) mice (Fig 7G).
Figure 7. A2AR expression is required for the emergence of TIGIT+ Treg cells in EAU model.
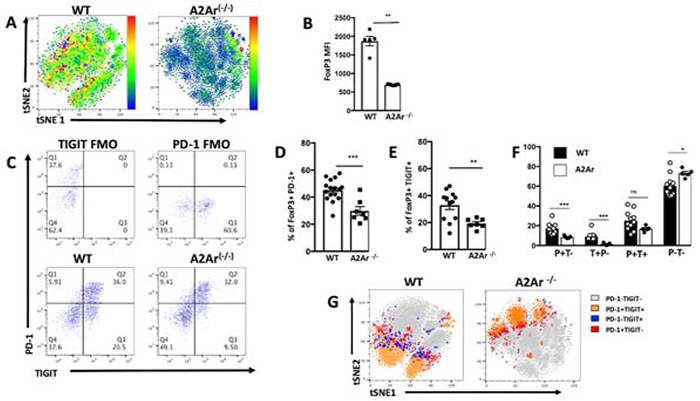
Post-EAU splenocytes from WT FoxP3-GFP or A2Ar(−/−) FoxP3-GFP mice were reactivated with IRBPp (1-20) for 48 hours and analyzed by flow cytometry. The tSNE plots of post-EAU CD4+GFP+ Treg cells from WTFoxP3-GFP or A2Ar(−/−) FoxP3-GFP mice were clustered based on the distribution and expression of FoxP3 protein are displayed in the representative Heatmaps (A). The mean ± SEM of FoxP3 expression (MFI) is shown in the summary data (B). Representative flow plots of TIGIT and PD-1 expression on post-EAU FoxP3+gated cells of WT and A2Ar(−/−) mice are shown with fluorescence minus one controls (FMO) (C). The mean ± SEM of the percentages of FoxP3+PD-1+ (D) or FoxP3+TIGIT+ (E) of WT or A2Ar(−/−) post-EAU CD4+ cells are shown. The mean ± SEM of the percentages of the TIGIT and PD-1 WT or A2Ar(−/−) post-EAU Treg subsets are shown in the bar graphs of the summary data (F). The gated CD4+GFP+ cells of WT FoxP3-GFP or A2Ar(−/−) FoxP3-GFP post-EAU splenocytes were clustered using tSNE based on the distribution and expression of PD-1 and TIGIT subsets are shown in the color-coded tSNE plots (G). Shown are 2-4 experiments, with 2-5 mice per experiment. Data were analyzed with one-way ANOVA with Tukey multiple-comparison test, * p<0.05, **p<0.01, ***p<0.001, ns not significant.
We have previously published that the induction of PD-1+ Tregs through a-MSH stimulation is significantly reduced in uveitis patients compared to healthy controls but not when stimulated through A2Ar with the A2Ar specific agonist, CGS21680 (CGS) [33], so we asked if the same mechanism induces TIGIT+ Tregs in uveitis patients. Our results showed that α-MSH stimulation induced a similar number of TIGIT+ Tregs in uveitis patients compared to healthy controls (Fig 8A, 8B). In contrast, we observed the opposite for TIGIT+ Tregs, with a significantly lower number of TIGIT+ Tregs induced by A2Ar stimulation in uveitis patients compared to healthy controls (Fig 8A, 8C). These observations demonstrate that PD-1+ Tregs and TIGIT+ Tregs are induced through different parts of the melanocortin-adenosinergic pathway in humans that may contribute to autoimmune uveitis.
Figure 8. TIGIT+ Treg cells are not induced by A2Ar stimulation fails in uveitis patients.
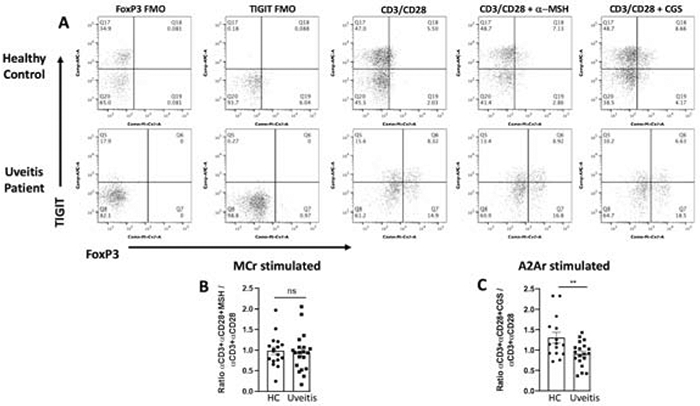
PBMCs from uveitis patients (n = 19) or healthy donors (n = 15-17) were activated with α-CD3/α-CD28 antibodies and treated with or without A2Ar agonist (CGS21680) or MC5r agonist (α-MSH) for 48 hrs. The cells were stained and analyzed by flow cytometry. Representative flow cytometry plots gated on CD4+CD25+ cells show TIGIT and FoxP3 expression, fluorescence minus one (FMO) controls are shown for healthy and uveitis patient (A). Summary data show the ratio of treated over untreated for healthy donor (HC) and patients (uveitis) following α-MSH treatment (B) or CGS21680 treatment (C). The results show the mean ± SEM of all subjects included in this study. Data were analyzed with unpaired student t-test **p<0.01, ns not significant.
4. Discussion
In this report, we present for the first time, the identification of TIGIT+ post-EAU Tregs, in vivo imaging of FoxP3+ Tregs entering the eye during EAU in A2Ar sufficient and deficient mice, and we demonstrate a clinical connection between A2Ar stimulation and induction of TIGIT+ Tregs. We found that post-EAU Tregs stably express FoxP3, and comparison of post-EAU FoxP3 Tregs with post-EAU exFoxP3 T cells found CD25, PD-1, PD-L1, GITR, 2B4 (CD244), LAG3, and TIGIT expression to be significantly greater in FoxP3 Tregs than exFoxP3 T cells. Co-staining for PD-1 and TIGIT revealed four populations, a PD-1 single positive, TIGIT single positive, a co-expressing PD-1 and TIGIT double-positive, and a double negative population. Adoptive transfer of each of these subsets into EAU recipient mice revealed that three of the populations had a different suppressive capacity, suggesting that at least three different post-EAU Treg subsets emerge in the spleen. We further demonstrate that FoxP3 expressing Tregs migrate into the eye during the onset of EAU, but the emergence of these Tregs is delayed in A2Ar(−/−) mice. Post-EAU mice deficient in A2Ar had significantly fewer TIGIT+FoxP3+CD25+CD4+ Tregs in the spleen compared with post-EAU wild-type mice. Stimulation of A2Ar also induced TIGIT+FoxP3+ Tregs in human PBMCs, but the number of TIGIT+FoxP3+ Tregs was significantly reduced in PBMCs from uveitis patients. These observations show that multiple Treg populations emerge during resolution of uveitis, the emergence of Tregs in the eye during uveitis is influenced by A2Ar expression, and a defect in the induction of TIGIT+ Tregs through A2Ar may contribute to chronic uveitis.
The dysfunction of Treg cells in the pathogenesis of autoimmune disease is well described [1, 2] and advancements in the field have identified numerous markers that define Tregs [7, 35-37]. Based on the differential expression of the numerous Treg markers, multiple different Treg subsets that correspond to different stages of disease and tissue sites have been defined [7, 35-37]. Here, based on TIGIT and PD-1 expression, we identify four subsets of FoxP3+ Tregs that are phenotypically and functionally different. Furthermore, the induction of these subsets seems to require different stimulation, either through melanocortin receptors or A2Ar. It has been demonstrated that Treg subsets with different developmental or tissue origins and phenotypes have different suppressive capacities [37, 38]. As such, these subsets may correspond to different phases of disease that require different functions.
Our analysis of Treg markers within each of the four TIGIT and PD-1 subpopulations provides potential explanations for the different suppressive capacities. The PD-1 single positive subset was the most suppressive and had a significantly greater percentage of CTLA+ cells compared to TIGIT single positive subset. In contrast, the TIGIT−PD-1− population that had no suppressive capacity had a significantly lower percentage of PD-L1+ cells compared to the three other subsets. However, because the TIGIT+PD-1+ population had a significantly greater percentage of TIM3+, LAG3+, and CTLA+ cells and had a similar suppressive capacity compared to the TIGIT single positive group suggests that suppression may be mediated by more than surface receptors. As such, additional investigation is necessary to determine a mechanistic explanation for the suppressive capacity of each of these subsets.
The immunosuppressive ocular microenvironment is maintained through multiple mechanisms. The eye is sequestered from a conventional immune response, and if this barrier is broken there are soluble factors and membrane bound factors to suppress inflammatory cells that may migrate into the eye [10]. If uveitis does occur, there are mechanisms such as Tregs [39] that are involved in resolution of inflammation, which also establishes long-term ocular antigen-specific regulatory immunity. Since there are already multiple mechanisms to suppress ocular inflammation, another explanation for the different Treg subsets that we identified is they may represent redundant mechanisms to re-establish and maintain the ocular immunosuppressive microenvironment.
Another important observation we report here is the role of A2Ar in the emergence of Tregs in the eye at the onset of EAU and in the induction of TIGIT+ Tregs. There are three possible explanations for the decrease in TIGIT+FoxP3+ Tregs in post-EAU mice. First, it has been shown that A2Ar stimulation facilitates permeability of the blood brain barrier [40], so it is possible that an A2Ar deficiency prevents Treg migration into the eye until a sufficient amount of retinal inflammation occurs. Second, it has been demonstrated that A2Ar-deficient mice still recover from EAU but have a significant reduction in post-EAU Tregs [23, 24], so the reduced number of TIGIT+ Tregs could be due to an overall reduction in Tregs. The third possibility is that A2Ar is necessary for the migration of Tregs into the eye at the onset of EAU that become the post-EAU Treg population found in the spleen, which is supported with our observation that the TIGIT+ Tregs are significantly decreased in the eye compared PD-1+ Tregs in the eye at the resolution of disease. However, these three explanations are not mutually exclusive of one another and further investigation is necessary to determine if one or more explanations are the case or not.
We also provide clinical relevance to our animal findings by demonstrating that stimulation of A2Ar induces TIGIT+FoxP3+ Tregs in PBMCs from healthy human volunteers. In contrast, the number of TIGIT+FoxP3+ Tregs induced through A2Ar stimulation was significantly reduced in uveitis patients. While we did not observe a difference in the number of TIGIT+ or PD-1+ Tregs when stimulated through melanocortin receptors, we did previously report a decrease in the number of PD-1+ Tregs in uveitis patients when stimulated through the melanocortin receptors [25, 33]. This difference could be due to the method of TCR stimulation, here we used α-CD3 and α-CD28 nonspecific stimulation, whereas we previously used tetanus toxoid as a means of antigen specific stimulation. Additional investigation and a larger sample size is necessary to answer this question, but our observations and our previous observations suggest that the different Treg subsets induced in humans respond to different signals of induction.
5. Conclusions
In summary, these observations identify a novel post-EAU TIGIT+ Treg subset that is distinct phenotypically and functionally and is dependent on the expression of A2Ar. Importantly, the stimulation of A2Ar on PBMCs was sufficient to induce TIGIT+ Tregs, that was reduced in uveitis patients. The importance of these findings is that multiple Treg subsets can be induced to provide remission to autoimmune uveitis and potentially other autoimmune diseases, and a defect in the induction of these Tregs through A2Ar stimulation may contribute to autoimmune uveitis.
Highlights:
TIGIT+ Tregs represent a novel subset of anti-uveitic Tregs
A2Ar is necessary for the induction of TIGIT+ Tregs in uveitis patients
FoxP3 Tregs emerge in the eye at the onset of retinal inflammation in an A2Ar-dependent manner
Acknowledgments
We would like to thank the Oklahoma Medical Research Foundation Core facility, Mark Dittmar and the Dean McGee Eye Institute Vivarium staff, the NEI P30 genotyping core, and NEI P30 live animal imaging core.
Funding: This work was supported by National Institutes of Health/National Eye Institute grants EY021725 (P30), EY024951 (DJL), EY029240 (DJL), and in part by an unrestricted Research to Prevent Blindness grant (New York, NY, USA).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Financial Interests
All other authors have no relevant competing financial interests.
Declarations of interest: None
Statement: All authors concur with the submission and all the material submitted in this manuscript has not been previously reported and is not under consideration for publication elsewhere.
References
- [1].Dominguez-Villar M, Hafler DA. Regulatory T cells in autoimmune disease. Nat Immunol, 2018;19:665–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wing JB, Tanaka A, Sakaguchi S. Human FOXP3(+) Regulatory T Cell Heterogeneity and Function in Autoimmunity and Cancer. Immunity, 2019;50:302–16. [DOI] [PubMed] [Google Scholar]
- [3].Godfrey VL, Wilkinson JE, Rinchik EM, Russell LB. Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc Natl Acad Sci U S A, 1991;88:5528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Godfrey VL, Wilkinson JE, Russell LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol, 1991;138:1379–87. [PMC free article] [PubMed] [Google Scholar]
- [5].DuPage M, Bluestone JA. Harnessing the plasticity of CD4(+) T cells to treat immune-mediated disease. Nat Rev Immunol, 2016;16:149–63. [DOI] [PubMed] [Google Scholar]
- [6].Bailey-Bucktrout SL, Martinez-Llordella M, Zhou X, Anthony B, Rosenthal W, Luche H et al. Self-antigen-driven activation induces instability of regulatory T cells during an inflammatory autoimmune response. Immunity, 2013;39:949–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Yadav M, Stephan S, Bluestone JA. Peripherally induced tregs - role in immune homeostasis and autoimmunity. Front Immunol, 2013;4:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol, 2009;10:1000–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Attias M, Al-Aubodah T, Piccirillo CA. Mechanisms of human FoxP3(+) Treg cell development and function in health and disease. Clinical and experimental immunology, 2019. [DOI] [PMC free article] [PubMed]
- [10].Streilein JW. Immunoregulatory mechanisms of the eye. Progress in retinal and eye research, 1999;18:357–70. [DOI] [PubMed] [Google Scholar]
- [11].Streilein JW. Ocular immune privilege: therapeutic opportunities from an experiment of nature. Nat Rev Immunol, 2003;3:879–89. [DOI] [PubMed] [Google Scholar]
- [12].Darrell RW, Wagener HP, Kurland LT. Epidemiology of uveitis. Incidence and prevalence in a small urban community. Arch Ophthalmol, 1962;68:502–14. [DOI] [PubMed] [Google Scholar]
- [13].Hwang DK, Chou YJ, Pu CY, Chou P. Epidemiology of uveitis among the Chinese population in Taiwan: a population-based study. Ophthalmology, 2012;119:2371–6. [DOI] [PubMed] [Google Scholar]
- [14].Suhler EB, Lloyd MJ, Choi D, Rosenbaum JT, Austin DF. Incidence and prevalence of uveitis in Veterans Affairs Medical Centers of the Pacific Northwest. Am J Ophthalmol, 2008;146:890–6 e8. [DOI] [PubMed] [Google Scholar]
- [15].Gritz DC, Wong IG. Incidence and prevalence of uveitis in Northern California; the Northern California Epidemiology of Uveitis Study. Ophthalmology, 2004;111:491–500; discussion [DOI] [PubMed] [Google Scholar]
- [16].Natkunarajah M, Kaptoge S, Edelsten C. Risks of relapse in patients with acute anterior uveitis. Br J Ophthalmol, 2007;91:330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Caspi RR. Animal models of autoimmune and immune-mediated uveitis. Drug Discovery Today: Disease Models, 2006;3:3–9. [Google Scholar]
- [18].Caspi RR, Roberge FG, Chan CC, Wiggert B, Chader GJ, Rozenszajn LA et al. A new model of autoimmune disease. Experimental autoimmune uveoretinitis induced in mice with two different retinal antigens. J Immunol, 1988;140:1490–5. [PubMed] [Google Scholar]
- [19].Chen J, Qian H, Horai R, Chan CC, Caspi RR. Mouse Models of Experimental Autoimmune Uveitis: Comparative Analysis of Adjuvant-Induced vs Spontaneous Models of Uveitis. Curr Mol Med, 2015;15:550–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Caspi RR, Silver PB, Luger D, Tang J, Cortes LM, Pennesi G et al. Mouse models of experimental autoimmune uveitis. Ophthalmic Res, 2008;40:169–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kitaichi N, Namba K, Taylor AW. Inducible immune regulation following autoimmune disease in the immune-privileged eye. J Leukoc Biol, 2005;77:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lee DJ, Taylor AW. Following EAU recovery there is an associated MC5r-dependent APC induction of regulatory immunity in the spleen. Invest Ophthalmol Vis Sci, 2011;52:8862–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Lee DJ, Taylor AW. Both MC5r and A2Ar Are Required for Protective Regulatory Immunity in the Spleen of Post-Experimental Autoimmune Uveitis in Mice. J Immunol, 2013;191:4103–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Lee DJ, Taylor AW. Recovery from experimental autoimmune uveitis promotes induction of antiuveitic inducible Tregs. J Leukoc Biol, 2015;97:1101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee DJ, Preble J, Lee S, Foster CS, Taylor AW. MC5r and A2Ar Deficiencies During Experimental Autoimmune Uveitis Identifies Distinct T cell Polarization Programs and a Biphasic Regulatory Response. Sci Rep, 2016;6:37790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol, 2011; 186:1338–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM et al. Vstm3 is a member of the CD28 family and an important modulator of T-cell function. Eur J Immunol, 2011;41:902–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nature immunology, 2009;10:48–57. [DOI] [PubMed] [Google Scholar]
- [29].Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity, 2014;40:569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Namba K, Kitaichi N, Nishida T, Taylor AW. Induction of regulatory T cells by the immunomodulating cytokines alpha-melanocyte-stimulating hormone and transforming growth factor-beta2. J Leukoc Biol, 2002;72:946–52. [PubMed] [Google Scholar]
- [31].Taylor AW, Alard P, Yee DG, Streilein JW. Aqueous humor induces transforming growth factor-beta (TGF-beta)-producing regulatory T-cells. Curr Eye Res, 1997;16:900–8. [DOI] [PubMed] [Google Scholar]
- [32].Muhammad F, Trivett A, Wang D, Lee DJ. Tissue specific production of MicroRNA-155 inhibits melanocortin 5 receptor dependent suppressor macrophages to promote experimental autoimmune uveitis. Eur J Immunol, 2019. [DOI] [PMC free article] [PubMed]
- [33].Muhammad F, Wang D, Montieth A, Lee S, Preble J, Foster CS et al. PD-1+melanocortin receptor dependent-Treg cells prevent autoimmune disease. Scientific Reports, 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Muhammad FY, Peters K, Wang D, Lee DJ. Exacerbation of autoimmune uveitis by obesity occurs through the melanocortin 5 receptor. J Leukoc Biol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity, 2016;44:989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hall BM, Verma ND, Tran GT, Hodgkinson SJ. Distinct regulatory CD4+T cell subsets; differences between naive and antigen specific T regulatory cells. Curr Opin Immunol, 2011;23:641–7. [DOI] [PubMed] [Google Scholar]
- [37].Schiavon V, Duchez S, Branchtein M, How-Kit A, Cassius C, Daunay A et al. Microenvironment tailors nTreg structure and function. Proc Natl Acad Sci U S A, 2019;116:6298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sharma A, Rudra D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front Immunol, 2018;9:883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Silver PB, Horai R, Chen J, Jittayasothorn Y, Chan CC, Villasmil R et al. Retina-specific T regulatory cells bring about resolution and maintain remission of autoimmune uveitis. J Immunol, 2015;194:3011–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bynoe MS, Viret C, Yan A, Kim DG. Adenosine receptor signaling: a key to opening the blood-brain door. Fluids Barriers CNS, 2015;12:20. [DOI] [PMC free article] [PubMed] [Google Scholar]