

To the Editor,
A novel kind of coronavirus has infected more than 1600 000 people, claimed over 100 000 lives, and spread to 212 countries and territories since December 2019. 1 The virus has been named severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) (provisionally termed novel coronavirus before), 2 and the pneumonia caused by it is coronavirus disease 2019. Though many scientists and researchers are trying to identify the animals harboring SARS‐CoV‐2, the intermediate host remains under the veil. To many people's surprise, the most pioneering host exploration was from Ji et al 4 that designated snake as the most likely intermediate host. 3
The paper by Ji et al 4 achieved indispensable importance in discovering SARS‐CoV‐2 is derived from a bat coronavirus and likely a recombination of two viruses. 4 Yet its result implied snakes as the most possible intermediate host of the virus seems unlikely. Considering viruses that transmit from poikilotherm to endotherm are rare, and the snake‐host speculation has arose widespread public attention, as well as referred in other studies, 5 we decided to take a close look at the host‐inferring method.
Snake was suspected to be the most possible source of the coronavirus by Ji et al 4 because of its closest distance of relative synonymous codon usage (RSCU) bias to SARS‐CoV‐2. This inferring methodology premises virus would evolve similar codon usage pattern to its hosts, which allows it to replicate more efficiently. Nevertheless, even though there were studies showing that RSCU bias between viruses and hosts are comparable, 6 none has suggested such codon usage similarity is necessary or sufficient for successful viral infection.
The reason under viral codon usage bias is far more complex than parasitic adaptation, which also involving mutation pressure, particular DNA/RNA or protein structure and genome size. 6 In fact, many studies have shown the RSCU of virus is not similar to that of its host. 7 , 8 Whether the host‐unlike virus simply has limited time for evolutionary host optimization, or its high mutation rates (especially RNA virus) outpaced the process of selection that drive such resemblance is in debate. 8 Therefore, attributing viral codon usage bias only to host adaptation and conducting host inference relies on it seems untenable.
Even if the codon usage bias is most similar between a particular virus and its host, the average RSCU of any virus should be selected to match the average RSCU of a vertebrate is in doubt. The codon usage frequency varies significantly among genes within the same organism, so as in viruses. 8 In a multicellular host, viruses are normally restricted to specific organ, tissue, or cell type. Thus, the RSCU of virus would be expected to resemble a particular RSCU calculated from expressed genes within the organ, tissue, or cell type of the host. More complicated, however, viral genes encoding structural proteins have more similar codon usage pattern to the host than other genes. 7 , 8 RSCU retrieved from Database such as Kazusa (http://www.kazusa.or.jp/codon/) presents only an average pattern, which does not reflect enormous intraspecific heterogeneity of codon usage, and undersampling of genes in some species may also introduce biases.
As for the reason why snakes share the most similar codon usage pattern with SARS‐CoV‐2, we believed that is because of the inherent AT(U)‐rich base compositions in both the genomes of the coronaviruses 7 and snakes. 9 Base composition, like GC3s (guanine‐cytosine [GC] content on the third codon position) value has been illustrated as a strong determinant in shaping codon usage both in viruses 8 , 10 and higher multicellular eukaryotes. 8 In a bid to test our speculation, we added viruses with known hosts and different AU3s values, as well as several host animals into Ji et al 4 sampling pool. Coding sequences of their genomes were obtained from GenBank and the RSCU similarity was calculated using the same method by Ji et al 4 (see detailed information in Supplementary Information Appendix 1). It turned out that viruses with high AU3s values are closer to animals have comparatively high AT3s values (herein N. atra, B. multicinctus, and M. condylurus) rather than AT3s low‐value species in RSCU distance (Figure 1). The result designated most investigated viruses (10/12) to infect snakes, regardless of their actual hosts.
Figure 1.
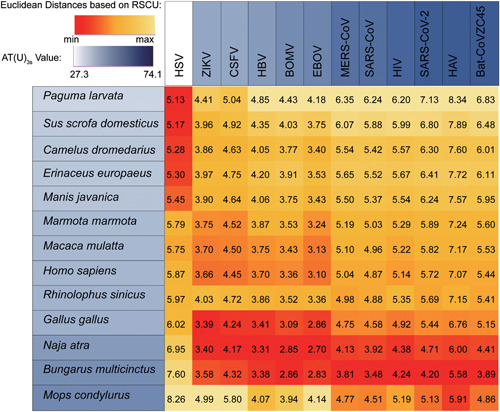
Heatmap and value of Euclidean distance between virus and potential hosts based on RSCU values. The distance for each virus between different animals was indicated by gradient from red to canary. The species were sorted by AT(U)3s value and indicated by gradient from white to dark blue. BOMV, bamboo mosaic virus; CSFV, classical swine fever virus; EBOV, ebola virus; HAV, hepatitis A virus; HBV, hepatitis B virus; HIV, human immunodeficiency virus; HSV, herpes simplex viruses; MERS‐CoV, Middle East respiratory syndrome coronavirus; RSCU, relative synonymous codon usage; SARS‐CoV, severe acute respiratory syndrome coronavirus; ZIKV, zika virus
In conclusion, the RSCU similarity analysis only links virus to animal(s) that possesses comparable GC‐content. We did not rule out the speculation that snakes would be the intermediate host of SARS‐CoV‐2, but we suspect if the possibility of these species was higher than any other creature in transmitting the virus. Scientific research that gives a quick response to public emergencies is imperative and appreciative, yet the methodology applied within has to be carefully examined.
Supporting information
Supplementary information
Supplementary information
REFERENCES
- 1. Coronavirus Disease (COVID‐2019) Situation Reports ‐ 83 . 2020. https://www.who.int/emergencies/disease/novel-coronavirus-2019/situation-reports. Accessed April 13, 2020.
- 2. Gorbalenya AE, Baker SC, Baric RS, et al. The species Severe acute respiratory syndrome‐related coronavirus: classifying 2019‐nCoV and naming it SARS‐CoV‐2. Nat Microbiol. 2020;5: 536–544. 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Callaway E, Cyranoski D. Why snakes probably aren't spreading the new China virus. One genetic analysis suggests reptilian reservoir—but researchers doubt that the coronavirus could have originated in animals other than birds or mammals. https://www.nature.com/articles/d41586-020-00180-8. Accessed February 22, 2020.
- 4. Ji W, Wang W, Zhao X, Zai J, Li X. Cross‐species transmission of the newly identified coronavirus 2019‐nCoV. J Med Virol. 2020;92(4):433‐440. 10.1002/jmv.25682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Z, Xiao X, Wei X, et al. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS‐CoV‐2. J Med Virol. 2020;92(6):595–601. 10.1002/jmv.25726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Andersen KG, Shapiro BJ, Matranga CB, et al. Clinical sequencing uncovers origins and evolution of Lassa virus. Cell. 2015;162(4):738‐750. 10.1016/j.cell.2015.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gu W, Zhou T, Ma J, Sun X, Lu Z. Analysis of synonymous codon usage in SARS coronavirus and other viruses in the Nidovirales. Virus Res. 2004;101(2):155‐161. 10.1016/j.virusres.2004.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bahir I, Fromer M, Prat Y, Linial M. Viral adaptation to host: a proteome‐based analysis of codon usage and amino acid preferences. Mol Syst Biol. 2009;5:311. 10.1038/msb.2009.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsubara K, Kuraku S, Tarui H, et al. Intra‐genomic GC heterogeneity in sauropsids: evolutionary insights from cDNA mapping and GC3 profiling in snake. BMC Genomics. 2012;13:604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao K, Liu WJ, Frazer IH. Codon usage bias and A+T content variation in human papillomavirus genomes. Virus Res. 2003;98(2):95‐104. 10.1016/j.virusres.2003.08.019 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary information
Supplementary information