

Graphical Abstract
Electrophilic aromatic substitution produces edge-specific modifications to CVD graphene and graphene nanoplatelets that are suitable for specific attachment of biomolecules
Electronic supplementary material
The online version of this article (10.1007/s10853-020-04662-y) contains supplementary material, which is available to authorized users.
Introduction
Graphene, a “two-dimensional” material made of sp2-hybridised carbon, is an attractive platform for nanomedicine, including drug delivery [1], theranostics [2], non-viral gene transfer [3], regenerative medicine [4, 5], sensors [6], and bioelectronics [7], because of its unique combination of properties including high carrier mobility [8], high yield strength [9, 10], and facile chemical modification [11]. Chemical modifications are useful for tuning graphene’s solvent dispersibility and for providing chemically reactive attachment points for further modifications, such as bonding to a matrix in a nanocomposite [12] or the attachment of biomolecules [13, 14].
Most existing covalent functionalisation of graphene family nanomaterials is based on grafting molecules through oxygen-containing functional groups of graphene oxide (GO), followed by chemical or thermal reduction to obtain reduced graphene oxide (rGO) [15, 16]. Graphene oxide (GO) is an oxidised, exfoliated form of graphite with a prevalence of oxygen-containing functional groups (carboxyl, hydroxyl and epoxide) on its exfoliated sheets. GO has become widely applied because it provides hydrophilic functional groups that allow it to form a stable dispersion in aqueous and polar solvents [17]. The oxidation process, however, generates defects on the GO sheets which disrupt π–π conjugation, leading to the loss of mechanical strength, as well as reduced electrical and thermal conductivity [18, 19]. Young’s modulus for GO is five times lower than that of single-layer graphene [20], and even the most conductive rGO has a carrier mobility ~ 103 times lower than pristine graphene [21]. GO-based materials are not well suited to applications that require on robust mechanical or electronic properties.
Functionalisation of pristine graphene can be achieved by both covalent and non-covalent interactions [15]. Pristine graphene has been predominantly modified through non-covalent methods because it possesses an extended π system which forms attractive hydrophobic and π–π interactions with aromatic molecules [15, 22]. Pristine graphene has been coated with pyrene [23, 24], pyridinium tribromide [25], triphenylene [26], and coronene [27], as well as biological surfactant molecules such as phospholipids and cholesterol [28]. Although these modifications overcome graphene’s problem of poor dispersibility in buffered aqueous media, surfactant molecules remain adsorbed on the surface and influence the biocompatibility [28] and conductivity [29] of the product.
Covalent modification of pristine graphene can be achieved through free radical addition to sp2 carbons on the basal plane using diazonium salts [30–37] or benzoyl peroxide [38] to form radicals. Graphene has also been covalently linked to dienes and dienophiles using the Diels–Alder reaction [39–47]. This reaction generally disrupts graphene’s extended π system and so degrades the product’s conductivity due to the conversion of sp2 carbons to sp3 carbons [15, 22, 33, 48], but some exceptions have been discussed based on DFT calculations [48, 49].
Edge-selective covalent functionalisation minimises chemical reactions on graphene’s basal plane and the attendant deterioration of engineering properties. Ball milling graphite in the presence of gases or gas mixtures has been reported to produce edge-specific functionalisation [50]. The technique produces reactive species (e.g. radicals, cations, and anions) that react with defects introduced in the graphite as it is being broken down by ball milling. The process has been used to introduce sulphonic acids [50], carboxylic acids [50–52], phosphonic acids [53], and halogens to the graphite [54]. However, ball milling process can generate a violent sparking reaction caused by active carbon species, metallic debris, and moisture in the air [55]. Ball milling also can introduce metallic residues from the balls, which require acidic work-up to remove [55]. An alternative method for achieving the edge-specific functionalisation of pristine graphene and GO is Friedel–Crafts acylation [12, 56, 57], where an acyl chloride with a Lewis acid such as AlCl3 is traditionally used. Benzoic acid derivatives with polyphosphoric acid and phosphorus pentoxide have been used for milder graphene acylations [12, 56, 57]. However, the presence of the carbonyl group which arises from Friedel–Crafts acylation may slow electron transfer between the graphene sheet and groups attached to its edges [58–60]. Other edge-functionalised graphene-type materials have been made from bottom-up methods such as Suzuki coupling of bromine-containing polyphenylene precursors followed by intramolecular oxidative cyclodehydrogenation [61]. Furthermore, edge-selective functionalisation of graphene monolayers treated by oxygen plasma can also be achieved electrochemically [52].
The current work extends the top-down method of electrophilic aromatic substitution to create five distinct types of edge-modified graphene with no intermediate carbonyl moiety. Our modifications significantly modified the graphene nanoparticles’ dispersibility. The synthetic utility of the directly attached reactive moieties was demonstrated by creating a “glycographene” through radical addition of allyl mannoside to edge-thiolated graphene. Chemical modifications were confirmed by FT-IR and XPS. Edge localisation was visualised on modified CVD graphene samples by scanning electron microscopy of gold nanoparticles attached to thiol groups, epifluorescence microscopy of a fluorescently tagged lectin–glycographene bioconjugate, and quenched stochastic optical reconstruction microscopy (qSTORM) [62–66], a super-resolution technique, of thiol-reactive fluorophores on edge-thiolated graphene.
Experimental methods
Materials
Graphene/graphite nanoplatelets (GNPs; grades C750, C300, and M25) were purchased from XG Sciences (Lansing, MI, USA). The C750 and C300 grades have nominal specific surface areas of 750 m2 g−1 and 300 m2 g−1, respectively, corresponding to 4–5 graphene layers for C750 and 8–9 layers for C300. The batches used in this work had BET specific surface areas of 794.9 ± 1.6 m2 g−1 and 268 m2 g−1. The M25 grade has a nominal lateral dimension of 25 µm. The specific surface area specified by the manufacturer is 120–150 m2 g−1, corresponding to about 17–22 graphene layers; the batch used in this work had a BET specific surface area of 55 m2 g−1. On receipt, the GNPs were portioned into 50-ml Falcon tubes and sealed with Parafilm.
Monolayer graphene produced by chemical vapour deposition (CVD) on copper was provided by 2-DTech (Manchester, UK) and transferred as ~ 1 cm2 sheets to a silicon wafer covered with 300 nm thermal oxide. Before use, CVD graphene was stored in a covered Petri dish under ambient conditions.
Graphene dispersions were also prepared by ultrasonic exfoliation of natural graphite flakes (Branwell Graphite, Ltd, Grade 2369). Following the method of Hernandez [67], 2 g graphite was sonicated in 500 ml N-methyl-2-pyrrolidone (NMP) at 37 Hz for 48 h in an ultrasonic bath (Elmasonic PH750EL). Remaining graphite was removed by cetrifugation (3 × 20 min at 4000 rpm). The supernatant was a graphene dispersion (ca. 0.4 g l−1). Graphene reaction dispersions were vacuum filtered through 0.02-µm Whatman Anodisc membrane filters to create graphene laminates. For analysis, samples were dried as laminates on the filter membrane or re-dispersed in water by sonication and freeze-dried (HETO PowderDry LL1500 Freeze Dryer, Thermo Electron Corporation). For long-term storage, the samples were kept in a desiccator at room temperature.
N,N-dimethylformamide (DMF, 99%) and methanol were purchased from Fisher Scientific, UK. Sodium borohydride (≥ 96%), iodine (≥ 99.8%) and triphenylphosphine (99%) were purchased from Sigma-Aldrich. Raney nickel catalyst (50% in water) and chlorosulphonic acid (1.75 g cm−3, ≥ 98%) were purchased from Merck Chemicals. All chemicals were used as supplied. Water was purified to a resistivity of 18.2 MΩ cm at 25 °C (Milli-Q).
SnakeSkin dialysis tubing (10 kDa MWCO, 35 mm dry ID) was purchased from Thermo Scientific. Disposable folded capillary cells for Zetasizer Nano Series were purchased from Malvern.
Synthesis of edge-modified graphene
Modifications to graphene nanoplatelets
Sulphonated graphene (G–SO3−, Scheme 1) was synthesised by suspending 500 mg GNPs in 10 ml neat chlorosulphonic acid at room temperature. The temperature was increased to 100 °C, and the suspension was stirred for 20 h. After cooling, the reaction mixture was poured into ice water and then neutralised using aqueous NaOH and universal pH paper. The material was dialysed by pouring ~ 100 ml of neutralised suspension into dialysis tubing and suspending it in ~ 2 l stirred deionised water. The water was changed 3 times at intervals of at least 8 h. The deionised suspension was filtered, resuspended in water, and then freeze-dried.
Scheme 1.

Synthesis of graphene sulphonate (G–SO3−) by electrophilic aromatic substitution, followed by its subsequent reduction to a thiol-containing form (G–SH), coupling to allyl mannoside, and selective binding of a ConA lectin tetramer (PDB: 5CNA)
Thiolated graphene (G–SH, Scheme 1) was synthesised by suspending 50 mg of graphene sulphonate (G-SO3) in 30 ml of toluene and sonication under N2 atmosphere for 15 min. Then, the reaction flask was connected to a condenser with nitrogen flow. 2.5 g of triphenylphosphine and 200 mg iodine were added into the mixture that was left stirring at 80 °C for 21 h. The product was filtered using vacuum filtration and 0.45-μm HV membrane filter and washed with toluene, acetone, 0.1 M sodium thiosulphate solution, and deionised water, followed by freeze-drying.
Modifications to CVD graphene
Before modification, samples were soaked in acetone for 30 min to remove residual PMMA and dried and then transferred to 20% v/v chlorosulphonic acid solution in DMF for 30 s at room temperature. Samples were then washed with deionised water and acetone to remove any remaining acid and hydrolysed by immersing in pH 10 NaOH solution for 15 min. These were washed with water and acetone and dried in a nitrogen stream, leaving CVD graphene sulphonate.
Thiolated CVD graphene was produced by immersing a sulphonated sample for 15 min in 10 ml anhydrous toluene containing 10 mM triphenylphosphine and 0.6 mM iodine. A nitrogen atmosphere was maintained throughout the procedure by flushing with a strong flow of dry house nitrogen. The sample was removed and washed with distilled water and acetone and dried as with sulphonate. Thiolated graphene that was not immediately subsequently functionalised was kept under vacuum to prevent thiol oxidation.
Gold nanoparticles (Aldrich, 20 nm diameter in 0.1 M phosphate-buffered saline, ~ 6 × 1011 particles ml−1) were bound to thiolated CVD graphene. The nanoparticle solution was diluted tenfold in phosphate-buffered saline (PBS pH 7.4, 0.01 M phosphate, 0.138 M NaCl, 2.7 mM KCl). A sample of modified CVD graphene was immersed in 10 ml of the diluted nanoparticle suspension overnight at ambient temperature. The sample was rinsed with water and acetone and dried in a nitrogen gas stream and stored at room temperature.
Glycographene (Scheme 1) was made by immersing thiolated CVD graphene in ethanol containing 150 mg (682 µmol) allyl mannoside and one spatula benzoyl peroxide overnight at 65 °C. The CVD graphene sample was rinsed with ethanol, water and acetone, dried in a nitrogen gas stream, and stored at ambient temperature. Concanavalin A labelled with fluorescein isothiocyanate (FITC-ConA) was dissolved in binding buffer (20 mM Tris, 500 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, pH 7.2) in a glass vial wrapped in aluminium foil. The CVD glycographene sample was added to the vial and incubated at 37 °C for 2 h. The sample was rinsed with water and dried in a nitrogen gas stream.
Characterisation
Raman spectra were recorded from 100 to 3200 cm−1 using a Renishaw inVia, with a 633-nm excitation laser set to 10% (0.89 mW) power or a 514-nm laser (0.176 mW). The energy resolution was 0.3 cm−1. Raman shifts were calibrated by setting the G (E2g) peak from silicon to 1581 cm−1 [68, 69]. The use of two excitation wavelengths led to positive shifts in the D and 2D peak positions when using the higher energy green excitation [69, 70]. Raman maps of modified CVD graphene samples were recorded on a Renishaw InVia with a 633-nm laser through a 50 × Leica NPLAN EPI objective (NA = 0.75). Spectra were acquired from 1000 to 3000 cm−1 every 0.5 µm (25 accumulations, 20 s exposure, 1%/0.016 mW laser power). Data were processed using WiRE 4.2 software to zero the baseline and remove cosmic rays. Nonlinear fits (SI) were applied to the peaks following the methods of Puech et al. [71]. Domain sizes were estimated from the relative intensity of the D band (i.e. ID/IG) using the Tuinstra–Koenig relationship (, Eq. S1) [72], the relative area of the D band (i.e. AD/AG) using the relationship from Cançado et al. (, Eq. S2) [73], and the HWHM of the D peak (, Eq. S3) [74]. The average spacing between point defects (LD) was estimated using the expression from Lucchese et al. (Eq. S4) [75].
X-ray photoelectron spectra (XPS) were acquired using a Kratos Axis Ultra with an Al Kα source (1486.6 eV) operated at 15 kV and 10 mA. The pressure of the vacuum chamber was below 5 × 10−8 mbar during measurements. Peaks were fit using CasaXPS with Shirley background correction. The C 1s energy was calibrated by fixing the binding energy for the sp2 component to 284.5 eV. The C 1s peak was fit as five components summarised in Table 1, which consistently fit the data adequately for all samples [76]. Uncertainty estimates in the sp3:sp2 C ratio were from Monte Carlo simulations within CasaXPS. Fits to 2p1/2 and 2p3/2 peaks were constrained to have identical FWHM values and an area ratio of 1:2.
Table 1.
Fit parameters used for deconvoluting the XPS spectra for C 1s peaks
| Chemical identity (binding energy) [76] | Line shapea | Binding energy constraint | FWHM constraint |
|---|---|---|---|
| C 1s sp2 (284.5 eV) | LA (1, 1.6, 50) | None | None |
| C 1s sp3 (284.8 eV) | GL (30) | BE (sp2) + 0.3 eV | None |
| C 1s C–O (285.5–286.5 eV) | GL (30) | BE (sp2) + 2 eV | Same as sp3 |
| C 1s C=O (287.5–288.9 eV) | GL (30) | BE (sp2) + 4 eV | Same as sp3 |
| C 1s π–π* (290–292 eV) | GL (30) | 290–292 eV | None |
aGL (30) is a symmetric lineshape that is 30% Lorentzian and 70% Gaussian. LA (1, 1.6, 50) is an asymmetric Lorentzian lineshape numerically convoluted with a Gaussian; at binding energies above the peak maximum, the Lorentzian function is taken to the 1.6 power [77]
FT-IR samples were prepared by mixing approximately 0.2 mg modified GNPs with 300 mg KBr (spectroscopic grade, 99%; Acros Organics) using an agate mortar and pestle then pressed at 10 tons from a hydraulic press for 5 min to obtain a sample disc. Transmission spectra (4000–400 cm−1, 32 scans, 4 cm−1) were acquired using a Nicolet 5700 FT-IR spectrometer in air. Background spectra were recorded every 10 min.
STORM images were taken using a previously described custom-built STORM system [64], consisting of an Olympus IX-71 inverted fluorescence microscope with Olympus UAPON 100XOTIRFM (NA = 1.49) TIRF oil immersion objective lens in an epi-illumination geometry. Sample movement was controlled using a motorised x–y stage (PRIOR HLD117) and a PRIOR ProScan III controller. The sample was illuminated by laser beams delivered to the back of the microscope using an optical fibre. A vibration motor was attached to the fibre to remove coherent artefacts in the final image due to laser speckle. Light from the sample was collected by the lens and incident on a Hamamatsu ORCA Flash v2 sCMOS camera. Up to 20000 individual images were acquired for each area with an exposure time of 10 ms. External and internal edges of thiolated CVD graphene were labelled with BODIPY FL L-cystine (Thermo Fisher, λex = 505 nm, λem = 512 nm, εmax = 265 mM−1 cm−1) and Alexa Fluor 647 maleimide (Thermo Fisher, λex = 651 nm, λem = 671 nm, εmax = 134 mM−1 cm−1). The graphene samples were immersed in 10 mL deionised water to which 10 mM dye stock in DMSO was added for 2 h at room temperature. After coupling, the samples were washed with water and acetone, dried under a nitrogen stream, and kept at 4 °C and away from light. Image data were processed using ImageJ with the ThunderSTORM plugin [78].
Epifluorescence images were collected on an Olympus BX51 upright microscope using UPlanFLN objectives and captured using a Coolsnap camera (Photometrics) through MetaVue software (Molecular Devices). A specific bandpass filter set for FITC (excitation BP480/40, dichroic Q505LP, emission 535/50) was used.
Scanning electron microscopy (SEM) images were acquired using an FEI Quanta 650 FEG-SEM operating at 1–5 kV. CVD graphene samples on SiO2/Si were mounted on 12.5-mm aluminium stubs. No surface coatings were applied.
Specific surface area was determined from the Brunauer–Emmett–Teller (BET) method using a Micromeritics Gemini V surface area and pore size analyser.
Thermogravimetric analysis (TGA) was performed on a TA Instruments Q500 thermogravimetric analyser. 1–3 mg of graphene sample was heated at 10 °C min−1 from 30 to 800 °C in a N2 atmosphere.
Zeta potential was determined using a Malvern Zetasizer Nano series. Aqueous graphene suspensions (0.05 mg ml−1) in deionised water were prepared and placed in disposable foldable capillary cells for testing. The measurement was repeated 6 times for each type of functionalised graphene.
Estimates of the interfacial electron transfer rate (k0) to pristine and edge-modified CVD graphene samples were carried out in a two-electrode set-up described Valota et al. [79] and Velický et al. [80]. The CVD graphene samples (Graphena) acted as the working electrode when wetted. Graphene was connected to a copper wire (99.9%, 0.15 mm diameter) using a two-part silver-loaded epoxy (RS Components Ltd) cured for 24 h. The connection was coated with a non-conductive epoxy resin (Araldite Rapid) to increase its robustness. An Ag|AgCl quasi-reference electrode was produced by partially exposing Ag in a PTFE-coated wire (99.99%, 0.15 mm diameter) and oxidising it in 0.5 M HCl. The reference was immersed in solution at the top of the pipette and connected to the potentiostat (PGSTAT302N, Metrohm Autolab). A two-electrode set-up, in which the counter and reference electrode are the same, was acceptable because of the small, reversible currents.
Micropipettes were created by pulling borosilicate glass with a micropipette puller (P-87, Sutter Instrument Co.) at 550 °C. The micropipette was filled with 5 mM potassium ferricyanide (99%, Sigma-Aldrich) in 6 M LiCl aqueous solution (99%, Sigma-Aldrich). A high concentration of LiCl was used to prevent droplet evaporation [80]. The filled micropipette was then connected to a pump to control droplet deposition (PV820 Pneumatic PicoPump, WPI) with argon (BOC Industrial Gases, 99.998%).
A camera with a microscope objective (N Plan Apo, Leica Microsystems) and light source were positioned parallel to the sample surface (Figure S7, Supporting Information). Droplet deposition was monitored and recorded using Infinity Analyze software (v.4.2) (Figure S8). A second camera above the sample provided low-resolution, live imaging of the sample to identify areas for deposition. After depositing a droplet, cyclic voltammetry was run using NOVA software (v. 1.11). An uncertainty of 24.4 mV was assigned according to the step size during the scan. Cyclic voltammograms (CVs) were conducted for seven scan rates in nine separate droplets. A set of measurements on a single droplet typically required ca. 30 min, an important consideration in light of the observations by Patel et al. that Fe(CN)4−6 adsorption on highly oriented pyrolytic graphic (HOPG) lowered electron transfer rates [59]. Typical CVs are shown in Figure S9. The Klingler–Kochi equation was used to calculate k0 values [81]. A diffusion coefficient of 1.84 × 10−6 cm2 s−1 was used based on previous measurements using the Randles–Ševčík equation [80]. Some electrowetting effects were observed (Figure S10), consistent with others’ reports [82, 83].
Results and discussion
Graphene/graphite nanoplatelets (GNPs) and monolayer CVD graphene were treated with chlorosulphonic acid and then hydrolysed with base to produce edge-modified graphene sulphonate (G–SO3−, Scheme 1). The corresponding edge-modified graphene thiol (G–SH) was produced by reduction with triphenyl phosphine and iodine.
Using a combination of analytic techniques was essential to establish not only the identity of the functional groups on the graphene, but also their location on the edges of the graphene.
FT-IR analysis (Fig. 1a) was best suited to confirming the presence of functional groups. Sulphur signatures are typically less pronounced in the IR spectrum. Nonetheless, the low-energy region contained the appropriate C–S and S=O vibrations for both the G–SO3− and the G–SH, and the G–SH showed a characteristic broad, weak S–H stretch at 2600 cm−1. The chemical presence of reactive sulphhydryl groups was further confirmed visually through an Ellman’s assay, in which the reagent becomes yellow coloured in the presence of free thiol groups through a thiol–disulphide exchange reaction. Figure 2 shows that G–SH and a cysteine positive control both produced a colour change, while G–SO3− did not.
Figure 1.

Characterisation of the chemical functionality on edge-modified C750 GNPs by a FT-IR and b XPS. XPS survey and C 1s scans are presented in Figure S3
Figure 2.

Ellman’s assay for the presence of free sulphhydryl (thiol) groups applied to edge-modified GNPs produced by ultrasonic exfoliation: a negative control containing 30 µl of graphene sulphonate and 10 µl Ellman’s reagent solution; b negative control containing 30 µl thiographene and 0 µl Ellman’s reagent solution; c 30 µl thiographene and 5 µl Ellman’s reagent solution; d 30 µl thiographene and 10 µl Ellman’s reagent solution; e positive control containing 30 µl 260 mM cysteine and 10 µl Ellman’s reagent solution. [Ellman’s reagent] = 10 mM; [graphene suspensions] = 0.5 g l−1
The XPS of sulphur-modified samples (Fig. 1b) showed the expected signatures for both sulphonate and thiolate groups. Both peaks were clearly asymmetric and could be well described by a deconvolution into S 2p1/2 and S 2p3/2 peaks. The sulphonate S 2p peaks appeared at higher binding energy (169.5 ± 0.2 eV and 168.1 ± 0.2 eV) than the thiol S 2p peaks (165.1 ± 0.2 eV and 164.0 ± 0.2 eV). The S 2p spectrum for G–SH showed that the reduction of the sulphonate to the thiol was incomplete (–SO3−/–SH = 2.8 in the example shown). Quantification of the XPS survey spectra for G–SO3− and G–SH gave a S:C atomic ratio of about 0.3:100 (Table 2).
Table 2.
Physicochemical parameters for the pristine C750 GNPs and edge-modified derivativesa
| Sample | Raman ID/IbG | LD/nmc | XPS sp3:sp2 atomic ratiod | XPS S:O:C atomic ratioe | TGA mass change 100–900 °Cf (%) | TGA mass loss peak(s) °Cf |
|---|---|---|---|---|---|---|
| G | 0.86 | 9 | 0.108 ± 0.011 | —:1.2:100 | − 10.1 | None |
| G–SO3− | 0.54 | 11 | 0.337 ± 0.008 | 0.33:11.7:100 | − 17.4 | 244 (br), 668 |
| G–SH | 0.69 | 10 | 0.763 ± 0.118 | 0.46:7.0:100 | − 21.5 | 221 (br), 356 (br), 447 |
aAdditional analyses are given in Table S1, Table S2, Table S5, and Table S6
bValues taken from spectra shown in Figure S4
cAverage distance between defects (Figure S4, Table S5, and Eq. S4)
dTaken from the deconvolution of C 1s scans shown in Figure S3
eTaken from the quantification of survey scans shown in Figure S3. A full analysis is given in Table S1
fTraces shown in Figure S17, br = broad
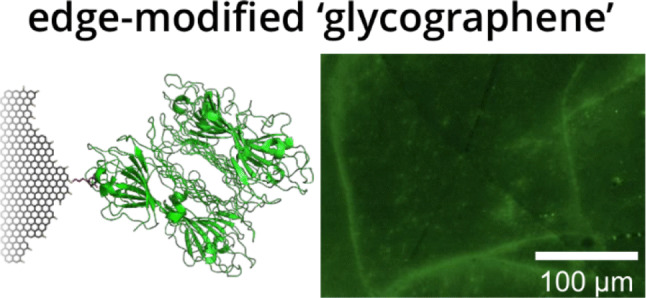
“Glycographene”, an edge-modified graphene bioconjugate, was synthesised through radical addition of allyl mannoside to G–SH (Scheme 1). This bioconjugate was used to highlight the edge specificity of the reactions. Fluorescently labelled ConA lectin (an antinutritional protein that selectively binds to α-D-mannosyl and α-D-glucosyl residues) was used to highlight the edge modification of glycographene. Figure 3 shows that while unmodified graphene adsorbs the lectin non-specifically (Fig. 3b), the lectin binds predominately to the edges of the glycographene (Fig. 3c) and can be displaced by adding excess methylmannoside substrate (Fig. 3d), which binds more strongly than the glycographene. These results are significant because they demonstrate reversible bioconjugation with graphene. This provides a platform for programmable assembly and disassembly of graphene-based nanostructures for regenerative medicine and wound healing. Additionally, surface groups and covalent functionalisation are known to affect carbon nanomaterials’ cell compatibility [28], biodistribution [84], and in vivo degradation [85, 86].
Figure 3.

Epifluorescence images of CVD graphene on a silicon wafer with a surface oxide layer: a pristine graphene, showing only background fluorescence, b pristine graphene incubated with FITC-labelled concanavalin (FITC-ConA) lectin, showing the lectin predominantly adsorbed on the basal surface, c mannose-terminated glycographene incubated with FITC-ConA, showing the lectin predominantly bound to the edges, d the same sample of glycographene after incubation with excess methylmannoside (an inhibitor of lectin–conjugate binding), displacing the FITC-ConA from the graphene surface
Gold nanoparticles were also used to highlight the position of surface thiol groups on CVD G–SH, exploiting the strong Au–S bond. The nanoparticles, which appear as bright circles on the high-magnification SEM image in Fig. 4a, were concentrated at the edges of the G–SH sheets. The basal plane of the graphene also showed some nanoparticle attachment, indicative of thiol modifications at internal edge defects. Figure 4b illustrates the concentration of the nanoparticles at the edges on a larger scale. The negative control using CVD G–SO3− (Fig. 4c) showed no concentration of nanoparticles at the edges. The modification of internal edge defects is further illustrated in the super-resolution fluorescence image in Fig. 4d. The maleimido group of the Alexa Fluor probe covalently bonds to thiols. The fluorophore is visible not only along the periphery of the flake, but also in straight-line segments on the interior of the flake, consistent with the presence of line defects where the grains of CVD graphene impinge on each other. Raman maps of another sample of BODIPY-labelled CVD G–SH (Figure S6) showed lower AD/AG values near modified edges than throughout the graphene layer.
Figure 4.

Labelled edge modification sites. a SEM image of gold nanoparticles concentrated on the edges of CVD G–SH. The bare SiOx/Si support is visible in the bottom right, b wider view SEM image of the same flake of CVD G–SH, c SEM image of gold nanoparticles distributed on CVD G–SO3− as a negative control. The lighter area in the top middle is the SiOx/Si support. The darker area in the bottom middle is where the torn area of CVD graphene folded over on itself, d super-resolution STORM image of CVD G–SH labelled with Alexa Fluor 647 maleimide (red) overlaid with the epifluorescence image of the same flake (greyscale)
The fluorescence from the graphene is initially unexpected. All fluorophores less than 10 nm from graphene or GO are expected to quench their fluorescence due to resonant energy transfer (RET) [87–89], an effect that has been used for contrast enhancement in STORM [66]. This RET is suppressed at edges and other defects in graphene [90]; however, the control experiment in which the dye conjugation procedure was repeated on CVD G–SO3− showed no fluorescence. The angle between the dipole in the fluorophore and that in the graphene sheet may be low, greatly reducing the resonant energy transfer [91].
The persistence of sp2 carbon in the GNPs after edge modification was deduced from deconvolution of the C 1s peak (Table 2 and Figure S3), taking advantage of the asymmetry of the sp2 peak due to the plasmon loss. The C 1s signals required an sp3 component to fit the products of chlorosulphonation and nitration, but there remained a clear asymmetry and π–π* shake-up features. These results are consistent with visible light and fluorescence microscopy images of CVD graphene, showing that chlorosulphonation damages the sheets. Some sp2 character is restored when G–SO3− is reduced to G–SH. Few other papers correctly use an asymmetric peak shape to fit the sp2 character. Both diazonium and Diels–Alder reactions attack the basal plane of graphitic materials [32], so the papers that included a high-resolution C 1s XPS plot showed that the spectra of their modified materials had no π–π* shake-up peak and generally showed a symmetric C 1s peak component at about 285 eV, shifted higher than unmodified graphene or graphite [30, 31, 45–47]. Also, fluorescent imaging of Diels–Alder–modified CVD graphene by Chang et al. [45] showed a uniform fluorescence across their samples, in contrast to the STORM and epifluorescence images shown in the present work. AFM measurements of Diels–Alder–modified CVD graphene also showed a uniform and thick layer consistent with saturated basal plane coverage of the modifier [46].
Raman analysis of the GNPs before and after edge modifications (Table 2 and Figure S4) did not show any consistent trends in the ID/IG ratio upon modification. There was a lot of variation in this value within single batches of pristine C750 GNPs and their edge-modified analogues, consistent with the Kovtun et al.’s [92] report of wide variation in commercially supplied graphene. The G peaks from the materials produced from GNPs showed a clear D′ peak (Figure S4 and Figure S19), caused by a one-phonon defect-assisted electron–defect scattering event from edges, grain boundaries, or internal defects [69]. GNPs produced by ultrasonic exfoliation and their sulphur derivatives had comparable ID/IG ratios and equally prominent D′ features (Figure S19 and Table S5). Larger GNPs (XG C300 and M25) showed a decreasing ID/IG ratio with increasing GNP size (Figure S23 and Table S7), consistent with the Tuinstra–Koenig relationship [72]. Raman analysis of the GNPs after sulphonation showed no consistent trends in ID/IG caused by the edge modification. A Raman map of sulphonated CVD graphene (Figure S6) showed that the ID/IG ratio was lowered at the edges, demonstrating that the chemical modifications complicate this simple metric for graphene order. This effect has previously been discussed for graphene with its basal plane modified by diazonium salts [93]. Metrics based on the relative areas of the D and G peaks, namely the average domain diameter, , and average distance between point defects, LD, were more consistent. Sets of modifications to commercial GNPs and graphene produced by mechanically exfoliating graphite in NMP showed little changes in and LD on modification relative to the source materials (Table S6). For CVD graphene, LD consistently dropped by about half and decreased to about 25–30% of the original value.
Sulphonated graphene (SGnP) has been produced by chemical exfoliation of graphite in chlorosulphonic acid. The work of Abdolmaleki et al. [94] reported a higher amount of sulphur compared to G–SO3− in the present study. The higher percentage of sulphur in SGnP indicated to the introduction of a significant number of new defects which could arise from the damage of graphene structure due to the fabrication process. This was evidenced by the Raman spectra, as the ID/IG ratio was 2.72 for SGnP, in comparison with a low defect (ID/IG < 1.2) of the edge-modified G–SO3− produced in this study, regardless of whether the GNPs or graphite exfoliated in NMP was used (Table S5). The Raman 2D peak shifts to lower energy with decreasing layer number when the number of layers is below ~ 10 [69], but no change in peak shape or shift in 2D peak position was observed for any modification except for C300 GNPs (Figure S4, Figure S19, Figure S23, and Table S7). This observation contrasts with other reports in which chlorosulphonic acid or methanesulphonic acid to exfoliate graphite [94–96].
Raman analysis of CVD graphene and edge-modified derivatives (Table 3 and Figure S5) produced more easily interpretable results. Unmodified CVD graphene had no detectable D peak, and a I2D/IG ratio of over 2 and a FWHM of 24 cm−1, indicative of single-layer graphene [70]. The I2D/IG ratio falls below 1 for bilayer graphene and drops further as the layer number increases. The spectra taken from the edges of the graphene sheet had a measurable but low ID/IG and lower I2D/IG ratio, consistent with the literature [97]. Modifications increased the ID/IG ratio, consistent with the more ragged appearance of the flake by visible light microscopy (Figure S5b, inset) and STORM (Fig. 4d), but the I2D/IG ratios remained above 2.5, confirming the persistence of single-layer graphene.
Table 3.
Peak height ratios based on Lorentzian fits to the Raman spectra of unmodified and edge-modified CVD graphene samples shown in Figure S5
| Sample | ID/IG | I2D/IG |
|---|---|---|
| G (Figure S5a) | 0 | 2.91 ± 0.04 |
| G–S–S–dye (Figure S5a) | 0.09 ± 0.01 | 2.95 ± 0.04 |
| G (centre) (Figure S5b) | 0 | 2.47 ± 0.25 |
| G (edge) (Figure S5b) | 0.10 ± 0.01 | 1.52 ± 0.01 |
| G–SH (Figure S5b) | 0.50 ± 0.01 | 2.72 ± 0.02 |
Uncertainty values are based on the standard error of the fit and the rules of propagation of uncertainty. Additional analyses (peak areas, La and LD) are given in Table S5 and Table S6
Thermogravimetric analyses (TGA) of the samples over a range of 100–900 °C (Table 2 and Figure S17) showed mass losses of between a fifth and a third of the initial mass, as well as distinctive peaks in the derivative traces. The reduced sample, G–SH, showed TGA greater mass loss than starting material, which we attributed to the contribution from strongly adsorbed organic solvent.
Electrochemical measurements of k0 on unmodified CVD graphene and CVD G–SO3− and dye-labelled G–SH showed no statistically significant difference (p = 0.01) between the unmodified samples and the dye-labelled G–SH (Fig. 5). The coefficient for G–SO3− was significantly different from the others. The sulphonation may increase the number of defects which are then healed in the subsequent reduction to the thiol or the SO3− groups may increase the rate of electron transfer.
Figure 5.

Semilog box-and-whisker representation k0 values for electron transfer of droplets deposited on the basal plane of unmodified and edge-modified CVD graphene. Data plotted on a linear y-axis shown in Figure S13, Figure S14 and Figure S16. Box: 25th, 50th, 75th‰; □ = mean; whiskers = 99% CI
Measurements of electron transfer rates to graphite and graphene have been the subject of considerable debate. For example, a ferricyanide probe of electron transfer to the basal plane of HOPG produced k0 < 10−9 cm s−1 [98], but later results from Patel et al. [59] showed that the ferricyanide probe can adsorb on the surface and impede electron transfer. The mean value of k0 for electron transfer to graphene/graphite varied by two orders of magnitude depending on the type of redox probe and measurement conditions [59, 80]. Basal plane kinetics on monolayer graphene have also been shown to be 2–8 times slower than on HOPG [99]. The CVD graphene is not pristine, and there is a contribution to electron transfer from defects. Polymer-free transfer and the use of hydrogenation to control defect density produced a k0 that stabilised around 1.5 × 10−4 cm s−1 as defect density increased [100]. Therefore, the CVD graphene used in the present analysis likely had residual surface PMMA.
The k0 measurements showed consistent relative changes on edge modification. Sulphonation increased the mean k0 for the basal plane to (1.9 ± 1.5) × 10−4 cm s−1 (Figure S14 and Figure S15). The large uncertainty in the result is likely due to inhomogeneity of the CVD surface (Figure S6). The mean k0 for the drops sitting at the edges of CVD G–SO3− was (3.6 ± 1.7) × 10−4 cm s−1 (Figure S14 and Figure S15). Electron transfer rates to the edges of single-layer graphene are expected to be two orders of magnitude greater than the basal plane [98]. The only twofold increase observed here is consistent with the hypothesis that some basal plane droplets will have contributions to their electron transfer rate from internal defects. For dye-labelled CVD G–SH, k0 reduced to (1.0 ± 0.5) × 10−5 cm s−1 (Figure S16), comparable to unmodified CVD graphene. Some of the decrease must be attributed to the bulky dye slowing edge electron transfer because the value of LD estimated from Raman spectra remained about half the value of unmodified CVD graphene, as it was for G–SH (Table S6). The electron transfer rates for the unfunctionalised CVD graphene and dye-labelled CVD graphene are comparable to those of the basal plane of HOPG exposed to air for > 60 min after exfoliation, based on comparable peak separations from scans at 100 mV s−1 [58].
The edge modifications produce marked changes in the graphenes’ dispersibility, consistent with the chemical changes (Figure S18 and Table S3). Sulphonation produces graphene that is water dispersible; reduction to the thiol creates a material dispersible only in toluene. Glycographene is moderately dispersible in water, making it a conceivable candidate for applications in biosensors or biomaterials. Contact angle measurements (Table S4) were consistent with these observations.
The edge sulphonation of larger GNPs also produced an unexpected trend. These were presumed to have a smaller amount of edge length for a given mass. Elemental analysis by XPS (Figure S22 and Table S7) showed the opposite trend; however, the largest flakes had the highest S/C ratio. This observation could be an artefact residual, unreacted chlorosulphonic acid trapped between the layers of the GNPs. Consistent with this, graphite has previously been intercalated by tosylate [96] and methanesulphonic acid [95].
The nitration of GNPs (G–NO2) was performed and reduced to aminographene (G–NH2). FT-IR (Figure S1) confirmed the presence of the nitrogen-containing functional groups. G–NO2 showed a distinct, sharp N–O vibration at 1385 cm−1 characteristic of nitrated aromatic systems. This signal disappeared when G–NO2 was reduced to form G–NH2, and distinctive N–H vibrations appeared from 3700 to 3300 cm−1 along with C–N stretches and N–H scissoring bands at lower energies.
The N 1s XPS spectra could be resolved into three peaks for both G–NO2 and G–NH2 (Figure S3). The relative amount of the nitro group (Eb = 406.9 ± 0.3 eV) decreases between the G–NO2 sample and its reduced G–NH2 analogue. The largest peak at 400.1 ± 0.2 eV can be assigned to organic nitrogen groups, including amines and nitrogen heterocycles [101–105], but tightly adsorbed NMP is likely the main contributor to this peak, consistent with its appearance in almost all samples produced by exfoliating graphite in NMP (Figure S20). This peak’s prominence in both spectra makes it impossible to infer the degree of nitro reduction in the G–NH2 sample. A small contribution at 402.8 ± 0.4 eV was attributed to nitrogen in an intermediate oxidation state, such as a hydroxyl amine. Nitration produced products with a greater heteroatom content (N:C around 0.5:100). Analysis of the G–NH2 was complicated by the presence of by-products from the nitro reduction over Raney nickel.
As a nitration caused too damaging to CVD graphene, epifluorescence of FITC-labelled GNPs M25 aminographene was an alternative method to highlight the specific edge modification in this study (Figure S2). A faint green fluorescence on the edge of GNP (Figure S2b) could be a possible dye attachment. Nevertheless, conventional fluorescence microscopy was not suitable to observe the edge nitration of GNPs due to a low fluorophore attachment.
Conclusions
Super-resolution microscopy of fluorophore-labelled edge-modified graphene, the sp3:sp2 C ratio, and estimated distance between defects (LD) from Raman are evidence of chemical functionalisation predominantly at the edges and internal defects. This straightforward and scalable method of edge-specific functionalisation creates a versatile platform material for biotechnological and biomedical applications of graphene, rather than graphene oxide or its derivatives. Ongoing and future work on these materials includes measurements of their electronic conductivity and interfacial electron transfer coefficients, applying them in nanoscale theronostics devices, using them as platforms for tissue engineering and regenerative medicine (that is, using the edge group to display specific cues in cell culture), synthesising MRI contrast agents (e.g. with Gd-DOTA–based side groups), creating multiple reactive groups (e.g. both thiol and amine) for bio-orthogonal functionalisation, and assessing whether the mechanical and electrical properties are maintained upon edge functionalisation.
Supplementary information
Supporting information available: synthesis of nitrographene and aminographene; Raman spectrum analysis methods; representative survey and C 1s XPS spectra; elemental analysis based on XPS survey spectra; Raman spectra; schematic for microelectrochemistry and representative measurements; thermogravimetric analysis of pristine and edge-modified GNPs; dispersibility and contact angle measurements; Raman comparison of GNP source for edge sulphonation; Raman and XPS analysis of GNP size on edge sulphonation. In compliance with the University of Manchester’s requirements and those of the funding agencies, these data for this work are available to download from https://data.mendeley.com/datasets/mwrvrcj639/draft?a=20782f0e-4a57-4056-a4e7-c24057beded6
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors acknowledge funding received from the UK’s Medical Research Council for a Graphene Bioscience Interdisciplinary Grand Challenges award (“Confidence in Concept 2012—The University of Manchester”, Grant reference MC_PC_12018) and the European Union’s Seventh Framework Programme FP7/2011–2015 (“GlycoBioM—Tools for the Detection of Novel Glyco-biomarkers”, Grant agreement number 259869). The European Union is not liable for any use that may be made of the information contained herein. For funding their doctoral studies, the authors acknowledge T.P.M.’s studentship through the UK’s Engineering and Physical Sciences Research Council (EPSRC) under Grant code EP/K502947/1; T.S.’s studentship from the Royal Thai Government; P.M.S.’s studentship from the UK’s Biotechnology and Biological Sciences Research Council (BBSRC) (BB/J014478/1); J.B.’s studentship from the EPSRC CDT in Science and Applications of Graphene and Related Nanomaterials (EP/L01548X/1); and T.M.’s studentship from the EPSRC’s NOWNANO DTC (EP/G03737X/1). F.F. thanks his parents and family for supporting his doctoral studies. The authors thank Pawin Iamprasertkun and Robert Dryfe for access and training related to the microelectrochemical measurements.
Author contributions
PMS, TS, MH, FF, JB, TPMN, and TMA all carried out wet-chemical modifications to GNPs and CVD graphene. PMS and MH provided all the results pertaining to edge-modified G–SO3− and G–SH on NMP-exfoliated GNPs; they also wrote the first drafts of the manuscripts. TS produced all the data and procedure related to edge modifications of commercially sourced GNPs (e.g. C750); she also contributed significantly to the writing of later paper drafts. MH produced the glycographene and the epifluorescence images thereof. MH and TPMN worked on gold nanoparticle attachment to thiolated GNPs; MH acquired the SEMs of them shown in the manuscript. FF carried out all the size dependence measurements of sulphonated commercially sourced GNPs. TMA carried out the initial work on Raman analysis of sulphonated CVD graphene. JB optimised the synthesis of CVD G–SO3− and G–SH and worked with HC and TAW to acquire qSTORM images on their custom-built system. JB also carried out the measurements of interfacial electron transfer rates and acquired the Raman map. AMS and RMJJ ran the initial XPS analyses on edge-modified graphene samples starting from NMP-exfoliated GNPs. SLF and CFB were responsible for providing funding and directing the project. CFB also carried out extensive XPS data analysis.
Data availability
In compliance with the University of Manchester’s requirements and those of bodies that funded this work, the data associated with this work are available to download from 10.17632/mwrvrcj639.1.
Compliance with ethical standards
Conflicts of interest
M.H., C.F.B., and S.L.F. are the inventors and assignees on a patent based on this work (US10173898B2). C.F.B. is a deputy editor-in-chief of J. Mater. Sci. and was blinded to all aspects of the peer review process.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Philippa M. Shellard and Thunyaporn Srisubin have contributed equally to this work.
Contributor Information
Sabine L. Flitsch, Email: sabine.flitsch@manchester.ac.uk
Christopher F. Blanford, Email: christopher.blanford@manchester.ac.uk
References
- 1.Quinn MD, Wang T, Al Kobaisi M, Craig VS, Notley SM. PEO-PPO-PEO surfactant exfoliated graphene cyclodextrin drug carriers for photoresponsive release. Mater Chem Phys. 2018;205:154–163. doi: 10.1016/j.matchemphys.2017.11.012. [DOI] [Google Scholar]
- 2.Orecchioni M, Cabizza R, Bianco A, Delogu LG. Graphene as cancer theranostic tool: progress and future challenges. Theranostics. 2015;5(7):710–723. doi: 10.7150/thno.11387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent M, de Lázaro I, Kostarelos K. Graphene materials as 2D non-viral gene transfer vector platforms. Gene Ther. 2016;24:123–132. doi: 10.1038/gt.2016.79. [DOI] [PubMed] [Google Scholar]
- 4.Bai RG, Ninan N, Muthoosamy K, Manickam S. Graphene: a versatile platform for nanotheranostics and tissue engineering. Prog Mater Sci. 2018;91:24–69. doi: 10.1016/j.pmatsci.2017.08.004. [DOI] [Google Scholar]
- 5.Qian Y, Zhao X, Han Q, Chen W, Li H, Yuan W. An integrated multi-layer 3D-fabrication of PDA/RGD coated graphene loaded PCL nanoscaffold for peripheral nerve restoration. Nat Commun. 2018;9(1):323. doi: 10.1038/s41467-017-02598-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park Y, Shim J, Jeong S, Yi GR, Chae H, Bae JW, Kim SO, Pang C. Microtopography-guided conductive patterns of liquid-driven graphene nanoplatelet networks for stretchable and skin-conformal sensor array. Adv Mater. 2017;29(21):1606453. doi: 10.1002/adma.201606453. [DOI] [PubMed] [Google Scholar]
- 7.McManus D, Vranic S, Withers F, Sanchez-Romaguera V, Macucci M, Yang HF, Sorrentino R, Parvez K, Son SK, Iannaccone G, Kostarelos K, Fiori G, Casiraghi C. Water-based and biocompatible 2D crystal inks for all-inkjet-printed heterostructures. Nat Nanotechnol. 2017;12(4):343–350. doi: 10.1038/nnano.2016.281. [DOI] [PubMed] [Google Scholar]
- 8.Castro Neto AH, Guinea F, Peres NMR, Novoselov KS, Geim AK. The electronic properties of graphene. Rev Mod Phys. 2009;81(1):109–162. doi: 10.1103/RevModPhys.81.109. [DOI] [Google Scholar]
- 9.Lee C, Wei X, Kysar JW, Hone J. Measurement of the elastic properties and intrinsic strength of monolayer graphene. Science. 2008;321(5887):385. doi: 10.1126/science.1157996. [DOI] [PubMed] [Google Scholar]
- 10.Papageorgiou DG, Kinloch IA, Young RJ. Mechanical properties of graphene and graphene-based nanocomposites. Prog Mater Sci. 2017;90:75–127. doi: 10.1016/j.pmatsci.2017.07.004. [DOI] [Google Scholar]
- 11.Liu J, Tang J, Gooding JJ. Strategies for chemical modification of graphene and applications of chemically modified graphene. J Mater Chem. 2012;22(25):12435–12452. doi: 10.1039/C2JM31218B. [DOI] [Google Scholar]
- 12.Liu K, Chen S, Luo Y, Jia D, Gao H, Hu G, Liu L. Edge-functionalized graphene as reinforcement of epoxy-based conductive composite for electrical interconnects. Compos Sci Technol. 2013;88:84–91. doi: 10.1016/j.compscitech.2013.08.032. [DOI] [Google Scholar]
- 13.Gong MG, Adhikari P, Gong YP, Wang T, Liu QF, Kattel B, Ching WY, Chan WL, Wu JZ. Polarity-controlled attachment of cytochrome c for high-performance cytochrome C/graphene van der Waals heterojunction photodetectors. Adv Funct Mater. 2018;28(5):7. doi: 10.1002/adfm.201704797. [DOI] [Google Scholar]
- 14.Loo AH, Bonanni A, Pumera M. Biorecognition on graphene: physical, covalent, and affinity immobilization methods exhibiting dramatic differences. Chem-Asian J. 2013;8(1):198–203. doi: 10.1002/asia.201200756. [DOI] [PubMed] [Google Scholar]
- 15.Georgakilas V, Otyepka M, Bourlinos AB, Chandra V, Kim N, Kemp KC, Hobza P, Zboril R, Kim KS. Functionalization of graphene: covalent and non-covalent approaches, derivatives and applications. Chem Rev. 2012;112(11):6156–6214. doi: 10.1021/cr3000412. [DOI] [PubMed] [Google Scholar]
- 16.Yang Q, Pan X, Clarke K, Li K. Covalent functionalization of graphene with polysaccharides. Ind Eng Chem Res. 2011;51(1):310–317. doi: 10.1021/ie201391e. [DOI] [Google Scholar]
- 17.Dreyer DR, Park S, Bielawski CW, Ruoff RS. The chemistry of graphene oxide. Chem Soc Rev. 2010;39(1):228–240. doi: 10.1039/B917103G. [DOI] [PubMed] [Google Scholar]
- 18.Liu L, Zhang J, Zhao J, Liu F. Mechanical properties of graphene oxides. Nanoscale. 2012;4(19):5910–5916. doi: 10.1039/C2NR31164J. [DOI] [PubMed] [Google Scholar]
- 19.Moradi O, Gupta VK, Agarwal S, Tyagi I, Asif M, Makhlouf ASH, Sadegh H, Shahryari-ghoshekandi R. Characteristics and electrical conductivity of graphene and graphene oxide for adsorption of cationic dyes from liquids: kinetic and thermodynamic study. J Ind Eng Chem. 2015;28:294–301. doi: 10.1016/j.jiec.2015.03.005. [DOI] [Google Scholar]
- 20.Suk JW, Piner RD, An J, Ruoff RS. Mechanical properties of monolayer graphene oxide. ACS Nano. 2010;4(11):6557–6564. doi: 10.1021/nn101781v. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Chen Y, Lacey SD, Xu L, Xie H, Li T, Danner VA, Hu L. Reduced graphene oxide film with record-high conductivity and mobility. Mater Today. 2018;21:186–192. doi: 10.1016/j.mattod.2017.1010.1008. [DOI] [Google Scholar]
- 22.Georgakilas V, Tiwari JN, Kemp KC, Perman JA, Bourlinos AB, Kim KS, Zboril R. Noncovalent functionalization of graphene and graphene oxide for energy materials, biosensing, catalytic, and biomedical applications. Chem Rev. 2016;116(9):5464–5519. doi: 10.1021/acs.chemrev.5b00620. [DOI] [PubMed] [Google Scholar]
- 23.Lee D-W, Kim T, Lee M. An amphiphilic pyrene sheet for selective functionalization of graphene. Chem Commun. 2011;47(29):8259–8261. doi: 10.1039/C1CC12868J. [DOI] [PubMed] [Google Scholar]
- 24.Schlierf A, Yang H, Gebremedhn E, Treossi E, Ortolani L, Chen L, Minoia A, Morandi V, Samorì P, Casiraghi C. Nanoscale insight into the exfoliation mechanism of graphene with organic dyes: effect of charge, dipole and molecular structure. Nanoscale. 2013;5(10):4205–4216. doi: 10.1039/C3NR00258F. [DOI] [PubMed] [Google Scholar]
- 25.Chen I-WP, Huang C-Y, Saint Jhou S-H, Zhang Y-W. Exfoliation and performance properties of non-oxidized graphene in water. Sci Rep. 2014;4:3928. doi: 10.1038/srep03928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Das S, Irin F, Ahmed HT, Cortinas AB, Wajid AS, Parviz D, Jankowski AF, Kato M, Green MJ. Non-covalent functionalization of pristine few-layer graphene using triphenylene derivatives for conductive poly (vinyl alcohol) composites. Polymer. 2012;53(12):2485–2494. doi: 10.1016/j.polymer.2012.03.012. [DOI] [Google Scholar]
- 27.Ghosh A, Rao KV, George SJ, Rao C. Noncovalent functionalization, exfoliation, and solubilization of graphene in water by employing a fluorescent coronene carboxylate. Chem Eur J. 2010;16(9):2700–2704. doi: 10.1002/chem.200902828. [DOI] [PubMed] [Google Scholar]
- 28.Raju APA, Offerman SC, Gorgojo P, Valles C, Bichenkova EV, Aojula HS, Vijayraghavan A, Young RJ, Novoselov KS, Kinloch IA, Clarke DJ. Dispersal of pristine graphene for biological studies. RSC Adv. 2016;6(73):69551–69559. doi: 10.1039/C6RA12195K. [DOI] [Google Scholar]
- 29.Shih CJ, Paulus GLC, Wang QH, Jin Z, Blankschtein D, Strano MS. Understanding surfactant/graphene interactions using a graphene field effect transistor: relating molecular structure to hysteresis and carrier mobillity. Langmuir. 2012;28(22):8579–8586. doi: 10.1021/la3008816. [DOI] [PubMed] [Google Scholar]
- 30.Sinitskii A, Dimiev A, Corley DA, Fursina AA, Kosynkin DV, Tour JM. Kinetics of diazonium functionalization of chemically converted graphene nanoribbons. ACS Nano. 2010;4(4):1949–1954. doi: 10.1021/nn901899j. [DOI] [PubMed] [Google Scholar]
- 31.Lomeda JR, Doyle CD, Kosynkin DV, Hwang W-F, Tour JM. Diazonium functionalization of surfactant-wrapped chemically converted graphene sheets. J Am Chem Soc. 2008;130(48):16201–16206. doi: 10.1021/ja806499w. [DOI] [PubMed] [Google Scholar]
- 32.Paulus GLC, Wang QH, Strano MS. Covalent electron transfer chemistry of graphene with diazonium salts. Acc Chem Res. 2013;46(1):160–170. doi: 10.1021/ar300119z. [DOI] [PubMed] [Google Scholar]
- 33.Huang P, Jing L, Zhu H, Gao X. Diazonium functionalized graphene: microstructure, electric, and magnetic properties. Acc Chem Res. 2013;46(1):43–52. doi: 10.1021/ar300070a. [DOI] [PubMed] [Google Scholar]
- 34.Peng C, Xiong Y, Liu Z, Zhang F, Ou E, Qian J, Xiong Y, Xu W. Bulk functionalization of graphene using diazonium compounds and amide reaction. Appl Surf Sci. 2013;280:914–919. doi: 10.1016/j.apsusc.2013.05.094. [DOI] [Google Scholar]
- 35.Greenwood J, Phan TH, Fujita Y, Li Z, Ivasenko O, Vanderlinden W, Van Gorp H, Frederickx W, Lu G, Tahara K, Tobe Y, Uji-i H, Mertens SFL, De Feyter S. Covalent modification of graphene and graphite using diazonium chemistry: tunable grafting and nanomanipulation. ACS Nano. 2015;9(5):5520–5535. doi: 10.1021/acsnano.5b01580. [DOI] [PubMed] [Google Scholar]
- 36.Qiu Z, Yu J, Yan P, Wang Z, Wan Q, Yang N. Electrochemical grafting of graphene nano platelets with aryl diazonium salts. ACS Appl Mater Interfaces. 2016;8(42):28291–28298. doi: 10.1021/acsami.5b11593. [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Li W, Wojcik M, Wang B, Lin L-C, Raschke MB, Xu K. Light-assisted diazonium functionalization of graphene and spatial heterogeneities in reactivity. J Phys Chem Lett. 2019;10(17):4788–4793. doi: 10.1021/acs.jpclett.9b02225. [DOI] [PubMed] [Google Scholar]
- 38.Liu H, Ryu S, Chen Z, Steigerwald ML, Nuckolls C, Brus LE. Photochemical reactivity of graphene. J Am Chem Soc. 2009;131(47):17099–17101. doi: 10.1021/ja9043906. [DOI] [PubMed] [Google Scholar]
- 39.Zhong X, Jin J, Li S, Niu Z, Hu W, Li R, Ma J. Aryne cycloaddition: highly efficient chemical modification of graphene. Chem Commun. 2010;46(39):7340–7342. doi: 10.1039/C0CC02389B. [DOI] [PubMed] [Google Scholar]
- 40.Sarkar S, Bekyarova E, Niyogi S, Haddon RC. Diels–Alder chemistry of graphite and graphene: graphene as diene and dienophile. J Am Chem Soc. 2011;133(10):3324–3327. doi: 10.1021/ja200118b. [DOI] [PubMed] [Google Scholar]
- 41.Bian S, Scott AM, Cao Y, Liang Y, Osuna S, Houk KN, Braunschweig AB. Covalently patterned graphene surfaces by a force-accelerated Diels–Alder reaction. J Am Chem Soc. 2013;135(25):9240–9243. doi: 10.1021/ja4042077. [DOI] [PubMed] [Google Scholar]
- 42.Sarkar S, Bekyarova E, Haddon RC. Chemistry at the Dirac point: Diels–Alder reactivity of graphene. Acc Chem Res. 2012;45(4):673–682. doi: 10.1021/ar200302g. [DOI] [PubMed] [Google Scholar]
- 43.Lazar I-M, Rostas AM, Straub PS, Schleicher E, Weber S, Mülhaupt R. Simple covalent attachment of redox-active nitroxyl radicals to graphene via Diels–Alder cycloaddition. Macromol Chem Phys. 2017;218(15):1700050. doi: 10.1002/macp.201700050. [DOI] [Google Scholar]
- 44.Zhang J, Wang W, Peng H, Qian J, Ou E, Xu W. Water-soluble graphene dispersion functionalized by Diels–Alder cycloaddition reaction. J Iran Chem Soc. 2017;14(1):89–93. doi: 10.1007/s13738-016-0960-5. [DOI] [Google Scholar]
- 45.Chang H-N, Sarkar S, Baker JR, Norris TB. Fluorophore and protein conjugated Diels–Alder functionalized CVD graphene layers. Opt Mater Express. 2016;6(10):3242–3253. doi: 10.1364/OME.6.003242. [DOI] [Google Scholar]
- 46.Li J, Li M, Zhou L-L, Lang S-Y, Lu H-Y, Wang D, Chen C-F, Wan L-J. Click and patterned functionalization of graphene by Diels–Alder reaction. J Am Chem Soc. 2016;138(24):7448–7451. doi: 10.1021/jacs.6b02209. [DOI] [PubMed] [Google Scholar]
- 47.Seo J-M, Baek J-B. A solvent-free Diels–Alder reaction of graphite into functionalized graphene nanosheets. Chem Commun. 2014;50(93):14651–14653. doi: 10.1039/C4CC07173E. [DOI] [PubMed] [Google Scholar]
- 48.Denis PA. Organic chemistry of graphene: the Diels–Alder reaction. Chem Eur J. 2013;19(46):15719–15725. doi: 10.1002/chem.201302622. [DOI] [PubMed] [Google Scholar]
- 49.Cao Y, Osuna S, Liang Y, Haddon RC, Houk KN. Diels–Alder reactions of graphene: computational predictions of products and sites of reaction. J Am Chem Soc. 2013;135(46):17643–17649. doi: 10.1021/ja410225u. [DOI] [PubMed] [Google Scholar]
- 50.Jeon I-Y, Choi H-J, Jung S-M, Seo J-M, Kim M-J, Dai L, Baek J-B. Large-scale production of edge-selectively functionalized graphene nanoplatelets via ball milling and their use as metal-free electrocatalysts for oxygen reduction reaction. J Am Chem Soc. 2013;135(4):1386–1393. doi: 10.1021/ja3091643. [DOI] [PubMed] [Google Scholar]
- 51.Kweon DH, Baek J-B. Edge-functionalized graphene nanoplatelets as metal-free electrocatalysts for dye-sensitized solar cells. Adv Mater. 2019;31(13):1804440. doi: 10.1002/adma.201804440. [DOI] [PubMed] [Google Scholar]
- 52.Yadav A, Iost Rodrigo M, Neubert TJ, Baylan S, Schmid T, Balasubramanian K. Selective electrochemical functionalization of the graphene edge. Chem Sci. 2019;10(3):936–942. doi: 10.1039/C8SC04083D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim M-J, Jeon I-Y, Seo J-M, Dai L, Baek J-B. Graphene phosphonic acid as an efficient flame retardant. ACS Nano. 2014;8(3):2820–2825. doi: 10.1021/nn4066395. [DOI] [PubMed] [Google Scholar]
- 54.Jeon I-Y, Choi H-J, Choi M, Seo J-M, Jung S-M, Kim M-J, Zhang S, Zhang L, Xia Z, Dai L. Facile, scalable synthesis of edge-halogenated graphene nanoplatelets as efficient metal-free eletrocatalysts for oxygen reduction reaction. Sci Rep. 2013;3:1810. doi: 10.1038/srep01810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jeon IY, Bae SY, Seo JM, Baek JB. Scalable production of edge-functionalized graphene nanoplatelets via mechanochemical ball-milling. Adv Funct Mater. 2015;25(45):6961–6975. doi: 10.1002/adfm.201502214. [DOI] [Google Scholar]
- 56.Jeon I-Y, Yu D, Bae S-Y, Choi H-J, Chang DW, Dai L, Baek J-B. Formation of large-area nitrogen-doped graphene film prepared from simple solution casting of edge-selectively functionalized graphite and its electrocatalytic activity. Chem Mater. 2011;23(17):3987–3992. doi: 10.1021/cm201542m. [DOI] [Google Scholar]
- 57.Chua CK, Pumera M. Friedel–Crafts acylation on graphene. Chem Asian J. 2012;7(5):1009–1012. doi: 10.1002/asia.201200096. [DOI] [PubMed] [Google Scholar]
- 58.Ji X, Banks CE, Crossley A, Compton RG. Oxygenated edge plane sites slow the electron transfer of the ferro-/ferricyanide redox couple at graphite electrodes. ChemPhysChem. 2006;7(6):1337–1344. doi: 10.1002/cphc.200600098. [DOI] [PubMed] [Google Scholar]
- 59.Patel AN, Collignon MG, O’Connell MA, Hung WOY, McKelvey K, Macpherson JV, Unwin PR. A new view of electrochemistry at highly oriented pyrolytic graphite. J Am Chem Soc. 2012;134(49):20117–20130. doi: 10.1021/ja308615h. [DOI] [PubMed] [Google Scholar]
- 60.Kaplan A, Yuan Z, Benck JD, Govind Rajan A, Chu XS, Wang QH, Strano MS. Current and future directions in electron transfer chemistry of graphene. Chem Soc Rev. 2017;46(15):4530–4571. doi: 10.1039/C7CS00181A. [DOI] [PubMed] [Google Scholar]
- 61.Keerthi A, Radha B, Rizzo D, Lu H, Diez Cabanes V, Hou IC-Y, Beljonne D, Cornil J, Casiraghi C, Baumgarten M, Müllen K, Narita A. Edge functionalization of structurally defined graphene nanoribbons for modulating the self-assembled structures. J Am Chem Soc. 2017;139(46):16454–16457. doi: 10.1021/jacs.7b09031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rust MJ, Bates M, Zhuang X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–796. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nahidiazar L, Agronskaia AV, Broertjes J, van den Broek B, Jalink K. Optimizing imaging conditions for demanding multi-color super resolution localization microscopy. PLoS One. 2016;11(7):18. doi: 10.1371/journal.pone.0158884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cox H, Georgiades P, Xu H, Waigh TA, Lu JR. Self-assembly of mesoscopic peptide surfactant fibrils investigated by STORM super-resolution fluorescence microscopy. Biomacromolecules. 2017;18(11):3481–3491. doi: 10.1021/acs.biomac.7b00465. [DOI] [PubMed] [Google Scholar]
- 65.Georgiades P, Allan VJ, Dickinson M, Waigh TA. Reduction of coherent artefacts in super-resolution fluorescence localisation microscopy. J Microsc. 2016;264(3):375–383. doi: 10.1111/jmi.12453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Li R, Georgiades P, Cox H, Phanphak S, Roberts IS, Waigh TA, Lu JR. Quenched stochastic optical reconstruction microscopy (qSTORM) with graphene oxide. Sci Rep. 2018;8(1):16928. doi: 10.1038/s41598-018-35297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hernandez Y, Nicolosi V, Lotya M, Blighe FM, Sun Z, De S, McGovern IT, Holland B, Byrne M, Gun’Ko YK, Boland JJ, Niraj P, Duesberg G, Krishnamurthy S, Goodhue R, Hutchison J, Scardaci V, Ferrari AC, Coleman JN. High-yield production of graphene by liquid-phase exfoliation of graphite. Nat Nanotechnol. 2008;3:563–568. doi: 10.1038/nnano.2008.215. [DOI] [PubMed] [Google Scholar]
- 68.Yoshikawa M, Nagai N. Vibrational spectroscopy of carbon and silicon materials. In: Chalmers JM, Griffiths PR, editors. Handbook of vibrational spectroscopy. New York: Wiley; 2002. pp. 2593–2620. [Google Scholar]
- 69.Ferrari AC, Basko DM. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat Nanotechnol. 2013;8:235–246. doi: 10.1038/nnano.2013.46. [DOI] [PubMed] [Google Scholar]
- 70.Ferrari AC, Meyer JC, Scardaci V, Casiraghi C, Lazzeri M, Mauri F, Piscanec S, Jiang D, Novoselov KS, Roth S, Geim AK. Raman spectrum of graphene and graphene layers. Phys Rev Lett. 2006;97(18):187401. doi: 10.1103/PhysRevLett.97.187401. [DOI] [PubMed] [Google Scholar]
- 71.Puech P, Kandara M, Paredes G, Moulin L, Weiss-Hortala E, Kundu A, Ratel-Ramond N, Plewa J-M, Pellenq R, Monthioux M. Analyzing the Raman spectra of graphenic carbon materials from kerogens to nanotubes: what type of information can be extracted from defect bands? C. 2019;5(4):69. doi: 10.3390/c5040069. [DOI] [Google Scholar]
- 72.Tuinstra F, Koenig JL. Raman spectrum of graphite. J Chem Phys. 1970;53(3):1126–1130. doi: 10.1063/1.1674108. [DOI] [Google Scholar]
- 73.Cançado LG, Takai K, Enoki T, Endo M, Kim YA, Mizusaki H, Jorio A, Coelho LN, Magalhães-Paniago R, Pimenta MA. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl Phys Lett. 2006;88(16):163106. doi: 10.1063/1.2196057. [DOI] [Google Scholar]
- 74.Puech P, Plewa J-M, Mallet-Ladeira P, Monthioux M. Spatial confinement model applied to phonons in disordered graphene-based carbons. Carbon. 2016;105:275–281. doi: 10.1016/j.carbon.2016.04.048. [DOI] [Google Scholar]
- 75.Lucchese MM, Stavale F, Ferreira EHM, Vilani C, Moutinho MVO, Capaz RB, Achete CA, Jorio A. Quantifying ion-induced defects and Raman relaxation length in graphene. Carbon. 2010;48(5):1592–1597. doi: 10.1016/j.carbon.2009.12.057. [DOI] [Google Scholar]
- 76.Ganguly A, Sharma S, Papakonstantinou P, Hamilton J. Probing the thermal deoxygenation of graphene oxide using high-resolution in situ X-ray-based spectroscopies. J Phys Chem C. 2011;115(34):17009–17019. doi: 10.1021/jp203741y. [DOI] [Google Scholar]
- 77.Fairley N (2009) CasaXPS manual 2.3.15 spectroscopy. Casa Software Ltd
- 78.Ovesný M, Křížek P, Borkovec J, Švindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014;30(16):2389–2390. doi: 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Valota AT, Toth PS, Kim Y-J, Hong BH, Kinloch IA, Novoselov KS, Hill EW, Dryfe RAW. Electrochemical investigation of chemical vapour deposition monolayer and bilayer graphene on the microscale. Electrochim Acta. 2013;110:9–15. doi: 10.1016/j.electacta.2013.03.187. [DOI] [Google Scholar]
- 80.Velický M, Bradley DF, Cooper AJ, Hill EW, Kinloch IA, Mishchenko A, Novoselov KS, Patten HV, Toth PS, Valota AT, Worrall SD, Dryfe RAW. Electron transfer kinetics on mono- and multilayer graphene. ACS Nano. 2014;8(10):10089–10100. doi: 10.1021/nn504298r. [DOI] [PubMed] [Google Scholar]
- 81.Klingler RJ, Kochi JK. Electron-transfer kinetics from cyclic voltammetry. Quantitative description of electrochemical reversibility. J Phys Chem. 1981;85(12):1731–1741. doi: 10.1021/j150612a028. [DOI] [Google Scholar]
- 82.Shih C-J, Wang QH, Lin S, Park K-C, Jin Z, Strano MS, Blankschtein D. Breakdown in the wetting transparency of graphene. Phys Rev Lett. 2012;109(17):176101. doi: 10.1103/PhysRevLett.109.176101. [DOI] [PubMed] [Google Scholar]
- 83.Ostrowski JHJ, Eaves JD. The tunable hydrophobic effect on electrically doped graphene. J Phys Chem B. 2014;118(2):530–536. doi: 10.1021/jp409342n. [DOI] [PubMed] [Google Scholar]
- 84.Wang JTW, Fabbro C, Venturelli E, Ménard-Moyon C, Chaloin O, Da Ros T, Methven L, Nunes A, Sosabowski JK, Mather SJ, Robinson MK, Amadou J, Prato M, Bianco A, Kostarelos K, Al-Jamal KT. The relationship between the diameter of chemically-functionalized multi-walled carbon nanotubes and their organ biodistribution profiles in vivo. Biomaterials. 2014;35(35):9517–9528. doi: 10.1016/j.biomaterials.2014.07.054. [DOI] [PubMed] [Google Scholar]
- 85.Sureshbabu AR, Kurapati R, Russier J, Ménard-Moyon C, Bartolini I, Meneghetti M, Kostarelos K, Bianco A. Degradation-by-design: surface modification with functional substrates that enhance the enzymatic degradation of carbon nanotubes. Biomaterials. 2015;72:20–28. doi: 10.1016/j.biomaterials.2015.08.046. [DOI] [PubMed] [Google Scholar]
- 86.Rajendra K, Fanny B, Julie R, Adukamparai Rajukrishnan S, Cécilia M-M, Kostas K, Alberto B. Covalent chemical functionalization enhances the biodegradation of graphene oxide. 2D Mater. 2018;5(1):015020. [Google Scholar]
- 87.Swathi RS, Sebastian KL. Resonance energy transfer from a dye molecule to graphene. J Chem Phys. 2008;129(5):054703. doi: 10.1063/1.2956498. [DOI] [PubMed] [Google Scholar]
- 88.Swathi RS, Sebastian KL. Long range resonance energy transfer from a dye molecule to graphene has (distance)−4 dependence. J Chem Phys. 2009;130(8):086101. doi: 10.1063/1.3077292. [DOI] [PubMed] [Google Scholar]
- 89.Chen Z, Berciaud S, Nuckolls C, Heinz TF, Brus LE. Energy transfer from individual semiconductor nanocrystals to graphene. ACS Nano. 2010;4(5):2964–2968. doi: 10.1021/nn1005107. [DOI] [PubMed] [Google Scholar]
- 90.Guo X, Amina Z, Haiyan N, Yuanfang Y, Weiwei Z, Zheng L, Xueao Z, Zhenhua N. Manipulating fluorescence quenching efficiency of graphene by defect engineering. Appl Phys Express. 2016;9(5):055502. doi: 10.7567/APEX.9.055502. [DOI] [Google Scholar]
- 91.Gaudreau L, Tielrooij KJ, Prawiroatmodjo GEDK, Osmond J, de Abajo FJG, Koppens FHL. Universal distance-scaling of nonradiative energy transfer to graphene. Nano Lett. 2013;13(5):2030–2035. doi: 10.1021/nl400176b. [DOI] [PubMed] [Google Scholar]
- 92.Kovtun A, Treossi E, Mirotta N, Scidà A, Liscio A, Christian M, Valorosi F, Boschi A, Young RJ, Galiotis C, Kinloch IA, Morandi V, Palermo V. Benchmarking of graphene-based materials: real commercial products versus ideal graphene. 2D Mater. 2019;6(2):025006. doi: 10.1088/2053-1583/aafc6e. [DOI] [Google Scholar]
- 93.Sampathkumar K, Diez-Cabanes V, Kovaricek P, del Corro E, Bouša M, Hošek J, Kalbac M, Frank O. On the suitability of Raman spectroscopy to monitor the degree of graphene functionalization by diazonium salts. J Phys Chem C. 2019;123(36):22397–22402. doi: 10.1021/acs.jpcc.9b06516. [DOI] [Google Scholar]
- 94.Abdolmaleki A, Mallakpour S, Mahmoudian M. Preparation and evaluation of edge selective sulfonated graphene by chlorosulfuric acid as an active metal- free electrocatalyst for oxygen reduction reaction in alkaline media. ChemSel. 2017;2(34):11211–11217. doi: 10.1002/slct.201702409. [DOI] [Google Scholar]
- 95.Wang Y, Shi Z, Fang J, Xu H, Ma X, Yin J. Direct exfoliation of graphene in methanesulfonic acid and facile synthesis of graphene/polybenzimidazole nanocomposites. J Mater Chem. 2011;21(2):505–512. doi: 10.1039/C0JM02376K. [DOI] [Google Scholar]
- 96.Wen P, Gong P, Mi Y, Wang J, Yang S. Scalable fabrication of high quality graphene by exfoliation of edge sulfonated graphite for supercapacitor application. RSC Adv. 2014;4(68):35914–35918. doi: 10.1039/C4RA04788E. [DOI] [Google Scholar]
- 97.Casiraghi C, Hartschuh A, Qian H, Piscanec S, Georgi C, Fasoli A, Novoselov KS, Basko DM, Ferrari AC. Raman spectroscopy of graphene edges. Nano Lett. 2009;9(4):1433–1441. doi: 10.1021/nl8032697. [DOI] [PubMed] [Google Scholar]
- 98.Davies TJ, Hyde ME, Compton RG. Nanotrench arrays reveal insight into graphite electrochemistry. Angew Chem Int Ed. 2005;44(32):5121–5126. doi: 10.1002/anie.200462750. [DOI] [PubMed] [Google Scholar]
- 99.Brownson DAC, Varey SA, Hussain F, Haigh SJ, Banks CE. Electrochemical properties of CVD grown pristine graphene: monolayer- vs. quasi-graphene. Nanoscale. 2014;6(3):1607–1621. doi: 10.1039/c3nr05643k. [DOI] [PubMed] [Google Scholar]
- 100.Jiang L, Fu W, Birdja YY, Koper MTM, Schneider GF. Quantum and electrochemical interplays in hydrogenated graphene. Nat Commun. 2018;9(1):793. doi: 10.1038/s41467-018-03026-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bae S, Kim H, Lee Y, Xu X, Park J-S, Zheng Y, Balakrishnan J, Lei T, Ri Kim H, Song YI, Kim Y-J, Kim KS, Özyilmaz B, Ahn J-H, Hong BH, Iijima S. Roll-to-roll production of 30-inch graphene films for transparent electrodes. Nat Nanotechnol. 2010;5:574. doi: 10.1038/nnano.2010.132. [DOI] [PubMed] [Google Scholar]
- 102.Balaji SS, Sathish M. Supercritical fluid processing of nitric acid treated nitrogen doped graphene with enhanced electrochemical supercapacitance. RSC Adv. 2014;4(94):52256–52262. doi: 10.1039/C4RA07820A. [DOI] [Google Scholar]
- 103.Bianco GV, Losurdo M, Giangregorio MM, Capezzuto P, Bruno G. Exploring and rationalising effective n-doping of large area CVD-graphene by NH3. Phys Chem Chem Phys. 2014;16(8):3632–3639. doi: 10.1039/C3CP54451F. [DOI] [PubMed] [Google Scholar]
- 104.Lin Y-C, Lin C-Y, Chiu P-W. Controllable graphene N-doping with ammonia plasma. Appl Phys Lett. 2010;96(13):133110. doi: 10.1063/1.3368697. [DOI] [Google Scholar]
- 105.Kumar NA, Nolan H, McEvoy N, Rezvani E, Doyle RL, Lyons MEG, Duesberg GS. Plasma-assisted simultaneous reduction and nitrogen doping of graphene oxide nanosheets. J Mater Chem A. 2013;1(14):4431–4435. doi: 10.1039/C3TA10337D. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
In compliance with the University of Manchester’s requirements and those of bodies that funded this work, the data associated with this work are available to download from 10.17632/mwrvrcj639.1.