

Abstract
Background:
Colonic atresias in the Fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) mouse model have been attributed to increased epithelial apoptosis and decreased epithelial proliferation at embryonic day (E) 10.5. We therefore hypothesized that these processes would co-localize to the distal colon where atresias occur (atretic precursor) and would be excluded or minimized from the proximal colon and small intestine.
Results:
We observed a global increase in intestinal epithelial apoptosis in Fgfr2IIIb−/− intestines from E9.5 to E10.5 that did not co-localize to the atretic precursor. Additionally, epithelial proliferations rates in Fgfr2IIIb−/− intestines were statistically indistinguishable to that of controls at E10.5 and E11.5. At E11.5 distal colonic epithelial cells in mutants failed to assume the expected pseudostratified columnar architecture and the continuity of the adjacent basal lamina was disrupted. Individual E-cadherin-positive cells were observed in the colonic mesenchyme.
Conclusions:
Our observations suggest that alterations in proliferation and apoptosis alone are insufficient to account for intestinal atresias and that these defects may arise from both a failure of distal colonic epithelial cells to develop normally and local disruptions in basal lamina architecture.
Keywords: Embryonic Development, Gene Expression, Mice, EMT, Basal Lamina, E-Cadherin, Laminin
1. Introduction
Intestinal atresias are a congenital interruption in the continuity of the intestinal tube. Intestinal atresias occur in roughly 1 in 10,000 live births though these rates vary depending on country ((ICBDSR), 2014). This defect results from a segmental loss of intestinal tissue during development. Atresias can present anywhere throughout the intestine and range in severity from a narrowing of the intestinal lumen to multiple regions of missing tissue (Dalla Vecchia LK, 1998; Nichol et al., 2011b; Takahashi et al., 2014; Lupo et al., 2017).
Surgical intervention in the neonatal period reestablishes intestinal continuity. However, the long-term morbidity following the defects can be severe. Individuals with repaired intestinal atresias are at higher risk of developing severe malnutrition issues related to the loss of intestine. This results in deficiencies of vitamins, minerals, macronutrients, fluids, and calories. Some patients have sufficient intestinal surface area, but motor function is severely impaired thereby affecting absorption of nutrients. In severe cases, patients require total parenteral nutrition (TPN), an intravenous form of nutrients which comes with the potential complications of liver failure, blood clots, and sepsis.
The developmental causes of intestinal atresia are not fully understood, though a number of hypotheses have been proposed. These include persistence of developmental epithelial overgrowth (Tandler, 1900), vascular occlusion (Louw, 1955; Barnard, 1956; Barnard, 1957), mechanical obstruction (Khen et al., 2004), and multiple genetic triggers (Celli, 2014; Fernandez et al., 2014). Animal models of intestinal atresia include ligation of vasculature or disruption of mesentery (Tovar et al., 1991), electrocoagulation resulting in bowel obstruction during development (Lopez de Torre et al., 1992), mutations in hedgehog protein signaling (Ramalho-Santos et al., 2000; Mo et al., 2001), or teratogen induction (Merei, 2002). In terms of genetic models, one of the best established mouse models of intestinal atresia is that of a Fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) mutation (De Moerlooze et al., 2000; Fairbanks et al., 2004; Kanard et al., 2005; Botham et al., 2012; Reeder et al., 2012a).
Based on observations using this model, it has been proposed that increased rates of apoptosis and decreased rates of proliferation in the embryonic epithelium at embryonic age (E)10.5 are potentially critical events in the formation of intestinal atresias (Fairbanks et al., 2006). However, apoptosis has also been shown to play an important role in normal organogenesis (Sukhotnik et al., 2005) and it remains uncertain the exact role cell death plays in either normal or abnormal intestinal development in this model.
We sought to characterize the location and timing of both apoptotic and proliferative epithelial activity during intestinal organogenesis in the Fibroblast growth factor receptor 2IIIb (Fgfr2IIIb) null mouse model (in the epithelium). Null embryos in this model have been shown to form a number of congenital defects (De Moerlooze et al., 2000). Of note for this study, these mutant mice present with distal colonic atresia with 100% penetrance across all homozygous mutant embryos (Fairbanks et al., 2006; Sala et al., 2006; Nichol et al., 2011a; Reeder et al., 2012a; Reeder et al., 2012b). Given the anatomic precision and fidelity of colonic atresia formation in this model, we focused on this distal colonic region as a discreet location where cellular differences during organogenesis would be of interest. We hypothesized that if loss of epithelium through apoptosis and decreased proliferation are mechanistically critical in atresias, then we would observe an increase in apoptotic activity and a concomitant reduction in proliferation that co-localizes to the intestinal segment where the atresia will occur (the distal colon, also termed the atretic precursor region).
2. Results
Apoptosis
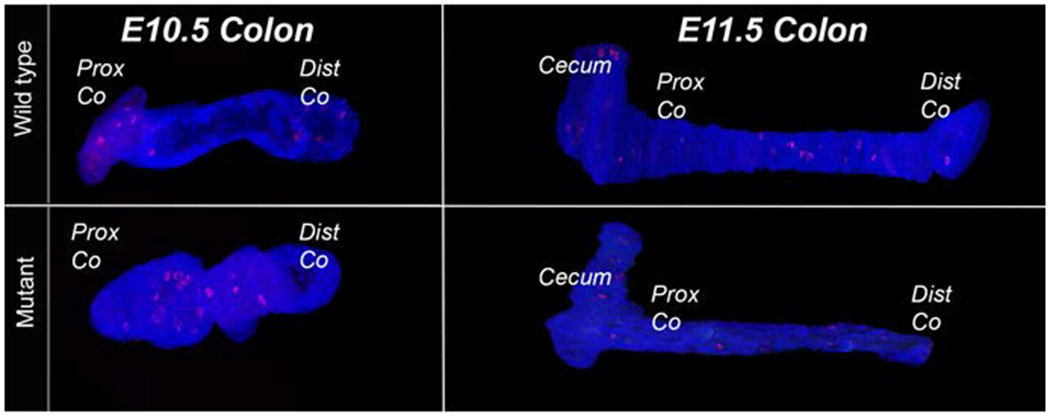
To determine the pattern of apoptosis throughout the intestinal epithelium, we created three-dimensional reconstructions of the intestinal epithelial cells using imaged sections of TUNEL (Terminal deoxynucleotidyl transferase (TdT) dUTP Nick-End Labeling) assay stained epithelial tissue. We collected wild type and Fgfr2IIIb null embryos between E9.5-E12.5. Samples were dehydrated, embedded in paraffin, microtome sectioned at 5μm, and subject to a TUNEL staining. The percentage of apoptotic epithelial cells per section was calculated using these images. The mesenchyme (where apoptosis is absent) was subtracted from the images for subsequent three-dimensional reconstruction of the intestinal epithelium. These resulting images of the epithelial tissue of the intestinal tracts allow for a representation of the spatial pattern of apoptosis.
Figure 1 shows three-dimensional reconstructions of epithelial apoptosis at all four time points. At the E9.5 and E10.5 time points the entirety of the tract is shown, whereas only the cecum and the entirety of the colon are shown in the E11.5 and E12.5 reconstructions. Apoptosis occurred throughout the entire intestinal epithelium in both control and null embryos at E9.5 (Figure 1A and 1A’) and was not restricted to, nor excessive, in the hindgut area that will later develop into the distal colon. At E10.5, apoptotic activity continued in both control and null embryos, though the pan-intestinal nature of the cell death is diminished compared to E9.5 (Figure 1B and 1B’). There was a temporal trend of decreasing apoptosis in the wild type intestinal epithelium in the colon with E9.5 embryos demonstrating the greatest amount of cell death with E12.5 embryos showing the least amount of cell death. Of note, apoptosis was not confined solely to the distal colon (the atretic precursor) in the null embryos at all-time points as would be predicted if apoptosis was the primary cause of atresia formation in this model.
Figure 1: 3D reconstruction of intestinal epithelium apoptotic cells using TUNEL stain.
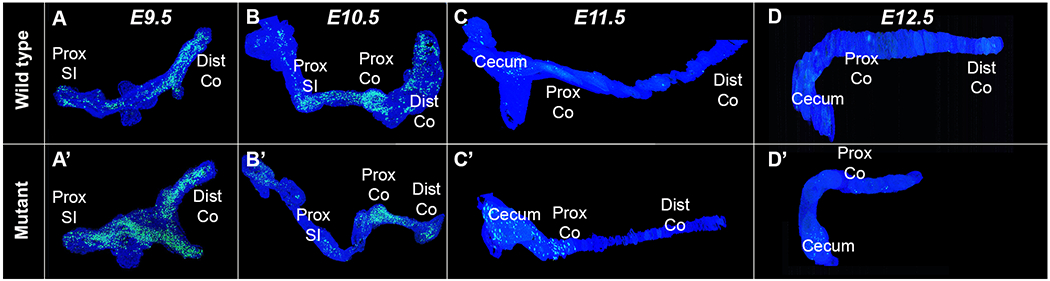
Three-dimensional reconstruction of intestinal tracts from control and Fgfr2IIIb mutant embryos aged E9.5-E12.5. Note that for E9.5-E10.5 the epithelial cells from the entirety of the intestinal tract are present, whereas only the cecum to distal colon are visualized in the E11.5-E12.5 reconstructions. Pan-epithelial apoptosis is present in both the control and null embryos at E9.5 (A and A’). At E10.5, there is a decrease in apoptosis globally, but a greater decrease in the distal colon of the null embryo (B and B’). By E11.5, apoptosis is largely absent outside of the developing cecal region (C and C’). At E12.5, little to no apoptosis is seen (D and D’). Note the shortened intestinal tract in D’, indicative of the distal colonic atresia and loss of the distal colon. Abbreviations: Prox= proximal, Dist= distal, SI= small intestine, Co = colon
Figure 2 depicts the quantification of TUNEL positive cells in the intestinal epithelium within 50μm-100μm regions of colon. This allows for an examination of cell death within the colon as a whole. In the Fgfr2IIIb nulls there was a marked increase in apoptosis occurring at E10.5, before a sharp overall decrease in apoptosis at E11.5 and E12.5 to levels below that seen at E9.5. Despite this eventual decrease in apoptosis, there was a statistically significant increase in overall epithelial apoptosis when comparing Fgfr2IIIb null mice to controls across E10.5-E12.5 with the greatest increase occurring at E10.5. Therefore, while apoptosis may play a role throughout atresia formation, the E10.5 time point would be a critical window for examining the role of apoptosis in atresia formation in this model.
Figure 2: Pooled TUNEL counts from all epithelial cells in 50-100 μm sections of colon.
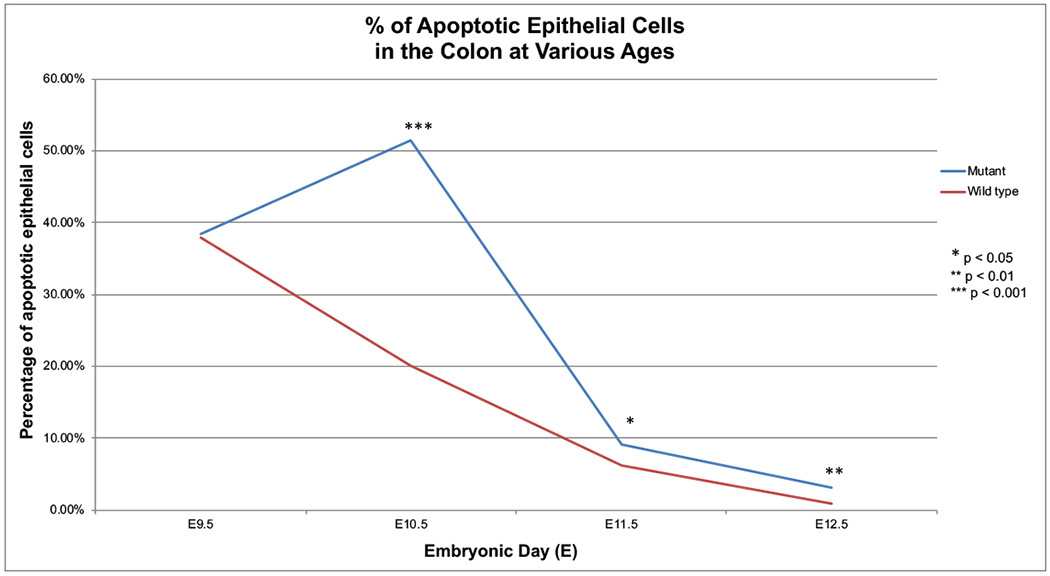
Evaluation of TUNEL positive cells within all the epithelial cells found in 50-100μm segments of colon across embryonic ages E9.5-E12.5 in wild type and Fgfr2IIIb−/− mutant intestines. TUNEL counts obtained using imaged 5μm sections. Pooling the apoptotic data across the epithelial cells allows for general synopsis of apoptosis within the colon.
When examining cellular death rates across discrete 5μm sections of epithelial tissue further we noted a lack of consistency in the percentage of apoptotic cells in the mutant intestinal tracts across the colon. A display of this lack of uniformity can be seen in Figure 3. These frequency distribution plots show the percentage of TUNEL positive stained epithelial cells per 5μm segment examined across 50-100μm regions of colon (with bins set at 10% intervals: 0-10%, 10-20%, etc.). While there was discrepancy in apoptosis rates across 5μm sections in both the controls and the mutant intestine, this variability was greatest in the null intestine. At E10.5 in the nulls, corresponding to the time point with the greatest cellular death, we note a large intersection variability in the apoptotic rates. This demonstrates that while the average rate of colonic epithelial apoptosis is indeed greater in the mutants than in the wild type epithelium, these rates can vary greatly from one 5μm segment of mutant colon to another. Within the E10.5 mutant epithelium, apoptosis can be extremely low (<10%) or almost complete (>90%) when comparing individual 5μm segments across the colon. This variability is not as pronounced in the wild type intestine across all ages examined.
Figure 3: Frequency distribution of apoptotic cells in 5μm sections.
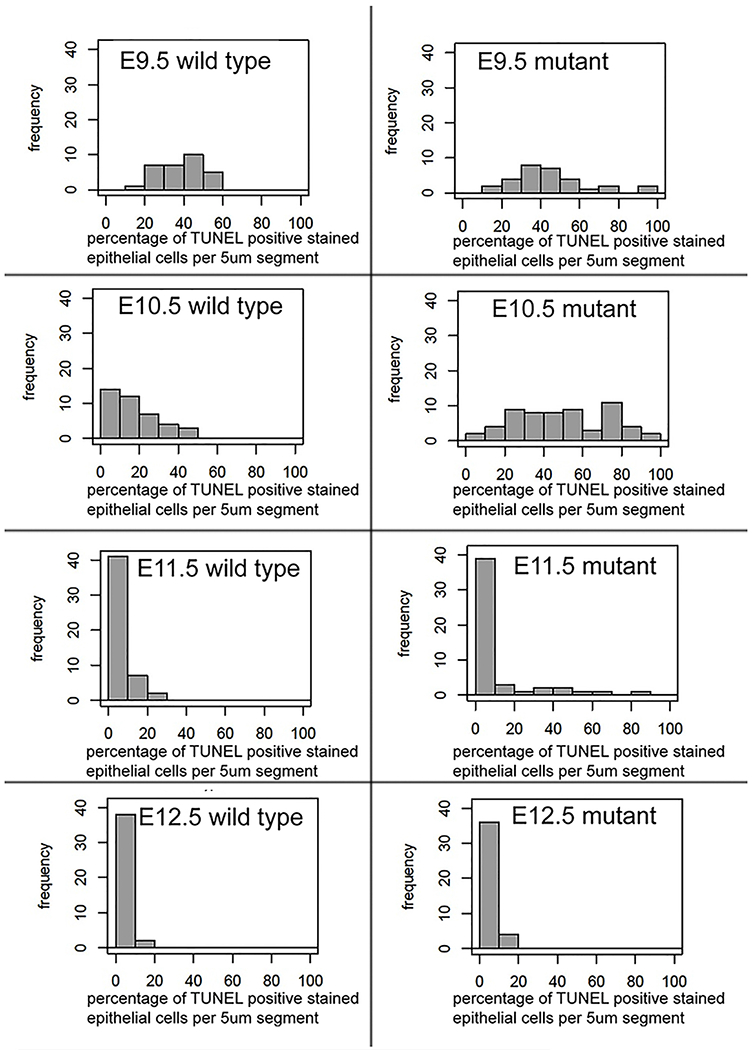
A high level of apoptotic variability occurs within the colon, with the greatest inconsistency in cell death across 5μm sections occurring in null embryos at E9.5-E10.5. Frequency distribution plots show the percentage of TUNEL positive stained epithelial cells per 5μm segment (with bins set at 10%: 0-10%, 10-20%, etc) across all 5μm sections counts from the colon at each stage.
To examine the atretic precursor region specifically, we analyzed apoptosis as a segmental event focusing on the distal and proximal colonic regions. We pooled TUNEL images to examine the population of cells within a segmental region as positively TUNEL stained versus unstained epithelial cells rather than calculating the average apoptotic cell percentage per discreet 5μm section (Table 1). Proximal colon was deemed as sections of colon occurring distal to the cecum. Distal colon sections were those sections from the end of our reconstruction moving several sections proximal to capture the most distal remainder of the colon. The distal colon segments did not overlap with proximal colon segments and neither segment reached beyond the halfway point of the colon reconstruction images. We conducted a statistical comparison of this segmental data using a 2 tailed Fisher’s exact test with a 95% confidence interval. We examined E10.5 and E11.5 intestinal tissue. By E12.5 the distal colon and its epithelium involutes making it impossible to accurately compare segmental apoptotic rates.
Table 1: Statistical analysis of apoptosis across age groups and colonic regions.
Examination of apoptotic rates using TUNEL staining results from 5 μm imaged sections in E10.5 and E11.5 wild type andF gfr2IIIb −/− mutant intestines. For statistical analysis, all epithelial cells across 50 μm colon regions were pooled together as a sample set so that differences could be determined between distal and proximal colon.
| Age | Wild type proximal colon mean apoptotic rate | Wild type distal colon mean apoptotic rate | P-value |
| E10.5 | 13.14% | 22.30% | <0.00001 |
| E11.5 | 10.71% | 1.67% | <0.00001 |
| Age | Fgfr2IIIb −/− mutant proximal colon mean apoptotic rate | Fgfr2IIIb −/− mutant distal colon mean apoptotic rate | P-value |
| E10.5 | 52.45% | 50.94% | not significant |
| E11.5 | 20.24% | 2.38% | <0.00001 |
| Age | Wild type proximal colon mean apoptotic rate | Fgfr2IIIb −/− mutant proximal colon mean apoptotic rate | P-value |
| E10.5 | 13.14% | 52.45% | <0.00001 |
| E11.5 | 10.71% | 20.24% | <0.001 |
| Age | Wild type distal colon mean apoptotic rate | Fgfr2IIIb −/− mutant distal colon mean apoptotic rate | P-value |
| E10.5 | 22.30% | 50.94% | <0.00001 |
| E11.5 | 1.67% | 2.38% | not significant |
The average apoptotic rates across wild type and mutant colons display a complex temporal and segmental pattern (Table 1). Within the wild type, the distal colon had more epithelial apoptosis than the proximal region at E10.5, but this pattern was reversed at E11.5. Within the Fgfr2IIIb null condition, there was no difference in the apoptotic rates between the proximal and distal colonic regions at E10.5. However, in the mutant colon at E11.5 there was a greater apoptotic rate in the epithelium of the proximal colon than in the distal colon. While the distal colon region in the Fgfr2IIIb mutant does exhibit a significant increase in apoptosis when compared to the wild type, this is a pan-colonic escalation in epithelial apoptosis in the null colon and did not co-localize specifically to the atretic precursor at either E10.5 or E11.5.
Proliferation
To determine if there were focal changes in the rate of proliferation that co-localized to the atretic precursor we assessed proliferation using anti-Phosphohistone H3 (PHH3) staining of 5μm segments of embryonic intestine at E10.5 and E11.5 in both wild type and Fgfr2IIIb null embryos. This approach labels cells actively mitosing, staining cells primarily in M phase, unlike a BrdU incorporation assay which can overestimate the proliferative rate since it incorporates during S phase and labels all resulting daughter cells.
A three-dimensional reconstructions of the PHH3 stained 5μm sections of E10.5 and E11.5 wild type and Fgfr2IIIb null colon epithelium were obtained in the same manner as the TUNEL three-dimensional reconstruction (Figure 4). These reconstructions display a low level of cellular proliferation across the length of the entire colon in all cases.
Figure 4: 3D reconstruction of PHH3 staining in epithelial cells of colon.
Three-dimensional reconstruction of colon epithelial tissue from control and Fgfr2IIIb mutant embryos aged E10.5-E11.5. Images reconstructed from 5μm PHH3 stained and images sections of tissue where the mesenchyme in the image was cropped out before reconstruction. There discreet staining found across the length of the epithelium in both the wild type and mutant conditions. Abbreviations: Prox= proximal, Dist= distal, Co = colon
Cellular proliferation rates were determined by calculating the percentage of PHH3 positive stained epithelial cells in stained 5μm sections of colon. Average rates were then compared using a Fisher’s exact test (Figure 5). We observed no statistically significant difference in rates of epithelial proliferation when wild type embryos were compared to null embryos at either E10.5 or E11.5. Furthermore, there was no statistically significant change in proliferation rates when comparing distal versus proximal regions within these ages despite the trend of a decrease in proliferation at the distal end of the colon in both the wild type and Fgfr2IIIb null epithelial cells (Table 2).
Figure 5: Epithelial cell proliferation in the colon at E10.5 and E11.5.
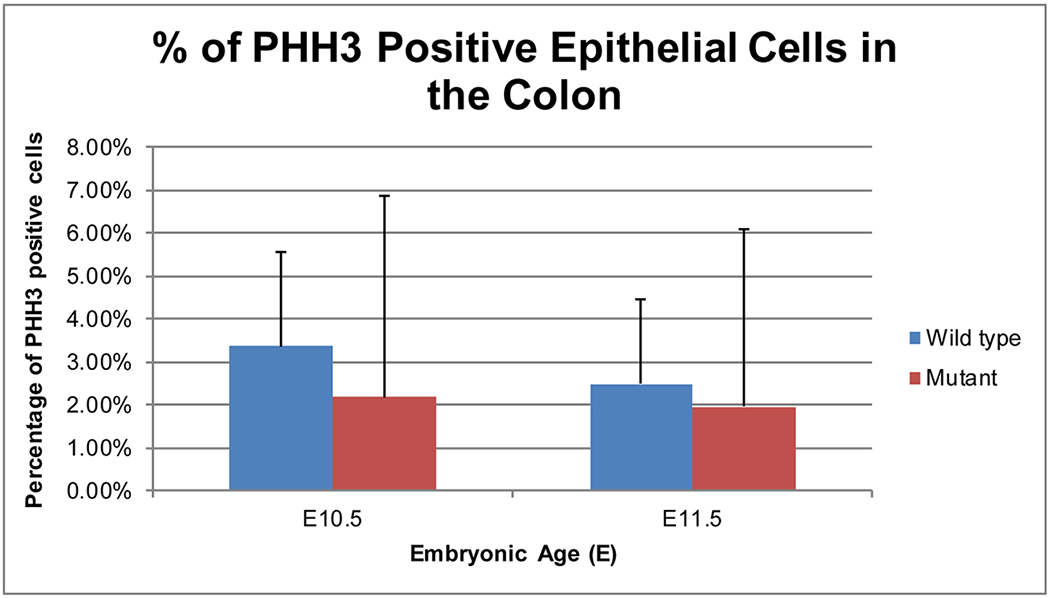
Mean percentage of PHH3 positive stained epithelial cells averaged across 5pm stained sections of colon on E10.5-E11.5 wild type and Fgfr2IIIb −/− mutant intestines. Using a Fisher’s exact test, there was no statistically significant difference found between conditions with a P-value 0.2429 at E10.5 and 0.518 at E11.5. n=3 embryos for E10.5 intestine and n=5 embryos for E11.5 intestine.
Table 2: Statistical analysis of cellular proliferation across age groups and colonic regions.
Examination of cellular proliferation rates as determined by PHH3 staining in E10.5 and E11.5 wild type and Fgfr2IIIb−/− mutant intestines across 50 μm colon segments. Epithelial cells within that region were pooled together as a set to examine differences between distal and proximal colon.
Counts were obtained from stained 5μm sections of intestine.
None of the comparisons yielded statistically significant results despite a trend of decreased cellular proliferation at the distal colon when compared to the proximal region in almost all cases.
| Age | Wild type proximal colon mean proliferation rate | Wild type distal colon mean proliferation rate | P-value |
| E10.5 | 3.89% | 2.75% | not significant |
| E11.5 | 2.60% | 2.31% | not significant |
| Age | Fgfr2IIIb −/− mutant proximal colon mean proliferation rate | Fgfr2IIIb −/− mutant distal colon mean proliferation rate | P-value |
| E10.5 | 3.33% | 0.98% | not significant |
| E11.5 | 2.62% | 1.27% | not significant |
| Age | Wild type proximal colon mean proliferation rate | Fgfr2IIIb −/− mutant proximal colon mean proliferation rate | P-value |
| E10.5 | 3.89% | 3.33% | not significant |
| E11.5 | 2.60% | 2.62% | not significant |
| Age | Wild type distal colon mean proliferation rate | Fgfr2IIIb −/− mutant distal colon mean proliferation rate | P-value |
| E10.5 | 2.75% | 0.98% | not significant |
| E11.5 | 2.31% | 1.27% | not significant |
E-cadherin (as a marker of epithelial identity) and cellular architecture
Given the lack of significant changes in proliferation or apoptosis specifically co-localizing to the atretic precursor we further examined the cellular architecture of the epithelial cells within this region prior to its involution at E12.5. Using E-cadherin staining of E11.5 distal colon, wild type intestine showed phenotypically normal, pseudostratified columnar epithelium with discrete staining along the plasma membrane (Figure 6A–C). Within the Fgfr2IIIb null distal colon at E11.5, E-cadherin staining is less consistent than in the wild type colon. At E11.5 the mutant, distal colon, epithelial cells fail to consistently maintain a discreet E-cadherin staining pattern at the outer edges of the epithelial cells (Figure 6D, F). In some mutant distal colon sections, we observe an increase in staining within the cytoplasm of the epithelial cells (Figure 6E). The overall cellular architecture of the epithelium in the distal colon of the Fgfr2IIIb null was disrupted, with the appearance of a mass of cells without discreet basal and apical structure and often lacking a lumen (Figure 6D–F). On rare occasions, we observed E-cadherin positive cells in the surrounding colonic mesenchyme in the Fgfr2IIIb null (arrow in Figure 6E) which we did not observe occurring in any of our stained sections of wild type embryos. Together, these data suggest a failure of epithelial differentiation into a pseudostratified, columnar epithelium during organogenesis in the Fgfr2IIIb null.
Figure 6: E-cadherin staining of distal colon.
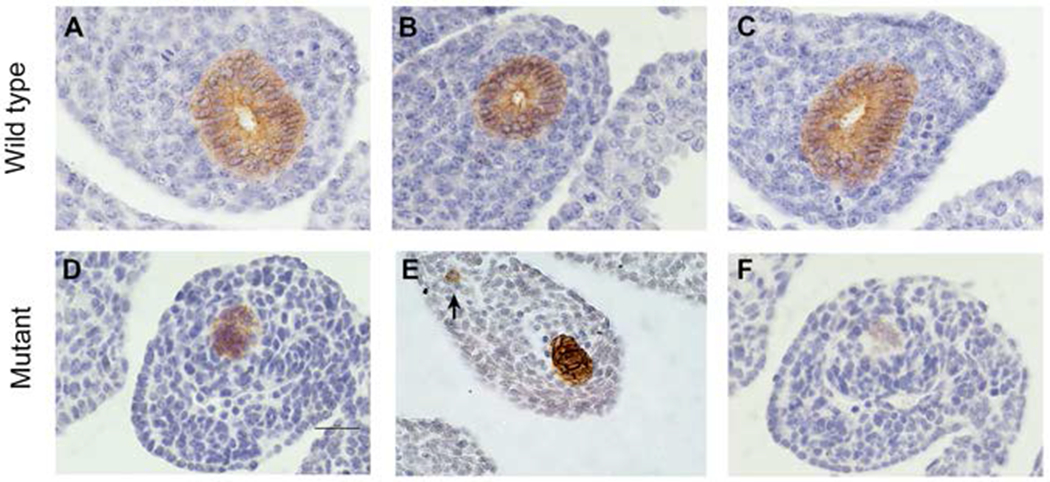
E-cadherin staining by immunohistochemistry on E11.5. Fgfr2IIIb−/− mutant and wild type distal colon. Within the wild type condition, there is a discreet staining of this epithelial cell marker at the cell borders and the epithelium retains a pseudostratified columnar organization (A-C). The Fgfr2IIIb−/− mutant the distal colon display a lack of pseudostratified columnar architecture often co-occurring with a loss of lumen (D-F), at times a diffuse pattern of E-cadherin staining (D,F), rare E-cadherin positive staining cells in the mesenchyme (indicated by the arrow in E), and near the termination of the atresia greatly diminished E-cadherin staining (F). Scale bar is 50 μm.
Basal lamina
Based on these observations, we examined a major structural component of the extracellular matrix surrounding the cells. Laminin serves a critical role in instructing polarity and identity of epithelial cells. Fluorescent staining of Laminin and E-cadherin in 5μm segments of intestine showed consistently intact laminin with few breaks in the basement membrane of wild type E11.5 colon (proximal and distal) and the proximal colon of an E11.5 Fgfr2IIIb null intestine (Figure 7A–C). However, the sections of intestine in the distal portion of the colon in the Fgfr2IIIb nulls showed a discontinuity in the laminin of the extracellular matrix with large segments demonstrating no laminin staining (Figure 7D). These abnormalities in the basal lamina are consistent with a breakdown in the cellular architecture of the epithelial cells in the atretic precursor.
Figure 7: Laminin and E-cadherin staining.
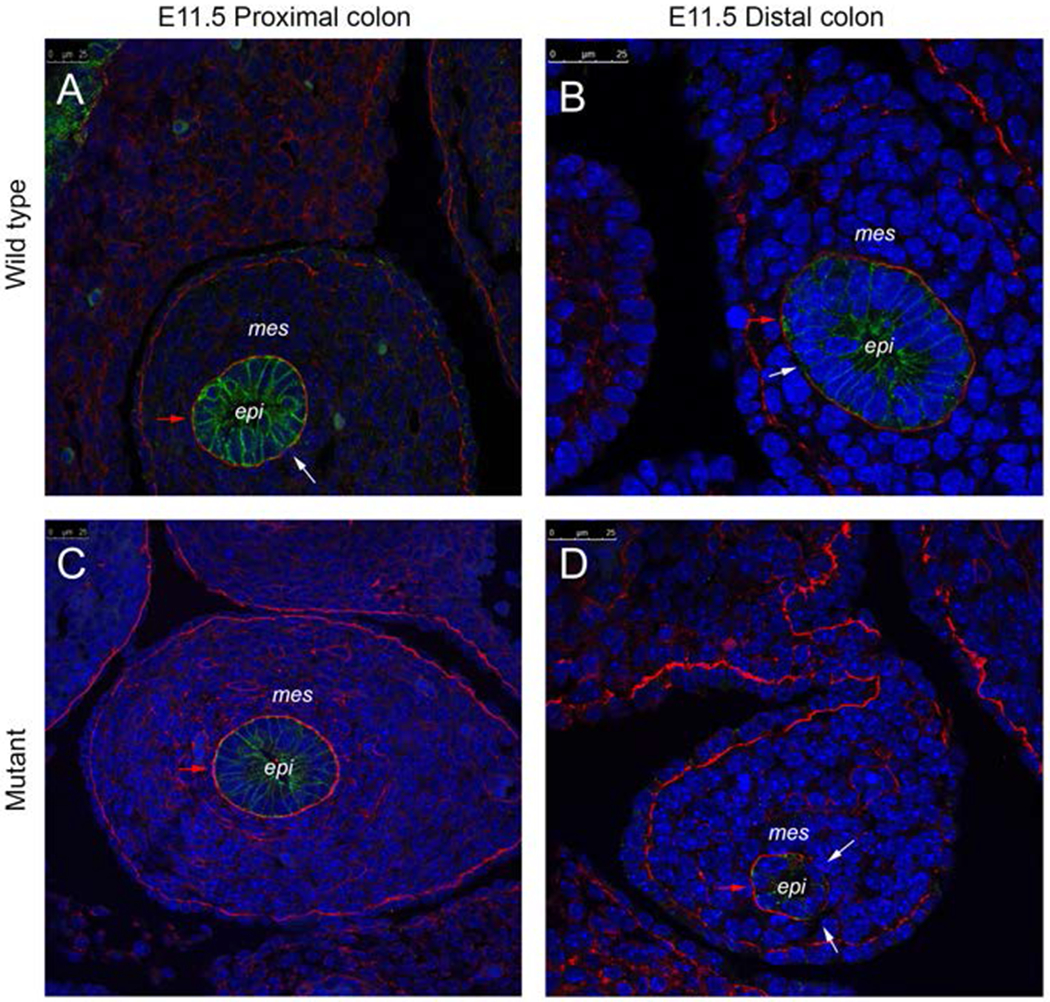
Laminin staining of extracellular matrix (red), E-cadherin staining of epithelial cells (green), and DAPI stain for nuclear material (blue) of the proximal colon of an E11.5 wild type intestine (A), distal colon of an E11.5 wild type intestine (B), the proximal colon of an E11.5 Fgfr2IIIb−/ −intestine (C), the distal colon at the atretic precursor region of an E11.5 Fgfr2IIIb−/ −intestine (D). Red arrow indicates an example section of intact laminin staining in each image. White arrows indicate breaks in laminin. Abbreviations: mes= mesenchyme, epi= epithelium. Scale bar is 25 μm.
Cellular identity markers
Based on the breaks found in the basal lamina and the cellular architecture and E-cadherin changes observed, we stained for markers consistent with a change in cellular identity or cellular movement. Alpha smooth muscle actin and vimentin are mesenchymal markers while β-catenin is upregulated during epithelial to mesenchymal transition (EMT). We examined wild type and Fgfr2IIIb null intestine at E11.5 as that is the stage during which changes in the basal lamina were observed and before the colon involutes at E12.5 (Figure 8).
Figure 8: Cellular identity/cellular movement markers.
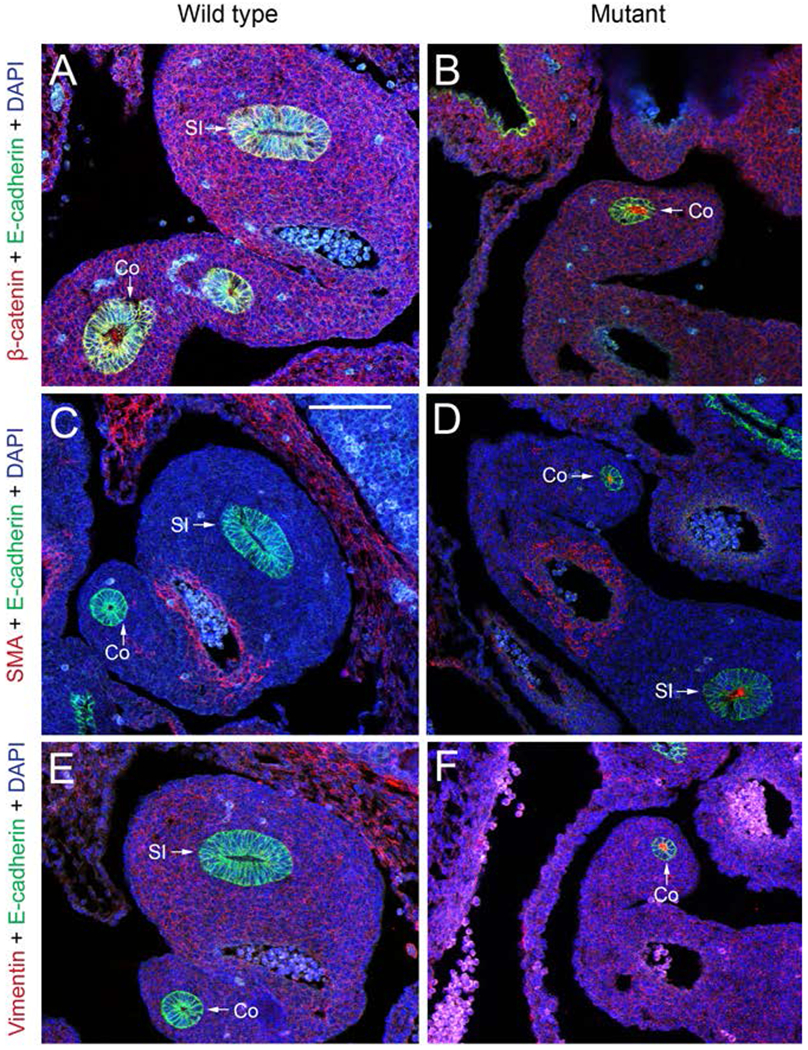
Immunostaining of cellular identity proteins in E11.5 wild type and Fgfr2IIIb−/ − intestine. Staining for β-catenin, alpha smooth muscle actin, and vimentin (red), E-cadherin staining of epithelial cells (green), and DAPI stain for nuclear material (blue). E-cadherin is an epithelial cell marker, alpha smooth muscle actin and vimentin are mesenchymal cell markers. β-catenin is upregulated during epithelial to mesenchymal transition. Abbreviations: Co= colon. SI= small intestine. Scale bar is 100 μm.
β-catenin was not observed in the wild type intestinal epithelium at E11.5 but was seen in the epithelium of Fgfr2IIIb null intestine at E11.5 (Figure 8 A–B). These β-catenin positive cells were not positive for E-cadherin suggesting that they lacked epithelial identity and were potential undergoing EMT (Figure 8B). No vimentin or alpha smooth muscle staining was found in the epithelial cells of the wild type intestinal tissue (Figure 8C, E), but punctate staining was seen for both of these proteins in the lumen of the intestine in the Fgfr2IIIb−/− nulls.
3. Discussion
Understanding the causes and mechanisms of intestinal atresia formation is critical to developing treatment strategies beyond surgical intervention for this developmental defect. We hypothesized that high rates of apoptosis and low rates of cellular proliferation would be restricted to, or disproportionate within, the epithelial cells in the atretic precursor region of Fgfr2IIIb null intestine across a crucial time course of intestinal organogenesis. However, our results indicate that apoptosis does not discreetly co-localize only to the atretic precursor prior to the involution of this region of intestine. Apoptosis is pan-epithelial across the colon of the Fgfr2IIIb null intestine at E10.5 despite the distal colon later forming an atresia with the proximal colon remaining intact. At E11.5 the Fgfr2IIIb null distal colon displays a sizable decrease in apoptosis despite its upcoming involution. Of interest, the mean apoptotic counts across 5μm sections displayed more inter-section variability in the Fgfr2IIIb null colons than in the wild type. This feature of the TUNEL staining suggests that a lack of this receptor may not affect every portion of the intestinal tract identically though it is unclear what causes the variability. The high rates of colonic epithelial apoptosis in the mutant versus the wild type across ages (E10.5-E12.5) does indicate that cell death is a major factor within this model but lack of co-localization of apoptosis to the atretic precursor region and a complex distribution pattern of apoptotic rates between conditions and embryonic ages suggests that apoptosis is not the sole regulatory factor and additional cellular processes warrant investigation.
We did not find statistically significant changes either across the colon as a whole when comparing conditions, or when comparing proximal versus distal segments in terms of proliferation. We did note a trend of decreasing proliferation from proximal to distal among colonic epithelial cells in both the wild type and the Fgfr2IIIb null intestines. We did not observe any statistically significant difference in epithelial proliferation despite looking during the same developmental window as such differences have been reported previously with BrdU staining (Fairbanks et al., 2006). The discrepancy between these two studies may be due to the stain chosen. BrdU incorporates into DNA at S phase and will label those cells and any resulting daughter cells while PHH3 only labels cells undergoing active mitosis, thus labeling a smaller population than BrdU. A small population of apoptotic cells have been shown to take up BrdU stain but not complete cellular division thereby allowing for an overestimation of cellular proliferation in an apoptotic cellular population when using this staining method (Kuan et al., 2004). This is of particular interest in our model due to the high percentage of cells undergoing apoptosis. Therefore, some of the cells visualized as BrdU positive may be indicative of DNA disruption as symptomatic of apoptosis rather than cells undergoing proliferation and account for the differences seen in cellular proliferation between our studies.
While increased apoptosis and a lack of the increased proliferation in the colonic epithelium necessary to overcome this cellular death must play a role in atresia formation, our study suggests that they cannot alone complete the picture of colonic atresia formation in this model. We observed several additional processes that were restricted to the atretic precursor. First, the presence of distorted epithelial architecture in the distal colon of Fgfr2IIIb nulls occurs at the atretic precursor. These cells do not exhibit the typical colonic pseudostratified columnar architecture, and instead clump together in a disorganized manner and appear to lose the apical-basal polarity. Second, and compatible with this first observation, E-cadherin staining was redistributed from the plasma membrane to the cytoplasm in these cells or disappearing. Note the lack of consistent E-cadherin antibody staining on the walls of the epithelial cells of the pre-atretic region of the Fgfr2IIIb null as compared to clear staining throughout the wild type and proximal colon epithelium of the Fgfr2IIIb null consistent with the observations seen in the distal colon when staining for E-cadherin using a HRP/DAB staining method. This is consistent with a loss of cellular polarity in which plasma membrane domains traffic intracellularly, causing a diffuse E-cadherin staining pattern (Low et al., 2000). Third, the basal lamina fails to maintain continuity at the far distal region of the colon. Together, these observations suggest that some of the distal colon epithelial cells are undergoing a dedifferentiation event in an environment that could allow movement out of the epithelial compartment.
Consistent with these observations of cellular identity loss and potential movement, rare E-cadherin stained cells were observed within the mesenchyme of the atretic precursor in Fgfr2IIIb nulls indicative of such a cellular movement. These were only seen very rarely (roughly once per intestinal tract) and never seen in any of the stained wild type intestinal tracts. Given this infrequency, we recognize this event may be de novo E-cadherin expression in the mesenchymal cells of this mutant rather than an indication of a past epithelial to mesenchymal transition (EMT) of those individual cells.
Our observed changes in E-cadherin expression within the colon are consistent with epithelial changes seen in similar models. Fibroblast growth factor 10 (Fgf10) is the primarily ligand for the Fgfr2IIIb receptor. A loss of this ligand results in colonic atresias and at E11.5 a reduced expression of Hnf3β which is a marker of the epithelium in endoderm-derived organs (Sala et al., 2006). Any disruption of the Fgfr2IIIb complex (either the ligand or the receptor itself) results in a loss of proper epithelial identity in the distal colon in these mouse models. A loss of E-cadherin has been shown to cause a failure of intestinal organogenesis. Within the small intestine, a conditional knockout of E-cadherin in the intestinal epithelium resulted in loss of proper intestinal morphology, decreased intestinal length, absent or blunted villi structure, and rounded rather than columnar epithelial cells at E18.5. Gene expression evaluation showed the greatest increase in markers consistent with cellular movement (Bondow et al., 2012).
For cells to exit the epithelial layer, there must be a corresponding disruption of the extracellular matrix around the epithelium to allow for the movement of these cells. When examining the extracellular matrix of E11.5 control and null embryos, we observed a breakdown of the laminin in the atretic precursor region of the null embryos, but no such large extensive breaks in the controls or in the proximal colons of null embryos. This suggests an environment that provides an opportunity for epithelial cells to leave their tissue layer in distal colon.
In order to further examine the possibility of dedifferentiation of epithelial cells in this model, we stained for markers of epithelial or mesenchymal identity. The expression of β-catenin in regions of distal colon epithelial cells in all of the Fgfr2IIIb null intestine embryos examined at E11.5 (n=3) along with a lack of E-cadherin expression in those same cells indicates that they have switched identity from epithelial to mesenchymal. The presence of punctate vimentin and alpha smooth muscle actin staining found in the lumen of the E11.5 Fgfr2IIIb null intestines could mean that cells who had lost epithelial identity in the past within this model had sloughed off from the basal lamina, with the remnants collecting in the lumen. It is unclear when this expression occurred and if it was concurrent with cells undergoing apoptosis or a different population of cells.
It is also worth noting that in reviewing our previous studies on duodenal atresia in this model, which occurs over a much shorter length of intestine, with less consistency (~50% incidence rate versus the 100% seen in the distal colon), and severity (either duodenal stenosis or atresia present versus 100% distal colonic atresia) we also observed a regional loss of the epithelial tube prior to the involution of the surrounding mesenchyme over approximately the same temporal window. This suggests that the mechanisms for atresias within both of these intestinal locations are likely similar.
The loss of a transmembrane tyrosine kinase receptor (Fgfr2IIIb) is clearly the inciting genetic event leading to the developmental deficits seen in this model. How that mutation consistently results in spatially specific colonic atresias and the steps preceding cell dedifferentiation, basal lamina disruption, and potential cell movement within those regions is unclear. An increase in overall epithelial apoptosis and a lack of increased cellular proliferation necessary to overcome cellular losses is present. Since these events are not restricted solely to the distal colon (the atretic precursor) and also occur in the proximal colon (which does not form an atresia) there must be other cellular events that cause the involution at the distal colon. Cellular-ECM interactions are complex and critical in both maturing ECM architecture and driving cellular differentiation. Thus, it is possible that a select group of epithelial cells critical for maintaining the functionality of the basal lamina is lost during the apoptotic period at E10.5. Alternatively, Fgfr2IIIb signaling may be critical for generating specific components of the ECM in the distal colonic region that are necessary to ensure progression of epithelial cells through normal development. Either scenario could disrupt normal development and result in epithelial cellular migration and atresias. There may also be a change in coordination of signaling molecules such as Wnt5a due to Fgfr2IIIb signaling disruption that prevents elongation and proper intestinal organogenesis (Cervantes et al., 2009; Kumawat and Gosens, 2016; Gao et al., 2018).
Based on our observations, we propose that atresias in this model result from both an increase in overall epithelial apoptosis and a failure of cells in the atretic precursor region to maintain proper epithelial differentiation. An increase in apoptosis and a lack of an increase in cellular proliferation to account for the cellular loss results in an unstable epithelial environment and causes disruption to the basal lamina. We suggest that this volatile environment may allow some epithelial cell to subsequently migrate out of the epithelium into the surrounding mesenchyme through breaks in the basal lamina. Such movement in other systems is well documented and termed epithelial to mesenchymal transition (EMT) (Lamouille et al., 2014). EMT has been shown to play a role in embryonic development (Carmona et al., 2013), and is associated with a loss of cell-cell adhesion and cellular polarity (Moreno-Bueno et al., 2008) as we have observed in this model. The exact role apoptosis plays in this process is not fully clear. Cellular death and the resulting chemical signals may be a factor in the failure of cells to progress properly during development, initiating or intensifying the breakdown of the basal lamina, act as a cue for cellular migration, or may impact all these processes. Other cells may simply lack the proper cues to maintain epithelial identity and slough off into the lumen of the null intestine. We therefore suggest that a combination of migration and a loss of adhesion and epithelial identity (separate from apoptosis) further adds to a loss of the epithelial tube and the involution of the surrounding mesenchyme.
4. Experimental Procedures
Animal care
IACUC approval for these studies was obtained from the University of Wisconsin School of Medicine and Public Health (P.F.N. protocol # M02258). All animals were maintained in a clean facility with ad libitum access to fresh food and water under a 12-hour alternating light/dark cycle.
Generation of embryos
Mice were bred on a C57BL6 background. Using the previously described HPRT-Cre system (Tang et al., 2002; Nichol et al., 2011a) Fgfr2IIIb −/−(null) embryos were generated by mating Fgfr2IIIbflox/+; HprtCre/+females with Fgfr2IIIbflox/flox males. Fgfr2IIIb flox/flox (control) embryos were generated within the same litters without the HPRT-Cre allele. Embryos were harvested at embryonic day (E) 9.5, E10.5, E11.5, and E12.5. Once harvested in cold PBS, tissue was taken for genotyping, and the embryos were fixed in a 4% paraformaldehyde solution at 4°C.
Apoptosis
Control and null embryos at E9.5-E12.5 were dehydrated, embedded in paraffin, microtome sectioned at 5μm, and subject to an apoptosis-detection assay (DeadEnd™ Fluorometric TUNEL System, Promega Corp., Madison, WI). Fluorescent images were obtained on a minimum of 3 embryos per condition on a Nikon® E600 microscope equipped with an Olympus® D973 camera. The total number of cells and number of cells staining positively for apoptotic activity were counted. The mesenchyme was subtracted from the images using Adobe Photoshop® to allow for a three-dimensional reconstruction of the epithelial cells of intestinal tracts using ImageJ (ImageJ, https://imagej.nih.gov/ij/).
Proliferation
Control and null embryos were harvested in the same fashion as with the apoptosis assay, at time points E10.5 and E11.5. Once sectioned, immunofluorescence was performed to detect phosphohistone-H3 (PHH3), a marker for active mitotic proliferation (M-phase). Rabbit polyclonal antibody against PHH3 was applied at 1:100 concentration (Sigma-Aldrich catalog #369A-15) overnight at 4°C followed by a one-hour room temperature incubation with goat anti-rabbit Alexa Fluor 594 at 1:250 concentration (Jackson ImmunoResearch catalog #111-585-144). Fluorescent images were obtained on a Nikon® E600 microscope equipped with an Olympus® D973 camera, and counts were conducted as described above for apoptotic staining. Fluorescent images were obtained on a minimum of 3 embryos per condition. For three-dimensional reconstruction of the colonic epithelial layer, the mesenchyme was subtracted from the images using Adobe Photoshop® and three-dimensional reconstruction of the epithelial cells of intestinal tracts was completed using ImageJ (ImageJ, https://imagej.nih.gov/ij/).
Immunohistochemistry
Laminin/E-cadherin/vimentin/alpha smooth muscle actin/β-catenin staining
Control and null embryos were harvested at E11.5, fixed in 4% paraformaldehyde, dehydrated through a series of escalating PBS-Tween/methanol steps, embedded in paraffin, sectioned at 5μm and subject to immunohistochemistry for E-cadherin, laminin, and DAPI. Antigen retrieval was performed using a heat induced citrate buffer (Thermo Fisher Scientific catalog #AP-9003-500). Mouse anti-E-cadherin antibody (BD Biosciences catalog #61081) and rabbit anti-laminin antibody (Sigma-Aldrich catalog #L9393) was applied at 1:100 concentration overnight at 4°C followed by one hour incubation in secondary antibodies at 1:200 concentration with goat anti-mouse Alexa Fluor 488 (Jackson ImmunoResearch catalog #155-545-146) and goat anti-rabbit Alexa Fluor 546 (Thermo Fisher Scientific catalog #A11035). A mounting media with DAPI was used for nuclear staining (Vector Laboratories catalog #H-1200). Subsequent slides were imaged on a confocal microscope (Leica® SP8 spectral confocal microscope fitted with a 20x oil immersion objective and images were captured using Leica® LASX 8 software).
Vimentin, alpha smooth muscle actin, and β-catenin antibody staining followed this same overall protocol. Mouse monoclonal anti-β-catenin was applied at 1:100 concentration (BD Transduction Labs catalog #610153). Mouse monoclonal anti-vimentin was applied at 1:250 concentration (Abcam catalog #20346). Mouse monoclonal anti-smooth muscle actin was applied at 1:200 concentration (Leica catalog #SMA-L-CE). Rabbit monoclonal anti-E-cadherin was applied at 1:200 concentration (Cell Signaling Technologies Catalog #3195S).
E-cadherin colorimetric detection using a DAB kit
Control and null embryos were harvested at E11.5, fixed, dehydrated, embedded in paraffin, sectioned at 5μm as described for laminin staining and subject to immunohistochemistry for E-cadherin. Antigen retrieval was performed using a heat induced citrate buffer, slides were rinsed in 3% hydrogen peroxide/PBS to block endogenous peroxidase, washed in 0.5% Triton X-100/PBS, nonspecific background staining blocked using 10%BSA incubation of 60 minutes, and incubated overnight at 4°C at 1:50 concentration using monoclonal mouse anti-E-cadherin antibody (BD Pharmingen catalog #610181). Slides were then incubated in Mach 2 goat anti-mouse IgG HRP conjugated polymer (Biocare Medical catalog #MHRP520H) for 40 minutes, developed with DAB (Dako’s DAB substrate-chromogen system catalog #K3466) for two minutes, washed, and counterstained with hematoxylin (Sigma-Aldrich catalog #HHS128). Slides were imaged on a Nikon® Eclipse 80i light microscope.
Acknowledgements
We would like to thank and acknowledge the work of Zach Erlichman for his valuable work of preparing some of the tissue samples for TUNEL staining and analysis and Drew Roenenburg for his assistance with E-cadherin/DAB counterstained slide preparation.
Grant Sponsors:
UW-Madison, Surgery Department, 233-AAA4927-539790
UW-Madison, UW Institute for Clinical and Translational Research (ICTR), 233-AAB8495-539790
Contributor Information
Anna Kowalkowski, Surgery Department, University of Wisconsin, Madison, WI, USA.
Krzysztof M. Zaremba, Surgery Department, University of Wisconsin, Madison, WI, USA
Andrew P. Rogers, Surgery Department, University of Wisconsin, Madison, WI, USA
Olivia R. Hoffman, Surgery Department, University of Wisconsin, Madison, WI, USA
Anne E. Turco, Department of Comparative Biosciences, University of Wisconsin, Madison, WI, USA
Peter F. Nichol, Surgery Department, University of Wisconsin, Madison, WI, USA
References
- (ICBDSR) ICFBDSAR. INTERNATIONAL CLEARINGHOUSE FOR BIRTH DEFECTS SURVEILLANCE AND RESEARCH Annual Report 2014. http://www.icbdsr.org/wp-content/annual_report/Report2014.pdf. Published 2014. Accessed September 10, 2018.
- Nichol PF, Reeder A, Botham R. Humans, mice, and mechanisms of intestinal atresias: a window into understanding early intestinal development. J Gastrointest Surg. 2011;15(4):694–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupo PJ, Isenburg JL, Salemi JL, et al. Population-based birth defects data in the United States, 2010-2014: A focus on gastrointestinal defects. Birth Defects Res. 2017;109(18):1504–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi D, Hiroma T, Takamizawa S, Nakamura T. Population-based study of esophageal and small intestinal atresia/stenosis. Pediatr Int. 2014;56(6):838–844. [DOI] [PubMed] [Google Scholar]
- Dalla Vecchia LK GJ, West KW, Rescorla FJ, Scherer LR, Engum SA. Intestinal atresia and stenosis: A 25-year experience with 277 cases. Archives of Surgery. 1998;133(5):490–497. [DOI] [PubMed] [Google Scholar]
- Tandler J Zur Entwicklungsgeschichte des menschlichen Duodenum in fruhen Embryonalstadien. Morphol Jahrb. 1900;no. 2:187. [Google Scholar]
- Barnard C, Louw J . The genesis of intestinal atresia. Minn Med. 1956;39(11):745. [PubMed] [Google Scholar]
- Barnard C The genesis of intestinal atresia. Surg Forum. 1957;7:393–396. [PubMed] [Google Scholar]
- Louw J, Barnard C. . Congenital intestinal atresia; observations on its origin Lancet. 1955;269(6899):1065–1067. [DOI] [PubMed] [Google Scholar]
- Khen N, Jaubert F, Sauvat F, et al. Fetal intestinal obstruction induces alteration of enteric nervous system development in human intestinal atresia. Pediatr Res. 2004;56(6):975–980. [DOI] [PubMed] [Google Scholar]
- Celli J Genetics of gastrointestinal atresias. Eur J Med Genet. 2014;57(8):424–439. [DOI] [PubMed] [Google Scholar]
- Fernandez I, Patey N, Marchand V, et al. Multiple intestinal atresia with combined immune deficiency related to TTC7A defect is a multiorgan pathology: study of a French-Canadian-based cohort. Medicine (Baltimore). 2014;93(29):e327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar JA, Suñol M, de Torre BL, Camarero C, Torrado J. Mucosal morphology in experimental intestinal atresia: Studies in the chick embryo. Journal of Pediatric Surgery. 1991;26(2):184–189. [DOI] [PubMed] [Google Scholar]
- Lopez de Torre B, Tovar JA, Uriarte S, Aldazabal P. The nutrition of the fetus with intestinal atresia: Studies in the chick embryo model. Journal of Pediatric Surgery. 1992;27(10):1325–1328. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Melton DA, McMahon AP. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development. 2000;127(12):2763. [DOI] [PubMed] [Google Scholar]
- Mo R, Kim JH, Zhang J, Chiang C, Hui CC, Kim PC. Anorectal malformations caused by defects in sonic hedgehog signaling. The American journal of pathology. 2001;159(2):765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merei JM. Embryogenesis of adriamycin-induced hindgut atresia in rats. Pediatric Surgery International. 2002;18(1):36–39. [DOI] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest J, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127(3):483–492.10631169 [Google Scholar]
- Fairbanks TJ, Kanard RC, De Langhe SP, et al. A genetic mechanism for cecal atresia: the role of the Fgf10 signaling pathway. J Surg Res. 2004;120(2):201–209. [DOI] [PubMed] [Google Scholar]
- Kanard RC, Fairbanks TJ, De Langhe SP, et al. Fibroblast growth factor-10 serves a regulatory role in duodenal development. J Pediatr Surg. 2005;40(2):313–316. [DOI] [PubMed] [Google Scholar]
- Reeder AL, Botham RA, Franco M, Zaremba KM, Nichol PF. Formation of intestinal atresias in the Fgfr2IIIb−/− mice is not associated with defects in notochord development or alterations in Shh expression. J Surg Res. 2012;177(1):139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botham RA, Franco M, Reeder AL, et al. Formation of duodenal atresias in fibroblast growth factor receptor 2IIIb−/− mouse embryos occurs in the absence of an endodermal plug. J Pediatr Surg. 2012;47(7):1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks TJ, Sala FG, Kanard R, et al. The fibroblast growth factor pathway serves a regulatory role in proliferation and apoptosis in the pathogenesis of intestinal atresia. J Pediatr Surg. 2006;41(1):132–136; discussion 132-136. [DOI] [PubMed] [Google Scholar]
- Sukhotnik I, Bernshteyn A, Mogilner JG. The basic biology of apoptosis and its implications for pediatric surgery. Eur J Pediatr Surg. 2005;15(4):229–235. [DOI] [PubMed] [Google Scholar]
- Sala FG, Curtis JL, Veltmaat JM, et al. Fibroblast growth factor 10 is required for survival and proliferation but not differentiation of intestinal epithelial progenitor cells during murine colon development. Dev Biol. 2006;299(2):373–385. [DOI] [PubMed] [Google Scholar]
- Nichol PF, Botham R, Saijoh Y, Reeder AL, Zaremba KM. A more efficient method to generate null mutants using Hprt-Cre with floxed alleles. J Pediatr Surg. 2011;46(9):1711–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeder AL, Botham RA, Zaremba KM, Nichol PF. Haploinsufficiency of retinaldehyde dehydrogenase 2 decreases the severity and incidence of duodenal atresia in the fibroblast growth factor receptor 2IIIb−/− mouse model. Surgery. 2012;152(4):768–775; discussion 775-766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CY, Schloemer AJ, Lu A, et al. Hypoxia-ischemia induces DNA synthesis without cell proliferation in dying neurons in adult rodent brain. J Neurosci. 2004;24(47):10763–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low SH, Miura M, Roche PA, Valdez AC, Mostov KE, Weimbs T. Intracellular Redirection of Plasma Membrane Trafficking after Loss of Epithelial Cell Polarity. Molecular Biology of the Cell. 2000;11(9):3045–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondow BJ, Faber ML, Wojta KJ, Walker EM, Battle MA. E-cadherin is required for intestinal morphogenesis in the mouse. Dev Biol. 2012;371(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes S, Yamaguchi TP, Hebrok M. Wnt5a is essential for intestinal elongation in mice. Dev Biol. 2009;326(2):285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Ajima R, Yang W, et al. Coordinated directional outgrowth and pattern formation by integration of Wnt5a and Fgf signaling in planar cell polarity. Development. 2018;145(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumawat K, Gosens R. WNT-5A: signaling and functions in health and disease. Cell Mol Life Sci. 2016;73(3):567–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15(3):178–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona R, Cano E, Mattiotti A, Gaztambide J, Munoz-Chapuli R. Cells derived from the coelomic epithelium contribute to multiple gastrointestinal tissues in mouse embryos. PLoS One. 2013;8(2):e55890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27(55):6958–6969. [DOI] [PubMed] [Google Scholar]
- Tang SH, Silva FJ, Tsark WM, Mann JR. A Cre/loxP-deleter transgenic line in mouse strain 129S1/SvImJ. Genesis. 2002;32(3):199–202. [DOI] [PubMed] [Google Scholar]